
Solvent-free organocatalytic preparation of cyclic organic carbonates under scalable continuous flow conditions†
Zhiguo
Wang‡
a,
Romaric
Gérardy
a,
Guillaume
Gauron
b,
Christian
Damblon
c and
Jean-Christophe M.
Monbaliu
 *a
*a
aCenter for Integrated Technology and Organic Synthesis, Research Unit MolSys, University of Liège, B-4000 Liège (Sart Tilman), Belgium. E-mail: jc.monbaliu@uliege.be
bCorning Reactor Technologies, Corning SAS, 7 bis, Avenue de Valvins CS 70156 Samois sur Seine, 77215 Avon Cedex, France
cCREMAN NMR Center, Research Unit MolSys, University of Liège, B-4000 Liège (Sart Tilman), Belgium
First published on 24th October 2018
Abstract
A solvent-free organocatalyzed process for the transesterification of dimethyl carbonate (DMC) with 1,2-diols under scalable continuous flow conditions is presented. Process parameters, such as temperature, residence time, DMC/glycerol molar ratio and catalyst loading are optimized for the carbonation of bio-based glycerol using 1,8-diazabicyclo(5.4.0)undec-7-ene (DBU) as a model organocatalyst. The catalytic performance of DBU is next compared with other homogeneous organic superbases including the proton sponge, Verkade's base, guanidines and phosphazenes. 2-tert-Butyl-1,1,3,3-tetramethylguanidine (Barton's base) stands as the most efficient organocatalyst, providing glycerol carbonate at 87% selectivity and 94% conversion within 2 minutes of residence time at 1 mol% loading. Representative examples of polystyrene-supported (PS) organic superbases of the amidine, guanidine and phosphazene-types are also considered as alternative heterogeneous catalysts. PS superbases typically enable up to 80 h of continuous operation with minor deactivation at elevated flow rates. The methodology is amenable to a library of other 1,2-diols, including biomass-derived substrates. Depending on the unique structural features of both substrates and products, either on-line IR or on-line NMR analytical procedures are implemented for real-time qualitative reaction monitoring. A final demonstrator showcases the transposition of the glycerol carbonation to a pilot-scale continuous flow reactor, affording the target cyclic carbonate with a 68.3 mol per day productivity (∼8 kg per day).
Introduction
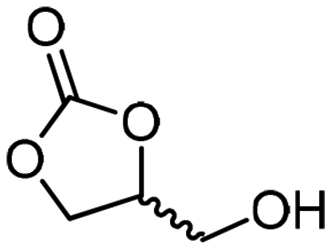
Glycerol (1) was highlighted by the US Department of Energy as one of the top value-added building blocks derived from biomass.1,2 A large stockpile of bio-based 1 is available worldwide as waste from the production of biodiesel, which stimulates intense research efforts for upgrading it to industrially relevant molecules.3,4 Numerous commodity chemicals, such as propanols, lactic acid, allyl alcohol, acetol, acrolein, acrylic acid, acrylonitrile, propene, aromatics, glycerol ethers and esters, as well as glycerol carbonate, can now be obtained through chemically upgrading 1.3–6Glycerol carbonate (2, 4-hydroxymethyl-2-oxo-1,3-dioxolane) is a biodegradable, non-toxic, non-flammable and water-soluble liquid with low vapor pressure. It comes with a broad range of applications, including as a building block for the preparation of commodity chemicals and polymers, as an organic solvent, and as a carrier for electrolytes in batteries, as well as an additive and humectant in cosmetic, personal care and health products.7–11 A recent survey highlighted that 2 stands as one of the most appealing targets to be produced in a glycerol biorefinery from the environmental and economic perspectives.12 Numerous procedures were reported for the direct preparation of glycerol carbonate from glycerol, including phosgenation, oxidative carbonylation with CO/O2, carboxylation with CO2, glycerolysis of urea, and transesterification with dialkyl/alkylene carbonates (Fig. 1).7–11
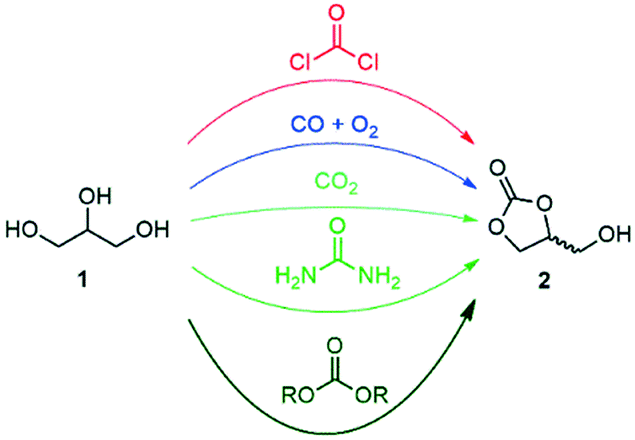 | ||
| Fig. 1 Synthetic methodologies for the transformation of glycerol into glycerol carbonate. | ||
The phosgene and carbon monoxide/oxygen routes suffer from safety and environmental concerns regarding industrial applicability. Although the carbon dioxide route is the most sustainable, typical processes come with low yields and require harsh conditions. The glycerolysis of urea is one of the most promising routes since urea is widely available, cheap and mostly CO2-sourced. However, the urea-based route produces corrosive ammonia, and the reaction has to be performed under vacuum or under a constant gas flow at high temperature, thus decreasing the overall cost-efficiency for industrial scale applications. Transesterification between 1 and organic carbonates – such as ethylene, diethyl or dimethyl carbonate – has received significant attention as well. These reagents have indeed a low toxicity profile, are widely available,13 and have been already successfully utilized for the production of 2 in high yields.7–11
A plethora of basic homogeneous and heterogeneous catalytic systems has been reported for the transesterification route,7–11 including organocatalysts. Typical organocatalysts can usually be removed more efficiently from highly polar reaction media than homogeneous mineral catalysts or metal cations resulting from the leaching of heterogeneous catalytic materials. Transesterification organocatalysts include amines and amidines,14–18N-heterocyclic carbenes and derivatives,19–22 and ammoniums and ionic liquids,23–30 as well as enzymes.31–36 Among the potential organocatalysts for this carbonation reaction, amidines, guanidines, proazaphosphatranes and phosphazenes are classes of typically non-nucleophilic, strong, non-ionic organic bases, endowed with high tolerance to moisture and heat, and high solubility in polar and apolar media. These advantages allow them to be useful as reagents and catalysts for organic transformations, and they have high potential for recyclability as well.37,38
Despite the many advantages of continuous flow manufacturing,6,39,40 only a few studies related to the synthesis of glycerol carbonate from glycerol have been conducted in flow reactors (Table 1).17,30,35,41–44 For instance, de Souza et al., reported a flow procedure for glycerol carbonate using 3 equiv. of dimethyl carbonate (DMC) and 1.5 mol% of 1,8-diazabicyclo(5.4.0)undec-7-ene (DBU) as the homogeneous catalyst under solvent-free conditions. A residence time of 8 min at 120 °C in the flow setup was necessary to reach 80% conversion in glycerol with 82% selectivity for the target cyclic carbonate.17 Selva et al. reported a catalyst-free flow process using high temperature (230–250 °C) and pressure (50 bar), as well as a large excess of DMC (10 equiv.).42 Methanol was used as the solvent in the process, and a residence time of 15 min enabled the preparation of glycerol carbonate in 83–92% selectivity at 78–94% glycerol conversion. Baxendale and co-workers prepared glycerol carbonate using 3.5 equiv. of DMC and 3 wt% of tetraethylammonium pipecolinate as the homogeneous catalyst. Glycerol was pumped as an ethanol solution with 30 min of residence time at 140 °C, affording 90% conversion and 85% selectivity. The authors also described the use of Ambersep® 900 hydroxide resin as the heterogeneous organocatalyst, which provided up to 97% conversion and 91% selectivity using 4 equiv. of DMC. Methanol was used as the solvent and the results were obtained within 4 min of residence time at 140 °C. The heterogeneous catalyst however did not tolerate solvent-free conditions and deactivation was noticed after 20 h of operation.30

|
In this contribution, we describe a solvent- and metal-free process under scalable continuous flow conditions for the upgrading of glycerol into glycerol carbonate. Process parameters – such as residence time, temperature, glycerol/DMC molar ratio and catalyst loading – were thoroughly assessed with DBU as the model homogeneous catalyst. A library of homogeneous organocatalysts, including the proton sponge, Verkade's base, guanidines and phosphazenes, was then investigated using the optimized process conditions. 2-tert-Butyl-1,1,3,3-tetramethylguanidine (Barton's base) appeared as the most promising catalyst, giving a 94% glycerol conversion with an 87% glycerol carbonate selectivity. This result was achieved at 1 mol% loading, 135 °C, with 2 min of residence time and 3 equiv. of DMC. To further improve the cost-efficiency and ease downstream purification, supported organocatalysts – such as polymer-bound DBU, P2-t-Bu phosphazene and tetramethylguanidine – were assessed as well. These heterogeneous organocatalysts proved to exhibit excellent stability over time, as conversion typically decreased by 13–15% after 80 h of operation. The methodology was extended to other 1,2-diols, including biomass-derived substrates, which were converted into the corresponding cyclic carbonates in good to excellent yields. Depending on the unique features of both substrates and products, either on-line IR or on-line NMR analytical procedures were implemented for convenient real-time qualitative reaction monitoring. Upon optimization of process parameters and organocatalysts, the methodology for glycerol carbonate was then applied to a commercial mesofluidic continuous flow reactor to assess the scalability using the best homogeneous catalyst (Barton's base). The conditions were then further applied to a pilot-scale continuous flow reactor, affording glycerol carbonate with a 68.3 mol per day productivity.
Experimental section
General information
Conversion and yield were determined by gas chromatography coupled with flame ionization detection (GC/FID) or high field 1H NMR spectroscopy with mesitylene as the internal standard. Structural identity was confirmed by 1H and 13C NMR spectroscopy conducted on a high field (400 MHz) Bruker Avance spectrometer in CDCl3 or d4-MeOD (ESI†). The chemical shifts are reported in ppm relative to TMS as the internal standard or to the solvent residual peak. GC conversions were determined using external calibration curves established using commercial standards of ethanediol, 1,2-propanediol, 1,2,3-propanetriol, 3-methoxy-1,2-propanediol, 3-tert-butoxy-1,2-propanediol, 1,2-butanediol, 2,3-butanediol, 3-butene-1,2-diol and 1,4-anhydroerythritol. GC yields were determined using external calibration curves established using commercial standards of 1,3-dioxolan-2-one, 4-methyl-1,3-dioxolan-2-one, 4-hydroxymethyl-1,3-dioxolan-2-one, 4-methoxymethyl-1,3-dioxolan-2-one, 4-ethyl-1,3-dioxolan-2-one, 4-vinyl-1,3-dioxolan-2-one and a synthesized sample of methyl ((2-oxo-1,3-dioxolan-4-yl)methyl) carbonate. Selectivity is defined as the ratio between the yield and the conversion. On-line IR reaction monitoring was carried out using a FlowIR™ from Mettler-Toledo. On-line NMR reaction monitoring was carried out using a 43 MHz Spinsolve™ carbon NMR spectrometer from Magritek®. Downstream pressures were regulated using dome-type backpressure regulators (7–11 bar, Zaiput Flow Technologies). Ethanediol, 1,2-propanediol, 1,2,3-propanetriol, 3-methoxy-1,2-propanediol, 3-tert-butoxy-1,2-propanediol, 1,2-butanediol, 2,3-butanediol, 3-butene-1,2-diol, 1,4-anhydroerythritol, dimethyl carbonate (DMC), 1,8-diazabicyclo(5.4.0)undec-7-ene (DBU), 2,8,9-trimethyl-2,5,8,9-tetraaza-1-phosphabicyclo[3.3.3]undecane (Verkade's base), 1,8-bis-(dimethylamino)-naphthalene (DMAN), 1,1,3,3-tetramethylguanidine (TMG), 2-tert-butyl-1,1,3,3-tetramethylguanidine (Barton's base), 1,8-bis(tetramethylguanidino)naphthalene (TMGN), imino-tris-(dimethylamino)phosphorane (P1-H), tert-butylimino-tris(dimethyl-amino)phosphorane (P1-t-Bu), tert-octylimino-tris(dimethylamino)-phosphorane (P1-t-Oct), tert-butylimino-tris(pyrrolidino)-phosphorane (BTPP), 2-tert-butylimino-2-diethylamino-1,3-dimethylperhydro-1,3,2-diazaphosphorine (BEMP), tetramethyl-(tris(dimethylamino)phosphoranylidene)-phosphorictriamid-ethyl-imine (P2-Et), polystyrene-supported phosphazene base P2-t-Bu (1.6 mmol g−1) and polystyrene-supported DBU (1.5–2.5 mmol g−1) were obtained from commercial sources and used as received. Polystyrene-supported TMG, methyl ((2-oxo-1,3-dioxolan-4-yl)methyl) carbonate and 2-benzyl-1,1,3,3-tetramethylguanidine (BnTMG) were prepared following literature procedures (ESI†).Experimental setup
 | ||
| Fig. 2 Simplified continuous flow setup for the homogeneous carbonation of glycerol. | ||
Typical runs
| Entry | Catalyst | Conv.b (%) | Selec.b (%) |
|---|---|---|---|
| a Conditions: 135 °C, 2 min residence time, 3 equiv. DMC, 1 mol% organocatalyst. b Conversion and yield were determined by GC/FID. c 2 mol% organocatalyst. | |||
| 1 | DBU | 89 | 79 |
| 2 | Verkade's base | 82 | 84 |
| 3c | DMAN | 9 | 67 |
| 4 | TMG | 68 | 83 |
| 5 | Barton's base | 94 | 87 |
| 6 | BnTMG | 89 | 81 |
| 7 | TMGN | 89 | 83 |
| 8 | P1-H | 88 | 85 |
| 9 | P1-t-Bu | 95 | 82 |
| 10 | P1-t-Oct | 95 | 83 |
| 11c | BTPP | 97 | 84 |
| 12 | BEMP | 93 | 82 |
| 13 | P2-Et | 93 | 80 |
|
|
|||||
|---|---|---|---|---|---|
| Entry | Conditions | Substrate | Product | Conv. (%) | Selec. (%) |
| Conditions A: 135 °C, 2 min residence time, 1 mol% catalyst, 7 bar. Conditions B: 160 °C, 4 min residence time, 2 mol% catalyst, 7 bar. Conditions C: 180 °C, 8 min residence time, 2 mol% catalyst, 11 bar.a Conversion and yield were determined by GC/FID.b Yield, determined by high-field 1H NMR in CDCl3 with mesitylene as the internal standard.c Conversion was determined by GC/FID and yield was determined by high-field 1H NMR in CDCl3 with mesitylene as the internal standard. | |||||
| 1a | A |
 1
1
|
 2
2
|
94 | 87 |
| 2a | A |
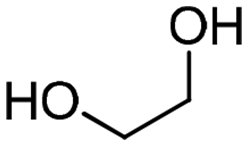 4a
4a
|
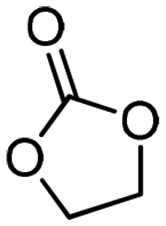 5a
5a
|
82 | 79 |
| 3a | A |
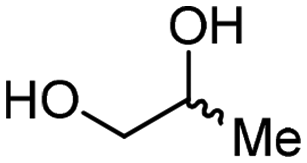 4b
4b
|
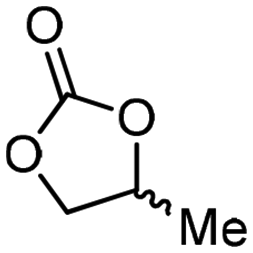 5b
5b
|
94 | 85 |
| 4 | C |
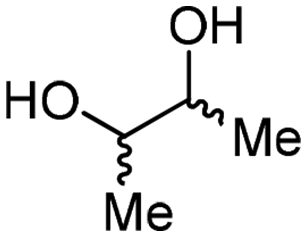 4c
4c
|
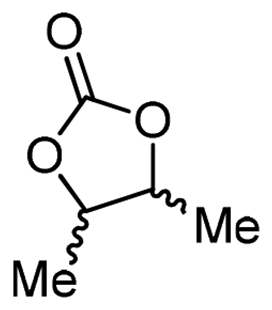 5c
5c
|
72b | |
| 5a | A |
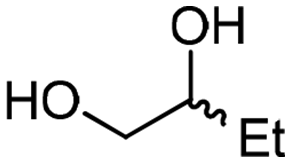 4d
4d
|
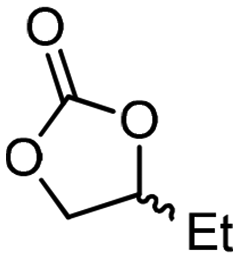 5d
5d
|
96 | 79 |
| 6a | B |
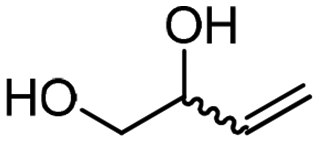 4e
4e
|
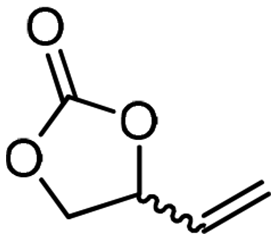 5e
5e
|
81 | 91 |
| 7a | A |
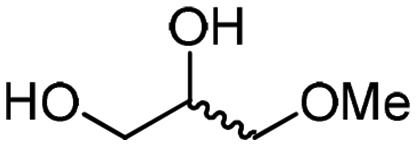 4f
4f
|
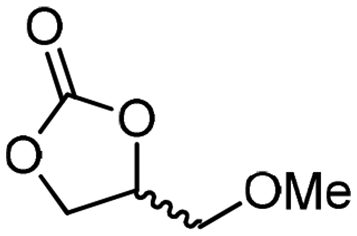 5f
5f
|
84 | 84 |
| 8c | B |
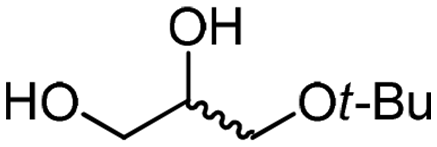 4g
4g
|
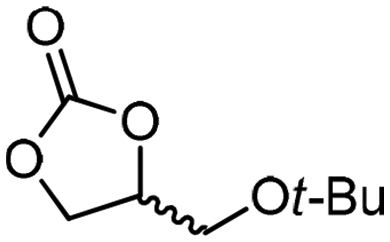 5g
5g
|
63 | 91 |
| 9c | B |
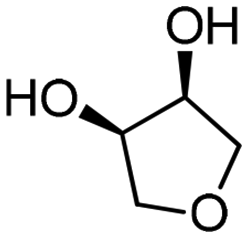 4h
4h
|
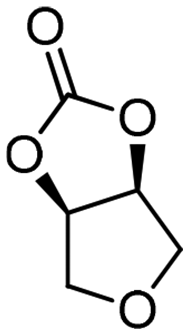 5h
5h
|
96 | 87 |
Results
Process optimization
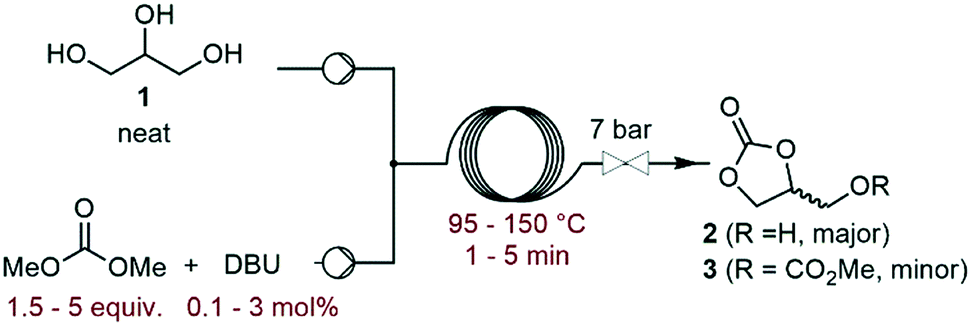
The investigation commenced with a thorough assessment of the parameters influencing the carbonation of neat glycerol (1) with dimethyl carbonate (DMC). DBU, a known organocatalyst for the carbonation of 1,15,17 was selected as the model catalyst. In a representative experiment, neat 1 and the solution of DBU in DMC were mixed through a T-mixer, and the biphasic mixture entered a heated capillary coil reactor, where it rapidly became homogeneous. A BPR set at 7 bar was inserted downstream the reactor to afford process temperatures above the boiling point of some components of the liquid phase (Fig. 2). The effluent of the reactor was analyzed by off-line GC/FID, and glycerol carbonate (2) was detected as the main product along with methyl ((2-oxo-1,3-dioxolan-4-yl)methyl) carbonate (3) as a minor impurity (ESI†).In a preliminary set of experiments, a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 DMC/glycerol molar ratio with 1 mol% of catalyst was reacted within 2 min of residence time, and the temperature was varied from 95 to 150 °C. The selectivity for 2 varied in the 70–79% range, while the conversion increased from 65% to 96% with the temperature (Fig. 3), and the selectivity for 3 increased from 0.7 to 6%. 135 °C was selected as the best temperature for further optimization, as it gave high conversion (89%) and selectivity (79%), while balancing the energy costs.
:1 DMC/glycerol molar ratio with 1 mol% of catalyst was reacted within 2 min of residence time, and the temperature was varied from 95 to 150 °C. The selectivity for 2 varied in the 70–79% range, while the conversion increased from 65% to 96% with the temperature (Fig. 3), and the selectivity for 3 increased from 0.7 to 6%. 135 °C was selected as the best temperature for further optimization, as it gave high conversion (89%) and selectivity (79%), while balancing the energy costs.
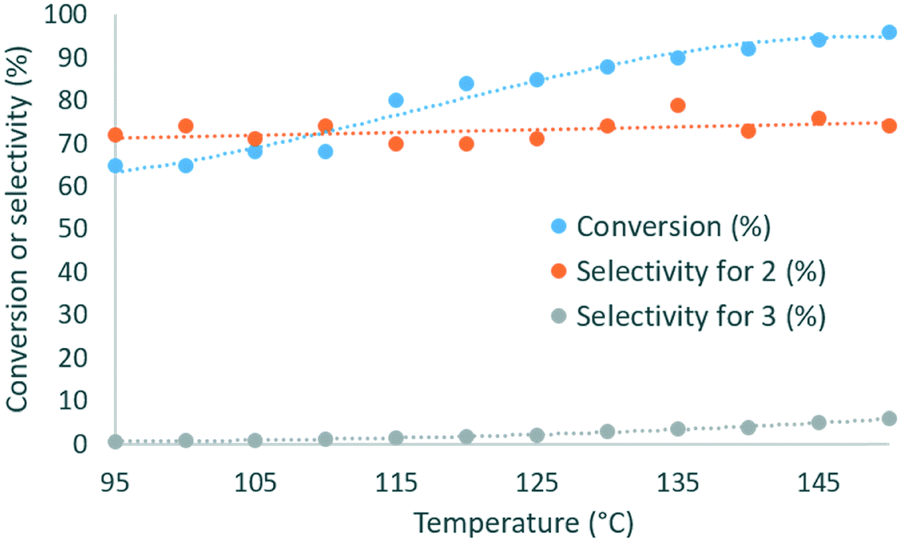 | ||
| Fig. 3 Evolution of the conversion of glycerol (blue dots) and of the selectivity for glycerol carbonate (2) (orange dots) and methyl ((2-oxo-1,3-dioxolan-4-yl)methyl) carbonate (3) (grey dots) as a function of the temperature. Conditions: 2 min residence time, 3 equiv. DMC, 1 mol% DBU. | ||
Next, the influence of the residence time was studied (ESI†). When it was increased from 1 to 2 min, the conversion improved from 78 to 89%. However, a further increase of up to 5 min led to a further increase in conversion (97%), while decreasing the productivity and selectivity for 2 (71–79% range). As the residence time increased, the selectivity for 3 increased from 1.7 to 6.8%. Therefore, the residence time was set at 2 min for further optimization. When the molar ratio between DMC and glycerol increased from 1.5 to 3, the conversion of 1 and selectivity for 2 improved from 80 to 89% and from 72 to 79%, respectively (ESI†). However, increasing the DMC amount up to 5 equiv. did not lead to any further improvement. The selectivity toward 3 followed a similar trend, evolving from 1.5 to 3.8% in the 1.5–3 molar ratio range, and then plateauing at 3.6–4% at a higher DMC excess. Finally, the catalytic loading of DBU was evaluated (Fig. 4).
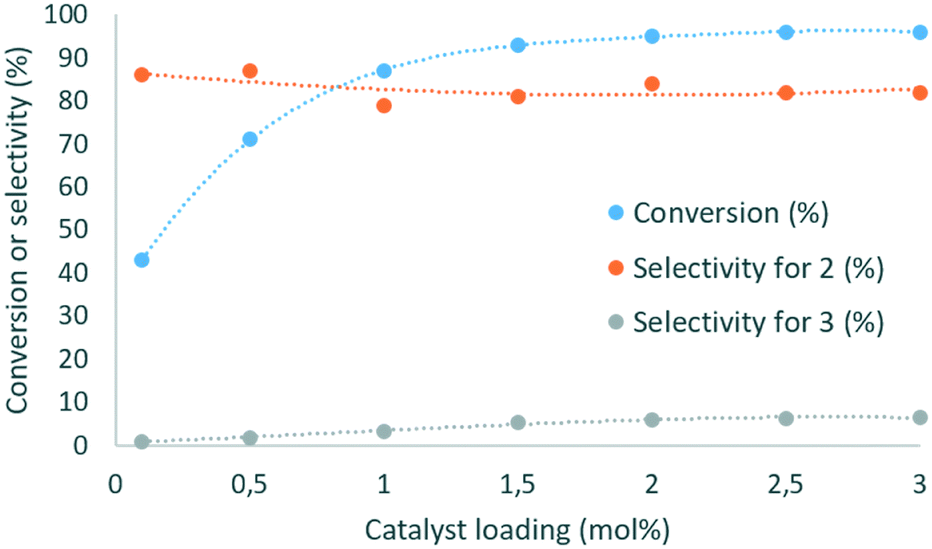 | ||
| Fig. 4 Evolution of the conversion of glycerol (blue dots) and of the selectivity for glycerol carbonate (2) (orange dots) and methyl ((2-oxo-1,3-dioxolan-4-yl)methyl) carbonate (3) (grey dots) as a function of the loading in DBU. Conditions: 135 °C, 2 min residence time, 3 equiv. DMC. | ||
This parameter had a critical impact on both conversion and selectivity. An increase from 0.1 to 1 mol% led to an increase in the conversion from 43 to 89%, and the selectivity for 2 decreased in the 79–87% range. At higher loading, the conversion was slightly improved (96% at 3 mol%), and the selectivity remained in the 79–82% range. The selectivity for 3 reached up to 5.3% as the catalyst loading increased from 0.1 to 1.5 mol%, and increased up to 6.6% with 3 mol% DBU. A similar trend was observed with Barton's base as the organocatalyst (ESI†).
Screening of homogeneous catalysts
With the optimized process conditions under homogeneous continuous flow conditions (135 °C, 2 min, 3 equiv. DMC), we assessed a library of organic superbases as potential catalysts (Fig. 5, Table 2). Catalyst loadings of 0.5, 1, 1.5 and 2 mol% were assessed for the catalysts depicted in Fig. 5 (see also Table 2). A loading of 1 mol% gave the best conversion/selectivity balance (ESI†). The library included proton sponge (DMAN), proazaphosphatrane (Verkade's base), guanidines (TMG, Barton's base, BnTMG and TMGN), and phosphazenes (P1-H, P1-t-Bu, P1-t-Oct, BTPP, BEMP and P2-Et).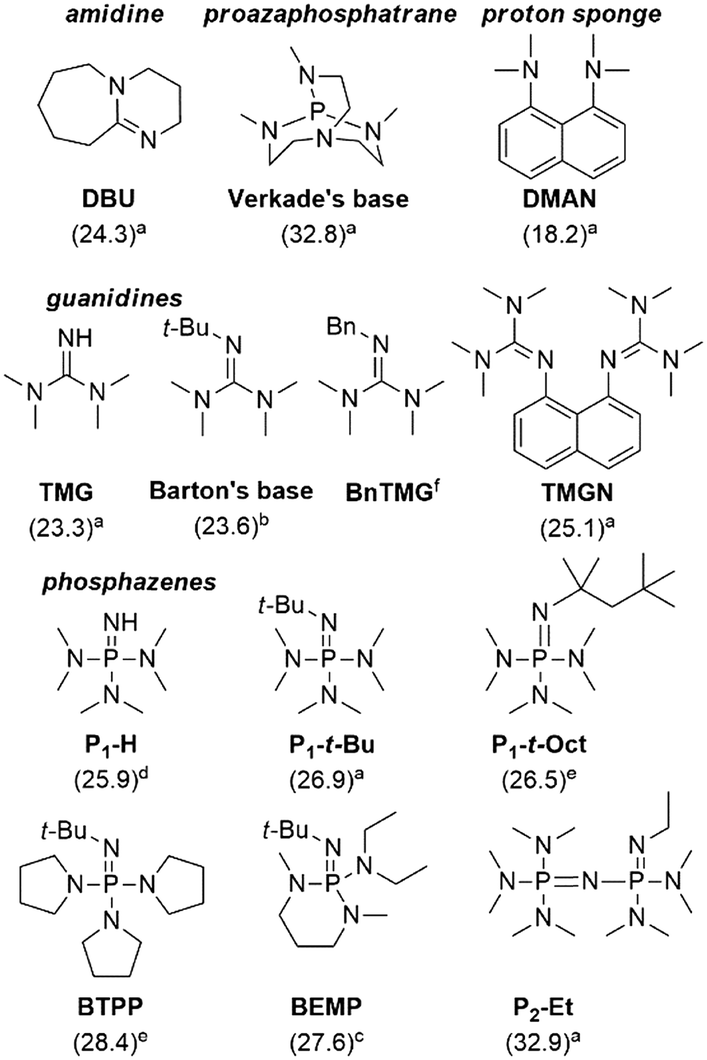 | ||
| Fig. 5 Structure of the organic bases assessed for the carbonation of glycerol. Literature values of the experimental pKBH+ in MeCN are given in parenthesis (a = from ref. 37, b = from ref. 45, c = from ref. 46, d = from reference ref. 47, e = from ref. 48, f = not available). | ||
The proton sponge (DMAN) was the least efficient catalyst, giving a mere 9% glycerol conversion (Table 2, entry 3). Its low pKBH+ value (18.2) accounted for such low conversion. TMG, with the second lowest pKBH+ value (23.3), gave a glycerol conversion of 68% (entry 4), while more basic catalysts gave conversions in the 82–97% range. Similar observations were made by Kelkar et al., who used amines and amidines as organocatalysts for the synthesis of 2 from 1 and DMC: the activity increased with the basicity of the nitrogen base, and amidines were thus better catalysts than amines.15 Similarly, Corma and co-workers assessed P1-t-Bu and P4-t-Bu phosphazenes for the transesterification of vegetable oils with methanol, and reported a direct correlation between the basicity and the activity of the catalyst.38 Our results indicate that in the 23.6–32.9 pKBH+ range, the basicity of the active site is not the only determining factor for activity. For example, Barton's base (pKBH+ = 23.6) is far more active (conv. = 94%) than Verkade's base (pKBH+ = 32.8, conv. = 82%) despite the higher basicity of the latter (entries 2 and 5). Overall, the best results were obtained with Barton's base (entry 5), and sterically hindered phosphazenes such as P1-t-Bu, P1-t-Oct, BTPP, BEMP and P2-Et (entries 9–13), affording glycerol conversions in the 93–97% range and selectivities toward 2 in the 80–87% range. The highest yield (82%) for glycerol carbonate was obtained with Barton's base, and this organocatalyst thus appeared as the most efficient.
Screening of heterogeneous catalysts
To further increase cost-efficiency of the process and ease downstream operation, we envisioned the utilization of supported organocatalysts. Resin-grafted DABCO (1,4-diazabicyclo[2.2.2]octane) and analogs were already reported as efficient heterogeneous catalysts for this reaction.16,18 Here, we assessed the heterogenized version of the best homogeneous catalysts above-mentioned, namely, polystyrene-supported P2-t-Bu (PS-P2-t-Bu) and polystyrene-supported TMG (PS-TMG). Polystyrene-supported DBU (PS-DBU) was utilized as a reference catalyst, as for the homogeneous process. PS-P2-t-Bu and PS-DBU were commercially available, while PS-TMG was prepared from TMG and Merrifield's resin according to a reported procedure (ESI†). In a representative experiment, neat glycerol was mixed with neat DMC through a T-mixer. The biphasic mixture was pre-heated in the first loop, and then reacted through a fixed-bed reactor packed with a dispersion of the PS-organocatalyst in glass beads. The reactor effluents were conveyed toward an on-line IR spectrometer that enabled continuous monitoring of the catalyst's performances over time (Fig. 6b and ESI†). Quantitative data for conversion and yield were obtained from periodic samples of the effluents of the reactor (GC/FID).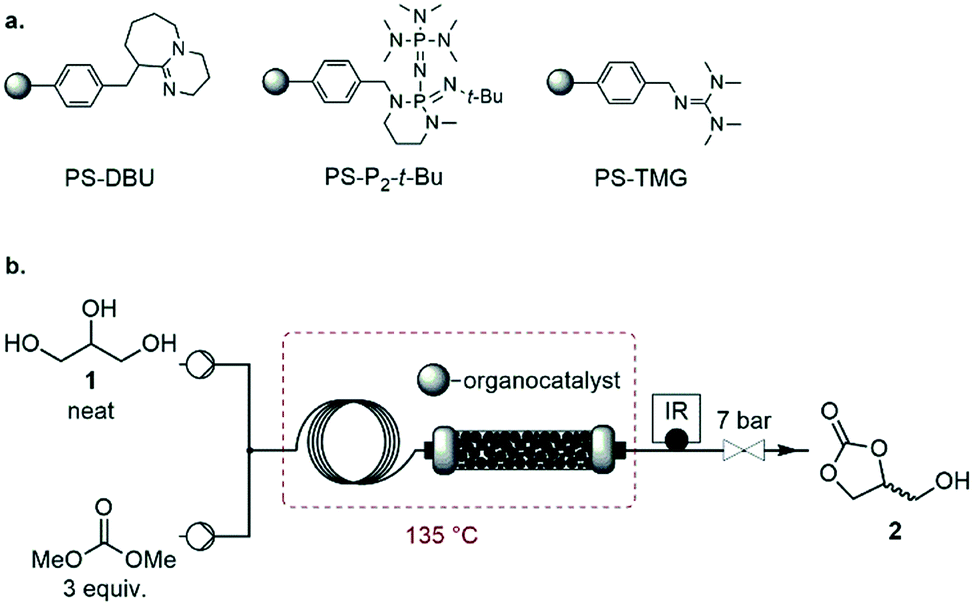 | ||
| Fig. 6 a. Structure of the active sites of the polystyrene-supported organocatalysts assessed for the heterogeneous carbonation of glycerol. b. Simplified continuous flow setup for the heterogeneous carbonation of glycerol. | ||
In the first set of experiments with PS-P2-t-Bu, the flow rate of 1 was varied while keeping the mass of the catalyst and the molar ratio DMC/1 constant. As expected, increasing the weight hourly space velocity (WHSV) from 6.9 to 24.3 h−1 led to a decrease in conversion from 98 to 85%, while the selectivity remained in the 80–86% range (Fig. 7). In addition to its ability to handle solvent-free conditions, PS-P2-t-Bu was also more active than the reported Ambersep® 900 hydroxide resin.30 Indeed, the latter gave 95% conversion in glycerol and 82% selectivity for 2 at 140 °C and 4 equiv. of DMC, using a WHSV of 2.25 h−1.30 However, when the WHSV was raised to 4.60 h−1, the conversion decreased to 84%.30
 | ||
| Fig. 7 Evolution of glycerol conversion (blue dots) and glycerol carbonate selectivity (orange dots) as a function of the WHSV (off-line GC/FID). The WHSV is defined as the mass of glycerol fed per hour and per mass of PS-P2-t-Bu. Conditions: 135 °C, 3 equiv. DMC. | ||
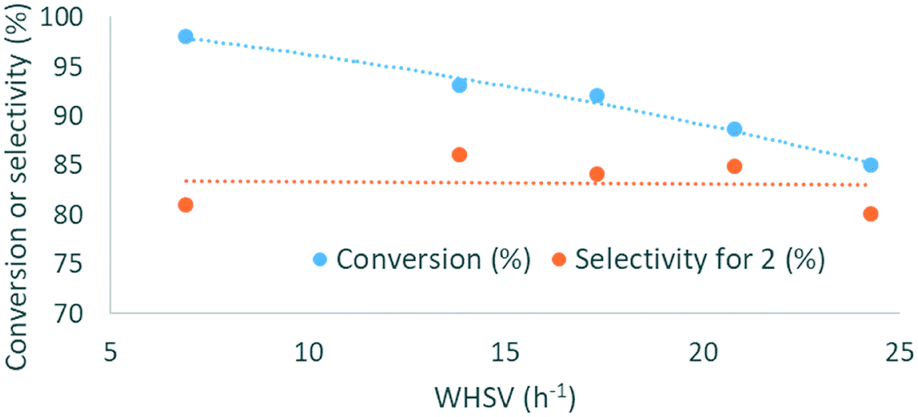
PS-P2-t-Bu, PS-TMG and PS-DBU were then evaluated at a steady WHSV of 20.8 h−1 for prolonged operation times (up to 80 h), to assess their stability. This WHSV was selected since it gave a good balance between conversion and productivity. Using PS-P2-t-Bu, the glycerol conversion decreased from 91% after 3 h of operation to 78% after 33 h and then remained steady for the next 50 h (Fig. 8a). The selectivity was not affected by the operation time, remaining in the 77–84% range. PS-TMG was less active and less stable than its phosphazene counterpart: glycerol conversion decreased from 89% after 1 h of operation to 74% after 25 h (Fig. 8b). The selectivity for 2 remained in the 65–71% range. PS-TMG was also assessed at a lower WHSV (10.5 h−1), and gave steady conversions (97–98%) and selectivities (64–74%) for over 47 h of operation (ESI†). PS-DBU gave a slightly lower selectivity for glycerol carbonate than PS-P2-t-Bu, although its stability over time was quite similar. Glycerol conversion decreased steadily from 93% after 2 h of operation to 78% after 73 h (Fig. 8c). Again, the selectivity for 2 was not affected by the operation time, and varied in the 68–78% range.
 | ||
| Fig. 8 On-line reaction monitoring showing the evolution of the conversion to glycerol carbonate by combining on-line IR qualitative monitoring (orange series, relative intensity of the characteristic vibration band at the 1403 cm−1 band) and off-line quantitative GC/FID (blue dots, conversion of 1). Conditions: 135 °C, 3 equiv. DMC, WHSV = 20.8 h−1, PS-supported organocatalyst = a. PS-P2-t-Bu, b. PS-TMG, c. PS-DBU. | ||
Substrate scope
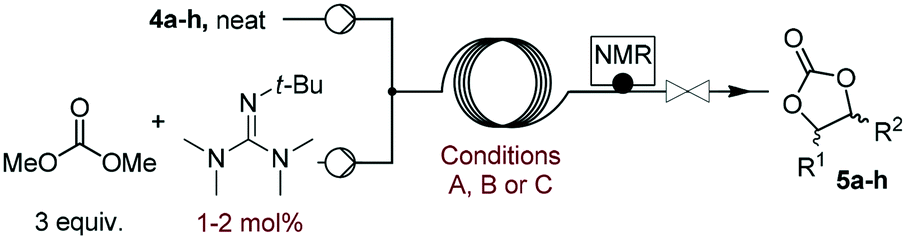
The scope of the methodology was extended to other liquid 1,2-diols (4a–h, Table 3). 1,2-Diols 4a–h notably included bio-based compounds such as ethylene glycol, 1,2-propanediol, 2,3-butanediol, glycerol tert-butyl and methyl ethers, as well as 1,4-anhydroerythritol. The corresponding cyclic carbonates 5a–h are also important chemicals with applications ranging from being additives in batteries and building blocks for the preparation of chemicals and polymers to potential fuel additives.49–54In a representative run, neat diol 4 was mixed with 3 equiv. of DMC and 1 mol% of Barton's base through a static T-mixer, and the mixture was reacted using the optimized operating conditions on glycerol (135 °C, 7 bar, 2 min or residence time) in a coil reactor (conditions A). The diols 4a, b, d, and f with comparable steric and electronic features to 1 underwent transesterification with DMC under conditions A with conversions in the 82–96% range and selectivities toward the corresponding carbonates 5a, b, d, and f in the 79–85% range (entries 2, 3, 5 and 7).
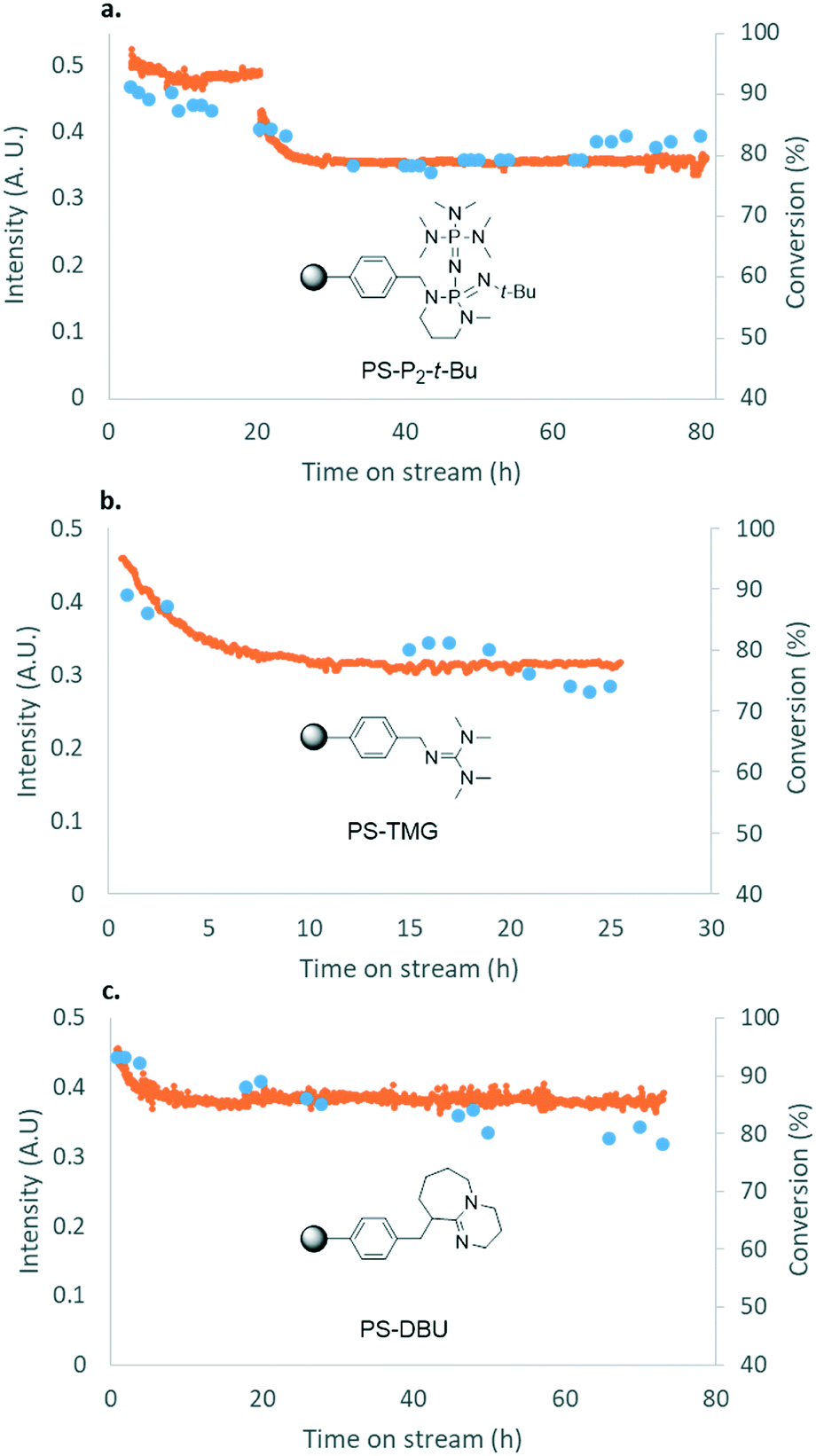
For some diols with higher steric hindrance or specific structural features, low conversions were obtained under conditions A, and reoptimization was required (ESI†). An on-line NMR spectrometer was positioned downstream the reactor for real-time monitoring of the process,55–57 ensuring fast reoptimization (ESI†). Samples were collected downstream, processed and analyzed by GC/FID to obtain quantitative information. Some substrates such as diols 4e, g, and h required 160 °C and a longer residence time (4 min) with 2 mol% of catalyst to reach a conversion in the 63–96% range and selectivity toward the corresponding carbonates 5e, g, and h in the 87–91% range.58 Interestingly, anhydroerythritol carbonate (5h) crystallized upon standing in a fridge at 4 °C overnight from the crude reactor effluent. After filtration and washing with diethyl ether, white needles of 5h were isolated, and their purity was confirmed by high-field NMR (ESI†). Substrate 4c was more demanding and required 8 min of residence time at 180 °C with 2 mol% of Barton's base to afford a 72% yield.
The latter reaction serves as an illustration of on-line low-field NMR optimization. Fig. 9 shows representative spectra acquired during the carbonation of 2,3-butanediol (4c) to 5c. Qualitative assessment of the reaction was carried out by comparing the relative intensities of a characteristic signal of 4c (0.6 ppm, light grey) and of 5c (1 ppm, dark grey).
 | ||
| Fig. 9 On-line NMR reaction monitoring showing the variation of the concentrations in 2,3-butanediol (4c) and 2,3-butylene carbonate (5c). The doublet at 0.6 ppm (light grey) was used to monitor the variation in the concentration of 4c. The doublet at 1 ppm (dark grey) was used to monitor the variation in the concentration of 5c. | ||
Scale-out
Two options were available for reaching the production scales under continuous flow conditions with the abovementioned results, either using a fixed-bed reactor packed with a heterogeneous catalyst or a flow reactor using a homogeneous catalyst. With the best performing heterogeneous catalyst (PS-P2-t-Bu), the long runs (see above) have emphasized that the conversion decreased from ∼90% to 80% for over 30 h and then plateaued at ∼80% for a production campaign of 80 h. With this in mind, plus the cost associated with such a heterogeneous catalyst, the homogenous option is arguably the most appealing option. Indeed, it affords a constant reaction profile and the low catalytic loading can be easily trapped downstream on an acidic heterogeneous material and recycled. The scalability of the homogeneous process was thus evaluated using commercial continuous flow glass reactors (Fig. 10) (Corning® Advanced-Flow™ G1LF and G1 reactors and the best performing homogeneous catalyst, i.e. Barton's base). Based upon the results obtained on the microscale, the best operating conditions were applied first in a mesofluidic reactor of 13 mL total internal volume featuring 5 fluidic modules (2.6 mL internal volume each, Corning® Advanced-Flow™ G1LF) fluidically connected in series and integrated with a heat exchanger. DBU was first selected as a homogeneous catalyst (1 mol%) for direct comparison with the microscale results. Process conditions were directly transposable from the microscale to mesoscale, affording 93% conversion, 74% yield and 79% selectivity toward glycerol carbonate with 2 min of residence time at 135 °C (7 bar). Increasing the residence time to 3 min left the conversion unaffected, although a slight increase in yield (80%) and selectivity (87%) was noticed. | ||
| Fig. 10 Photograph of the Corning® Advanced-Flow™ G1LF for the production of glycerol carbonate. I1: inlet for preheated glycerol; I2: inlet for DMC and Barton's base; H: inlet and outlet for the thermofluid; FM: fluidic module; O: reactor outlet; BPR: back pressure regulator. | ||
Aiming for larger productivity, the 2.6 mL internal volume fluidic modules were changed to pilot-scale 8 mL internal volume fluidic modules, thus giving a flow setup of 40 mL total internal volume (Corning® Advanced-Flow™ G1). The same operating parameters (135 °C, 7 bar, 2 min, 1 mol% DBU, 3 equiv. DMC) afforded 92% conversion, 73% yield and 80% selectivity toward glycerol carbonate, thus giving consistent results without reoptimization. The best operating conditions on the pilot scale were next assessed with Barton's base as the homogeneous catalyst. With 3 min of residence time, 97% conversion was obtained, while with only 2 min of residence time, up to 98% conversion with 80% selectivity was obtained for glycerol carbonate. Under these conditions, a 68.3 mol per day productivity (∼8 kg per day) for glycerol carbonate was obtained with a reactor featuring a L × l × h 90 × 30 × 40 cm footprint (frame included, Fig. 10).
Conclusion
This work illustrates a solvent-free organocatalyzed continuous flow process for the transesterification of dimethyl carbonate with various 1,2-diols. A palette of homogeneous organic superbases such as the proton sponge, DBU, Verkade's base, guanidines and phosphazenes were assessed as catalysts for the transesterification on glycerol as a model substrate under microfluidic conditions. 2-tert-Butyl-1,1,3,3-tetramethylguanidine (Barton's base) was identified as the most efficient organocatalyst, affording glycerol carbonate with 87% selectivity and 94% conversion within 2 minutes of residence time at 1 mol% loading under microfluidic conditions at 135 °C under 7 bar of counter-pressure. These conditions surpass all reported homogeneous procedures so far. Alternatively, supported organic superbases were next evaluated as heterogeneous catalysts under flow conditions. Under heterogeneous conditions, microfluidic operation could be maintained for up to 80 h with elevated flow rates, and the conversion typically dropped from 10–15% through minor catalyst deactivation. Upon optimization, the homogeneous strategy was successfully expanded to a library of other 1,2-diols, including biomass-derived substrates. Depending on the unique features of both substrates and products, either on-line IR or on-line NMR analytical procedures were implemented for real-time qualitative reaction monitoring. The most valuable process from the industrial perspective, i.e. the transesterification toward glycerol carbonate, was next applied under mesofluidic conditions. In less than one day, the microfluidic conditions were successively applied to lab-scale and then finally pilot-scale continuous flow reactors without reoptimization, affording the target cyclic carbonate with a productivity of 68.3 mol per day (∼8 kg per day).Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the University of Liège (Welcome Grant WG-13/03, JCMM). ZW acknowledges the Department of Education (Hubei Province, China) and the Hubei Polytechnic University for their financial support.Notes and references
- US Department of Energy, Top Value Added Chemicals from Biomass Volume I - Results of Screening for Potential Candidates from Sugars and Synthesis Gas, 2004 Search PubMed.
- J. J. Bozell and G. R. Petersen, Green Chem., 2010, 12, 539–554 RSC.
- S. Bagheri, N. M. Julkapli and W. A. Yehye, Renewable Sustainable Energy Rev., 2015, 41, 113–127 CrossRef CAS.
- M. Anitha, S. K. Kamarudin and N. T. Kofli, Chem. Eng. J., 2016, 295, 119–130 CrossRef CAS.
- N. N. Tshibalonza and J.-C. M. Monbaliu, Green Chem., 2017, 19, 3006–3013 RSC.
- R. Gérardy, N. Emmanuel, T. Toupy, V.-E. Kassin, N. N. Tshibalonza, M. Schmitz and J.-C. M. Monbaliu, Eur. J. Org. Chem., 2018, 2301–2351 CrossRef.
- J. R. Ochoa-Gómez, O. Gómez-Jiménez-Aberasturi, C. Ramírez-López and M. Belsué, Org. Process Res. Dev., 2012, 16, 389–399 CrossRef.
- M. O. Sonnati, S. Amigoni, E. P. Taffin de Givenchy, T. Darmanin, O. Choulet and F. Guittard, Green Chem., 2013, 15, 283–306 RSC.
- W. K. Teng, G. C. Ngoh, R. Yusoff and M. K. Aroua, Energy Convers. Manage., 2014, 88, 484–497 CrossRef CAS.
- P. U. Okoye and B. H. Hameed, Renewable Sustainable Energy Rev., 2016, 53, 558–574 CrossRef CAS.
- A. Galadima and O. Muraza, Waste Biomass Valoriz., 2017, 8, 141–152 CrossRef CAS.
- G. M. Lari, G. Pastore, M. Haus, Y. Ding, S. Papadokonstantakis, C. Mondelli and J. Pérez-Ramírez, Energy Environ. Sci., 2018, 11, 1012–1029 RSC.
- G. Fiorani, A. Perosa and M. Selva, Green Chem., 2018, 20, 288–322 RSC.
- J. R. Ochoa-Gómez, O. Gómez-Jiménez-Aberasturi, C. Ramírez-López and B. Maestro-Madurga, Green Chem., 2012, 14, 3368–3376 RSC.
- M. K. Munshi, S. M. Gade, M. V. Mane, D. Mishra, S. Pal, K. Vanka, V. H. Rane and A. A. Kelkar, J. Mol. Catal. A: Chem., 2014, 391, 144–149 CrossRef CAS.
- F. S. H. Simanjuntak, J. S. Choi, G. Lee, H. J. Lee, S. D. Lee, M. Cheong, H. S. Kim and H. Lee, Appl. Catal., B, 2015, 165, 642–650 CrossRef CAS.
- D. O. Nogueira, S. P. De Souza, R. A. C. Leão, L. S. M. Miranda and R. O. M. A. De Souza, RSC Adv., 2015, 5, 20945–20950 RSC.
- Y. Wan, Y. Lei, G. Lan, D. Liu, G. Li and R. Bai, Appl. Catal., A, 2018, 562, 267–275 CrossRef CAS.
- P. U. Naik, L. Petitjean, K. Refes, M. Picquet and L. Plasseraud, Adv. Synth. Catal., 2009, 351, 1753–1756 CrossRef CAS.
- B. Hervert, P. D. McCarthy and H. Palencia, Tetrahedron Lett., 2014, 55, 133–136 CrossRef CAS.
- J. A. Stewart, R. Drexel, B. Arstad, E. Reubsaet, B. M. Weckhuysen and P. C. A. Bruijnincx, Green Chem., 2016, 18, 1605–1618 RSC.
- A. Axelsson, A. Antoine-Michard and H. Sundén, Green Chem., 2017, 19, 2477–2481 RSC.
- R. A. Grey, US Pat., 5091543, 1992 Search PubMed.
- S. M. Gade, M. K. Munshi, B. M. Chherawalla, V. H. Rane and A. A. Kelkar, Catal. Commun., 2012, 27, 184–188 CrossRef CAS.
- C. Chiappe and S. Rajamani, Pure Appl. Chem., 2012, 84, 755–762 CAS.
- M. K. Munshi, P. S. Biradar, S. M. Gade, V. H. Rane and A. A. Kelkar, RSC Adv., 2014, 4, 17124–17128 RSC.
- M. K. Munshi, S. M. Gade, V. H. Rane and A. A. Kelkar, RSC Adv., 2014, 4, 32127–32133 RSC.
- Y. Zhou, F. Ouyang, Z.-B. Song, Z. Yang and D.-J. Tao, Catal. Commun., 2015, 66, 25–29 CrossRef CAS.
- Z. I. Ishak, N. A. Sairi, Y. Alias, M. K. T. Aroua and R. Yusoff, Chem. Eng. J., 2016, 297, 128–138 CrossRef CAS.
- S. Van Mileghem, W. M. De Borggraeve and I. R. Baxendale, Chem. Eng. Technol., 2018, 41, 2014–2023 CrossRef CAS.
- M. Tudorache, L. Protesescu, S. Coman and V. I. Parvulescu, Green Chem., 2012, 14, 478–482 RSC.
- H. Jung, Y. Lee, D. Kim, S. O. Han, S. W. Kim, J. Lee, Y. H. Kim and C. Park, Enzyme Microb. Technol., 2012, 51, 143–147 CrossRef CAS PubMed.
- K. Lanjekar and V. K. Rathod, J. Environ. Chem. Eng., 2013, 1, 1231–1236 CrossRef CAS.
- K. A. Cushing and S. W. Peretti, RSC Adv., 2013, 3, 18596–18604 RSC.
- R. A. C. Leão, S. P. De Souza, D. O. Nogueira, G. M. A. Silva, M. V. M. Silva, M. L. E. Gutarra, L. S. M. Miranda, A. M. Castro, I. I. Junior and R. O. M. A. De Souza, Catal. Sci. Technol., 2016, 6, 4743–4748 RSC.
- Y. Du, J. Gao, W. Kong, L. Zhou, L. Ma, Y. He, Z. Huang and Y. Jiang, ACS Omega, 2018, 3, 6642–6650 CrossRef CAS PubMed.
- Superbases for Organic Synthesis: Guanidines, Amidines, Phosphazenes and Related Organocatalysts, ed. T. Ishikawa, John Wiley & Sons, Ltd, Chichester, UK, 2009 Search PubMed.
- M. Cerro-Alarcón, A. Corma, S. Iborra and J. P. Gómez, Appl. Catal., A, 2008, 346, 52–57 CrossRef.
- R. L. Hartman, J. P. Mcmullen and K. F. Jensen, Angew. Chem., Int. Ed., 2011, 50, 7502–7519 CrossRef CAS PubMed.
- M. B. Plutschack, B. Pieber, K. Gilmore and P. H. Seeberger, Chem. Rev., 2017, 117, 11796–11893 CrossRef CAS PubMed.
- M. G. Álvarez, M. Plíšková, A. M. Segarra, F. Medina and F. Figueras, Appl. Catal., B, 2012, 113–114, 212–220 CrossRef.
- S. Guidi, R. Calmanti, M. Noè, A. Perosa and M. Selva, ACS Sustainable Chem. Eng., 2016, 4, 6144–6151 CrossRef CAS.
- S. E. Kondawar, R. B. Mane, A. Vasishta, S. B. More, S. D. Dhengale and C. V. Rode, Appl. Petrochem. Res., 2017, 7, 41–53 CrossRef CAS.
- G. M. Lari, A. B. L. de Moura, L. Weimann, S. Mitchell, C. Mondelli and J. Pérez-Ramírez, J. Mater. Chem. A, 2017, 5, 16200–16211 RSC.
- M. Stańczyk-Dunaj and A. Jarczewski, Pol. J. Chem., 2005, 6, 1025–1032 Search PubMed.
- I. Novak, X. Wei and W. S. Chin, J. Phys. Chem. A, 2001, 105, 1783–1788 CrossRef CAS.
- Z. Glasovac, M. Eckert-Maksić and Z. B. Maksić, New J. Chem., 2009, 33, 588–597 RSC.
- R. Schwesinger, J. Willaredt, H. Schlemper, M. Keller, D. Schmitt and H. Fritz, Chem. Ber., 1994, 127, 2435–2454 CrossRef CAS.
- J. H. Clements, Ind. Eng. Chem. Res., 2003, 42, 663–674 CrossRef CAS.
- B. Schäffner, F. Schäffner, S. P. Verevkin and A. Börner, Chem. Rev., 2010, 110, 4554–4581 CrossRef PubMed.
- H. Blattmann, M. Fleischer, M. Bähr and R. Mülhaupt, Macromol. Rapid Commun., 2014, 35, 1238–1254 CrossRef CAS PubMed.
- C. Martín, G. Fiorani and A. W. Kleij, ACS Catal., 2015, 5, 1353–1370 CrossRef.
- G. Rokicki, P. G. Parzuchowski and M. Mazurek, Polym. Adv. Technol., 2015, 26, 707–761 CrossRef CAS.
- H. Büttner, L. Longwitz, J. Steinbauer, C. Wulf and T. Werner, Top. Curr. Chem., 2017, 375, 50 CrossRef PubMed.
- P. Giraudeau and F.-X. Felpin, React. Chem. Eng., 2018, 3, 399–413 RSC.
- N. Emmanuel, C. Mendoza, M. Winter, C. R. Horn, A. Vizza, L. Dreesen, B. Heinrichs and J.-C. M. Monbaliu, Org. Process Res. Dev., 2017, 21, 1435–1438 CrossRef CAS.
- R. Gérardy, M. Winter, C. R. Horn, A. Vizza, K. Van Hecke and J.-C. M. Monbaliu, Org. Process Res. Dev., 2017, 21, 2012–2017 CrossRef.
- N. N. Tshibalonza, R. Gérardy, Z. Alsafra, G. Eppe and J.-C. M. Monbaliu, Green Chem., 2018, 20, 5147–5157 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Details of the fluidic reactors, components and experimental and analytical procedures. See DOI: 10.1039/c8re00209f |
| ‡ Current address: Hubei Key laboratory of Mine Environmental Pollution Control and Remediation, School of Chemistry and Chemical Engineering, Hubei Polytechnic University, Huangshi, 435003, Hubei, China. |
| This journal is © The Royal Society of Chemistry 2019 |