
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNew insights into the crystallization of polymorphic materials: from real-time serial crystallography to luminescence analysis†‡
P.
Lindenberg
a,
L. Ruiz
Arana
a,
L. K.
Mahnke
 a,
P.
Rönfeldt
a,
N.
Heidenreich
ab,
G.
Doungmo
a,
N.
Guignot
c,
R.
Bean
d,
H. N.
Chapman
def,
D.
Dierksmeyer
b,
J.
Knoska
de,
M.
Kuhn
b,
J.
Garrevoet
b,
V.
Mariani
d,
D.
Oberthuer
d,
K.
Pande
d,
S.
Stern
d,
A.
Tolstikova
de,
T. A.
White
d,
K. R.
Beyerlein
*d and
H.
Terraschke
*a
a,
P.
Rönfeldt
a,
N.
Heidenreich
ab,
G.
Doungmo
a,
N.
Guignot
c,
R.
Bean
d,
H. N.
Chapman
def,
D.
Dierksmeyer
b,
J.
Knoska
de,
M.
Kuhn
b,
J.
Garrevoet
b,
V.
Mariani
d,
D.
Oberthuer
d,
K.
Pande
d,
S.
Stern
d,
A.
Tolstikova
de,
T. A.
White
d,
K. R.
Beyerlein
*d and
H.
Terraschke
*a
aInstitut für Anorganische Chemie, Christian-Albrechts-Universität zu Kiel, Max-Eyth-Str. 2, 24118 Kiel, Germany. E-mail: hterraschke@ac.uni-kiel.de
bDESY Photon Science, Notkestr. 85, 22607 Hamburg, Germany
cSynchrotron SOLEIL, L'Orme des Merisiers, Saint-Aubin, BP 48, 91192 Gif-sur-Yvette Cedex, France
dCenter for Free-Electron Laser Science, Deutsches Elektronen-Synchrotron DESY, Notkestraße 85, 22607 Hamburg, Germany
eDepartment of Physics, University of Hamburg, Luruper Chaussee 149, 22607 Hamburg, Germany
fCentre for Ultrafast Imaging, Luruper Chaussee 149, 22761 Hamburg, Germany
First published on 10th July 2019
Abstract
Detailed analysis of reaction mechanisms by in situ techniques are important for detecting metastable intermediates, analysing polymorphic transitions and thereby for the discovery of new compounds. This article presents the first combination of serial crystallography with in situ luminescence and X-ray diffraction (XRD) measurements to monitor the synthesis of [Eu(phen)2(NO3)3] (phen = 1,10-phenanthroline). In a batch reaction, it is found that this complex is polymorphic, crystallizing into two distinct monoclinic structures. We track the evolution of the synthesis conditions for these phases using in situ XRD combined with real time measurements of pH value, ion conductivity, infrared (IR) spectroscopy and in situ luminescence analysis of coordination sensors (ILACS). However, in a flow reactor a different combination of phases is produced. A serial crystallography experiment utilizing a nanofocused synchrotron X-ray beam to identify individual crystallites reveals the simultaneous formation of the two phases, as well as, a third unknown phase. This showcases the feasibility of phase detection on an individual crystallite level to track the synthesis of new materials.
1. Introduction
Due to the constant pursuit of modern chemistry to develop new functional materials, the application of in situ analysis methods has gained in importance in the last years.1–4 These characterization techniques deliver information about how reaction parameters (temperature, reactant concentration, or pH) influence the processes culminating in the final product formation, like induction time, ion desolvation, crystal growth and phase transitions.5 Such studies are driven by the desire for rational synthesis of new materials, and are often aimed at detecting the formation of reaction intermediates, which could show interesting properties but would remain undetected by ex situ analysis techniques.Here, we investigate the mechanism of formation for different polymorphs of the highly luminescent complex [Eu(phen)2(NO3)3].6 Such lanthanide complexes have been intensively studied for lighting,7,8 cell labelling9–11 and medical treatment applications.12,13 This compound has been found to crystallize into two different phases, both belonging to the monoclinic space group C2/c with Z = 4.14–18 Phase 1 (CSD ECABOZ02) has the unit cell parameters a = 9.5153(10) Å, b = 15.4546(10) Å, c = 17.1763(10) Å, β = 93.451(10)° and V = 2521.3(3) Å3. However, phase 2 (CSD ECABOZ01) crystallizes with parameters a = 11.1555(10) Å, b = 17.9698(10) Å, c = 13.0569(10) Å, β = 100.507(10)°, and V = 2572.1(3) Å3.18
To track their formation, we detail experiments including synchrotron-based X-ray diffraction analysis and in situ measurements of pH value, ion conductivity, infrared spectroscopy and luminescence.
The in situ luminescence analysis of coordination sensors (ILACS)19 approach is particularly sensitive to the local environment of lanthanide ions (Ln) due to its influence on their spectroscopic properties.20–22 These coordination sensors (CS) are incorporated into the analysed substance during synthesis and changes to the coordination environment are monitored by in situ luminescence measurements. The spectroscopic influence of the ligand field by trivalent europium ions makes them one of the most powerful coordination sensors.23–25 This approach complements synchrotron-based in situ X-ray diffraction methods by characterizing species in solution or amorphous phases, and can also be flexibly performed in common university laboratories.26,27
Serial crystallography is an emergent X-ray technique that is designed to interrogate the structure of individual microcrystallites.28,29 This is done using a bright, tightly focused X-ray source and collecting a large set of short-exposure diffraction patterns. A common approach is to stream a crystal slurry into the X-ray focus, and use a large 2D detector to collect diffraction images continuously for later sorting and analysis. While it was originally developed to mitigate room temperature radiation damage30,31 and study time-resolved protein dynamics,32–35 such an approach is also amenable to wet chemical materials synthesis studies. In this case, the reactants are mixed together in a microfluidic flow reactor and the product is delivered into the X-ray focus. Mixing serial crystallography has already been used to study chemically induced dynamics in protein crystals,36–38 however, its use as an in situ crystallization probe has not been previously demonstrated.
The article proceeds with a description of the experimental setups used to monitor the formation of [Eu(phen)2(NO3)3], followed by a discussion of the results. First, the syntheses performed in a common batch reactor are tracked with in situ spectroscopy and powder diffraction. Then, the product phases using a flow reactor are investigated with ILACS and serial crystallography.
2. Experimental part
2.1. Materials
For the [Eu(phen)2(NO3)3] synthesis, a simplified co-precipitation synthesis has been applied, in which an ethanolic solution of Eu(NO3)3·6H2O (99.9%, Sigma-Aldrich Chemie GmbH, Taufkirchen, Germany) was initially added to the reactor. Afterwards, a solution of 1,10-phenanthroline (phen) (99%, Alfa Aesar GmbH & Co KG, Karlsruhe, Germany) and acetylacetone (acac) (>99%, Merck, Darmstadt, Germany) in ethanol was added to the reactor under magnetic stirring, during which the in situ characterization techniques have been performed. Table 1 supplies detailed information on the volumes, concentrations and temperatures applied for single experiment types. All in situ batch experiments have been performed with an addition rate of 0.5 mL min−1 of the ligand to metal solutions.| Experiment number | I | II | III | IV | V | VI | VII | VIII | IX | X | XI | XII |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Setup | A | A | B | B | B | B | B | D | D | D | C | B |
| Beamline | P09 | P09 | — | — | — | — | — | — | — | — | PSICHÉ | P06 |
| X-ray energy/keV | 23 | 23 | — | — | — | — | — | — | — | — | EDXRD | 11 |
| Temperature | RT | RT | 60 °C | 60 °C | RT | 60 °C | RT | RT | RT | RT | RT | RT |
| Excitation wavelength/nm | 395 | 395 | 395 | 365 | 395 | 395 | 395 | — | — | — | — | 365 |
| Eu3+ solution | ||||||||||||
| Eu3+ concentration/M | 0.09 | 0.09 | 0.02 | 0.02 | 0.02 | 0.02 | 0.02 | 0.02 | 0.02 | 0.02 | 0.11 | 0.06 |
| Volume of water/mL | — | — | — | — | — | — | — | — | — | — | 30 | — |
| Volume of ethanol/mL | 40 | 40 | 40 | 40 | 40 | 40 | 40 | 5 | 5 | 5 | — | 2.5 |
| Ligand solution | ||||||||||||
| Phen concentration/M | 0.72 | 0.72 | 0.18 | 0.18 | 0.18 | 0.18 | 0.18 | 0.02 | 0.04 | 0.02 | 1.35 | 0.06 |
| Acac concentration/M | — | 2.15 | 0.53 | 0.53 | 0.53 | — | — | — | — | — | — | — |
| Volume of water/mL | — | — | — | — | — | — | — | — | — | 2.5 | 2.5 | 1.25 |
| Volume of ethanol/mL | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 2.5 | 2.5 | 1.25 |
Eu![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :phen:acac molar ratio :phen:acac molar ratio |
1:1:0 |
1:1:3 |
1:1:3 |
1:1:3 |
1:1:3 |
1:1:0 |
1:1:0 |
1:1:0 |
1:2:0 |
1:1:0 |
1:2:0 |
1:1:0 |
| Polymorphic phase | 1 | 2 | 2 | 2 | 2 | 1 | 1 | 1 | 1 | 2 | 2 | 1 + 2 |
2.2. In situ measurement assemblies for batch reactor
The same batch reactor geometry was used for the measurements taken at SOLEIL. However, there a white X-ray beam in the energy range of 20–80 keV was used for energy dispersive XRD (EDXRD) with a Ge solid state detector (Canberra Industries, France) at a 2θ angle of 4°.
2.3. Ex situ [Eu(phen)2(NO3)3] synthesis
Additional ex situ experiments have been carried out for confirming the influence of reaction parameters such as Eu:phen molar ratio and the presence of water in the reaction system on the formation of the different [Eu(phen)2(NO3)3] polymorphic phases. Hence, 5 mL of a ligand solution were added dropwise to the europium(III) nitrate solution and stirred for 30 minutes. The different solution compositions are listed in Table 1 (Exps. VIII–X). Afterwards, the formed solid material was centrifuged and dried at 80 °C for two hours.
For confirming the formation of the products, ex situ XRD measurements have been performed using a STOE Stadi-p X-ray powder diffractometer (STOE & Cie GmbH, Darmstadt, Germany) with a DECTRIS® MYTHEN 1 K detector (DECTRIS, Baden-Daettwil, Switzerland) with Cu Kα1 radiation (λ = 1.54056 Å) and Ge monochromator.
2.4. In situ serial crystallography and ILACS experiments with flow reactor
To study the early stages of the reaction, a simultaneous serial crystallography and ILACS experiment was conducted at beamline P06 of the PETRA III synchrotron. A schematic of the measurement geometry is shown in Fig. 1. The sample was synthesized in a capillary-based flow reactor by mixing the Eu3+ with the 1,10-phenantrholine solutions using equal flow rates of 10 μL min−1 (exp. XI, Table 1). The output capillary had in inner diameter of 200 μm and a length of 30 cm. Assuming laminar flow of the solutions, a diffusion time of approximately 5 seconds would be necessary for them to fully mix in a capillary of this diameter. The output of the mixer was then transported to a 12 μm thick polyimide tape that was drawn between two rollers of a tape drive at a rate of 0.05 mm s−1. This device transported the formed crystals into the interaction region, where they were probed with UV and X-ray light. The deposition onto the tape allowed for control of the exposure time independent of the reaction flowrates and avoided clogging of the reactor by using a short transport capillary. The in-capillary and on-tape transport time of the solutions to the interaction region were 3 seconds and 2 minutes, respectively. The solutions were mixed upon deposition onto the tape by the caused turbulence, and the reaction time was taken to be the on-tape transport time of 2 minutes. Shortly after deposition onto the tape the turbulence ceased and further mixing during crystal growth was dictated by diffusion. This is in contrast to the experiments carried out in a batch reactor, where the solutions were continuously mixed throughout the growth process. | ||
| Fig. 1 Simultaneous serial X-ray crystallography and ILACS setup. Two reactant solutions are depicted as flowing into a black mixing union and pink product crystals in a transport tube. The output is deposited onto a yellow polyimide tape, which is being drawn onto the roller on the right. Along the way the crystals are exposed to light from a UV lamp and an X-ray beam that is focused normal to the tape surface. The fluorescence is captured by a spectrometer and the X-ray diffraction pattern is collected on a large 2D detector. | ||
The deposited solutions spread out into a stream that covered the 1 cm wide tape. As the crystals passed through the interaction region, 2D X-ray diffraction images and UV spectra were simultaneously recorded. The X-ray energy was 11 keV and the focus diameter was around 500 nm. At this energy, the flux was expected to be on the order of 1010 photons per second. Once the solution began to flow, X-ray diffraction images were continuously recorded on an Eiger 4 M detector at a repetition rate of 5 Hz with an integration time of 200 ms. The X-ray detector was positioned in transmission geometry at a distance of 95 mm. This corresponded to a 2θ angular coverage of about 45 degrees and a maximum resolution around 1 Angstrom at the edge of the detector. As individual crystallites were seen in a live inline camera, the tape drive was translated vertically to intercept them with the X-ray beam. From the speed of the tape drive, crystallites on the tape translated 10 μm through the beam in this exposure time, which is comparable to the average crystallite size. However, the small X-ray beam size compared to the wide solution stream means that less than 1 percent of the synthesized crystallites were measured.
Simultaneously, fluorescence spectra of the sample on the tape were detected by an optical fiber connected to the same EPP2000 spectrometer and 365 nm UV LEDs used in the in situ reactor-based XRD experiments described in section 2.2. The streaming reaction was carried out for 1 hour. The collected X-ray diffraction images were processed by the Offline Data Analysis program,44 which performs background subtraction and looks for sharp Bragg spots in the frame. Those that contained a diffraction pattern from a crystal (a “hit”) were saved for later processing. A total of 413 hit images were found in the collected data and were later analysed.
3. Results and discussions
3.1. In situ luminescence, XRD and light transmission measurements
Two different ligand solutions have been used to monitor potential metal–ligand exchange processes during the formation of the [Eu(phen)2(NO3)3]. Experiment I (Table 1) was performed adding a 1,10-phenanthroline solution to the reactor containing an europium(III) nitrate solution, while experiment II used a 1,10-phenanthroline solution additionally containing acetylacetone. For both experiments, before the addition of the ligand solution, the characteristic emission peaks assigned to the 5D0 → 7F0–4 Eu3+ electronic transitions were observed (Fig. 2). Initially, the emission intensity was rather low, due to the quenching effect caused by the ethanol molecules in the coordination environment of Eu3+ within the solvation shell.45 For experiment I (Fig. 2a and b), upon addition of the phen solution, the luminescence intensity slightly decreased, most probably due to the increase of the solution volume and consequent dilution. This hypothesis is confirmed through the analysis of the normalized emission spectrum (Fig. S6‡) collected at, for instance, 0 and 1 minute after the addition of the phen solution. Even though the luminescence intensity at t = 1 min was slightly lower, the emission spectrum at this reaction time was identical to the one of Eu3+ in ethanol at t = 0 min, indicating that the Eu3+ coordination environment remained the same. The decrease of the emission intensity up to t = 1 min caused by a probable dilution effect is also indicated by monitoring the transmitted intensity of the excitation lamp through the solution (Fig. S5,‡λ = 386 nm, pink curve). Since the excitation light source was placed outside the reactor and its intensity was measured by the optical fiber submersed inside, the increase or decrease of the lamp intensity delivers information about the transmittance and consequently about the turbidity of the solution. Therefore, the increase of the lamp intensity for t = 0–1 min indicates a decrease in the turbidity of the solution, caused by the initial dilution effect. At t > 1 min, the intensity of the lamp rapidly decreased, indicating an increase of the solution turbidity and the formation of the solid product.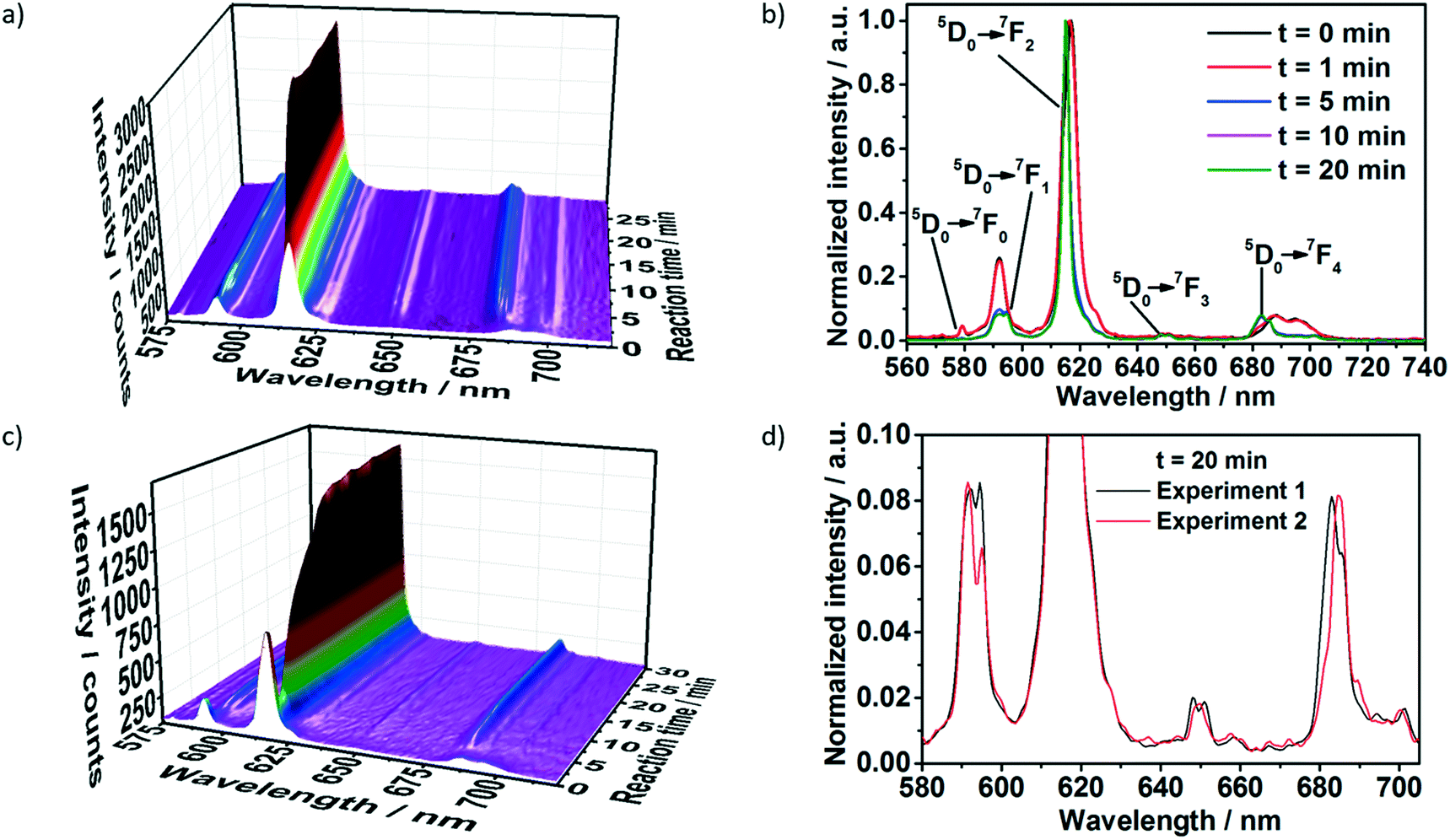 | ||
| Fig. 2 In situ luminescence measurements (λex = 395 nm) during formation of [Eu(phen)2(NO3)3] a) and b) without acac (experiment I, Table S1‡) and c) with acac (experiment II, Table S1‡), measured simultaneously to in situ XRD at the P09 DESY beamline. d) Comparison of in situ emission spectra recorded at t = 20 min for phase 1 (experiment I, Table S1‡) and phase 2 (experiment 2, Table S1‡) of [Eu(phen)2(NO3)3]. | ||
The delayed nucleation of the product is also clearly observed by the drastic increase of the emission intensity (Fig. 2a). As explained in our previous work,19 the increase of the emission intensity is caused by the desolvation process, in which the ethanol molecules present in the Eu3+ solvation shell are gradually exchanged by the phen molecules during the formation of [Eu(phen)2(NO3)3]. Due to the antenna effect, the organic ligand absorbs the excitation light and transfers it to the Eu3+ ions, increasing its emission intensity.46 The change in the profile of the peaks assigned to different transitions of the Eu3+ spectra also indicate the formation of the product, showing the presence of two different coordination environments for the Eu3+ ions before and after t = 1 min (Fig. 2b). One example is the presence of a peak assigned to the 5D0 → 7F0 transition within the spectrum of Eu3+ in ethanol and its absence in the spectrum assigned to [Eu(phen)2(NO3)3]. In addition, the splitting of the 5D0 → 7F1 peak, the intensity ratio between the 5D0 → 7F2 and 5D0 → 7F1 transitions as well as the position of the 7D0 → 7F4 transition completely change after t = 1 min, due to the incorporation of Eu3+ ions within the [Eu(phen)2(NO3)3] complex. The stabilization of the emission intensity at t ≈ 10 min indicates the end of the reaction.
Interestingly, for experiment II, upon the addition of the acac and phen solution to the reactor, the emission intensity first assigned to Eu3+ ions in ethanol strongly decreased until t = 2.5 min and increased afterwards (Fig. 2c). In contrast to the measurements carried out without acac (Fig. 2a), this decrease at the emission intensity is not caused by dilution and an increase of the solution volume. Instead, the decrease in the intensity of the transmission of the excitation light (Fig. S7‡) indicates an increase of the turbidity of the solution, probably caused by the formation of a reaction intermediate. The decrease of the emission intensity at λex = 395 nm implies that the intermediate cannot be excited at this wavelength. A possible explanation for this phenomenon would be the formation of [Eu(acac)3(phen)] as a reaction intermediate until t = 2.5 min, which excitation energy lies below 320 nm.47 After t = 2.5 min, the emission intensity drastically increased up to t ≈ 14 min, indicating the formation of a new compound. The emission increased at a slower rate up to t ≈ 22 min, when it stabilized, indicating the end of the reaction.
Comparing the emission spectra of the final compounds formed with and without the presence of acac (Fig. S8‡), the differences in the degeneracy of the 5D0 → 7FJ (J = 1–4) Eu3+ states imply the formation of two distinct compounds for these two reactions.
The in situ luminescence results are confirmed by the simultaneously recorded in situ XRD analysis shown in Fig. 3. For experiment I, the addition of the phen solution caused the rise of the Bragg reflections at e.g. 3.77, 3.99, 4.33 and 4.58° 2θ, assigned to phase 1 of [Eu(phen)2(NO3)3] (Fig. 3a and b). The intensity of these reflections e.g. at 4.3° 2θ start to rise at t = 1 min, confirming the formation of the product detected by in situ luminescence analysis (Fig. S5‡). Interestingly, the increase of the emission intensity for this compound occurs faster than the increase of the intensity of the XRD reflection, indicating that the complex is also formed in solution in parallel to the crystalline material detected by in situ XRD. In contrast, for experiment II, the increase of emission intensity started at t = 2.5 min coinciding with the increase of the simultaneously measured in situ XRD (Fig. S6‡), indicating a different crystallization behaviour than for experiment I. The in situ XRD measurements (Fig. 3c) also confirm this hypothesis, in which reflections e.g. at 3.45, 3.60, 3.94, 4.39 and 4.58° 2θ are assigned to phase 2 of [Eu(phen)2(NO3)3] (Fig. 3d), induced by the addition of acac. Most likely, the crystallinity of the possible reaction intermediate detected by the strong decrease on emission intensity (Fig. 2c) and by the decrease on light transmission (Fig. S6‡) was too low to be detected by in situ XRD, often observed for small crystallites.
 | ||
| Fig. 3 In situ XRD measurements during formation of [Eu(phen)2(NO3)3] without acac (a) and (b), experiment I, phase 118 and (b) with acac (c) and (d), experiment II, phase 2,18 measured simultaneously to in situ luminescence at the P09 DESY beamline. | ||
3.2. In situ measurements of luminescence, IR and pH value
To further investigate the experimental conditions leading to the formation of the two [Eu(phen)2(NO3)3] polymorphic phases, experiments III to VII (Table 1) have been carried out in the presence and absence of acac, characterized by in situ measurements of luminescence, pH value, ion conductivity and IR spectroscopy. Fig. S7‡ shows, for instance, the influence of the temperature on the formation of [Eu(phen)2(NO3)3], in the presence of acac (experiments III and IV) and without acac (experiments VI and VII). Similar to the evolution observed in experiments I and II, the in situ emission spectra in the presence of acac at 25 °C and 60 °C show an initial decrease of the emission intensity, indicating the formation of a reaction intermediate. As explained in the experimental section, an increased concentration is necessary for experiments I and II (Fig. 2 and 3), for improving the signal-to-noise ratio for the in situ XRD experiments. The decrease on concentration from experiments I and II (0.9 M) to experiments III–VII (0.2 M), caused a delay on the crystallization of the product 2 from t = 2.5 min to t = 7 min, indicated by the increase of the emission intensity, as previously observed for [Eu(bipy)2(NO3)3].48 Interestingly, the time-dependent emission spectra show no significant difference on the crystallization behaviour when the temperature was increased from 25 to 60 °C (Fig. S7‡). However, at 60 °C, the emission intensity is strongly influenced by temperature quenching effects,20 indicated by the increased of the emission intensity after cooling the solution down from 60 °C to room temperature (Fig. 4). | ||
| Fig. 4 Time-dependence of the volume of added ligand solution (phen and acac, green curve) in comparison to the time-dependence of the temperature (red curve), emission intensity of the 5D0 → 7F1 Eu3+ transition (orange curve) and IR absorption of nitrate vibrations at 1294 cm−1 (blue curve) during formation of phase 2 at 60 °C (experiment III, λex = 395 nm). | ||
For experiment III at 60 °C, the crystallization process began after 7 minutes, indicated by the increase of emission intensity by in situ luminescence analysis (Fig. 4). This is additionally confirmed by in situ IR measurements (Fig. S8‡) monitoring the nitrate vibrations at 1294 cm−1.49 The IR absorption remained constant during the first 6 minutes and increased afterwards. This behaviour is expected for the partial crystallization of the [Eu(phen)2(NO3)3] product on the tip of the IR sensor, increasing the nitrate concentration in the measured range in comparison to the rest of the solution. This hypothesis is confirmed by the time-dependence of the IR intensity at 843 cm−1, assigned to the vibrations of the 1,10-phenanthroline ligand50 (Fig. S9‡). At 843 cm−1, the IR intensity first slowly increased, due to the addition of the ligand solution, then rapidly increased after t ≈ 6 min, due to the crystallization of solid material on the sensor tip, identified as [Eu(phen)2(NO3)3] by the in situ XRD analysis (Fig. 3c). Monitoring the vibrations of the acetylacetone ligand e.g. at 1365 cm−1 (ref. 51 and 52) indicates that the IR sensor is not completely covered by the solid material (Fig. S12‡). Signal is still detected in solution because it is not fully incorporated into the solid material, as also shown in the in situ XRD measurements (Fig. 3c and S10‡). The IR intensity of the acac vibrations support the hypothesis of the formation of a reaction intermediate before the crystallization of 2, discussed in relation to Fig. 1b and 2b. In this experiment, the acac IR intensity slowly increased before t = 6 min, most probably, due to the uptake of the acac molecules for forming the reaction intermediate in parallel to the addition of the ligand solution. After t ≈ 7 min, the crystallization of 2 overcomes the formation of the reaction intermediate, the acac molecules are no longer consumed, and the IR intensity at 1365 cm−1 increased due to the further addition of the ligand solution.
A further indication for the formation of an acac-containing reaction intermediate prior to the crystallization of 2 is given by comparing the in situ emission intensity excited at 395 nm (Fig. 4) with the one excited at 365 nm. The excitation at the 7F0 → 5L6 Eu3+ transition at 395 nm is able to excite directly the Eu3+ ions in the ethanolic solvation sphere. Therefore, the decrease of the respective emission intensity indicates the uptake of the europium ions from the solution and the incorporation into the reaction intermediate, before the formation of the solid product 2. Since [Eu(phen)2(NO3)3] is also excited at 395 nm (Fig. S11‡), the further increase of the emission intensity can be assigned to the product formation and the consequent antenna effect, as confirmed by the synchrotron-based in situ XRD measurements (Fig. 3). In contrast, recording in situ emission spectra by exciting the phen ligand at 365 nm (Fig. S12‡), the emission intensity remained constant, because the wavelength of 365 nm is not able to efficiently excite the Eu3+ ions23,53 in solution or the acac-containing intermediate, as [Eu(acac)3phen] is excited by wavelengths shorter than 320 nm.47
In order to further investigate the formation of phases 1 and 2, the in situ analysis of pH value (Fig. S13‡) has been carried out. For experiments VI and VII, the addition of basic phen solution to the reactor containing solvated lanthanide nitrates initially caused the pH to increase. Upon the formation of the [Eu(phen)2(NO3)3] product, the pH stopped increasing, despite the further addition of the ligand, due to the uptake of the phen molecules of the solution for the complex formation, as described in our previous works.19,54 Like the results shown by the in situ luminescence measurements (Fig. S7‡), the simultaneously recorded in situ pH values also confirm that the increase of temperature from 25 °C to 60 °C do not significantly influence the product formation.
Interestingly, for experiments III and V, the pH decreased in three main phases (Fig. S13‡), which can also be recognized on the respective in situ luminescence spectra. For t < 2.5 min, the pH value abruptly decreased upon the addition of the ligand solution, because of the acidic nature of acetylacetone (Hacac) due to the presence of the active methylene hydrogen.55 Then, the pH reduction slowed at t = 2.5–7.5 min, due to the uptake of the acetylacetone molecules from the solution for the formation of the possible [Eu(acac)3phen] intermediate, as explained above. At t > 7.5 min, the pH decreased even slower, which could be explained by the release of acac into the solution and recombination to Hacac as well as the uptake of the NO3− ions from the solution for the formation of the [Eu(phen)2(NO3)3] complex. This difference in the pH values might explain the formation of different [Eu(phen)2(NO3)3] phases with and without acac.
3.3. Influence of the solvent and Eu![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :phen molar ratio on the [Eu(phen)2(NO3)3] formation
:phen molar ratio on the [Eu(phen)2(NO3)3] formation
The influence of the Eu-to-phen molar ratio and the presence of water in the reaction system on the crystallization of [Eu(phen)2(NO3)3] have also been explored.
Experiments II–V with the acac ligand mentioned above applied a Eu:phen:acac molar ratio 1:1:3, originally attempting to synthesize the [Eu(acac)3phen], and experiments I, VI and VII without acac applied a Eu:phen:acac molar ratio 1:1:0 for comparison, resulting in the two [Eu(phen)2(NO3)3] phases (Table 1). For this reason, it was important to clarify in the next step, if the application of the stoichiometric Eu:phen:acac molar ratio 1:2:0 for the formation of [Eu(phen)2(NO3)3] presents an influence on the formation of the polymorphic phase. Fig. S14‡ shows however that phase 1 has been obtained using both the 1:1:0 and the 1:2:0 Eu:phen:acac molar ratio. In contrast, the addition of water in the reaction system causes the formation of the polymorphic phase 2 instead of 1.
Synthesis of [Eu(phen)2(NO3)3] in water-containing reaction medium (Exp. IX) was additionally monitored by in situ energy dispersive XRD at the SOLEIL synchrotron facility, demonstrating the direct formation of phase 2, without the previous formation of phase 1 (Fig. 5) or a reaction intermediate, due to the absence of the acac molecules. The above presented results raises the question why the formation of phase 2 is favoured instead of phase 1 upon the use of additives such as acetylacetone or water in the reaction medium. Most probably, phase 2 is the more stable polymorph being formed, even at the lowered pH values caused by the addition of acetylacetone to the reaction medium. Additional indications that phase 2 is more stable are delivered by two additional experiments. In the first one, 50 mg of each polymorph is added to 1 mL ethanol for one hour. After centrifuging and drying, the peaks assigned to the phase 1 significantly decreased and the reflections assigned to the phase 2 are dominant (Fig. S15‡). In a last experiment, XRD analysis of phase 1 (Exp. VIII) and phase 2 (Exp. X) were performed three years after their preparation. After this time, phase 1 had been partially converted to the phase 2, while the phase 2 sample remained stably pure.
 | ||
| Fig. 5 a) In situ energy dispersive XRD measurements performed during the synthesis of [Eu(phen)2(NO3)3] in water-containing reaction medium (Exp. IX), b) in comparison to the calculated diffraction pattern of 2.18 | ||
3.4. In situ serial crystallography
The single crystallite sensitivity of serial crystallography was used to gain a different view into the role that water plays in the synthesis process, (Exp. XII of Table 1). The 413 diffraction images collected during the experiment previously described, were identified according to the presence of the known structures of [Eu(phen)2(NO3)3]. Insight into the phases formed in the early stage of this reaction was initially gained by studying the peaks present in the merged powder pattern, shown in Fig. 6. Peaks belonging to both phase 1 and 2 phases can be identified in the region from 0.1 to 0.3 1/A. In contrast to the smooth growth of phase 2 found in the in situ reactor study of experiment XI (Fig. 5), the two minute reaction time in such a flow reactor is found to be more complex and polymorphic. | ||
| Fig. 6 Merged powder plot (exp. XII). The merged powder plot made from the integrated spot intensities measured in the serial crystallography experiment is compared to the peaks from the 1 and 2 phases. | ||
To obtain the merged powder pattern, each hit image was analysed using the indexamajig program of the CrystFEL analysis package56,57 to find all of the diffraction spots and integrate them. A histogram was then made from this list of spot positions and intensities and normalized by the number of pixels contributing to each bin. Such a merging and normalization results in a pattern that resembles a typical powder diffraction pattern.58 The difference being that the merged serial powder pattern does not have contributions from the background or peak broadening due to crystallite size and strain, as they are removed during spot integration. A pattern related to the average structure factor (F-pattern) can also be obtained by normalizing the serial data by the number of spots contributing to each bin in the histogram. This representation removes effects of preferred orientation and point group reflection multiplicities and allows for clear visualization of high angle peaks.
As the intensity of the binned powder pattern was unreliable, a standard powder diffraction quantitative phase analysis was not possible. Instead, an image classification algorithm was developed to quantify the phase fractions by counting the number of images that contained peaks belonging to each of the phases.
The algorithm compared the spot list for each pattern to that of the two known phases and calculated a similarity score for each, Si. This was computed using a Gaussian-based penalty relationship for the observed spot positions
| Si = Πp exp (−εp2/2σ2), |
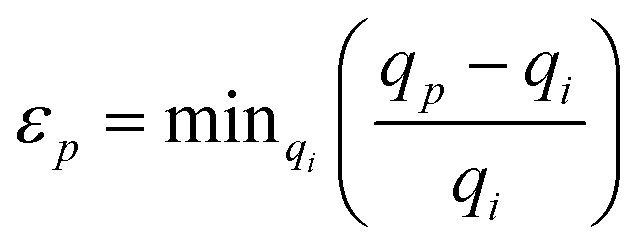 , with qp being the position of the p-th peak in the image, and qi is a peak from the list belonging to the i-th phase. The variable σ determines the allowable precision of the spot position. When all of the spots in an image are found to match a phase this metric will have a value of 1, while if a spot is found to not agree, it will tend to 0. Such penalty functions are the basis for Bayesian quantitative phase analysis algorithms.59
, with qp being the position of the p-th peak in the image, and qi is a peak from the list belonging to the i-th phase. The variable σ determines the allowable precision of the spot position. When all of the spots in an image are found to match a phase this metric will have a value of 1, while if a spot is found to not agree, it will tend to 0. Such penalty functions are the basis for Bayesian quantitative phase analysis algorithms.59
The similarity scores for each image are plotted in Fig. 7a. It can be seen that the images tend to cluster into the four corners of the plot. This was used to designate four zones and classify the images as either 1, 2, mixed, or no match. A total of 63 (23%), 86 (31%), 88 (31%), 42 (15%) images were found to belong to each classification respectively. Then these sorted images were used to generate four different powder patterns that are shown in Fig. 7b.
 | ||
| Fig. 7 Sorted powder plots (exp. XII). (a) The similarity scores of the hit images for phases 1 and 2 are plotted as open black rings. Those patterns that fell into the depicted areas were designated as 1, 2, mixed or no match. (b) The merged diffraction patterns for each of these groups is then shown, with the colouring corresponding to that used in (a). In each case, the intensities are normalized to depict the F-patterns. Also, green and red vertical dashed lines depict the diffraction peaks from phases 1 and 2 respectively, while peaks found to not agree with either are marked with a cross. | ||
The sorted patterns for phases 1 and 2 in Fig. 7b primarily only contain peaks from each respective phase indicating the algorithm reliably sorted the images belonging to the two phases. The mixed phase pattern predictably contains peaks that can be ascribed to either phase. Interestingly, the “no match” powder pattern contains many peaks that are slightly shifted with respect to the assumed phases, suggesting the presence of crystallites with slightly different unit cell parameters. A few strong peaks were also found that do not agree with either phase and are indicated in the figure by crosses. Notably, these peaks were found in 10 images at different positions around the diffraction ring, indicating they are not artefacts from bad pixels. However, it is hard to classify the unknown phase without indexing the images, as many other peaks from this structure overlap with those of the 1 and 2 phases.
This phase analysis found that phase 2 is most prevalent at this early reaction time, which agrees with the measurements made in a reactor (Exp. X–XI). However, it is also clear that the serial measurement also found a sizable fraction of phase 1, and possibly a third product. A complete picture of the reaction kinetics requires more measurements at different reaction times, mixing rates and water concentrations.
Fluorescence measurements were also made to test the possibility of performing ILACS simultaneously with the serial crystallography experiments. Fig. S16‡ shows the emission spectrum (λex = 365 nm) recorded during this synthesis of [Eu(phen)2(NO3)3] in flow regime (Exp. XII) at the P06 DESY beamline, integrated during 5 s. This spectrum presents the typical 5D0 → 7F0–4 Eu3+ transitions between 575 and 704 nm and the multiple peaks assigned to the 5D0 → 7F0 between 575 and 585 nm indicate the presence of multiple crystallographic sites for the Eu3+ cation, suggesting the formation of multiple phases, in agreement to the serial crystallographic results. However, additional experiments with a better spectral resolution must be performed to confirm this hypothesis.
4. Conclusions
In contrast to that expected from complexation processes involving well-defined moieties, such as single metal cations and organic ligands, the outcome of the reaction mixing the Eu3+ with 1,10-phenantholine molecules is everything but simple (Scheme 1). However, in the present article, we combine broad variety on characterization techniques, including synchrotron-based in situ X-ray diffraction, in situ luminescence analysis of coordination sensors (ILACS), serial crystallography and real-time measurements of pH value and infrared spectroscopy for understanding the mechanism behind this reaction. | ||
| Scheme 1 Schematic representation of synthesis conditions leading to the formation of [Eu(phen)2(NO3)3] phase 1 (represented in the magenta color) or phase 2 (represented in the green color), besides the third unidentified phase detected in flow reactor serial crystallography experiments and the intermediate [Eu(acac)3phen] (represented in the blue color). | ||
In summary, within a batch reaction system, the application of ethanol as solvent, under different temperatures with stoichiometric and non-stoichiometric metal-to-ligand ratios lead to the formation of phase 1 of the [Eu(phen)2(NO3)3] complex. The application of additives such as water or acetylacetone on the other hand, leads to the formation of phase 2. Within experiments with acac, the formation of an intermediate, most probably [Eu(acac)3(phen)] has been identified by real time luminescence and infrared spectroscopy. A flow reaction experiment performed at early reaction times in the presence of water found a predominance of phase 2, but also detected the simultaneous formation of phase 1 and a third non-identified phase. This showcases how achieving the single-crystallite detection limit of in situ characterization can lead to new insights into materials synthesis.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the German Research Foundation (DFG) within the Priority Program 1415 and project TE 1147/1-1, the German Electron Synchrotron (DESY), the MATsynCell consortium and the Daimler Benz Foundation (32-11/15) for the financial support. Thanks to Dr. Milan Köppen for the development of the in situ data helper software used for analysing the DESY, luminescence and IR data. Dr. A. K. Inge for the help analysing the SOLEIL data. To Marvin Radke for the pictures of the In situ Crystallization Cell at Kiel University and the setup used at DESY. Also thanks to the ILACS Group for the enriching discussions and Prof. Wolfgang Bensch for providing the equipment needed to perform the experiments. Parts of this research were carried out at PETRA III at DESY, a member of the Helmholtz Association (HGF). We would like to also thank Dr. J. Strempfer and Dr. S. Francoual for assistance in using the beamline P09. We thank Alke Meents for assistance with the tape drive during the P06 experiment.References
- N. Pienack and W. Bensch, Angew. Chem., Int. Ed., 2011, 50, 2014–2034 CrossRef CAS.
- N. Pienack, L. Ruiz Arana, W. Bensch and H. Terraschke, Crystals, 2016, 6, 157 CrossRef.
- N. Heidenreich, U. Rütt, M. Köppen, A. Ken Inge, S. Beier, A.-C. Dippel, R. Suren and N. Stock, Rev. Sci. Instrum., 2017, 88, 104102 CrossRef CAS.
- H. Embrechts, M. Kriesten, K. Hoffmann, W. Peukert, M. Hartmann and M. Distaso, J. Phys. Chem. C, 2018, 122, 12267–12278 CrossRef CAS.
- N. Pienack, P. Lindenberg, G. Doungmo, N. Heidenreich, F. Bertram, M. Etter, M. T. Wharmby and H. Terraschke, Z. Anorg. Allg. Chem., 2018, 644, 1902–1907 CrossRef CAS.
- I. Georgieva, N. Trendafilova, T. Zahariev, N. Danchova and S. Gutzov, J. Lumin., 2018, 202, 192–205 CrossRef CAS.
- R. Reyes, M. Cremona and H. F. Brito, Opt. Mater., 2018, 84, 631–635 CrossRef CAS.
- S. SeethaLekshmi, A. R. Ramya, M. L. P. Reddy and S. Varughese, J. Photochem. Photobiol., C, 2017, 33, 109–131 CrossRef CAS.
- J. Yuan and G. Wang, J. Fluoresc., 2005, 15, 559–568 CrossRef CAS.
- R. D. Teo, J. Termini and H. B. Gray, J. Med. Chem., 2016, 59, 6012–6024 CrossRef CAS.
- M. E. Mesquita, S. S. Nobre, M. Fernandes, R. A. S. Ferreira, S. C. G. Santos, M. O. Rodrigues, L. D. Carlos and V. de Zea Bermudez, J. Photochem. Photobiol., A, 2009, 205, 156–160 CrossRef CAS.
- J. B. M. Resende Filho, P. R. Santos, J. A. Vale, W. M. Faustino, D. S. Farias, H. F. Brito, M. C. F. C. Felinto and E. E. S. Teotonio, J. Braz. Chem. Soc., 2017, 28, 2281–2290 CAS.
- J. B. M. Resende Filho, J. C. Silva, J. A. Vale, H. F. Brito, W. M. Faustino, J. G. P. Espínola, M. C. F. C. Felinto and E. E. S. Teotonio, J. Braz. Chem. Soc., 2014, 25, 2080–2087 CAS.
- Y. Fan and P. Yang, J. Phys. Chem., 1996, 100, 69–74 CrossRef CAS.
- A. G. Mirochnik, B. V. Bukvetskii, P. A. Zhikhareva and V. E. Karasev, Russ. J. Coord. Chem., 2001, 27, 443–448 CrossRef CAS.
- F. Werner, K. Tada, A. Ishii, M. Takata and M. Hasegawa, CrystEngComm, 2009, 11, 1197–1200 RSC.
- Y.-Q. Zheng, L.-X. Zhou, J.-L. Lin and S.-W. Zhang, Z. Anorg. Allg. Chem., 2001, 627, 1643–1646 CrossRef CAS.
- G. G. Sadikov, A. S. Antsyshkina, I. A. Kuznetsova and M. N. Rodnikova, Crystallogr. Rep., 2006, 51, 47–52 CrossRef CAS.
- H. Terraschke, L. Ruiz Arana, P. Lindenberg and W. Bensch, Analyst, 2016, 141, 2588–2594 RSC.
- W. M. Yen, H. Yamamoto and S. Shionoya, Phosphor Handbook, Laser and Optical Science and Technology, CRC Press, Boca Raton, 2006 Search PubMed.
- T. Wylezich, R. Böttcher, A. D. Sontakke, V. Castaing, B. Viana, A. Pöppl and N. Kunkel, J. Phys. Chem. C, 2019, 123, 5031–5041 CrossRef CAS.
- M. Suta and C. Wickleder, J. Lumin., 2019, 210, 210–238 CrossRef CAS.
- K. Binnemans, Coord. Chem. Rev., 2015, 295, 1–45 CrossRef CAS.
- J.-C. G. Bünzli, Acc. Chem. Res., 2006, 39, 53–61 CrossRef.
- C. Görller-Waldrand and K. Binnemans, in Handbook on the Physics and Chemistry of Rare Earths, Elsevier Science B. V., Amsterdam, 1998, vol. 25, pp. 101–264 Search PubMed.
- J. Ströh, L. Ruiz Arana, P. Polzin, I. V. Eliani, P. Lindenberg, N. Heidenreich, C. S. Cunha, S. Leubner and H. Terraschke, Z. Anorg. Allg. Chem., 2019, 645, 537–543 CrossRef.
- L. Ruiz Arana, J. Olchowka and H. Terraschke, Z. Naturforsch., B: J. Chem. Sci., 2019, 74, 147–152 CAS.
- H. Chapman, P. Fromme, A. Barty, T. White, R. Kirian and A. Aquila, et al. , Nature, 2011, 470, 73–77 CrossRef CAS.
- S. Boutet, L. Lomb, G. Williams, T. Barends, A. Aquila and R. Doak, et al. , Science, 2012, 337, 362–364 CrossRef CAS.
- R. Kirian, T. White, J. Holton, H. Chapman, P. Fromme and A. Barty, et al. , Acta Crystallogr., Sect. A: Found. Crystallogr., 2011, 67, 131–140 CrossRef CAS.
- L. Lomb, T. Barends, S. Kassemeyer, A. Aquila, S. Epp and B. Erk, et al. , Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 214111 CrossRef.
- A. Aquila, M. Hunter, R. Doak, R. Kirian, P. Fromme and T. White, et al. , Opt. Express, 2012, 20, 2706–2716 CrossRef CAS PubMed.
- C. Kupitz, S. Basu, I. Grotjohann, R. Fromme, N. Zatsepin and K. Rendek, et al. , Nature, 2014, 513, 261–265 CrossRef CAS PubMed.
- K. Pande, C. Hutchison, G. Groenhof, A. Aquila, J. Robinson and J. Tenboer, et al. , Science, 2016, 352, 725–729 CrossRef CAS.
- T. Barends, L. Foucar, A. Ardevol, K. Nass, A. Aquila and S. Botha, et al. , Science, 2015, 350, 445–450 CrossRef CAS.
- D. Oberthuer, J. Knoška, M. Wiedorn, K. Beyerlein, D. Bushnell and E. Kovaleva, et al. , Sci. Rep., 2017, 7, 44628 CrossRef.
- K. Beyerlein, D. Dierksmeyer, V. Mariani, M. Kuhn, M. I. Sarrou and A. Ottaviano, et al. , IUCrJ, 2017, 4, 769–777 CrossRef CAS.
- J. Olmos, S. Pandey, J. Martin-Garcia, G. Calvey, A. Katz and J. Knoska, et al. , BMC Biol., 2018, 16, 59 CrossRef.
- H. Terraschke, M. Rothe and P. Lindenberg, Rev. Anal. Chem., 2018, 37, 20170003 Search PubMed.
- N. Schell, A. King, F. Beckmann, T. Fischer, M. Müller and A. Schreyer, Mater. Sci. Forum, 2013, 772, 57–61 Search PubMed.
- J. Strempfer, S. Francoual, D. Reuther, D. K. Shukla, A. Skaugen, H. Schulte-Schrepping, T. Kracht and H. Franz, J. Synchrotron Radiat., 2013, 20, 541–549 CrossRef CAS.
- A. Hammersley, FIT2D, ESRF, France, 2014 Search PubMed.
- H. Terraschke, M. Rothe, A.-M. Tsirigoni, P. Lindenberg, L. Ruiz Arana, N. Heidenreich, F. Bertram and M. Etter, Inorg. Chem. Front., 2016, 4, 1157–1165 RSC.
- V. Mariani, A. Morgan, C. Yoon, T. Lane, T. White and C. O'Grady, et al. , J. Appl. Crystallogr., 2016, 49, 1073–1080 CrossRef CAS.
- A. Beeby, I. M. Clarkson, R. S. Dickins, S. Faulkner, D. Parker, L. Royle, A. S. de Sousa, J. A. Gareth Williams and M. Woods, J. Chem. Soc., Perkin Trans. 2, 1999, 493–504 RSC.
- H. C. Streit, M. Adlung, O. Shekhah, X. Stammer, T. Ladnorg, H. Gliemann, M. Franzreb, C. Wöll and C. Wickleder, ChemPhysChem, 2012, 13, 2699–2702 CrossRef CAS.
- M. D. McGehee, T. Bergstedt, C. Zhang, A. P. Saab, M. B. O'Regan, G. C. Bazan, V. I. Srdanov and A. J. Heeger, Adv. Mater., 1999, 11, 1349–1354 CrossRef CAS.
- P. Polzin, I. V. Eliani, J. Ströh, M. Braun, N. Ruser, N. Heidenreich, P. Rönfeldt, F. Bertram, C. Näther, S. Wöhlbrandt and H. Terraschke, Phys. Chem. Chem. Phys., 2018, 20, 7428–7437 RSC.
- B. H. Stuart, Infrared Spectroscopy: Fundamentals and Applications, John Wiley & Sons, Ltd, Hoboken, 2005 Search PubMed.
- M. M. Campos-Vallette, R. E. Clavijo, F. Mendizabal and W. Zamudio, Vib. Spectrosc., 1996, 37, 37–44 CrossRef.
- T. A. Kovacs, M. C. F. C. Felinto, T. B. Paolini, B. Ali, L. K. O. Nakamura, E. E. S. Teotonio, H. F. Brito and O. L. Malta, J. Lumin., 2018, 193, 98–105 CrossRef CAS.
- J. Čeponkus, R. Platakytė, V. Šablinskas and A. Gutierrez Quintanilla, Chemija, 2018, 29, 1–15 CrossRef.
- K. Binnemans, Chem. Rev., 2009, 109, 4283–4374 CrossRef CAS.
- L. Ruiz Arana, P. Lindenberg, H. Said, M. Radke, N. Heidenreich, C. dos Santos Cunha, S. Leubner and H. Terraschke, RSC Adv., 2017, 7, 52794–52800 RSC.
- R. K. Sodhi and S. Paul, Catal. Surv. Asia, 2018, 22, 31–62 CrossRef CAS.
- T. White, R. Kirian, A. Martin, A. Aquila, K. Nass and A. Barty, et al. , J. Appl. Crystallogr., 2012, 45, 335–341 CrossRef CAS.
- T. White, V. Mariani, W. Brehm, O. Yefanov, A. Barty and K. Beyerlein, et al. , J. Appl. Crystallogr., 2016, 49, 680–689 CrossRef CAS.
- K. Beyerlein, C. Jooss, A. Barty, R. Bean, S. Boutet and S. Dhesi, et al. , Powder Diffr., 2015, 30, S25–S30 CrossRef CAS.
- A. Mikhalychev and A. Ulyanenkov, J. Appl. Crystallogr., 2017, 50, 776–786 CrossRef CAS.
Footnotes |
| † These results have been partially reported as posters and oral presentations at the conferences “18. Vortragstagung Fachgruppe Festkörperchemie und Materialforschung”, September 19th–21st 2016, Innsbruck, Austria (doi 10.1002/zaac.201605097) and “3rd joint workshop of MATsynCELL and C3”, October 6th–7th 2016, Uppsala, Sweden. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c9re00191c |
| This journal is © The Royal Society of Chemistry 2019 |