
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydrocarbon production by continuous hydrodeoxygenation of liquid phase pyrolysis oil with biogenous hydrogen rich synthesis gas
Klara
Treusch
 ab,
Anna Magdalena
Mauerhofer
c,
Nikolaus
Schwaiger
*a,
Peter
Pucher
b,
Stefan
Müller
c,
Daniela
Painer
a,
Hermann
Hofbauer
c and
Matthäus
Siebenhofer
a
ab,
Anna Magdalena
Mauerhofer
c,
Nikolaus
Schwaiger
*a,
Peter
Pucher
b,
Stefan
Müller
c,
Daniela
Painer
a,
Hermann
Hofbauer
c and
Matthäus
Siebenhofer
a
aInstitute of Chemical Engineering and Environmental Technology, Graz University of Technology, Austria. E-mail: nikolaus.schwaiger@tugraz.at
bBDI-BioEnergy International GmbH, Austria. E-mail: klara.treusch@bdi-bioenergy.com
cInstitute of Chemical, Environmental and Bioscience Engineering, TU Wien, Austria
First published on 7th February 2019
Abstract
This paper presents a beneficial combination of biomass gasification and pyrolysis oil hydrodeoxygenation for advanced biofuel production. Hydrogen for hydrodeoxygenation (HDO) of liquid phase pyrolysis oil (LPP oil) was generated by gasification of softwood. The process merges dual fluidized bed (DFB) steam gasification, which produces a hydrogen rich product gas and the HDO of LPP oil. Synthesis gas was used directly without further cleaning and upgrading, by making use of the water gas-shift (WGS) reaction. The water needed for the water gas-shift reaction was provided by LPP oil. HDO was successfully performed in a lab scale over 36 h time on stream (TOS). Competing reactions like the Boudouard reaction and Sabatier reaction were not observed. Product quality was close to Diesel fuel specification according to EN 590, with a carbon content of 85.4 w% and a residual water content of 0.28 w%. The water-gas shift reaction was confirmed by CO/CO2-balance, high water consumption and 28% less hydrogen consumption during HDO.
Introduction
In the mid-1990s, the first climate agreement was published in the Kyoto protocol,1 initiating several actions in climate policy. In 2015, the Paris agreement2 from the United Nations Framework Convention on Climate Change was a big step forward to support renewable energy intentions. The main target of this agreement is to hold climate change significantly below 2 °C above the pre-industrial level; it was signed by 196 member states worldwide. In parallel, the European Union developed different directives, the most important one being the renewable energy directive 2009/28/EC3 (RED) in 2009, followed by a recast, directive (EU) 2018/2001 called RES,4 in December 2018. These are just the most important directives. All agreements target a common objective: to mitigate climate change by reducing greenhouse gas emissions. This ambitious goal can only be achieved if all feasible sources for renewable energy production are exploited. From this point of view, the concept of biofuel production via the bioCRACK process and subsequent hydrodeoxygenation of liquid phase pyrolysis (LPP) oil with synthesis gas (syngas) from renewable feed has been developed. It combines two major pathways for biomass liquefaction: indirect liquefaction through gasification5 with subsequent synthesis and direct liquefaction through pyrolysis6 and hydrodeoxygenation.Liquid phase pyrolysis
LPP is less common than flash or fast pyrolysis.6–10 As the name implies, a liquid heat carrier is used for heat transfer during the pyrolysis process. The process may be performed with or without a catalyst, under inert gas or hydrogen atmosphere and under atmospheric or elevated pressure, depending on the boiling point of the heat carrier to assure its liquid state. Klaigaew et al.11 performed LPP of Giant Leucaena in hexane with an initial nitrogen pressure of 10 bar in the presence of a metal oxide catalyst in the temperature range of 325 to 400 °C with different holding times. Ratanathavorn et al.12 used decane as a heat carrier with an initial hydrogen pressure of 5 to 30 bar, also in the presence of different heterogeneous catalysts in the temperature range of 250 to 400 °C. Szabó et al.13 performed LPP of wheat straw and poplar in n-hexadecane under inert atmosphere at about 350 °C and a maximum pressure of 20 bar. They observed a negligible effect of different catalysts. Schwaiger et al.14,15 performed various experiments with spruce wood in a mixture of n-alkanes with a boiling range of 410 to 440 °C without a catalyst under nitrogen atmosphere at ambient pressure. He observed a partial degradation of the heat carrier oil as an unwanted effect. This effect can be used when the cracking of less valuable streams of crude refining is combined with LPP of biomass.Based on that, the bioCRACK process was developed by BDI-BioEnergy International GmbH. In this process, biomass is pyrolysed in a heavy oil fraction from crude oil refining. Different to other pyrolysis technologies like fast pyrolysis, non-polar biomass constituents, which are formed during the cracking step, are then dissolved in this heavy oil fraction. The extraction of nonpolar cracking products from biomass into the cracked and residual heavy oil fractions produces a pyrolysis oil of low organic load and high water content, which is called liquid phase pyrolysis oil (LPP oil). A detailed discussion of the bioCRACK process can be found in the publications of Ritzberger et al.,16 Schwaiger et al.17 and Treusch et al.18
Dual fluidized bed steam gasification
The dual fluidized bed (DFB) steam gasification is a thermo-chemical conversion process for the production of a hydrogen-rich gas from solid fuels like biomass. Based on the conventional DFB steam gasification process,5,19–22 the Sorption Enhanced Reforming (SER) process with reduced gasification temperatures of 600–700 °C was developed. The SER process aims to generate a product gas with high hydrogen contents of up to 75 w% db (dry basis) and in situ carbon dioxide transfer from the product gas into the flue gas.23 The DFB reactor system combines a gasification reactor (GR) and a combustion reactor (CR), which are connected via a loop seal. Through the loop seal, unconverted fuel from the GR, so called char, is transported to the CR, which is fluidized with air. In the CR, char is burnt and provides heat energy, which is transferred to the GR with the bed material, so that the overall endothermic gasification reactions take place. Fig. 1 displays the fundamental principle of SER with sorption active bed material for the transfer of CO2 from the GR to the CR.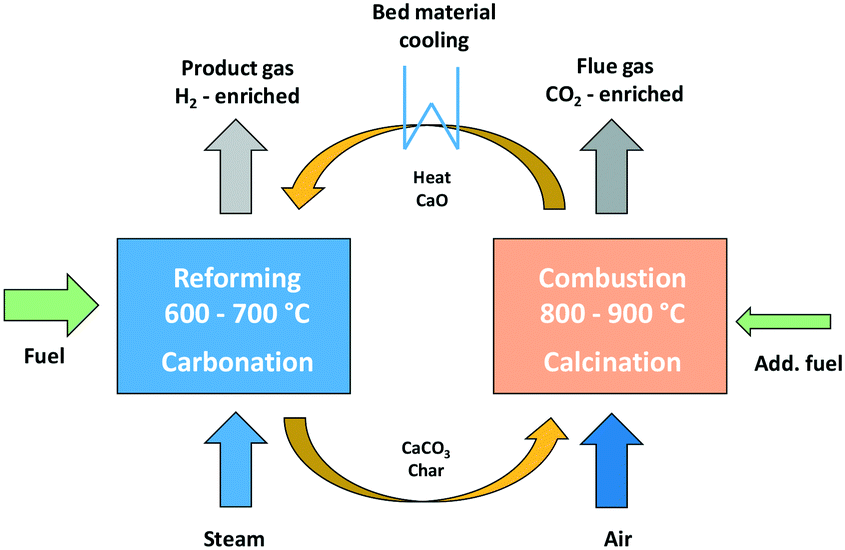 | ||
| Fig. 1 Fundamental principle of the sorption enhanced reforming process.22 | ||
For the SER process, limestone (CaCO3) is used as bed material. Due to the high temperatures in the CR (800–900 °C), calcination of CaCO3 to calcium oxide (CaO), shown in eqn (1), takes place, and CO2 is released. The release of CO2 during calcination reaction in the CR results in a CO2-enriched flue gas.
| CaCO3 ↔ CaO + CO2 | (1) |
| CaO + CO2 ↔ CaCO3 | (2) |
The temperature level in the GR allows carbonation and CO2 is adsorbed from the product gas to react with CaO, according to eqn (2). Further information about sorption enhanced reforming can be found in literature.22,24
Hydrodeoxygenation of pyrolysis oil
Hydrodeoxygenation is a technology for pyrolysis oil upgrading. The technology is not yet established on an industrial scale. During hydrodeoxygenation, oxygen is removed from pyrolysis oil with hydrogen by the formation of water and hydrocarbons. HDO, especially of fast pyrolysis oil, is well investigated und published.25–34 Usually, a two-step process is applied, whereas the first step is used to stabilize the most reactive components in pyrolysis oil, especially aldehydes and ketones,30 in order to prevent coking, whereas in the second step it is fully hydrodeoxygenated.32–38 The process is reported either in batch27,28,39–41 or in continuous29,30,32,34–38,42–45 operation mode. Not only hydrodeoxygenation of pyrolysis oil is reported, but also of differently liquefied biomass, such as solvolysed biomass, as reported by Rezzoug and Capart,46 Kunaver et al.47 and Grilc et al.48–50 The latter ones liquefied biomass through solvolysis and acidolysis in glycerol, diethylene glycol and p-toluenesulfonic acid. The resulting liquefied biomass has a composition close to fast pyrolysis oils, with a comparably high carbon content and therefore high gross calorific value of over 20 MJ kg−1. HDO of several bio oils with different biomass origin has been established, the hydrogen source is usually not mentioned, suggesting that the hydrogen is not of biogenous origin.HDO of LPP oil may be performed at a high liquid hourly space velocity (LHSV), making integration in oil refinery processes feasible. The influence of the LHSV on HDO performance between 0.5 to 3 h−1 was investigated.51 The highest HDO rate was achieved at LHSV 0.5 and 1 h−1 with an oxygen content of nearly 0 w% and a H/C ratio of nearly 2. The influence of the temperature was studied in the range of 350 to 400 °C.52 It was observed, that in this temperature range the difference in product quality is minor. HDO fuel fractions contained less than 0.15 w% water and showed similar properties for different operation temperature. At LHSV between 0.5 and 1 h−1, the influence of the temperature between 350 and 400 °C on product quality can be neglected.
Hydrodeoxygenation with synthesis gas
The application of synthesis gas for hydrodeoxygenation of pyrolysis oil is until now a poorly explored field of research. The underlying basis is an in situ water-gas shift (WGS) reaction53 according to eqn (3), with water contained in pyrolysis oil. LPP oil contains about 60 w% water. The reaction is exothermic. Reaction starts above 200 °C. A pressure of 25 to 30 bar is typically applied.| CO + H2O ↔ CO2 + H2 | (3) |
In 2014, Steele et al.54 from the Mississippi State University registered a patent application describing the upgrading of bio-oil with synthesis gas. A pressure range between 20 and 83 bar as well as a temperature range between 200 and 350 °C was suggested. Tanneru and Steele55 as well as Luo et al.56 described the hydrodeoxygenation of oxidized flash pyrolysis oil, following this patent. Both groups performed the experiments discontinuously in two steps, whereas synthesis gas was only used in the first step. Tanneru and Steele used a mixture of H2 and CO in different proportions at a pressure of 68.9 bar in both steps. The temperature was 340 °C in the first step, 400 °C in the second step. Nickel on different support materials was used as catalyst. Residence time was 90 min in the first and 150 min in the second step. Luo et al. hydrodeoxygenated a fractionated oxidized flash pyrolysis oil with a syngas composed of 18 vol% H2, 22 vol% CO, 11 vol% CO2, 2 vol% CH4 and 47 vol% N2 in the first step. The partially upgraded pyrolysis oil was then fully hydrogenated with pure hydrogen in a second step. They applied 55 bar and 360 °C in the first step and 96 bar and 425 °C in the second step. Residence time was 120 min in each step. Wijayapala et al.,57 also from Mississippi State University, investigated the HDO of two model compounds, guaiacol and furfural, in a batch reactor with a residence time of 240 min. Experiments were performed with two different synthesis gas mixtures, 50/50 synthesis gas and bio synthesis gas consisting of 18 vol% H2, 23 vol% CO and 46 vol% N2, at 40 bar and different temperatures.
So far, only batch experiments with oxidized flash pyrolysis oil or model compounds have been performed.55–57 Until now, only one research group has investigated this topic. They concluded that HDO with synthesis gas could only be successfully performed with oxidized bio-oil. In this paper, the continuous hydrodeoxygenation of untreated liquid phase pyrolysis oil with synthesis gas from biomass gasification is investigated. The application of LPP oil is especially auspicious due to the surpassingly high water content of about 60 w%. As water cannot simply be added to pyrolysis oil, this makes the usage of LPP oil unique.
Combined biofuel production
The combined biofuel production route was composed of three processes carried out by Graz University of Technology in cooperation with BDI-Bioenergy International GmbH and TU Wien. TU Graz and BDI-BioEnergy International GmbH carried out the pyrolysis and the hydrodeoxygenation step, TU Wien provided the H2-rich product gas for the hydrodeoxygenation step from a SER experiment carried out in the 100 kWth DFB steam gasification pilot plant at TU Wien. In Fig. 2, a flow sheet of the combined processes, the so-called “biofuel production route”, is shown. Thus, a 100% biogenous liquid fuel can be produced.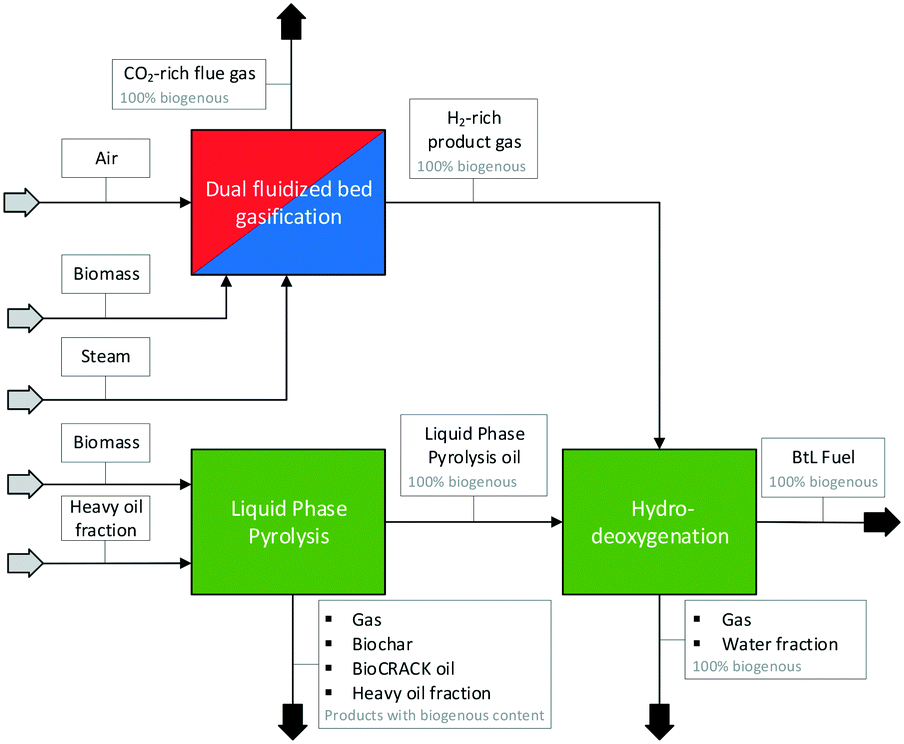 | ||
| Fig. 2 Combined biofuel production route of TU Wien and TU Graz; dual fluidized bed gasification, liquid phase pyrolysis and hydrodeoxygenation. | ||
Dual fluidized bed steam gasification
The H2-rich product gas was produced through the sorption enhanced reforming process. The SER test run was carried out in the 100 kWth pilot plant at TU Wien with softwood as fuel and limestone as bed material. A scheme is shown in Fig. 3. In the lower gasification reactor (bubbling bed), temperatures of about 630 °C were applied, in the upper gasification reactor (counter-current column), temperature was about 670 °C. The steam to fuel ratio was set to 0.8 kg per kgdaf. More information about the test run can be found elsewhere.24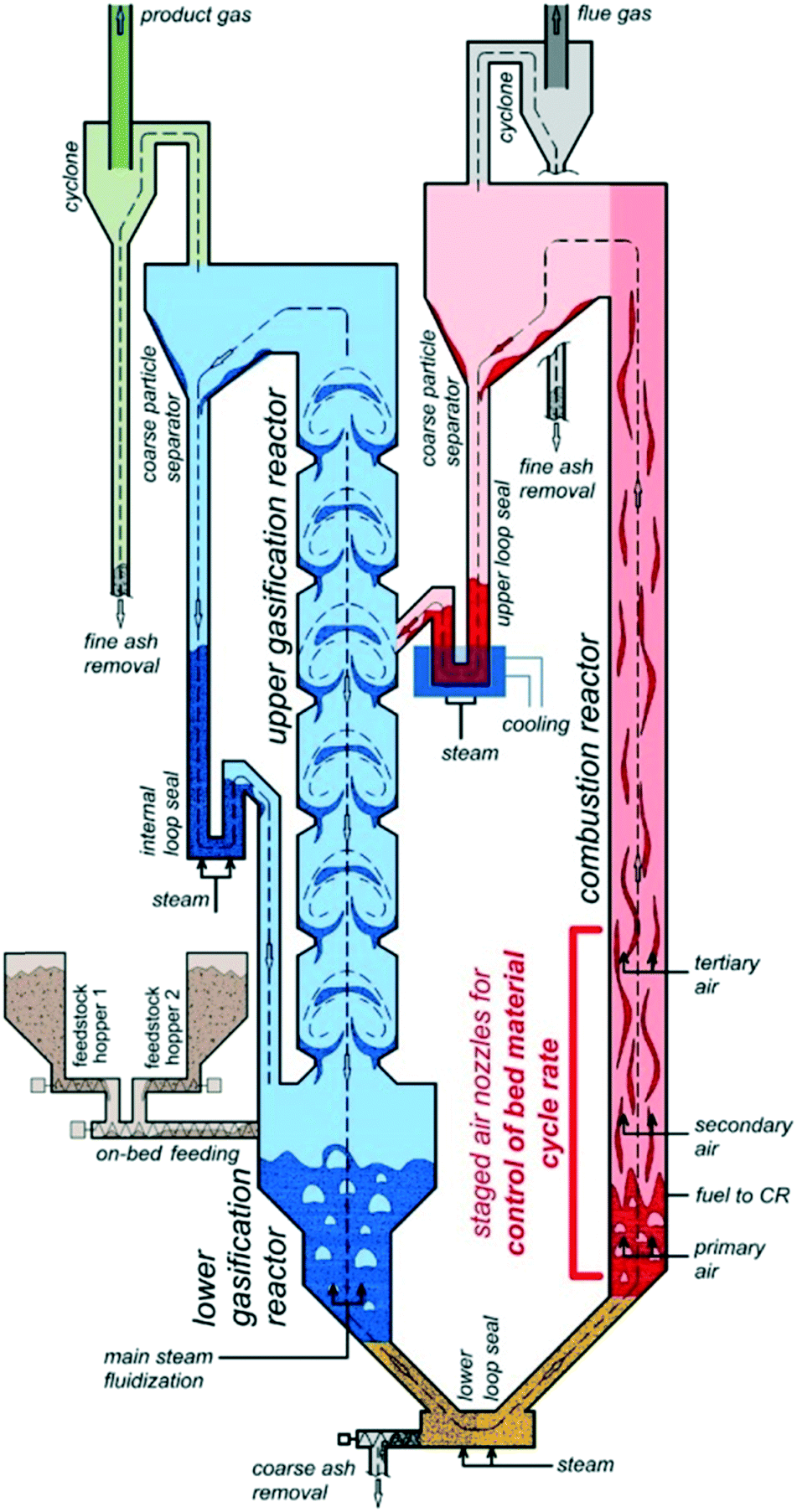 | ||
| Fig. 3 Schematic plant concept of the 100 kWth dual fluidized bed steam gasification pilot plant.22 | ||
Validation of process data
Based on the process data, which were recorded during the test campaign, mass and energy balances were calculated with the process simulation tool IPSEpro. For the validation of measured data with IPSEpro, a model library, which was developed at TU Wien, was used.58,59 For the evaluation of the presented test campaign, the following key figures were selected. The steam to fuel ratio φSF expresses the mass of steam used as fluidization agent and the mass of water in the fuel, it is related to the mass of dry and ash-free fuel (see eqn (4)). In eqn (5), the steam-related water conversion XH2O is given. XH2O describes the amount of water consumed for e.g. CO and H2 formation, it is related to the sum of water, which is fed to the gasification reactor. The product gas yield PGY (see eqn (6)) is defined as the ratio of dry product gas to dry and ash-free fuel fed to the gasification reactor. The cold gas efficiency ηCG displayed in eqn (7) presents the chemical energy content of gaseous components in the tar- and char-free product gas to the chemical energy in the fuel fed to the gasification reactor. All values are based on the lower heating value. eqn (8) displays the overall cold gas efficiency ηCG,o. It describes the amount of chemical energy in the product gas related to the fuel fed to the gasification and additional fuel fed to the combustion reactor minus apparent heat losses.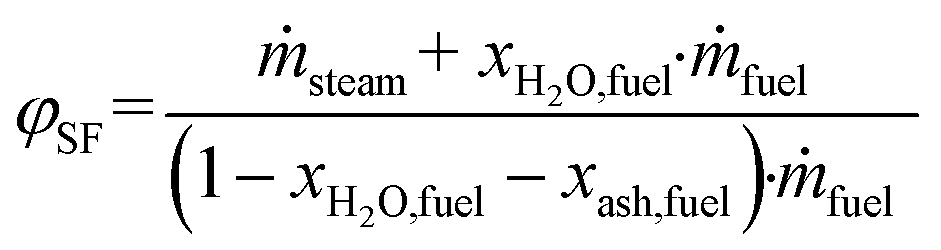 | (4) |
ṁ steam = mass flow of steam fed to GR in kg s−1.
ṁ fuel = mass flow of fuel introduced into GR in kg s−1.
X H2O,fuel = weight percent of water in the fuel in w%.
X ash,fuel = weight percent of ash in the fuel in w%.
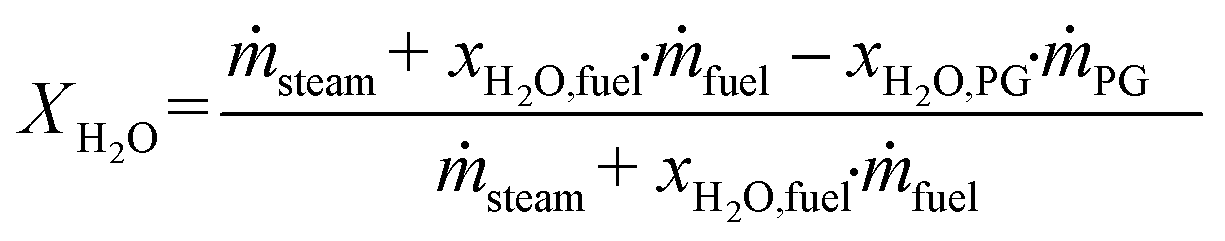 | (5) |
ṁ PG = mass flow product gas kg s−1.
X H2O,PG = weight percent of water in the product gas in w%.
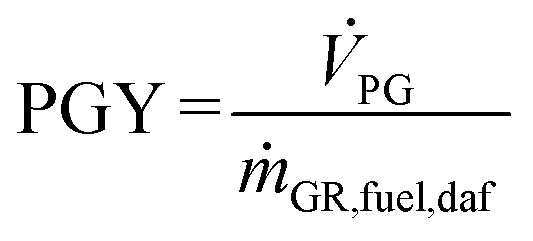 | (6) |
![[V with combining dot above]](https://www.rsc.org/images/entities/i_char_0056_0307.gif) PG = dry volumetric product gas flow in m3 sstp−1.
PG = dry volumetric product gas flow in m3 sstp−1.
ṁ GR,fuel,daf = mass flow dry an ash-free fuel fed to GR in kg s−1.
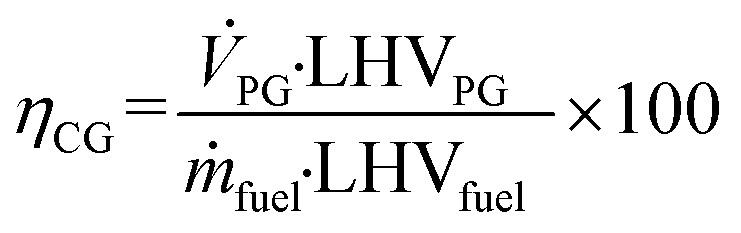 | (7) |
LHVPG = Lower heating value of product gas in MJ kgdb−1.
LHVfuel = Lower heating value of fuel in MJ kgdb−1.
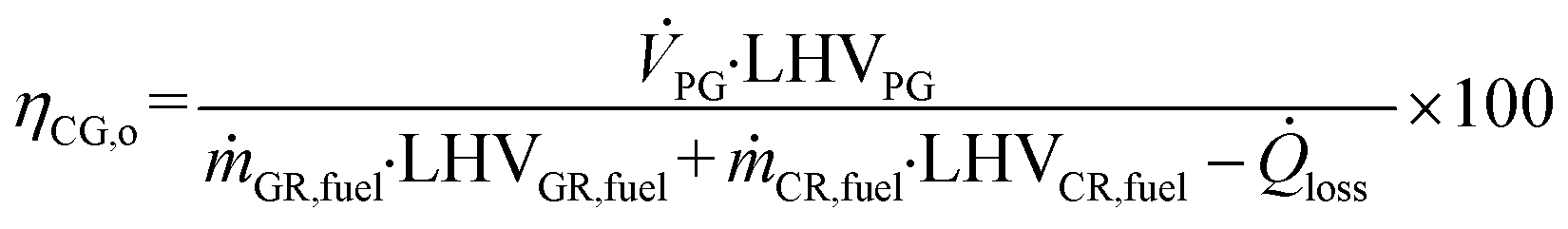 | (8) |
ṁ GR,fuel = mass flow fuel fed to GR in kg s−1.
ṁ CR,fuel = mass flow fuel fed to CR in kg s−1.
![[Q with combining dot above]](https://www.rsc.org/images/entities/i_char_0051_0307.gif) loss = heat loss in kW.
loss = heat loss in kW.
Materials
For the DFB steam gasification experiment, softwood pellets with an ash content of 0.2 w% and a diameter of 6 mm according to the Austrian standard ÖNORM M 7135 were used as fuel. The proximate and ultimate analysis of softwood pellets can be seen in Table 1. Limestone was used as bed material in the DFB steam gasification experiment. The composition and further properties of limestone can be found in Table 2.| Parameter | Unit | Softwood pellets |
|---|---|---|
| Ash content | [w%db] | 0.2 |
| Carbon (C) | [w%db] | 50.7 |
| Hydrogen (H) | [w%db] | 5.9 |
| Nitrogen (N) | [w%db] | 0.2 |
| Sulphur (S) | [w%db] | 0.005 |
| Chloride (Cl) | [w%db] | 0.005 |
| Oxygen (O) | [w%db] | 43.0 |
| Volatiles | [w%db] | 85.4 |
| Fixed C | [w%db] | 14.6 |
| Water content | [w%] | 7.2 |
| LHV (dry) | [MJ kgdb-1] | 18.9 |
| LHV (moist) | [MJ kg−1] | 17.4 |
| Parameter | Unit | Limestone |
|---|---|---|
| a Particle density after full calcination. | ||
| Al2O3 | [w%] | — |
| CaCO3 | [w%] | 95–97 |
| Fe2O3 | [w%] | — |
| K2O | [w%] | — |
| MgCO3 | [w%] | 1.5–4.0 |
| Na2O | [w%] | — |
| SiO2 | [w%] | 0.4–0.6 |
| Trace elements (<0.4 per element) | [w%] | ≤ 3.1 |
| Hardness | [Mohs] | 3 |
| Sauter mean diameter | [mm] | 0.382 |
| Particle density | [kg m−3] | 2650, 1500a |
Analytical methods
During gasification experiments, the pilot plant operation control was ensured with a programmable logic controller (PLC). Data of all flow rates, temperatures, pressures and gas compositions were measured and recorded continuously. The main gas components H2, CO, CO2 and CH4 were recorded online with a Rosemount NGA2000 measuring device. C2H4 was determined with a Perkin Elmer ARNEL – Clarus 500 gas chromatograph every 12 to 15 min. Before analysis, the product gas had to be cleaned to protect the measurement equipment from contaminants. For this purpose, it was filtered with a glass wool filter and washed with rapeseed methyl ester (RME) to eliminate condensable components like water and tar. A more detailed explanation of the measurement, equipment and procedure is given by Kolbitsch et al.60Dual fluidized bed gasification results
The product gas composition as well as relevant process indicating key figures of the SER test run at TU Wien are displayed in Table 3. During the test run, the steam to fuel ratio was set to 0.8 kg kgdaf−1. It was possible to generate a product gas with 70 w% hydrogen. The water conversion rate XH2O as well as the product gas yield PGY lie in a good range compared to other SER test runs (see literature22). Cold gas efficiencies of about 70–73% could be reached, which are typical values for this DFB gasification system.61 Based on these results, a test gas bomb from Air Liquide was transferred to TU Graz with the gas composition given in Table 4.| Product gas composition | SER test run | |
|---|---|---|
| H2 | [vol%db] | 70.3 |
| CO | [vol%db] | 8.2 |
| CO2 | [vol%db] | 5.3 |
| CH4 | [vol%db] | 14.0 |
| C2H4 | [vol%db] | 1.14 |
| C2H6 | [vol%db] | 1.14 |
| Performance indicating key parameters | SER test run | |
|---|---|---|
| X H2O | [kgH2O/kgH2O] | 0.29 |
| PGY | [m3stp,db kgfuel,daf-1] | 0.91 |
| η CG | [%] | 73.1 |
| η CG,o | [%] | 70.5 |
| Product gas composition | Test gas bomb | |
|---|---|---|
| H2 | [vol%db] | 70.5 |
| CO | [vol%db] | 8 |
| CO2 | [vol%db] | 5.5 |
| CH4 | [vol%db] | 14 |
| C2H4 | [vol%db] | 1 |
| C2H6 | [vol%db] | 1 |
Hydrodeoxygenation of LPP oil
For HDO, a plug flow reactor of Parr instrument GmbH with an inner diameter of 3/8 inch, a heated zone of 12 inch, specified for operation at 200 bar and 550 °C, with a maximum working pressure of 180 bar was used. The reactor was heated by a single zone external electric heater. Temperature was detected by an internal thermowell with a thermocouple with 3 measurement points. The reactor was fed from the top with both gaseous and liquid reactants. Gas flow was controlled by a mass flow controller, type Bronkhorst high – tech B.V., with a bypass valve for flushing the reactor in the startup phase of experiments. The liquid feed, sulfidation agent and LPP oil, was pumped through the reactor with two high pressure pumps. The reaction products were cooled down to 3 °C directly after leaving the reactor with a cooling thermostat. Afterwards, they were collected in two product vessels of Parr Instrument GmbH. The pressure was regulated by a Swagelok pressure regulating valve. Outlet gas flow was measured by a drum-type gas meter of Dr.-Ing. Ritter Apparatebau GmbH & Co. KG. A scheme of the whole setup is shown in Fig. 4.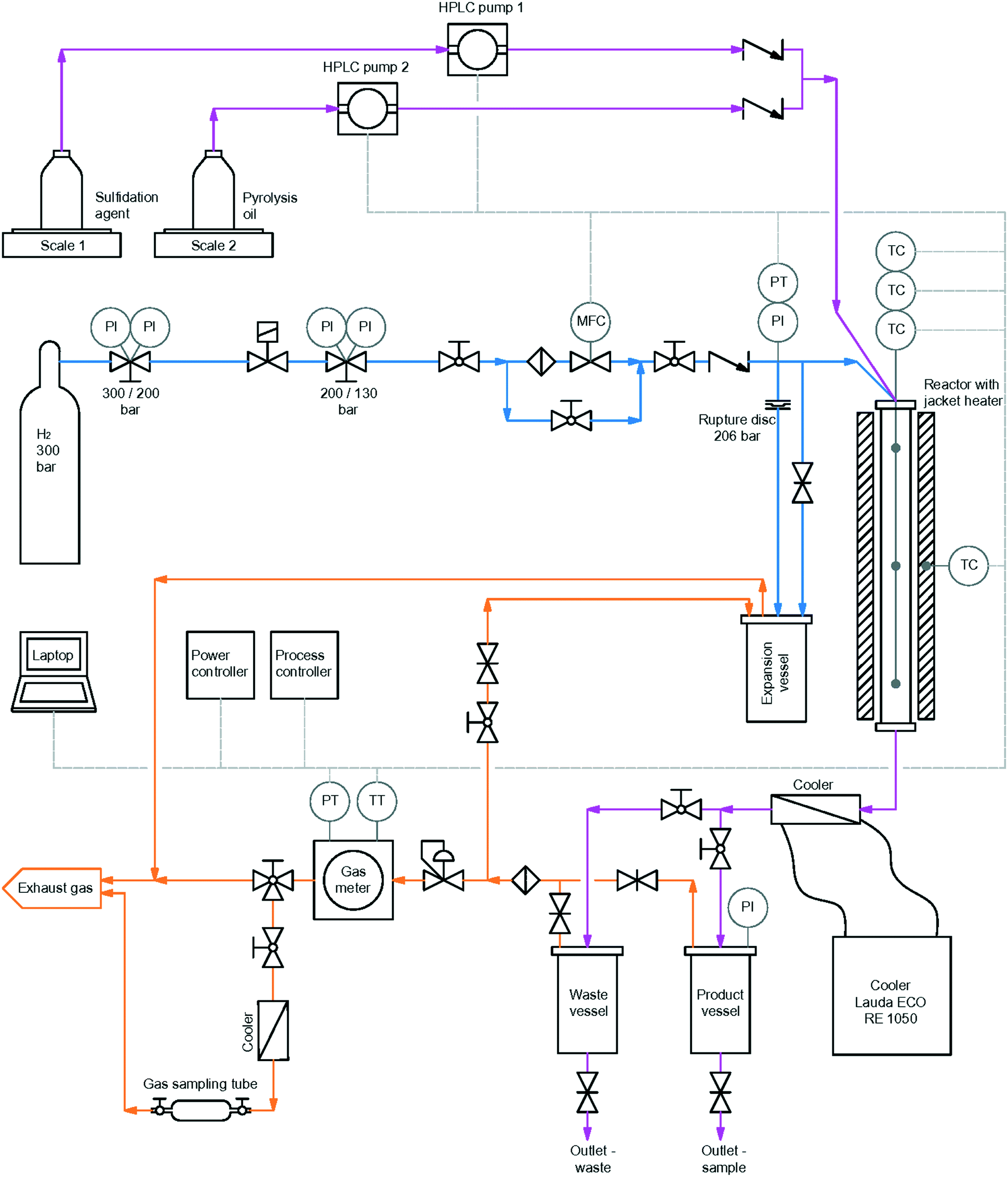 | ||
| Fig. 4 Schematic overview of the reactor set-up for HDO of LPP oil with H2 bottle.62 | ||
Experiments were performed at 120 bar. HDO with syngas was performed at 350 °C and LHSV 0.5 h−1, a reference experiment with pure hydrogen was performed at 400 °C and LHSV 1 h−1. Gas flow rate was 1 L minstp−1 for hydrogen HDO. The HDO experiment with synthesis gas will be referred to as syngas HDO in this work. To increase the water to synthesis gas ratio and therefore enhance WGS reaction, the gas flow for syngas HDO was set to 0.265 L minstp−1, which results in a hydrogen flow of 0.187 L minstp−1. The catalyst was sulfided in situ with a flow rate of 0.18 ml min−1, whereas a temperature program was started to slowly heat up the reactor to 400 °C. In the temperature range of 150 to 350 °C, the temperature was increased with a rate of 100 °C h−1. Then the temperature was increased to 400 °C and sulfidation was carried out for 3 h at this temperature. Afterwards, the pump with the sulfidation agent was stopped, the HDO temperature was adjusted and LPP oil pumping was started. In order to provide enough sulphur during HDO, di-tert-butyl-disulfide (DTBDS) equivalent to 1000 ppm of sulphur was added to LPP oil. After 5 hours of lead time, 36 hours of steady state operation mode started. Samples were taken after 12, 28 and 36 hours during syngas feed and every 12 hours in the reference experiment with hydrogen. Therefore, the experiments were divided into three periods. Gas sampling was performed every 4 hours.
Materials
For HDO, LPP oil from the bioCRACK16,18 pilot plant was used. It was produced by liquid phase pyrolysis of spruce wood at 375 °C. A cobalt molybdenum on aluminium oxide catalyst from Alfa Aesar with a particle size of 200–600 μm was used in sulfided form.Inline sulfidation was performed with 35 w% DTBDS in an iso-paraffine mixture of C15 to C20 alkanes.
During sulfidation, hydrogen 5.0 was used, during HDO synthesis gas was used as hydrogen supply. The high pressure synthesis test gas bomb was provided by Airliquide Austria GmbH, based on the TU Wien gasification results, the composition is shown in Table 4. Detailed information of the catalyst can be found in Table 5. The composition and important parameters of LPP oil are listed in Table 6.
| Catalyst | |
|---|---|
| Supplier | Alfa Aesar |
| Cobalt oxide [w%] | 4.4 |
| Molybdenum oxide [w%] | 11.9 |
| Surface area [m2 g−1] | 279 |
| Batch number | 45![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 579 579 |
| LPP oil | ||
|---|---|---|
| Properties | Unit | Value |
| Water content | [w%] | 57.0 |
| Lower heating value | [MJ kg−1] | 7.4 |
| Density | [kg m−3] | 1092 |
| Viscosity | [mPa s] | 3.5 |
| Carbon content | [w%] | 22.3 |
| Hydrogen content | [w%] | 9.4 |
| Oxygen content (balance) | [w%] | 67.8 |
| Nitrogen content | [w%] | <1 |
Analytical methods
The ultimate analysis of all streams was carried out by a vario MACRO CHN-analyzer, “Elementar Analysensysteme GmbH”. The water content of the aqueous product phase was determined by a gas-phase chromatograph, type Agilent 7890A, with a TCD-detector and a HP-INNOWAX column, 30 m × 0.530 mm × 1 μm. The boiling range of the hydrocarbon product phase was determined by a gas-phase chromatograph, type Agilent 7890A, with a FID-detector and a Restek-column MXT-2887, 10 m × 0.53 mm × 2.65 μm. The total acid number (TAN) was quantified by titration. The water content of the oil fraction was determined by Karl-Fischer-titration with a Schott Titro Line KF-Titrator and Hydranal titration reagent. Density was measured by a digital viscometer, SVM 3000, Anton Paar GmbH. The composition of the hydrocarbon product phase was determined by a gas-phase chromatograph with a quadrupole mass spectrometer (GC-MS), type Schimadzu GCMS QP 2010 Plus, with a VF-1701 MS column, 60 m × 0.25 mm × 0.25 μm.The composition of the gas phase was analysed by a micro gas-phase chromatograph, type Agilent 3000A, with a TCD-detector, a molecular sieve column and a plot u column. Sulfur content, micro carbon residue, H-NMR, metal screening and surface area of catalysts were determined externally by the “Centralni ispitni laboratorij” of INA industrija nafte d.d. Sulfur content was determined according to ASTM D 2622:2016, micro carbon residue according to HRN EN ISO 10370:2014, catalyst surface area according to ASTM D 3663 modified: 2015 and metal screening was performed by wave dispersive X-ray.
The viscometer has a density reproducibility of 0.0005 g cm−3 in the observed density range. GC-MS analysis was used for semi-quantitative determination only. Simulated distillation was performed once. All other internal analyses were performed with 3-fold determination with a maximum deviation of 0.5%, from which the average was built and displayed.
Hydrodeoxygenation of LPP oil results and discussion
For HDO of LPP oil with synthesis gas, a gas to LPP oil weight-ratio of 1 to 1.6 was applied. In the high carbon ratio of synthesis gas to LPP oil in the balance given in Table 7, one can see the high portion of carbon that was fed with the synthesis gas. Assuming, that no carbon is transferred from gas to liquid phase, the carbon yield of the hydrocarbon product phase with respect to LPP oil amounts 44 w%, which is in the range of LPP oil HDO with hydrogen.51,52| Unit | Mass balance | Carbon balance | |
|---|---|---|---|
| LPP oil | [w%] | 61.27 | 43.63 |
| Synthesis gas | [w%] | 38.73 | 56.37 |
| Hydrocarbon product phase | [w%] | 6.56 | 19.13 |
| Aqueous phase | [w%] | 42.64 | 3.80 |
| Gaseous phase | [w%] | 44.44 | 61.53 |
| Coking | [w%] | 0.93 | 1.14 |
| Balance inaccuracy | [w%] | 5.44 | 14.39 |
Properties of the HDO fuel fraction
The impact of syngas on the HDO of LPP oil was the main challenge. The results of the syngas HDO experiments are compared to an experiment with pure hydrogen, referred to as hydrogen HDO in this work, at 400 °C and LHSV 1 h−1, since it had been observed in former experiments, that the temperature in the range of 350 to 400 °C and the LHSV in the range of 0.5 to 1 h−1 has a minor impact on the product quality and technical feasibility. The lower amount of hydrogen applied for HDO might be reflected in the product composition; on the other hand, the in situ generated hydrogen could improve HDO. Competing reactions, as discussed later, might also influence the product quality. Through HDO with synthesis gas, organic components in LPP oil were hydrophobized to a degree that led to phase separation. An overview of the product quality can be found in Table 8. Beside the elemental composition, typical fuel parameters such as density, micro carbon residue and metal content are listed. Results of the H-NMR analysis can be found in Table 10. Through water separation and HDO, the water content from LPP oil was decreased drastically to below 0.5 w%. On the other hand, the water fraction contained about 95 w% of water. Carbon content as well as the organic hydrogen content was significantly increased from 22.3 w% to 85.4 w%. LPP oil itself contained about 35 ppm sulphur. Through addition of a sulfidation agent for a more effective HDO, the sulphur content was increased. Compared to the product of hydrogen HDO, slightly inferior product quality was achieved. In both cases a very low micro carbon residue was found in the product compared to EN 590 (ref. 63) standard Diesel. Native LPP oil has a high acid number of 80.8 mg KOH g−1. In the products, no acid was found.| Parameter | Unit | LPP oil | HDO syngas | HDO hydrogen | Diesel |
|---|---|---|---|---|---|
| a Determined by elemental analysis: Diesel with HVO additives. | |||||
| C | [w%] | 22.3 | 85.4 | 86.6 | 86.3a |
| H | [w%] | 9.4 | 11.9 | 12.8 | 13.7a |
| N | [w%] | <1 | <1 | <1 | <1a |
| S | [mg kg−1] | 34.6 | 132.3 | 42.4 | 10 (ref. 63) |
| O (by difference) | [w%] | 67.8 | 2.3 | 0.0 | 0.0a |
| Water content | [w%] | 56.96 | 0.28 | 0.02 | ≤0.02 (ref. 63) |
| TAN | [mg g−1] | 80.8 | 0.0 | 0.0 | n.a. |
| Density | [kg m−3] | 1092 | 877.2 | 831.1 | 820–845 (ref. 63) |
| Lower heating value (equation of Boie64) | [MJ kg−1] | 7.4 | 40.7 | 42.2 | 43.2a |
| Micro carbon residue | [w%] | n.a. | 0.04 | <0.01 | ≤0.30 (ref. 63) |
| Metal content | [w%] | Ni: 0.006 | No metals found | No metals found | n.a. |
Despite the high corrosivity, catalyst leaching can be excluded as metals were neither found in the organic nor aqueous product phase, controlled by wave dispersive X-ray analysis.
Concerning density and heating value, the quality of diesel was not fully met with syngas HDO. Semi-quantitative GC-MS analysis of the products is shown in Table 9. The most significant molecules found in LPP oil are listed. For comparison, the total ion content peak areas were normalized with the internal standard flouranthene.
| HDO gas | Syngas | Hydrogen | ||||
|---|---|---|---|---|---|---|
| Molecule | Period 1 | Period 2 | Period 3 | Period 1 | Period 2 | Period 3 |
| n.d. = not detected. | ||||||
| Pentane | 22.2 | 20.2 | n.d. | 49.1 | 43.7 | 30.7 |
| 2-Methylpentane | 12.5 | 9.2 | 5.1 | 31.2 | 24.7 | 14.8 |
| Cyclopentane | 16.8 | 17.6 | n.d. | 48.1 | 41.4 | 30.5 |
| Hexane | 14.2 | 9.0 | 4.3 | 42.3 | 35.1 | 22.2 |
| Methylcyclohexane | 52.7 | 27.2 | 19.8 | 67.0 | 46.9 | 32.9 |
| Ethylcyclopentane | 27.3 | 20.6 | 14.4 | 40.4 | 36.7 | 29.0 |
| 1-Butanol | n.d. | 6.7 | 12.9 | n.d. | n.d. | n.d. |
| 1-Ethyl-3-methylcyclopentane | 14.1 | 10.5 | 9.1 | 18.5 | 18.2 | 14.4 |
| Toluoene | 8.3 | 5.0 | 5.0 | 37.2 | 18.2 | 13.6 |
| Propylcyclopentane | 18.6 | 17.3 | 16.1 | 27.8 | 26.0 | 20.7 |
| Ethylcyclohexane | 46.8 | 30.7 | 25.7 | 55.0 | 40.8 | 35.4 |
| 1-Ethylcyclohexene | n.d. | 9.7 | 12.9 | n.d. | n.d. | 6.8 |
| 1-Methyl-2-propylcyclopentane | 16.3 | 11.6 | 10.5 | 22.3 | 20.2 | 15.0 |
| 1-Ethyl-4-methylcyclohexane | 15.6 | n.d. | n.d. | 20.5 | 15.8 | 9.2 |
| 2-Methyl-1-pentanol | n.d. | n.d. | 12.1 | n.d. | n.d. | n.d. |
| Propylcyclohexane | 47.7 | 25.1 | 18.2 | 60.2 | 43.1 | 29.8 |
| 1-Hexanol | n.d. | n.d. | 12.8 | n.d. | n.d. | n.d. |
| 2-Butyl-1-octanol | 18.1 | 10.6 | 7.1 | n.d. | 30.0 | 19.1 |
| Tetrahydro-2-furanmethanol | 95.0 | 114.7 | 128.9 | 26.2 | 25.4 | 26.5 |
| 3-Methylphenol | n.d. | 8.8 | 9.2 | n.d. | n.d. | 6.9 |
| 3-Ethylphenol | n.d. | 6.3 | 7.7 | n.d. | n.d. | 15.9 |
| 4-Propylphenol | n.d. | 16.1 | 16.9 | n.d. | n.d. | 16.7 |
LPP oil mainly contains oxygenated components, such as guaiacols, levoglucosan, organic acids, hydroxyacetone and phenolics.17 The hydrodeoxygenated products are to a big part composed of acyclic and cyclic alkanes, alkenes as well as some acyclic alcohols and phenols. Generally, the amount of alkanes is higher in products of HDO with pure hydrogen, whereas experiments performed with syngas consisted of more alkenes and phenols. With ongoing experiment, more alcohols and phenols were found, such as 1-butanol, 2-methyl-1-pentanol, 1-hexanol and 2-butyl-1-octanol. Several substituted phenols were found in products of syngas HDO already after the 2nd period, whereas in hydrogen HDO they were not present until the 3rd period of the experiment. Big amounts of tetrahydro-2-furanmethanol were found in the hydrocarbon and aqueous product phases of syngas HDO, as it is soluble in both, polar and non-polar, liquids.
H-NMR results in Table 10 show nearly the same aliphatic hydrogen content, but an interesting difference in the aromatic- and olefinic hydrogen content. Whereas HDO with syngas seems to be more effective concerning cyclic alkene saturation, olefins are more likely to be saturated when pure hydrogen is applied. All in all the differences are minor, but with about 0.02% of phenolic or olefinic hydrogen in the product of pure hydrogen HDO these structures can assumed to be fully hydrogenated in contrast to when syngas is applied.
| H aromatic | H phenolic or olefinic | H aliphatic | |
|---|---|---|---|
| Unit | [mol%] | [mol%] | [mol%] |
| HDO syngas | 4.62 | 0.83 | 94.55 |
| HDO hydrogen | 4.90 | 0.02 | 95.06 |
The boiling range (Fig. 5) shows more high boiling components in the product of syngas HDO, than for pure hydrogen based HDO. For sure, the higher content of alcohols and other oxygen containing high boiling components is remarkable. Less cracking reactions occurred, because of more competing reactions and the lower reaction temperature.
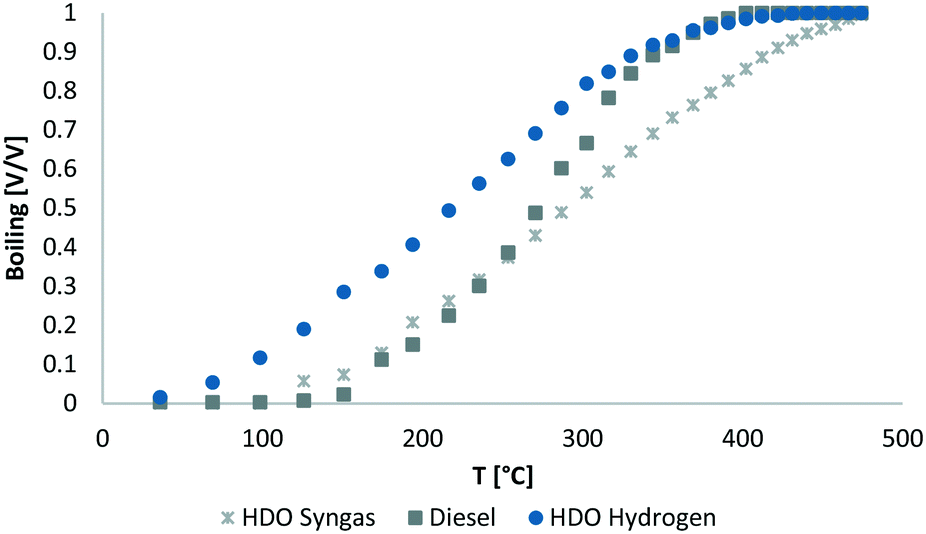 | ||
| Fig. 5 Boiling range of syngas HDO product phase compared to hydrogen HDO product phase and Diesel. | ||
Water-gas shift reaction
During the WGS reaction shown in eqn (3), CO is converted into CO2 with a stoichiometric ratio of 1. Competing reactions to be considered might be the Boudouard reaction or the Sabatier reaction shown in eqn (9) and (10). According to the Boudouard reaction,53 two molecules of CO form one molecule of CO2 and solid carbon. The stoichiometric ratio is therefore 2:1 for gaseous components in this reaction. The chemical equilibrium is shifted towards CO2 with decreasing temperature (below 400 °C) and increasing pressure, as it is an exothermic and volume increasing reaction. At room temperature, CO is fully resistant as a metastable molecule due to the low reaction rate.| 2CO ↔ C + CO2 | (9) |
In the Sabatier reaction, the stoichiometric ratio of CO to CH4 is 1. This reaction takes place at pressures below 50 bar and at 250 to 300 °C over Nickel catalysts.53 As formation of methane from CO and H2 goes along with the formation of water, a high excess of water should shift the chemical equilibrium composition to the left side of the reaction and therefore prevent methanation of CO. In the opposite direction, the reaction might also produce hydrogen from methane and water. This reaction though takes place at higher temperatures (700–830 °C in the presence of a catalyst).
| CO + 3H2 ↔ CH4 + H2O | (10) |
The WGS reaction is exothermic with 41.2 kJ mol−1,53 which amounts 1.47 MJ kg−1(CO) compared to 0.84 MJ kg−1 (LPP oil)65 for the exothermic HDO of LPP oil. For the adjusted gas and LPP oil flow for HDO with synthesis gas, the energy output of HDO amounts about 8.4 kJ h−1, the additional energy output of the WGS amounts 2.4 kJ h−1. During the switch from hydrogen for sulfidation to synthesis gas for HDO, a temperature increase of about 20 °C was observed.
The key figures confirming WGS reaction are listed in Table 11. These are the hydrogen consumption, CO/CO2 ratio and CH4 production. The hydrogen consumption was decreased by the usage of synthesis gas by about 28 w%. According to stoichiometry, the CO/CO2 ratio during Boudouard reaction is 2:1, whereas during the HDO experiment with synthesis gas a ratio of 1:1.08 was observed, which comes close to the ratio of 1:1 for WGS reaction. Therefore, Boudouard reaction can be excluded, although at low temperatures and at high pressure the chemical equilibrium is on the right hand side of solid carbon and CO2. The molar ratio of CO consumed for methane production was 44.9, which is far off the stoichiometric ratio of 1 during Sabatier reaction. The increase in methane was about 1.22 w%. It was shown, that during HDO of LPP oil methane and CO2, as well as CO and ethylene, ethane, propane and butane, are produced.51 The additional methane and CO2 production, which was observed, is attributed to HDO side reactions. This means, that no methane was produced by hydrogenation of CO, thus the Sabatier reaction can be excluded.
| H2 consumption/LPP oil | [mg g−1] | HDO hydrogen | 16.91 |
| HDO syngas | 12.25 | ||
| CO(consumed)/CO2(produced) | [mol/mol] | HDO syngas | 1.08 |
| CO(consumed)/CH4(produced) | [mol/mol] | HDO syngas | 44.92 |
In Fig. 6, the outlet gas composition of HDO of the main components is compared to the inlet gas composition (synthesis gas). TOS is defined as time on stream. In the first 8 hours, the gas phase composition is not stable yet, as the hydrogen used for sulfidation was not fully replaced due to the low gas feed. Therefore, the balance period was made for the experimental time span of 12–36 hours. CO was nearly fully converted, merely about 0.5 vol% were detected in the outlet gas stream of HDO, whereas the portion of CO2 increased from 5.5 to about 14 vol%. The net hydrogen content increased slightly, whereas the net methane content was nearly untampered with about 14 to 16 vol%.
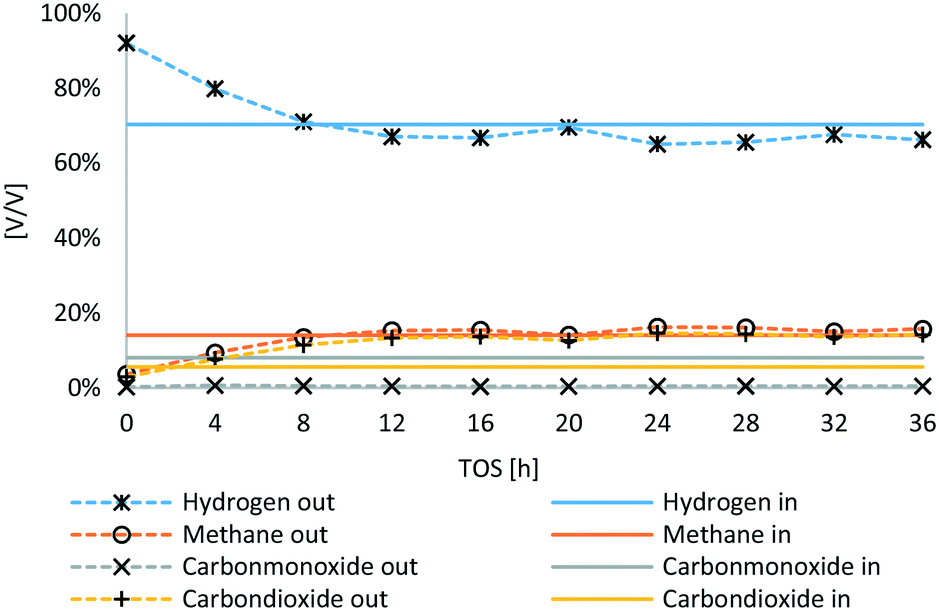 | ||
| Fig. 6 Gas phase composition of HDO inlet gas (synthesis gas) and outlet gas. | ||
Subsequently, the water balance was observed, as water is on the one hand produced during HDO and on the other hand consumed during WGS reaction. Following the reaction stoichiometry, the amount of consumed water was calculated for the case that all CO is transferred into CO2 by WGS reaction according to eqn (11).
| m(Theroetical WGS consumed H2O) = n(COin)·M(H2O) | (11) |
m(Theoretical WGS consumed H2O) = mass of water consumed by WGS reaction if 100% of the introduced CO is consumed in g.
n(COin) = molar amount of fed CO in mol.
M(H2O) = molar mass of consumed water in g mol−1.
With respect to LPP oil, 3 to 7 w% of water was produced when syngas was applied, whereas it was 14 to 19 w% when hydrogen was applied, as shown in Fig. 7. In comparison to HDO with pure hydrogen, 68.6 w% less water was produced. Considering that water was consumed by WGS reaction when syngas was applied, a higher amount of water must have been originally produced by HDO. This would amount about 65.8 w% more water, assuming that all CO was converted into hydrogen and CO2 by WGS reaction. With respect to LPP oil, this would mean that between 9.6 to 10.3 w% of water are consumed. This sums up to 13 and 17 w% of water, which must have been produced, before consumed for WGS reaction, which is comparable with the results of hydrogen HDO and again confirms the conversion of water and CO through the WGS reaction.
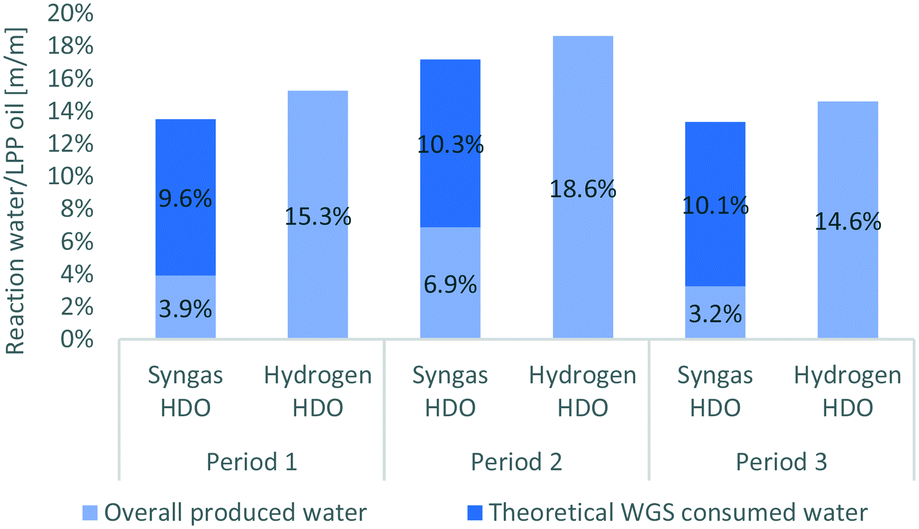 | ||
| Fig. 7 Formation and consumption of water during HDO using syngas over the WGS reaction compared to HDO with pure hydrogen. | ||
Catalyst deactivation
A major obstacle in HDO of pyrolysis oil is catalyst deactivation through coke deposition. Table 12 shows the surface area of fresh catalyst compared to that of the used catalysts from syngas and hydrogen HDO. A clear decrease in surface area after HDO can be observed. According to Olarte et al.,29 plugs usually consist of inorganic constituents and polymerized bio-oil condensation products. As LPP oil doesn't contain appreciable amounts of inorganic matter in contrast to fast pyrolysis oils7 (see metal content in Table 8), the main factor for coke formation is unstable organic matter. Additionally, due to the usage of synthesis gas, a high load of organic matter is introduced to the reactor. While it is highly unlikely that alkanes and alkenes with a small chain length (methane, ethane and ethylene) condense in the reactor and form coke, a main factor could be the Boudouard reaction. If this reaction takes place even in small amounts, the coke formed could still lead to plugging and force the system to break down due to a high pressure drop. It was therefore concluded, that a high excess of water is necessary to force the reaction equilibrium to the side of hydrogen and CO2 and to suppress Boudouard reaction. This was achieved by the high water content of LPP oil and a nearly triple fold LPP oil to hydrogen ratio compared to previous experiments.| ASTM D3663 | [m2 g−1] |
|---|---|
| Fresh | 239 |
| HDO syngas | 131 |
| HDO hydrogen | 141 |
The surface area of the catalyst was reduced by 45% in the experiment with synthesis gas, compared to 41% in an experiment with hydrogen. The difference is negligible considering measurement uncertainty. For more information, the amount of organic matter and carbon on the catalyst were determined. For the determination of combustibles, catalysts were incinerated in a muffle type furnace at 550 °C for 48 h. Thus, the catalyst in the main reaction zone in the middle of the reactor was analysed. The results were quite surprising. The amount of combustibles is the same on both catalysts, but less carbon seemed to be deposited on the catalyst in HDO with syngas, as shown in Table 13. The net carbon content of the coke was about 54 w% in hydrogen HDO but only 38 w% in syngas HDO. A big part of the combustibles is most probably sulphur from sulfidation. About 7 w% is contributed by hydrogen. The difference is assumed to be oxygen. Origin of coke deposition cannot be allocated to Boudouard reaction or standard coking from HDO side reactions only by catalyst analyses.
| Unit | Syngas HDO | Hydrogen HDO | |
|---|---|---|---|
| Combustibles | [w%] | 24.4 | 24.8 |
| Carbon content | [w%] | 9.3 | 13.4 |
| Carbon content/combustibles | [w%] | 38.2 | 54.2 |
Coke deposition has a huge impact on product quality, as less catalyst surface is available if plugs are formed. A decreasing product quality was observed for all experiments with LPP oil,51,52 whereas the rate is different. In Fig. 8, the oxygen content as well as the water content of products from syngas HDO is compared to that of hydrogen HDO. In the first period of both experiments, the oxygen content was zero and water content below 0.1 w%. This indicates the same degree of HDO at the start of the experiment. On the one hand, this can be described by switch from hydrogen to syngas. After 5 hours of lead-time, there was still a higher amount of hydrogen in the system, as can be observed in the product gas composition from HDO experiments in Fig. 6. On the other hand, this might also indicate, that on a freshly sulfided catalyst, syngas and pure hydrogen are more or less equally effective. According to Sheu et al.,66 deoxygenation is a function of the oxygen content and partial hydrogen pressure. This would explain the lower oxygen removal due to the lower hydrogen to LPP oil ratio during Syngas HDO. Grilc et al.48–50 observed comparable oxygen removal when using nitrogen as process gas, conducted with higher decarbonylation and decarboxylation reactions resulting in lower liquid product yield. They also observed high HDO rates for molybdenum sulphide catalysts. In order to enhance the water gas shift reaction, the water to CO ratio was increased, compared to hydrogen based HDO experiments, and therefore only about 35 w% of pure hydrogen was available per g LPP oil for HDO compared to the reference experiment with hydrogen.
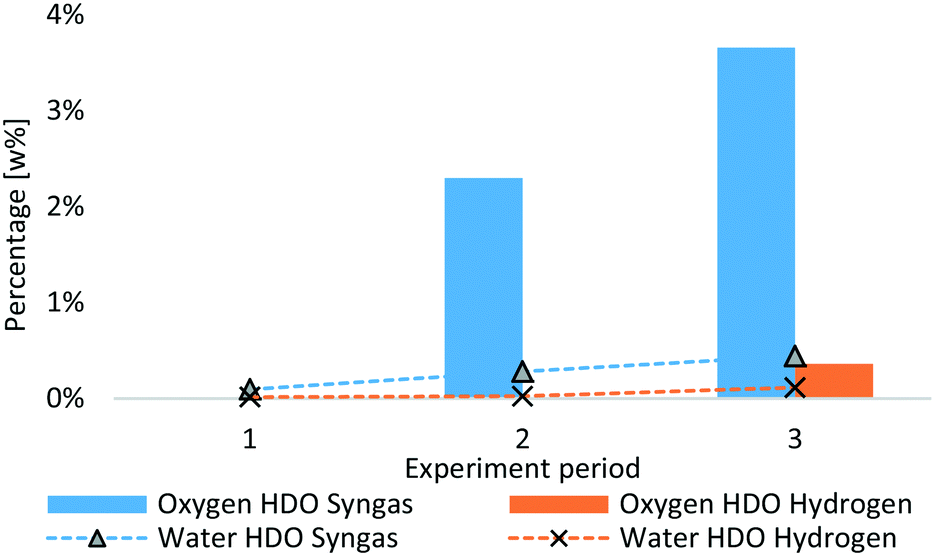 | ||
| Fig. 8 Oxygen and water content of HDO products from syngas and hydrogen experiment. | ||
According to Wijayapala et al., HDO with synthesis gas seems to be slower, but delivers competitive conversions compared to H2 (ref. 57). Therefore, an even lower LHSV might be necessary for the same product quality than with hydrogen.
After 36 h TOS, the oxygen content of the product from syngas HDO, determined by difference, amounts 3.7 w%, whereas the oxygen content of the product from hydrogen experiment amounts 0.4 w%. The increasing oxygen content originates mainly in alcohols, as shown in Table 9, which also facilitates water absorption.
Summary and conclusions
By dual fluidized bed steam gasification of softwood, a biogenous synthesis gas with a high hydrogen content of about 70 vol% was produced. A gas with the composition of the synthesis gas was directly used for HDO of LPP oil, produced by LPP of spruce wood. HDO of LPP oil was then performed successfully for 36 h in a first experiment. Pre-treatment of LPP oil was not necessary due to the high water content and negligible particle content. The quality of LPP oil concerning fuel standards was increased significantly. Although high coke deposition can be excluded, product quality still decreased over time. Compared to HDO with pure hydrogen, a slightly lower degree of hydrodeoxygenation was achieved. The reason for this might be the lower hydrogen to LPP oil ratio, which was 37 w% compared to hydrogen HDO, in order to enhance WGS reaction. The consumption of hydrogen for HDO was reduced by 28 w%, while nearly all the CO was consumed for hydrogen production according to stoichiometry. Competing reactions like the Boudouard reaction or Sabatier reaction were not observed. Reasons for that are on the one hand the reaction conditions, especially the temperature when discussing the Sabatier reaction, and on the other hand the surpassingly high water content of LPP oil, which supresses the Boudouard reaction. A synthesis gas with a higher CO content and lower CO2 content might lead to lower or even zero hydrogen consumption for advanced biofuel production from liquid phase pyrolysis oil.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the Austrian Research Promotion Agency (FFG) under the scope of the Austrian Climate and Energy Fund. Furthermore, this work was supported by the European Union's Horizon 2020 research and innovation programme. The authors want to acknowledge Mario Lukasch, Samir Reiter, Thomas Sterniczky, Sarah Koller and Daniela Grosinger for their outstanding work with HDO experiments, as well as Johannes Schmid, Josef Fuchs and Florian Benedikt for their excellent work regarding the DFB steam gasification process.Notes and references
- United Nations, Kyoto Protocol to the United Nations Framework Convention on Climate Change, United Nations, 1998 Search PubMed.
- UNFCCC, ADOPTION OF THE PARIS AGREEMENT: Proposal by the President to the United Nations Framework Convention on Climate Change, 2015, 21932, 1–32 Search PubMed.
- in Official Journal of the European Union, Brussels, Belgium, 2009, pp. 16–62 Search PubMed.
- O. F. T. H. E., Council, 2018, 2018 Search PubMed.
- H. Hofbauer, in Encyclopedia of Sustainability Science and Technology, Springer, 2017, pp. 459–478 Search PubMed.
- A. V. Bridgwater, D. Meier and D. Radlein, Org. Geochem., 1999, 30, 1479–1493 CrossRef CAS.
- A. V. Bridgwater and G. V. C. Peacocke, Renewable Sustainable Energy Rev., 2000, 4, 1–73 CrossRef CAS.
- S. Czernik and A. V. Bridgwater, Energy Fuels, 2004, 18, 590–598 CrossRef CAS.
- A. V. Bridgwater, J. Anal. Appl. Pyrolysis, 1999, 51, 3–22 CrossRef CAS.
- A. V. Bridgwater, Biomass Bioenergy, 2011, 38, 68–94 CrossRef.
- K. Klaigaew, P. Wattanapaphawong, N. Khuhaudomlap, N. Hinchiranan, P. Kuchontara, K. Kangwansaichol and P. Reubroycharoen, Liquid Phase Pyrolysis of Giant Leucaena Wood to Bio-Oil over NiMo/Al2O3 Catalyst, Elsevier B.V., 2015, vol. 79 Search PubMed.
- W. Ratanathavorn, C. Borwornwongpitak, C. Samart and P. Reubroycharoen, Chem. Technol. Fuels Oils, 2016, 52, 360–368 CrossRef CAS.
- B. Szabó, M. Takács, A. Domján, E. Barta-rajnai and J. Valyon, J. Anal. Appl. Pyrolysis, 2018, 1–9 Search PubMed.
- N. Schwaiger, V. Witek, R. Feiner, H. Pucher, K. Zahel, A. Pieber, P. Pucher, E. Ahn, B. Chernev, H. Schroettner, P. Wilhelm and M. Siebenhofer, Bioresour. Technol., 2012, 124, 90–94 CrossRef CAS PubMed.
- N. Schwaiger, R. Feiner, K. Zahel, A. Pieber, V. Witek, P. Pucher, E. Ahn, P. Wilhelm, B. Chernev, H. Schröttner and M. Siebenhofer, BioEnergy Res., 2011, 4, 294–302 CrossRef.
- J. Ritzberger, P. Pucher and N. Schwaiger, Chem. Eng. Trans., 2014, 39, 1189–1194 Search PubMed.
- N. Schwaiger, D. C. Elliott, J. Ritzberger, H. Wang, P. Pucher and M. Siebenhofer, Green Chem., 2015, 17, 2487–2494 RSC.
- K. Treusch, J. Ritzberger, N. Schwaiger, P. Pucher and M. Siebenhofer, R. Soc. Open Sci., 2017, 4, 171122 CrossRef CAS PubMed.
- A. M. Mauerhofer, F. Benedikt, J. C. Schmid, J. Fuchs, S. Müller and H. Hofbauer, Energy, 2018, 157, 957–968 CrossRef CAS.
- F. Benedikt, J. Fuchs, J. C. Schmid, S. Müller and H. Hofbauer, Korean J. Chem. Eng., 2017, 34, 2548–2558 CrossRef CAS.
- F. Benedikt, J. C. Schmid, J. Fuchs, A. M. Mauerhofer, S. Müller and H. Hofbauer, Energy, 2018, 164, 329–343 CrossRef CAS.
- J. Fuchs, J. C. Schmid, F. Benedikt, S. Müller, H. Hofbauer, H. Stocker, N. Kieberger and T. Bürgler, Energy, 2018, 162, 35–44 CrossRef CAS.
- S. Koppatz, C. Pfeifer, R. Rauch, H. Hofbauer, T. Marquard-Moellenstedt and M. Specht, Fuel Process. Technol., 2009, 90, 914–921 CrossRef CAS.
- J. C. Schmid, J. Fuchs, F. Benedikt, A. M. Mauerhofer, S. Müller, H. Hofbauer, H. Stocker, N. Kieberger and T. Bürgler, 2017.
- S. Oh, H. Hwang, H. S. Choi and J. W. Choi, Fuel, 2015, 153, 535–543 CrossRef CAS.
- S. Oh, H. S. Choi, I.-G. Choi and J. W. Choi, RSC Adv., 2017, 7, 15116–15126 RSC.
- C. Boscagli, C. Yang, A. Welle, W. Wang, S. Behrens, K. Raffelt and J. D. Grunwaldt, Appl. Catal., A, 2017, 544, 161–172 CrossRef CAS.
- C. Boscagli, K. Raffelt and J. D. Grunwaldt, Biomass Bioenergy, 2017, 106, 63–73 CrossRef CAS.
- M. V. Olarte, A. H. Zacher, A. B. Padmaperuma, S. D. Burton, H. M. Job, T. L. Lemmon, M. S. Swita, L. J. Rotness, G. N. Neuenschwander, J. G. Frye and D. C. Elliott, Top. Catal., 2016, 59, 55–64 CrossRef CAS.
- D. C. Elliott, Energy Fuels, 2007, 21, 1792–1815 CrossRef CAS.
- D. Carpenter, T. Westover, D. Howe, S. Deutch, A. Starace, R. Emerson, S. Hernandez, D. Santosa, C. Lukins and I. Kutnyakov, Biomass Bioenergy, 2017, 96, 142–151 CrossRef CAS.
- P. A. Meyer, L. J. Snowden-Swan, K. G. Rappé, S. B. Jones, T. L. Westover and K. G. Cafferty, Energy Fuels, 2016, 30, 9427–9439 CrossRef CAS.
- D. C. Elliott, T. R. Hart, G. G. Neuenschwander, L. J. Rotness and A. H. Zacher, Environ. Prog. Sustainable Energy, 2009, 28, 441–449 CrossRef CAS.
- M. V. Olarte, A. B. Padmaperuma, J. R. Ferrell, E. D. Christensen, R. T. Hallen, R. B. Lucke, S. D. Burton, T. L. Lemmon, M. S. Swita, G. Fioroni, D. C. Elliott and C. Drennan, Fuel, 2017, 620–630 CrossRef CAS.
- D. Howe, T. Westover, D. Carpenter, D. Santosa, R. Emerson, S. Deutch, A. Starace, I. Kutnyakov and C. Lukins, Energy Fuels, 2015, 29, 3188–3197 CrossRef CAS.
- D. Carpenter, T. Westover, D. Howe, S. Deutch, A. Starace, R. Emerson, S. Hernandez, D. Santosa, C. Lukins and I. Kutnyakov, Biomass Bioenergy, 2016, 96, 142–151 CrossRef.
- G. Kim, J. Seo, J. W. Choi, J. Jae, J. M. Ha, D. J. Suh, K. Y. Lee, J. K. Jeon and J. K. Kim, Catal. Today, 2017, 0–1 Search PubMed.
- K. Routray, K. J. Barnett and G. W. Huber, Energy Technol., 2017, 5, 80–93 CrossRef CAS.
- S. Oh, H. S. Choi, I.-G. Choi and J. W. Choi, RSC Adv., 2017, 7, 15116–15126 RSC.
- S. Cheng, L. Wei, J. Julson and M. Rabnawaz, Energy Convers. Manage., 2017, 150, 331–342 CrossRef CAS.
- F. De Miguel Mercader, P. J. J. Koehorst, H. J. Heeres, S. R. A. Kersten and J. A. Hogendoorn, AIChE J., 2011, 57, 3160–3170 CrossRef CAS.
- J. Neumann, N. Jäger, A. Apfelbacher, R. Daschner, S. Binder and A. Hornung, Biomass Bioenergy, 2016, 89, 91–97 CrossRef CAS.
- I. Kim, A. A. Dwiatmoko, J. W. Choi, D. J. Suh, J. Jae, J. M. Ha and J. K. Kim, J. Ind. Eng. Chem., 2017, 56, 74–81 CrossRef CAS.
- W. Yin, A. Kloekhorst, R. H. Venderbosch, M. V. Bykova, S. A. Khromova, V. A. Yakovlev and H. J. Heeres, Catal. Sci. Technol., 2016, 6, 5899–5915 RSC.
- J. Michl, J. Neumann, H. Rottengruber and M. Wensing, Appl. Therm. Eng., 2016, 98, 502–512 CrossRef CAS.
- S. A. Rezzoug and R. Capart, Appl. Energy, 2002, 72, 631–644 CrossRef CAS.
- M. Kunaver, E. Jasiukaityte and N. Čuk, Bioresour. Technol., 2012, 103, 360–366 CrossRef CAS PubMed.
- M. Grilc, B. Likozar and J. Levec, Biomass Bioenergy, 2014, 63, 300–312 CrossRef CAS.
- M. Grilc, B. Likozar and J. Levec, Appl. Catal., B, 2014, 150–151, 275–287 CrossRef CAS.
- M. Grilc, G. Veryasov, B. Likozar, A. Jesih and J. Levec, Appl. Catal., B, 2015, 163, 467–477 CrossRef CAS.
- K. Treusch, N. Schwaiger, K. Schlackl, R. Nagl, A. Rollett, M. Schadler, B. Hammerschlag, J. Ausserleitner, A. Huber, P. Pucher and M. Siebenhofer, React. Chem. Eng., 2018, 258–266 RSC.
- K. Treusch, N. Schwaiger, K. Schlackl, R. Nagl and P. Pucher, Front. Chem., 2018, 6(297), 1–8 Search PubMed.
- A. F. Holleman, E. Wiberg and N. Wiberg, Holleman-Wiberg: Lehrbuch der Anorganischen Chemie, de Gruyter, Berlin, 102nd edn., 2007 Search PubMed.
- P. H. Steele, S. K. Gajjela, T. E. Mlsna, C. U. Pittman and F. Yu, Pat. US 2014/0073827A1, 2014 Search PubMed.
- S. K. Tanneru and P. H. Steele, Renewable Energy, 2015, 80, 251–258 CrossRef CAS.
- Y. Luo, E. B. Hassan, V. Guda, R. Wijayapala and P. H. Steele, Energy Convers. Manage., 2016, 115, 159–166 CrossRef CAS.
- R. Wijayapala, A. G. Karunanayake, D. Proctor, F. Yu, C. U. Pittman and T. E. Mlsna, in Handbook of Climate Change Mitigation and Adaptation, ed. W.-Y. Chen, T. Suzuki and M. Lackner, Springer New York, New York, NY, 2016, pp. 1–34 Search PubMed.
- S. Müller, J. Fuchs, J. C. Schmid, F. Benedikt and H. Hofbauer, Int. J. Hydrogen Energy, 2017, 42, 29697–29707 CrossRef.
- T. Pröll and H. Hofbauer, Int. J. Chem. React. Eng., 2008, 6, A89 Search PubMed.
- M. Kolbitsch, Doctoral thesis, TU Wien, 2016 Search PubMed.
- S. Müller, Doctoral thesis, TU Wien, 2013 Search PubMed.
- M. Lukasch, Master thesis, Graz University of Technology, 2018 Search PubMed.
- EN 590, 2004 Search PubMed.
- K.-H. Grote and J. Feldhusen, Dubbel Taschenbuch für Maschinenbau, Springer, 22nd edn., 2007 Search PubMed.
- K. Schlackl, Master thesis, Graz University of Technology, 2016 Search PubMed.
- Y.-H. E. Sheu, R. G. Anthony and E. J. Soltes, Fuel Process. Technol., 1988, 19, 31–50 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2019 |