
A two-step telescoped continuous flow switchable process leading to nitriles, diaziridine or hydrazine derivatives†
Audun
Drageset
,
Nils Åge
Frøystein
,
Karl Wilhelm
Törnroos
 and
Hans-René
Bjørsvik
*
and
Hans-René
Bjørsvik
*
Department of Chemistry, University of Bergen, Allégaten 41, N-5007 Bergen, Norway. E-mail: hans.bjorsvik@kj.uib.no; Web: http://folk.uib.no/nkjhn/index.html Tel: +47 55 58 34 52
First published on 1st November 2018
Abstract
Primary and benzylic alcohols were cost-effectively transformed into their corresponding nitriles using a classical batch approach and a continuous flow process implemented on a multi-jet oscillating disk (MJOD) reactor platform. The alcohols as substrates were treated with a (2,2,6,6-tetra-methylpiperidin-1-yl)oxidanyl (TEMPO) free radical as the pre-catalyst with 1,3-dichloro-5,5-dimethylhydantoin (DCH) as the terminal oxidant to produce their corresponding carbonyl compounds. The reaction was conducted at a reaction temperature of 35 °C and a flow reactor residence time of 5 min. This alcohol to carbonyl oxidation step was telescoped with a subsequent step that involved treatment with aqueous ammonium hydroxide and 1,3-diiodo-5,5-dimethylhydantoin (DIH) at a reaction temperature of 65 °C with concomitant oxidation to nitrile using a flow reactor residence time of 15 min. A solvent exchange process was conducted in-between the two synthetic reaction steps by means of an in-line extraction process with ethyl acetate, a step that was concatenated by using an in-line liquid–liquid separation process using a hold-up tank. The second synthetic step was revealed to be tuneable, since four distinct products might be produced at varying degrees of selectivity. When DIH was used as the terminal oxidant, a high selectivity towards the original target nitrile was achieved, but if DIH was replaced with DCH as the terminal oxidant, 1,2-di((E)-benzylidene)hydrazine and two different stereoisomers of the 1,3,5-triazabicyclo[3.1.0]hexane scaffold were produced. The selectivity towards the various products was highly influenced by the reaction temperature. A scope and limitation study of the nitrile process with an assortment of primary alcohols as substrates provided excellent yields which revealed good functional group tolerance.
Introduction
Nitriles are essential intermediates for the synthesis of numerous important products in industry and society comprising pharmaceuticals, agrochemicals, advanced materials, and an assortment of other fine and commodity chemicals.1 Various synthetic methods have been developed for the production of compounds bearing the nitrile functionality,2 including amine oxidation,3 dehydration of amides and aldoximes,4 and ipso-substitution of halides with the cyano group.5 However, several of these methods are saddled with various drawbacks including harsh reaction conditions, toxic reagents, and production of side (waste) streams that result in unfavourable green metrics.6 Developments during the last few years have however afforded methods that permit the production of nitriles from cheap starting materials such as alcohols and aldehydes by means of processes that demand somewhat mild and environmentally benign processing conditions.7 Some of these caught our interest especially7a,c due to the use of TEMPO [(2,2,6,6-tetra-methylpiperidin-1-yl)oxidanyl free radical] as the pre-catalyst with DIH (1,3-diiodo-5,5-dimethylhydantoin) as the terminal oxidant. We have previously used TEMPO as the pre-catalyst for the oxidation of alcohols to their corresponding carbonyl compounds,8 and we have developed methods for the production of DIH in a highly efficient continuous flow process,9 in which ultimately DIH was used as the iodination reagent for a variety of N-heterocyclic compounds, in particular, for imidazole.10 In this context, we have also assessed its analogous reagents 1,3-dichloro-5,5-dimethylhydantoin (DCH) and 1,3-dibromo-5,5-dimethylhydantoin (DBH), respectively, as chlorination and bromination reagents. This spurred us to investigate TEMPO with DCH11 as the terminal oxidant in a Montanari-type oxidation process.12 From a process point of view, this was of huge interest as DCH emerges as a significantly cheaper reagent than DIH.13 Adaption and implementation of such procedures as continuous flow processes might additionally enhance the overall process efficiency, throughput, and economy.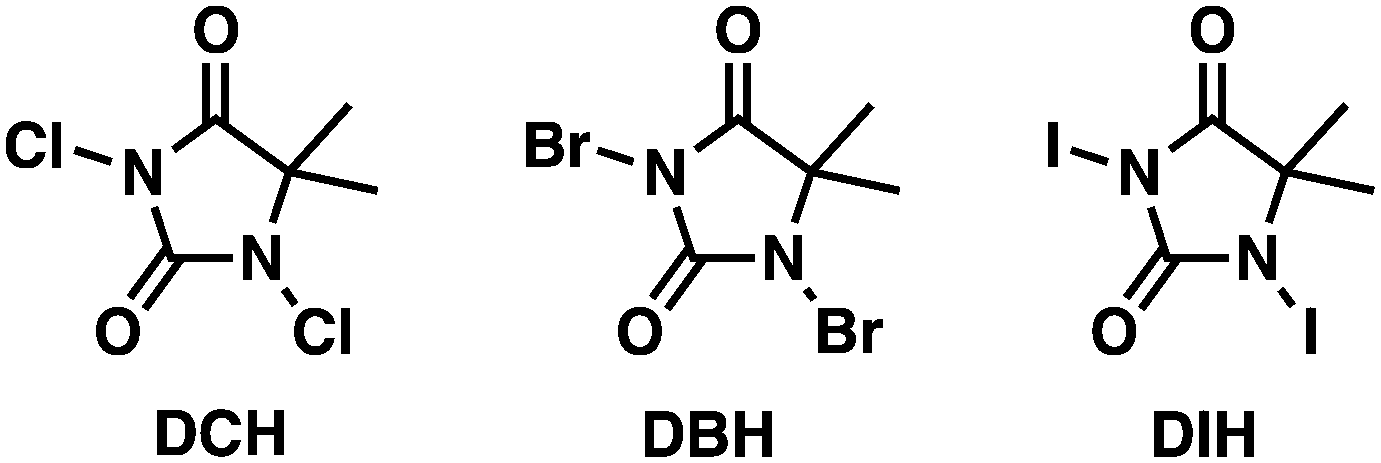
We have previously experienced that batch protocols, in general, can smoothly be transformed into continuous flow processes by means of our MJOD continuous flow reactor platform.14 The resulting processes demand usually for a substantial shorter processing time (reactor residence time), which of course is also advantageous for an enhanced mass throughput and will thus contribute to an overall improvement of the process economy.
Results and discussion
A modified Montanari-type oxidation of alcohols using TEMPO as the pre-catalyst and DCH as the terminal oxidant was developed with high yield (Scheme 1). Acetonitrile was used as solvent due to its high ratio moxidant × (Vsolvent)−1 [g mL−1]. Further investigations were conducted under continuous flow conditions using our multi-jet oscillating disk (MJOD) continuous flow reactor platform previously designed, developed, and described by our group.15 A brief description of the MJOD flow reaction rig used in this study is included in the ESI.†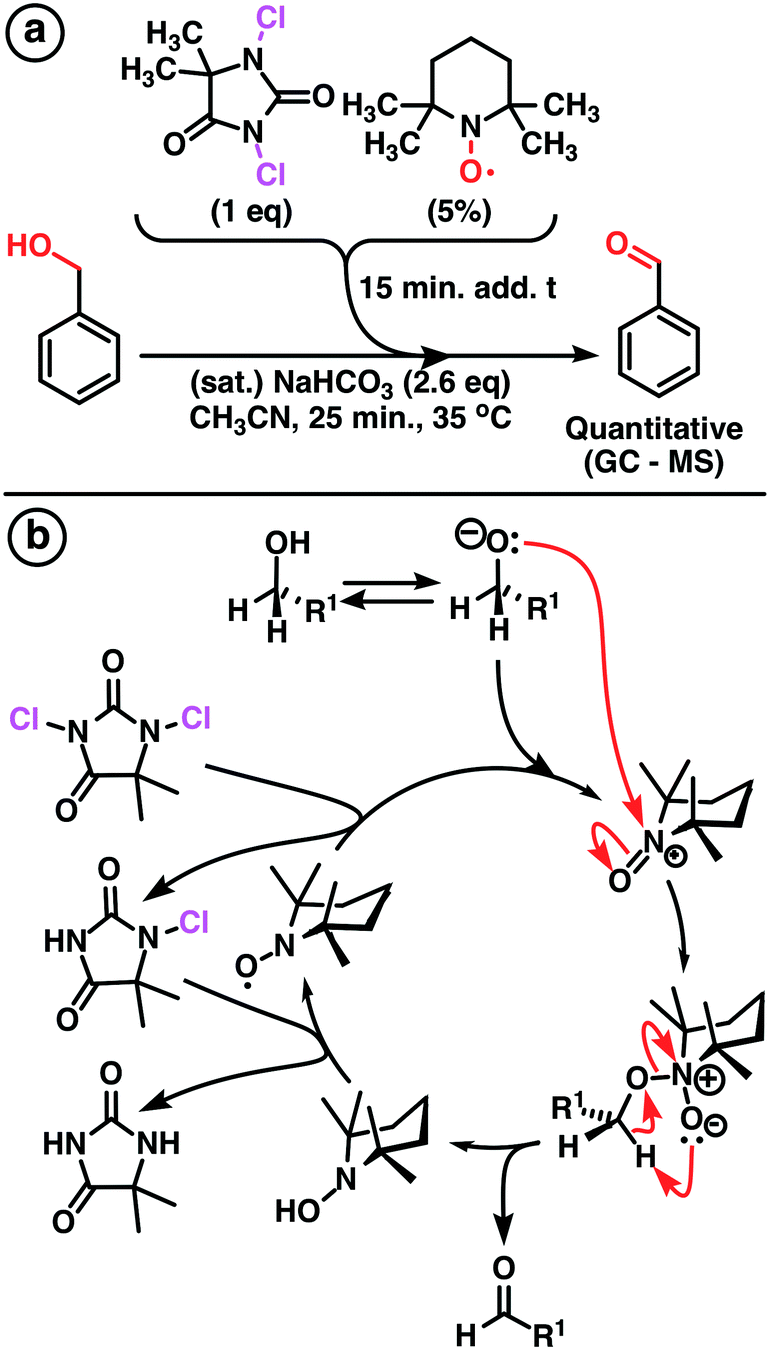 | ||
| Scheme 1 a) Oxidation of benzyl alcohol, a modified Montanari-type oxidation process utilizing TEMPO as the pre-catalyst and DCH as the terminal oxidant. b) Reaction mechanism-modified Montanari-type oxidation using 1,3-dichloro-5,5-dimethylhydantoin as the terminal oxidant. | ||
The oxidation was carried out at a significantly increased rate using a reactor residence time of 5 min producing target aldehydes with high throughput (19.2 g h−1) and excellent yield (95%).
Togo and collaborators16 reported a method that utilized mild conditions for the oxidative conversion of aldehydes to nitriles using ammonium hydroxide together with an oxidant (I2 or DIH). DIH is an expensive research reagent compared with its corresponding bromo and chloro alternatives (DCH and DBH). We have previously developed a continuous flow process for multi-kilogram laboratory scale production of DIH9 making the reagent more accessible, but we wanted to investigate whether the two other 1,3-dihalo-5,5-dimethylhydantoin reagents could be used. Screening trials were conducted using DCH, DBH, or DIH, respectively (Scheme 2).
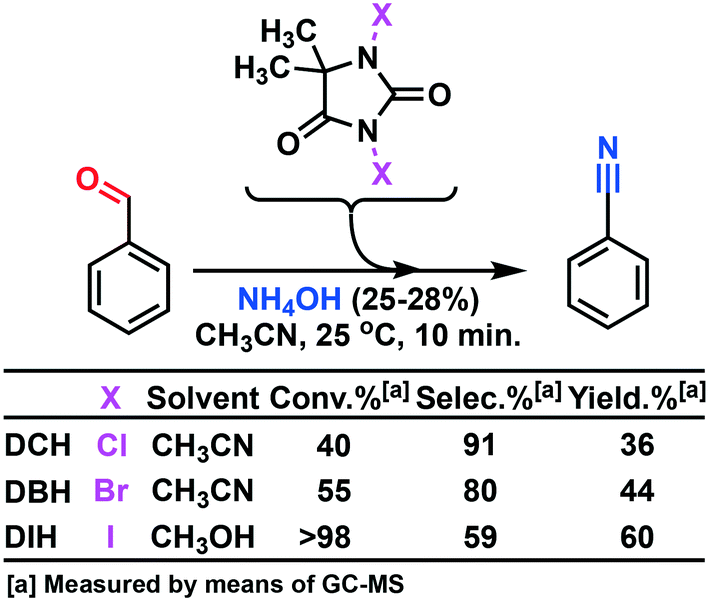 | ||
| Scheme 2 Screening experiment using the three distinct DXH (X = Cl, Br, I) oxidants for the oxidative conversion of aldehydes to nitriles in the presence of ammonium hydroxide under flow conditions (see Fig. 1 for the flow reactor set-up). | ||
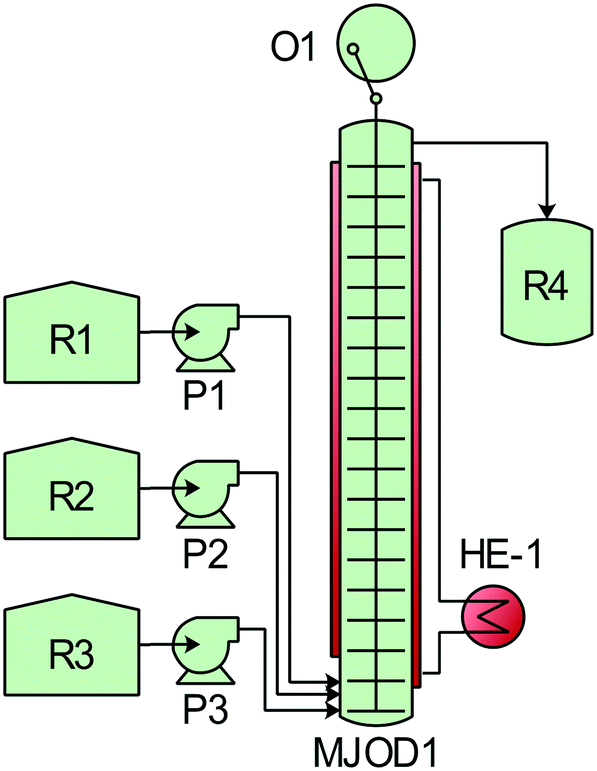 | ||
| Fig. 1 Multi-jet oscillating disk (MJOD) reactor set-up for the modified Montanari oxidation of alcohols. The MJOD1 continuous flow reactor was preheated at 35 °C using heat exchanger HE-1. The internal shaft was oscillated at a frequency of f = 1.5 Hz using cam engine O1. The alcohol was placed in reservoir R1. DCH and TEMPO were dissolved in acetonitrile and transferred to R2. A saturated solution of sodium bicarbonate was placed in reservoir R3. The reagents were pumped from the various reservoirs into the MJOD1 continuous flow reactor using their corresponding pumps P1–P3 with preprogramed flow rates resulting in a total residence time of 5 min. The post reaction mixture was collected in reservoir R4. | ||
The screening experiments, revealed a superior reactivity for DIH, but with a low selectivity. DCH afforded low conversion, but at a high selectivity. Based on cost aspects, DCH was our first choice as the oxidant for the second step of the outlined process. Utilizing the identical oxidant in both steps would decrease the amount of preparation, and DCH is a cheap reagent compared to DIH. Further investigation of the oxidation using DCH as the terminal oxidant revealed a series of competing reactions. In this context, an extended reaction time and an increased reaction temperature gave rise to three various side products, as shown in Scheme 3 and Fig. 2.
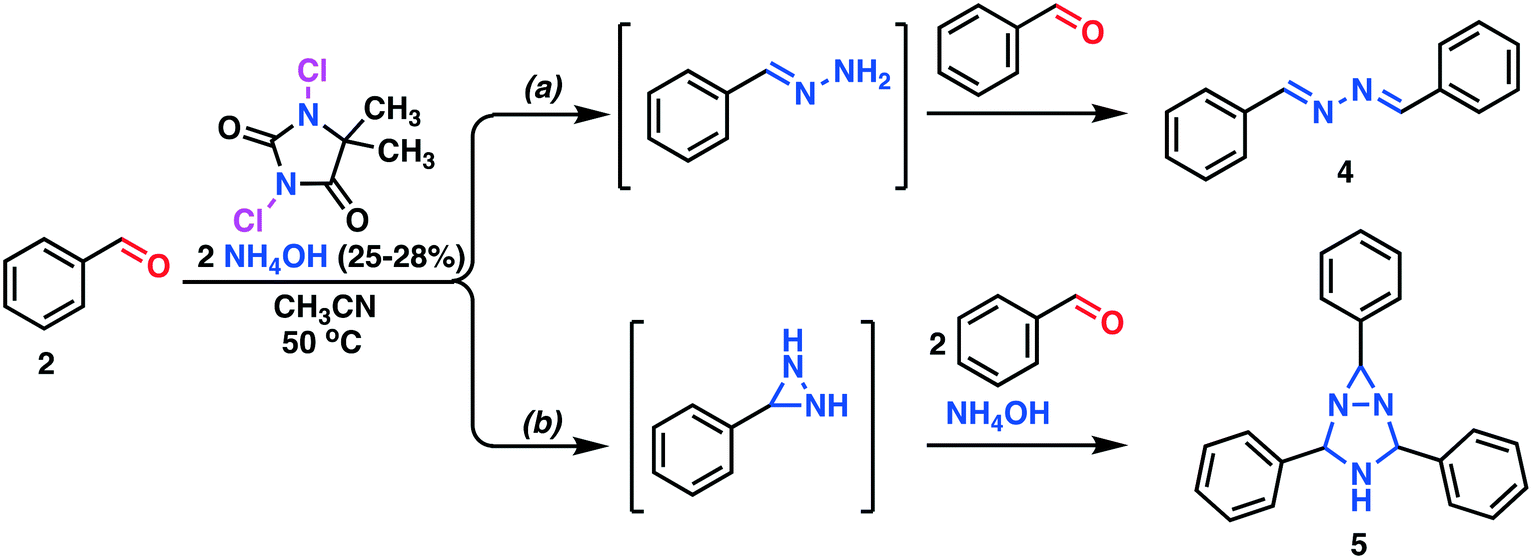 | ||
| Scheme 3 Competing side reactions. a) The Bayer ketazine process17 leading to the formation of 1,2-di((E)-benzylidine)hydrazine. b) The Schmitz diaziridine reaction18 leading to the formation of 2,4,6-triphenyl-1,3,5-triazabicyclo[3.1.0]hexane 5. | ||
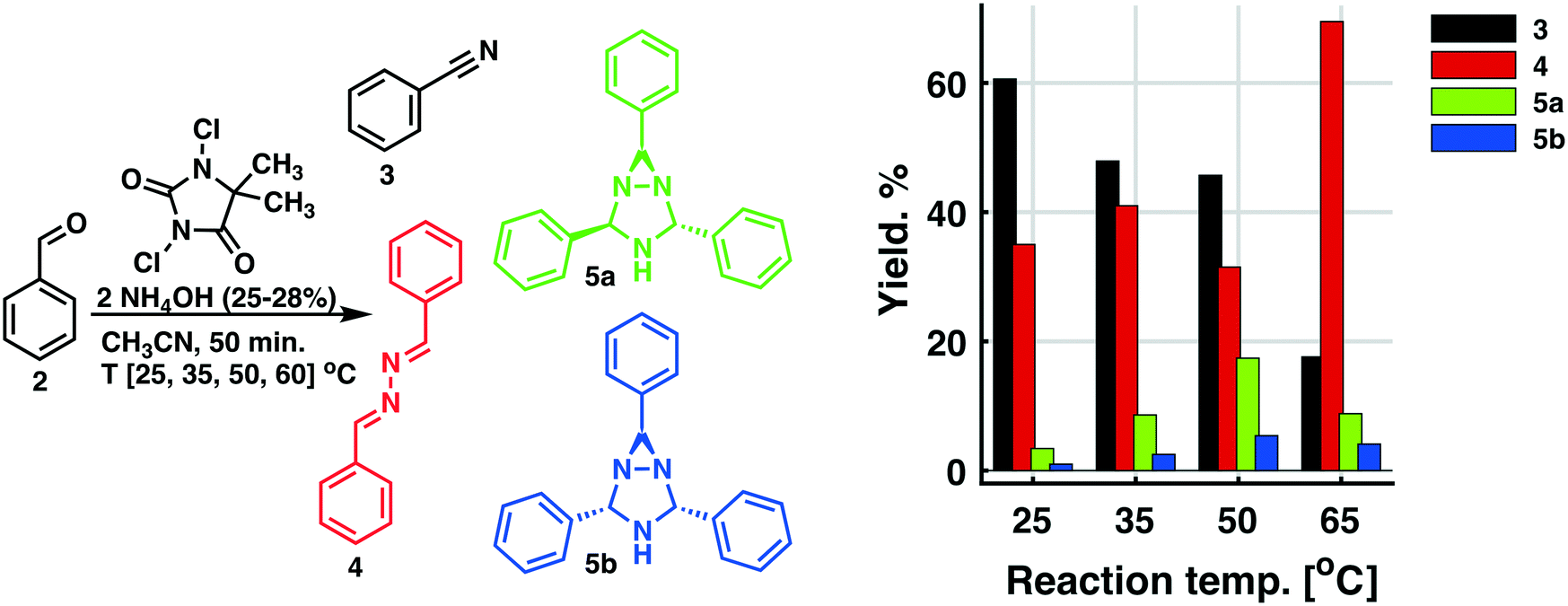 | ||
| Fig. 2 Reaction conducted in a glass reactor (glass flask). When aldehyde 2 is oxidized with DCH as the terminal oxidant with an excess amount of NH4OH present, three distinct side products 4, 5a, and 5b are formed. The reaction temperature has large influence on the course of the reaction. | ||
The three side products were isolated from the post-reaction mixture by crystallization. Compound 5 crystallized as white needles during the reaction and could be isolated by filtration of the post-reaction mixture. The white needle crystals of 5 was recrystallized and investigated by means of NMR and HRMS. The HRMS analysis revealed a molecule with composition C21H19N3. Its basic structure was determined using 1D 1H and 13C NMR spectra together with information from 2D 1H–13C HSQC, 1H–13C HMBC,19 and 1H–1H NOESY spectra. A detailed study of proton splitting patterns and Karplus-type relationships between vicinal (3JHH) coupling constants and dihedral angles together with the interpretation of NOESY cross peaks in terms of interproton distances revealed the structural conformation of 5a (Fig. 2). The structure elucidated by NMR was later confirmed with X-ray crystallography (Fig. 3). The crude crystals isolated from the post-reaction mixture were analyzed by NMR and the spectral data matched the literature spectra.20 Compound 4 co-crystallized with 5,5-dimethylhydantoin as yellow crystals. The structure of compound 4 was confirmed by means of X-ray crystallography, see Fig. 3.
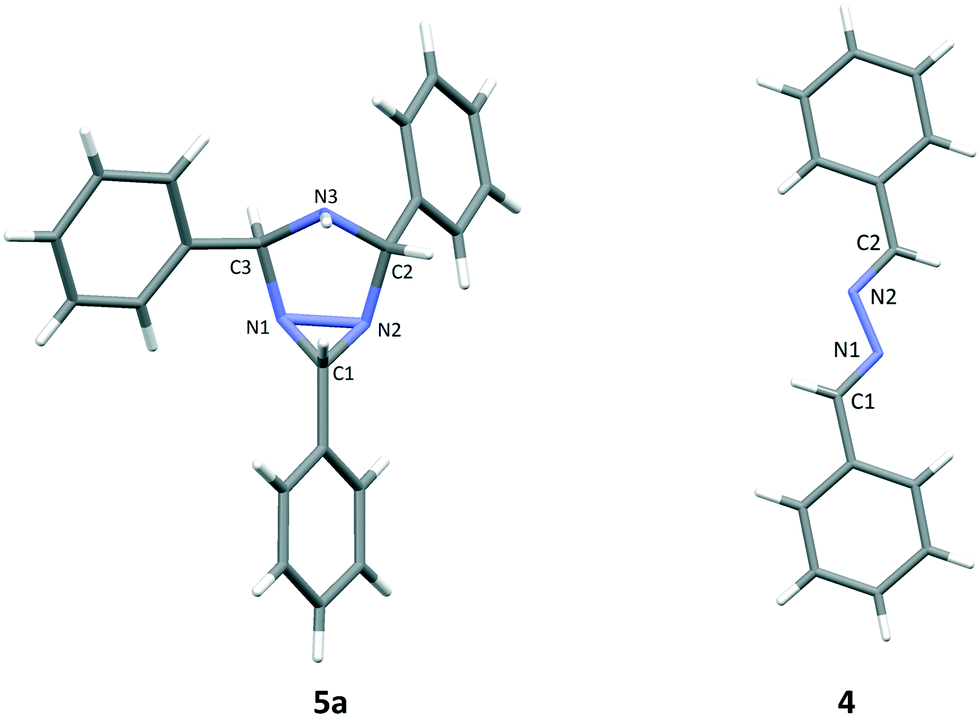 | ||
| Fig. 3 X-ray structure of compounds 5a and 4. Color coding: C, dark gray; N, blue; H, white. Selected geometrical parameters for compound 5a: bond lengths: C1–N1 1.515 Å, N1–N2 1.494 Å, C1–N2 1.437 Å, N2–C2 1.504 Å, C2–N3 1.447 Å. Bond angles: ∠N1–C1–N2 60.7, ∠C1–N1–N2 57.0. Torsion angle: ∠C1–N2–C2–N3 −43.9. Selected geometrical parameters for compound 4: bond lengths: C1–N1 1.282 Å, N1–N2 1.411 Å, N2–C2 1.282 Å. Bond angles: ∠C1–N1–N2 111.3, ∠N1–N2–C2 111.3. Torsion angle: ∠C1–N1–N2–C2 179.9. | ||
A concomitant low reaction temperature and prolonged reaction time favored the formation of target nitrile 3. However, the low flow rate of the two-phase systems was proven to be difficult to operate in a stable manner.
Therefore, we abandoned the use of DCH as the terminal oxidant in the second reaction step. Oxidation with DIH, Scheme 4, was optimized and performed in a continuous flow process. The model substance benzaldehyde 2 was smoothly converted into the target product benzonitrile 3 with high yield (90%) and a throughput of 5.1 g h−1.
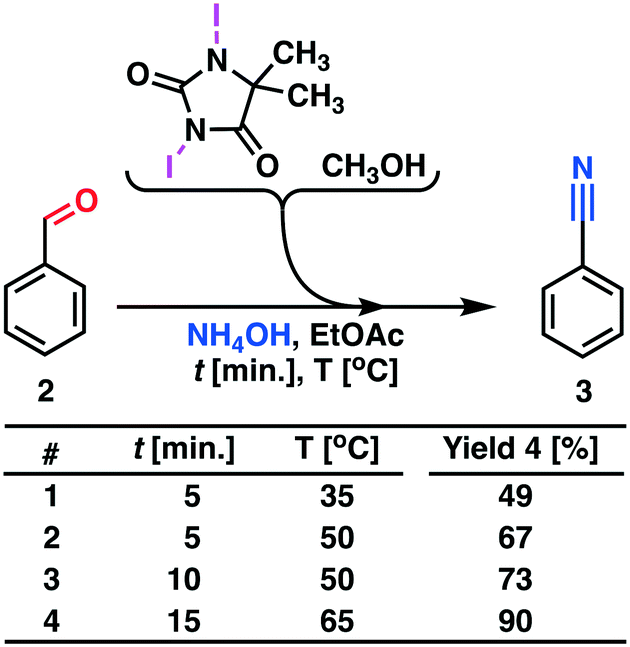 | ||
| Scheme 4 Process development for the oxidative conversion of aldehydes to nitriles. Process description: the reactor set-up shown in Fig. 4 was utilized. The MJOD1 reactor was heated to 65 °C using heat exchanger HE-1 and the internal shaft was oscillated at a frequency of f = 2 Hz using a cam engine driven by electric motor O1. Benzaldehyde 2 dissolved in ethyl acetate was transferred to reservoir R1 and DIH was dissolved in methanol and transferred to reservoir R2. A solution of ammonium hydroxide was transferred to reservoir R3. The reagent reservoirs R1–R3 were connected to their corresponding pumps P1–P3 and injected into the MJOD1 continuous flow reactor at preprogramed flow rates giving a total residence time of 15 min. The crude reaction product was collected in R4 and analyzed by GC-MS. | ||
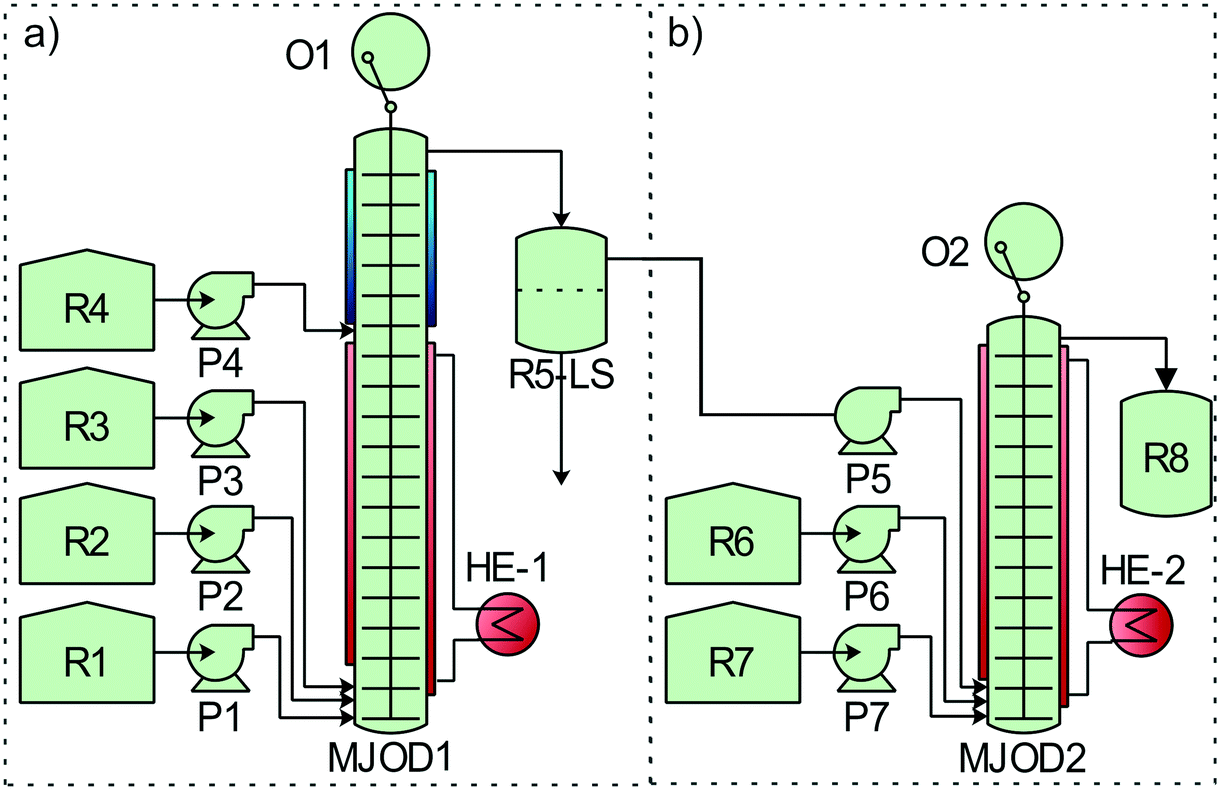 | ||
| Fig. 4 Process flow diagram of the telescoped continuous flow process for the production of nitriles. a) The MJOD1 reactor was heated at 35 °C using heat exchanger HE-1. The internal shaft was oscillated at a frequency of f = 1.5 Hz using a cam mechanism that was driven by electric motor O1. The substrate (the alcohol) was dissolved in acetonitrile and transferred to reservoir R1. The oxidant DCH and the free radical pre-catalyst TEMPO were dissolved in acetonitrile and transferred to reservoir R2. Reservoir R3 was charged with a saturated aqueous solution of sodium bicarbonate. The reagent reservoirs R1–R3 were connected to their corresponding pumps P1–P3 that supplied the reagents to the MJOD1 continuous flow reactor at adjusted flow rates that afforded a reactor residence time of 5 min. At a certain distance from the bottom of the MJOD1 flow reactor, the reaction mixture meets a stream of the extracting solvent (ethyl acetate, reservoir R4) fed by pump P4. This in-line ethyl acetate extraction was performed at a flow rate giving an extraction time of 1.5 min. The mixture of the post-reaction mixture with the extracting solvent was collected in the liquid–liquid separator R5-LS where the separated aquatic phase was discarded. b) The MJOD2 reactor was pre-heated to 65 °C using heat exchanger HE-2 and the internal shaft of MJOD2 was oscillated at a frequency of f = 2 Hz using a cam mechanism driven by electric motor O2. DIH in methanol was added to reservoir R6 and reservoir R7 was filled with an ammonium hydroxide solution. The organic layers from the liquid–liquid separator R5-LS was pumped together with the reagents from reservoir R6 and R7 into the MJOD2 reactor using predetermined flow rates giving a total reactor residence time of 15 min. The crude reaction mixture was collected in the reservoir R8. | ||
The two high yielding oxidation steps developed and implemented on the MJOD continuous flow reactor platform were telescoped with the goal to reduce the overall processing time. However, in order to realize this, it was necessary to perform a solvent exchange process. An in-line extraction process was included at the upper part of the flow reactor MJOD1 in order to facilitate the solvent exchange (water/acetonitrile to ethyl acetate). We have previously successfully used this in-line extraction process in the synthesis of iodoimidazole.21 Ethyl acetate was placed in reservoir R4 and pumped into MJOD1 using pump P4. The post-reaction mixture was transferred from the output section, that is at the top of the MJOD1 flow reactor to a liquid–liquid separator (R5-LS) that also operates as the hold-up tank. The organic phase was pumped (P5) from the R5-LS separator directly into the continuous flow reactor MJOD2 for the subsequent reaction steps of the process. A complete process block diagram is displayed in Fig. 4.
Scope and limitation
A scope and limitation study was conducted using both a classical batch approach and the MJOD continuous flow reactor platform (the process set-up shown in Fig. 4). The modified Montanari oxidation step afforded target products in excellent yield (95–98%, measured for 2b, d, f, g, h, and i, Table 1) with both electron-donating and electron-withdrawing substituents on the aromatic ring. As expected, phenolic substrate 1e afforded only trace amounts of aldehyde 2e, due to the formation of the phenoxyl radical (oxygen-centered radical). The second oxidation step with DIH and NH4OH afforded good to excellent yield (67–98%) for electron-withdrawing and weakly withdrawing substituents, while strong withdrawing groups (3i) afforded low yield (25%).Conclusion
We have designed and developed two distinct continuous flow processes using mild reaction conditions for the production of aldehydes and nitriles. Both processes afforded excellent yields (90–95%) and throughputs of 19.2 g h−1 and 5.1 g h−1, respectively. The two processes were seamlessly telescoped to afford a continuous flow process producing nitriles from their corresponding alcohols in good to excellent yields (67–98%). In addition, we explored other reaction pathways that were discovered during this investigation and the various side-products were structure elucidated and characterized. Interestingly, one of the side products (1,3,5-triazabicyclo[3.1.0]hexane derivative (5a)) that was isolated has previously been reported in the literature, but it was then synthesized at cryogenic-like reaction temperatures,17 which is in huge contrast to our findings. In fact, our synthesis afforded substantial quantities (>20%) of the triazabicyclo derivative 5a at a reaction temperature of 50 °C. Further investigation and optimization of this reaction will be undertaken in the future.Experimental
General methods
GC-MS analyses were conducted with a capillary gas chromatograph furnished with a fused silica column (25 m length, 0.20 mm i.d., 0.33 μm film thickness) using a helium pressure of 200 kPa, splitless/split injector and flame ionization detector.NMR
The 1H NMR spectra were recorded on Bruker Biospin AV500 and AV600 spectrometers operating at 500 MHz and 600 MHz respectively. The AV600 was equipped with a cryogenic probehead. The 13C NMR spectra were recorded at 125 MHz and 150 MHz. 2D 1H–13C HSQC, 1H–13C HMBC,19 and 1H–1H NOESY experiments were recorded on the AV600 spectrometer. The NOESY mixing time was 2 s. The chemical shifts were referenced to the deuterated solvents used for the NMR experiments.X-ray
IR
IR analysis was performed with a Nicolet iS50R FTIR. The spectra were recorded in the wavelength range ν 5000–50 cm−1.Flash chromatography
Flash chromatography was preformed utilizing a Reveleris® flash chromatography system. Detection: ELSD (2-propanol as solvent) and UV, λ 200–600 nm, detectors. Solvents: hexane and ethyl acetate. Columns used for small samples: silica flash cartridge (Reveleris™ SRS) 12 g, flow rate of 28 mL min−1. Large samples: silica flash cartridge (Reveleris™ SRS) 80 g, flow rate of 55 mL min−1.The MJOD continuous flow reactor
The multi-jet oscillating disk (MJOD) continuous flow reactor platform was previously developed in our laboratory and described in detail.14 A short account follows herein. Fig. 5 shows the specifications of MJOD1 and MJOD2 continuous flow reactors used in the integrated continuous flow processing mini-plant depicted in the process flow diagram of Fig. 4.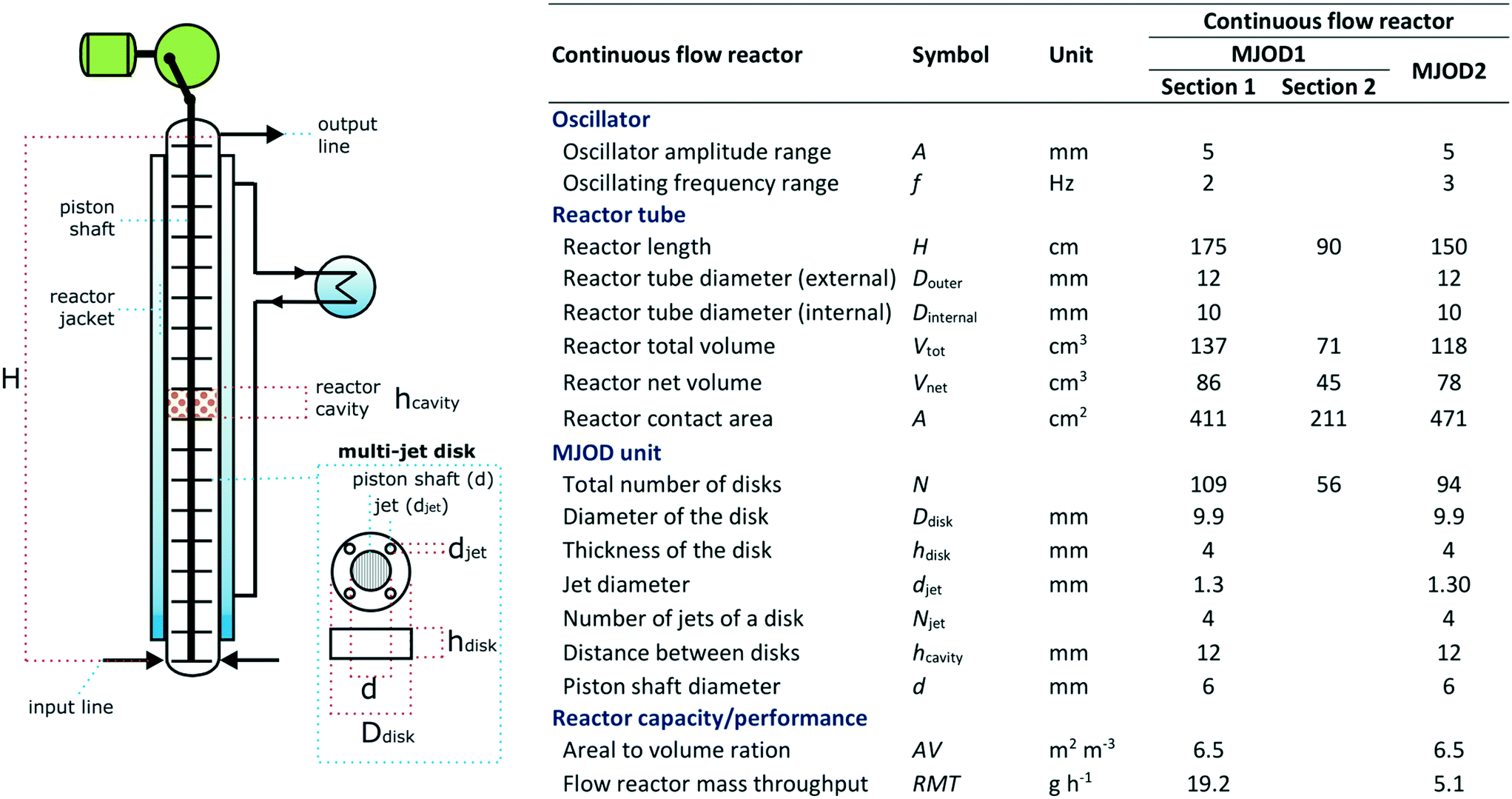 | ||
| Fig. 5 Specifications for the MJOD reactors as integral parts of the continuous flow production mini-plant for the conversion of aldehydes to their corresponding nitriles. | ||
The MJOD continuous flow reactor is a linear tubular reactor that is made of stainless steel. The reactor tube is placed in a vertical direction and furnished with an internal oscillating piston rod. The oscillating motion, that is a vertical up-down movement at amplitude A and frequency f is delivered by a speed adjustable electrical motor. The piston rod is equipped with multiple (n) multi-jet disks and is made of Teflon®, whereof each is furnished with four jets and every disk is separated with a disk spacer (hcavity). The entire length (H) of the reactor tube is encapsulated by a heating/cooling jacket.
The reagents and substrate solutions are transferred to the various reservoirs (RN) and connected to their corresponding feeding pumps (PN). The pump rate is adjusted to provide the desired reactor residence time and mole equivalents between the reagents. Every reactor inlet is fitted with a one-way valve in order to prevent “kick-back” from the reactor. The tubing connecting P2 to MJOD1 was fitted with an extra T-connector for the removal of build-up of precipitates in the reactor inlet. Pressurized solvent was flushed through the line in order to prevent clogging.
Synthesis of benzaldehyde (2a)
A magnet stirrer bar and acetonitrile (1 mL) were transferred to a reactor vial (6 mL) that was sealed. Benzyl alcohol 1a (0.10 mL, 0.10 g, 0.9 mmol) and a solution of NaHCO3 (2 mL, 0.22 g, 2.65 mmol) were then added to the stirred vial at a temperature of 35 °C. 1,3-Dichloro-5,5-dimethylhydantoin (0.211 g, 1,0 mmol) and (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (0.007 g, 0.045 mmol) were dissolved in acetonitrile (3 mL) and added to the vial over a period of 15 min by means of a syringe pump. After the addition, the reaction mixture was stirred for another 25 min at 35 °C. The reaction mixture was then poured into a brine (5 mL) solution and extracted with ethyl acetate (4 × 5 mL). The organic layers were combined and washed with brine (5 mL), and dried over sodium sulfate, whereupon the solvent was removed under reduced pressure. Benzaldehyde was achieved in a quantitative yield according to the GC-MS analysis. GC-MS (EI) m/z (%): 107.06 (6.4), 106.05 (78.8), 105.02 (86.0), 78.05 (15.7), 77.05 (100).Continuous flow synthesis of benzaldehyde (2a)
The continuous flow MJOD1 reactor was heated using HE-1 that was fixed at a temperature of 35 °C. The mixing oscillator O1 was adjusted to a frequency of f = 1.5 Hz. Neat benzyl alcohol 1a (3 mL, 3.12 g, 29 mmol) was transferred to reservoir R1 (Vtot = 3.0 mL). 1,3-Dichloro-5,5-dimethylhydantoin (5.74 g, 29 mmol) and (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO) (0.276 g, 1.77 mmol, 6 mol%) were dissolved in acetonitrile (45 mL) and added to reservoir R2 (Vtot = 45 mL). A solution of saturated NaHCO3 (70 mL) was added to reservoir R3 (Vtot = 70 mL). Reservoirs R1, R2, and R3 were connected to their corresponding pumps and pumped into the MJOD reactor using pump rates of P1 = 0.33 mL min−1, P2 = 5.0 mL min−1, and P3 = 7.67 mL min−1 that resulted in a residence time of 5 min. The crude reaction mixture was collected in a hold-up tank (R4-HT). The crude reaction mixture was added to a brine solution (25 mL) and then extracted with ethyl acetate (3 × 50 mL). The collected organic layers were combined and washed with brine (3 × 10 mL), and dried over sodium sulfate, and finally the solvent was removed under reduced pressure. The isolated crude product was analyzed by means of GC-MS that revealed a yield of 95% of benzaldehyde 2a (2.92 g) at quantitative conversion. GC-MS (EI) m/z (%): 107.06 (6.4), 106.05 (78.8), 105.02 (86.0), 78.05 (15.7), 77.05 (100).Oxidant screening and synthesis of benzonitrile (3a)
The MJOD2 continuous flow reactor was heated (HE-2) at 25 °C. The oscillation frequency (O2) was adjusted at 2 Hz. Benzaldehyde (3.0 mL, 3.1 g, 29 mmol) was diluted in acetonitrile (22 mL) and transferred to reservoir R5 (Vtot = 25 mL). The oxidant DXH 1 eq. (29 mmol) was dissolved in acetonitrile (95 mL) (DIH was dissolved in MeOH (95 mL)) and transferred to reservoir R6 (Vtot = 95 mL). A solution of NH4OH (conc. 25–28%) (25 mL) was transferred to reservoir R7 (Vtot = 25 mL). The reservoirs were connected to feeding pumps. The reagents were pumped into the input section of the MJOD reactor at the following rates: P5 = 5.9 mL min−1, P6 = 1.55 mL min−1, and P7 = 1.55 mL min−1, which afforded a residence time of 10 min. After 20 min of reaction run (that correspond to two net reactor volumes), the samples were collected and analyzed using GC-MS. DCH: 40.5% conversion, 36.4% yield. DBH: 54.2% conversion, 44.0% yield. DIH: quantitative conversion, 59.6% yield.General procedure for the experimental design – oxidation of benzaldehyde with DCH
The MJOD continuous flow reactor was cooled at x2 ∈ [0, 5, 10] °C and the mixing oscillator O1 was set to frequency x3 ∈ [1, 1.5, 2] Hz. Benzaldehyde (3.0 mL, 3.1 g, 29 mmol) was diluted in acetonitrile (92 mL) and transferred to reservoir R1. 1,3-Dichloro-5,5-dimethylhydantoin (5.72 g, 29 mmol) was dissolved in acetonitrile (25 mL) and transferred to reservoir R2. Reservoir R3 was charged with NH4OH (25–28%) (25 mL). Reservoirs R1, R2, and R3 were connected to their corresponding pumps (P1–P3) that pumped the reagents into the input section of the MJOD reactor to assure three distinct reactor residence times x1 ∈ [30 min {P1 = 2 mL min−1, P2 = 0.5 mL min−1, P3 = 0.5 mL min−1}, 45 min {P1 = 1.33 mL min−1, P2 = 0.34 mL min−1, P3 = 0.34 mL min−1}, 60 min {P1 = 1 mL min−1, P2 = 0.25 mL min−1, P3 = 0.25 mL min−1}]. After a run corresponding to a net reactor volume, the samples were collected and analyzed by means of GC-MS.Synthesis of compounds 4 and 5
Benzaldehyde 1a (5.56 g, 52 mmol), acetonitrile (30 mL), ammonium hydroxide (25–28%) (40 mL), and a magnetic stirrer bar were added to a round bottom flask (150 mL). The flask was sealed with a septum and heated to 50 °C under vigorous stirring. 1,3-Dichloro-5,5-dimethylhydantoin (DCH) (10.825 g, 54.8 mmol) was dissolved in acetonitrile (40 mL), and the DCH solution was added to the round bottom flask via a syringe pump over a period of 30 min. The reaction mixture was then stirred for further 30 min before it was cooled to room temperature and further cooled on ice. The precipitates as crystals, a mixture of 5a and 5b, were filtered off (0.462 g, 1.48 mmol). Recrystallization from dichloromethane afforded pure 5a. The mother liquid was extracted with ethyl acetate (10 mL × 3), whereupon the organic layers were combined and left over night to crystallize. The crystals were filtered and washed with cold ethyl acetate. The crystals were analyzed with X-ray crystallography revealing that compound 4 co-crystalized with 5,5-dimethylhydantoin that is the reduced oxidant DCH.Screening the formation of side products 4 and 5 (Fig. 3) and general procedure
Benzaldehyde (0.1 mL, 0.9 mmol), acetonitrile (3 mL) and a magnet were added to a reaction glass vial (8 mL) that was capped and flushed with argon. The reaction vial was then heated at target temperature T ∈ [25, 35, 50, 65] °C under vigorous stirring. 1,3-Dichloro-5,5-dimethylhydantoin (0.197 g, 1 mmol) was dissolved in acetonitrile (1 mL) and transferred to a syringe. Ammonium hydroxide (conc. 25–28%, 1 mL) was transferred to a syringe. The DCH and ammonium hydroxide solutions were added to the reaction vial by means of a syringe pump over a period of 10 min. After that, the reaction mixture was stirred for another 50 min. The solvent was then removed using a vacuum-assisted vortex concentration set-up. The crude product was analyzed with 1H-NMR.1,2-Di((E)-benzylidene)hydrazine (4)
1H-NMR (600 MHz, CDCl3) δ: 8.67 (s, 2H), 7.86–7.84 (m, 4H), 7.48–7.44 (m, 6H). 13C-NMR (150 MHz, CDCl3) δ: 162.1, 134.1, 131.2, 128.8, 128.6. m/z HRMS (EI+) found: [M + H] (12C141H1314N2): 209.10685, calculated: 209.10787.(2R,4R)-2,4,6-Triphenyl-1,3,5-triazabicyclo[3.1.0]hexane (5a)
1H-NMR (600 MHz, (CD3)2SO) δ: 7.86–7.67 (m, 2H), 7.55–7.53 (m, 2H), 7.42–7.38 (m, 4H), 7.37–7.30 (m, 7H) 5.62 (d, j = 7.11 Hz, 1H) 4.96 (d, j = 12.36 Hz, 1H) 4.54 (q, j = 7.10 Hz, 12.33 Hz, 1H) 3.70 (s, 1H). 13C-NMR (150 MHz, (CD3)2SO) δ: 141.1, 136.9, 135.8, 128.5, 128.3, 128.2, 128.1, 128.0, 127.7, 127.3, 127.0, 126.9, 80.72, 80.69, 50.1. m/z HRMS (EI+) found: [M + H] (12C211H2014N3): 314.16334, calculated: 314.16572. FT-IR, ν (cm−1): 3270, 3084, 3060, 3026, 2926, 2882, 1603, 1493, 1449, 1402, 1349, 1247, 997, 900, 736, 706, 693.(2R,4S,6S)-2,4,6-Triphenyl-1,3,5-triazabicyclo[3.1.0]hexane (5b)
1H-NMR (500 MHz, CDCl3) δ: 7.80–7.28 (m, 15H) 5.57–5.53 (m, 2H), 3.19 (s, 1H). Mixture of 5a and 5b (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1).
:1).
Synthesis of benzonitrile 3a
The MJOD2 reactor was heated to 65 °C (HE-2) and oscillator O2 was adjusted at f = 2 Hz. Benzaldehyde 2a (1.5 mL, 1.5 g, 14.7 mmol) was diluted in ethyl acetate (53.5 mL) and added to reservoir R5 (Vtot = 55 mL). The oxidant 1,3-diiodo-5,5-dimethylhydantoin (6.5 g, 17 mmol) was suspended in MeOH (25 mL) and added to reservoir R6 (suspension was agitated while feeding) (Vtot = 25 mL). A solution of NH4OH (25–28%) (15 mL) was added to reservoir R7 (Vtot = 15 mL). The reservoirs were connected to their corresponding pumps and injected into the MJOD reactor (P5 = 3.43 mL min−1, P6 = 1.58 mL min−1, P7 = 0.94 mL min−1) resulting in a residence time of t = 15 min. The crude product was collected in reservoir R8 and extracted with ethyl acetate (3 × 50 mL), and the organic layers were combined, and washed with brine (3 × 10 mL) and sodium thiosulfate solution. The solution was then dried over sodium sulfate and the organic solvent was removed under reduced pressure to afford benzonitrile 3a (1.35 g, 13.2 mmol, 90%). GC-MS (EI) m/z (%): 104.07 (7.5), 103.03 (100), 77.06 (7), 76.05 (56.5), 75.05 (11.3).General continuous flow protocol (Table 1) and synthesis of benzonitrile (3a)
|
|
||||||
|---|---|---|---|---|---|---|
| Batch | Flow | Batch | ||||
| a Reaction time (t) step 1: batch (25 min), continuous flow (5 min). b Reaction temp. (T) step 2: batch (60 °C), continuous flow (65 °C). c Reaction time (t) step 2: batch (30 min), continuous flow (15 min). d Calculated yield from NMR and GC-MS of the crude product. | ||||||
|
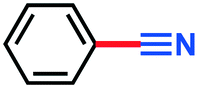
2a | — | >98% |
|
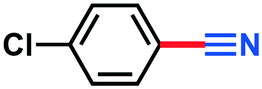
2h | >98% |
| 3a | — | 90% | 3h | 93% | ||
|
2b | >98% | 96% |
|
2i | 95% |
| 3b | 93% | 84% | 3i | 25%d | ||
|
2c | — | 95% |
|
3j | 67% |
| 3c | — | 80% | ||||
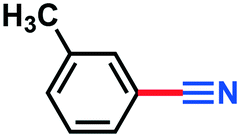
|
2d | >98% | — |
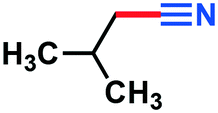
|
3k | 95% |
| 3d | 91% | — | ||||
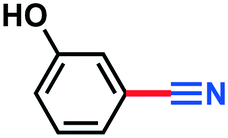
|
2e | Trace | — |
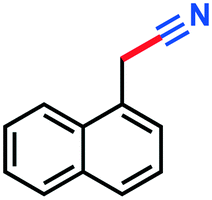
|
3l | 64% |
| 3e | 0% | — | ||||
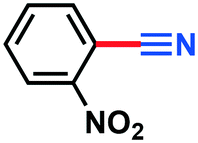
|
2f | >98% | — |
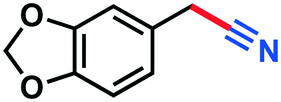
|
3m | 48% |
| 3f | 92% | — | ||||
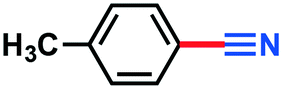
|
2g | >98% | — |
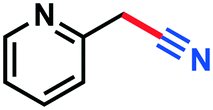
|
3n | 42% |
| 3g | 94% | — |
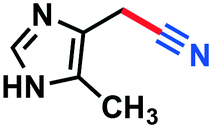
|
3o | n.d. | |
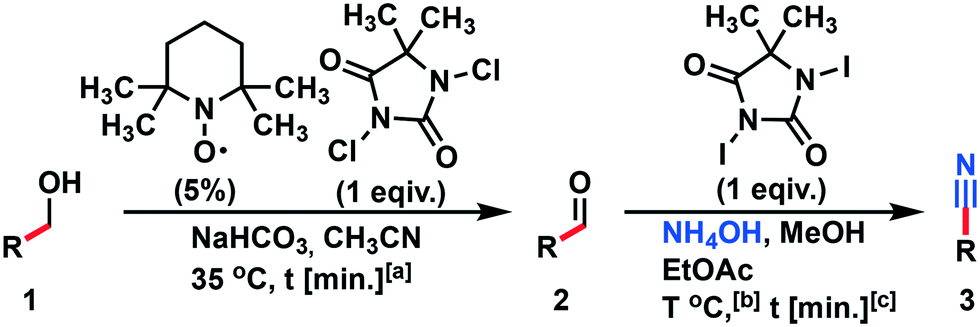
The MJOD1 reactor was heated at a temperature of 35 °C (HE-1). Oscillator O1 was adjusted at a frequency f = 1.5 Hz. Benzyl alcohol 1a (29 mmol) was transferred to reservoir R1 and diluted with acetonitrile (Vtot = 6.0 mL). 1,3-Dichloro-5,5-dimethylhydantoin (6.8 g, 34.4 mmol) and (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO) (0.22 g, 1,4 mmol, 5 mol%) were dissolved in acetonitrile (45 mL) and added to reservoir R2 (Vtot = 40 mL). A solution of saturated NaHCO3 (70 mL) was added to reservoir R3 (Vtot = 70 mL). Reservoirs R1, R2 and R3 were connected to their corresponding pumps and injected to the MJOD reactor (P1 = 0.67 mL min−1, P2 = 4.48 mL min−1, P3 = 7.80 mL min−1) resulting in a residence time of t = 5 min. Ethyl acetate (45 mL) was added to reservoir R4 and pumped into the MJOD1 reactor (P4 = 5.0 mL min−1) resulting in a 1.4 min extraction time. The crude reaction mixture was collected in liquid separator R5-LS. The organic phase was analyzed using GC-MS. The MJOD2 reactor was heated at 65 °C (HE-2) and the oscillator (O2) was adjusted at a frequency of f = 2 Hz. 1,3-Diiodo-5,5-dimethylhydantoin (6.5 g, 17 mmol) was suspended in MeOH (25 mL) and added to reservoir R6 (suspension was agitated while feeding) (Vtot = 25 mL). A solution of NH4OH (25–28%) (15 mL) was added to reservoir R7 (Vtot = 15 mL). The reservoirs were connected to their corresponding pumps and injected into the MJOD reactor (P5 = 3.43 mL min−1, P6 = 1.58 mL min−1, P7 = 0.94 mL min−1) resulting in a residence time of 15 min. The crude mixture was collected in reservoir R8 and extracted with ethyl acetate (3 × 50 mL), and the organic layers were combined, and washed with brine (3 × 10 mL) and sodium thiosulfate solution. The solution was then dried over sodium sulfate and the organic solvent was removed under reduced pressure yielding benzonitrile 3a. The crude reaction mixture was then purified utilizing flash chromatography (hexane
:ethyl acetate, 9.5:0.5 → 5.0:5.0) yielding pure benzonitrile 3a.
Benzonitrile (3a)
2.69 g, 90% yield, GC-MS (EI) m/z (%): 104.07 (7.5), 103.03 (100), 77.06 (7), 76.05 (56.5), 75.05 (11.3).3-Nitrobenzonitrile (3b)
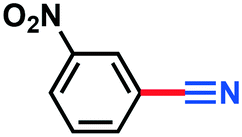
3.60 g, 84% yield, 1H-NMR (500 MHz, CDCl3) δ: 8.54–8.53 (m, 1H), 8.50–8.47 (m, 1H), 8.02–7.98 (m, 1H), 7.76–7.72 (t, j = 8.18 Hz, 1H). 13C-NMR (125 MHz, CDCl3) δ: 148.2, 137.6, 130.7, 127.5, 127.2, 116.5, 114.2. GC-MS (EI) m/z (%): 147.97 (30.7), 102.00 (100), 90.03 (21), 76.04 (27.6), 75.05 (56).3-Chlorobenzonitrile (3c)
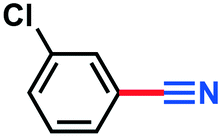
3.18 g, 80% yield, 1H-NMR (500 MHz, CDCl3) δ: 7.64 (s, 1H), 7.60–7.55 (m, 2H), 7.45–7.41 (t, j = 8.08 Hz, 1H), 13C-NMR (125 MHz, CDCl3) δ: 135.3, 133.3, 131.9, 130.5, 130.3, 117.5, 114.0, GC-MS (EI) m/z (%): 140.02 (4.3), 138.99 (31.8), 138.0 (9.3) 136.98 (100), 102.04 (44.6), 76.05 (18.8) 75.07 (39.4).General batch procedure (Table 1) and synthesis of benzonitrile (3a)
A magnetic stirrer bar and acetonitrile (1 mL) were transferred to a reaction vial (6 mL). Benzyl alcohol (1 mmol) was then added, capped, and heated at 35 °C. 1,3-Dichloro-5,5-dimethylhydantoin (0,236 g, 1.2 mmol) and (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (0.007 g, 0.045 mmol) were dissolved in acetonitrile (3 mL) and added dropwise to the vial over a period of 10 min utilizing a syringe pump. The reaction mixture was then left for stirring for another 20 min. The crude mixture was afterwards poured into a brine solution (2 mL) and extracted with ethyl acetate (2 mL × 3). The organic phase was placed in a round bottom flask (15 mL) together with a magnetic stirrer bar. The flask was capped with a septum and heated at 60 °C. 1,3-Diiodo-5,5-dimethylhydantoin (0.478 g, 1.2 mmol) was dissolved in methanol (4 mL) and placed in a syringe. A solution of NH4OH (25–28%) (1 mL) was added to another syringe. Both the ammonium hydroxide and 1,3-diiodo-5,5-dimethylhydantoin solutions were injected to the reaction flask over a period of 10 min utilizing syringe pumps. After that, the reaction mixture was left to be stirred for another 20 min, whereupon the reaction mixture was quenched with sodium thiosulfate and extracted with ethyl acetate (5 mL × 3). The organic layers were combined and washed with brine (3 mL × 2) and dried over sodium sulfate before the solvents were removed under reduced pressure using a rotary evaporator. The crude reaction mixture was then purified utilizing flash chromatography (hexane:ethyl acetate, 9.5:0.5 → 5.0:5.0) that afforded pure benzonitrile.
3-Nitrobenzonitrile (3b)
0.137 g, 93% yield, 1H-NMR (500 MHz, CDCl3) δ: 8.54–8.53 (m, 1H), 8.50–8.47 (m, 1H), 8.02–7.98 (m, 1H), 7.76–7.72 (t, j = 8.18 Hz, 1H). 13C-NMR (125 MHz, CDCl3) δ: 148.2, 137.6, 130.7, 127.5, 127.2, 116.5, 114.2. GC-MS (EI) m/z (%): 147.97 (30.7), 102.00 (100), 90.03 (21), 76.04 (27.6), 75.05 (56).3-Methylbenzonitrile (3d)
0.106 g, 91% yield, 1H-NMR (500 MHz, CDCl3) δ: 7.47–7.44 (m, 2H), 7.42–7.39 (m, 1H), 7.36–7.32 (m, 1H), 2.39 (s, 3H), 13C-NMR (125 MHz, CDCl3) δ: 139.2, 133.6, 132.5, 129.3, 129.0, 119.0, 112.3, 21.5. GC-MS (EI) m/z (%): 118 (9.3), 117.08 (100), 116.06 (63.2), 91.09 (28.0), 90.09 (67.1), 76.07 (6.9), 75.07 (8.5).2-Nitrobenzonitrile (3f)
0.135 g, 91% yield, 1H-NMR (500 MHz, CDCl3) δ: 8.37–8.32 (m, 1H), 7.96–7.92 (m, 1H), 7.86–7.81 (m, 2H). 13C-NMR (125 MHz, CDCl3) δ: 143.3, 130.3, 128.9, 128.3, 120.3, 109.6, 102.9. GC-MS (EI) m/z (%): 122.03 (4.4), 121.02 (68.3), 104.02 (24.4), 93.04 (46.6), 77.06 (31.8), 76.05 (64.3), 74.05 (21.4), 65.05 (100).4-Methylbenzonitrile (3g)
0.110 g, 94% yield, 1H-NMR (500 MHz, CDCl3) δ: 7.55 (d, j = 8.11 Mz, 2H), 7.28 (d, j = 7.55 Hz, 2H), 2.44 (s, 3H). 13C-NMR (125 MHz, CDCl3) δ: 143.7, 132.1, 129.8, 119.2, 109.3, 21.8. GC-MS (EI) m/z (%): 118 (8.1), 177.05 (91.8), 91.07 (17.3), 90.06 (52.0), 89.06 (36.2), 63.06 (26.5).4-Chlorobenzonitrile (3h)
0.128 g, 93% yield, 1H-NMR (500 MHz, CDCl3) δ: 7.61–7.58 (m, 2H), 7.48–7.45 (m, 2H). 13C-NMR (125 MHz, CDCl3) δ: 139.6, 133.4, 129.7, 118.0, 110.8. GC-MS (EI) m/z (%): 138.99 (33.5), 138.0 (14.4), 138.98 (100), 122.09 (14.1), 102.02 (44.0), 76.03 (13.0), 75.03 (26.4).4-Methoxybenzonitrile (3i)
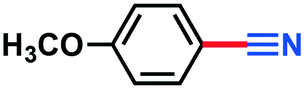
0.033 g, 25% calculated yield, the product isolated as the mixture of nitrile and aldehyde. 1H-NMR (500 MHz, CDCl3) δ: 7.59–7.57 (m, 2H), 6.95–6.93 (m, 2H), 3.85 (s, 3H).Cinnamonitrile (3j)
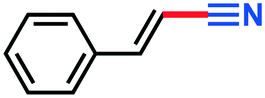
0.086 g, 67% yield, 1H-NMR (500 MHz, CDCl3) δ: 7.37–7.28 (m, 5H + residual CHCl3), 5.77–5.74 (d, j = 16.54 Hz, 2H). 13C-NMR (125 MHz, CDCl3) δ: 150.7, 133.6, 131.4, 129.3, 127.6, 118.3, 96.6. GC-MS (EI) m/z (%): 130.09 (10.1), 129.07 (100), 103.0 (26.9), 102.08 (67.0), 78.07 (27.0), 78.07 (29.6), 75.07 (20.0), 74.06 (14.0), 63.09 (21.6).3-Methylbutanenitrile (3k)
0.079 g, 95% yield, 1H-NMR (500 MHz, CDCl3) δ: 2.38–2.37 (d, j = 6.27 Hz, 2H), 2.22–2.14 (m, 1H), 1.22–1.21 (d, j = 6.60 Hz, 6H). 13C-NMR (125 MHz, CDCl3) δ: 119.1, 26.6, 26.2, 22.1.1-Naphthonitrile (3l)
Crude yield calculated from NMR and GC-MS: 0.097 g, 64%, 1H-NMR (500 MHz, CDCl3) δ: 8.26–8.24 (m, 1H) (nitrile signal), 10.41 (s, 1H) (aldehyde signal). GC-MS (EI) m/z (%):154.1 (12.4), 153.1 (100), 127.0 (5.3), 126.0 (17.9), 100.0 (3.0), 99.0 (3.1), 76.3 (5.8), 63.0 (7.3).Benzo[d][1,3]dioxole-5-carbonitrile (3m)
0.071 g, 48%, 1H-NMR (500 MHz, CDCl3) δ: 7.66–7.64 (m, 1H), 7.47–7.46 (m, 1H), 6.83 (d, J = 8.18 Hz, 1H), 6.03 (s, 2H). 13C-NMR (125 MHz, CDCl3) δ: 151.6, 125.3, 124.2, 109.5, 108.0, 101.7. GC-MS (EI) m/z (%): 148.0 (5.7), 147.0 (62.7), 146.0 (100), 90.0 (4.1), 89.0 (8.2), 88.0 (6.0), 63.0 (11.3), 62.0 (17.2).Picolinonitrile (3n)
0.044 g, 42%, 1H-NMR (500 MHz, CDCl3) δ: 8.58 (m, 1H), 8.22–8.18 (m, 1H), 7.88–7.84 (m, 1H), 7.47–7.44 (m, 1H). 13C-NMR (125 MHz, CDCl3) δ: 148.33, 137.32, 137.27, 126.51, 122.52, 122.39. GC-MS (EI) m/z (%): 105.0 (7.5), 104.0 (100), 78.0 (7.9), 77.0 (40.8), 76.0 (13.2), 75.0 (7.8), 52.0 (9.1), 51.0 (17.3), 50.0 (18.0).Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledged the Swiss-Norwegian beamlines at the ESRF, Grenoble, France and Dr. D. Chernyshov for assistance on beamline BM1A. A. D. gratefully acknowledges the Department of Chemistry at the University of Bergen for the research fellowship.Notes and references
- (a) P. Pollak, G. Romeder, F. Hagedorn and H.-P. Gelbke, Ullmann's Encyclopedia of Industrial Chemistry, 2000, Nitriles, DOI:10.1002/14356007.a17_363; (b) F. F. Fleming, L. Yao, P. C. Ravikumar, L. Funk and B. C. Shook, J. Med. Chem., 2010, 53, 7902–7917 CrossRef CAS PubMed; (c) G. Yan, Y. Zhang and J. Wang, Adv. Synth. Catal., 2017, 359, 4068–4105 CrossRef CAS.
- (a) K. Friedrich and K. Wallenfels, Chemistry of the Cyano Group, ed. Z. Rappoport, Wiley, London, U.K, 1970 Search PubMed; (b) A. J. Fatiadi, in Preparation and Synthetic Applications of Cyano Compounds, Wiley, New York, 1983 Search PubMed; (c) J. S. Miller and J. L. Manson, Acc. Chem. Res., 2001, 34, 563–570 CrossRef CAS PubMed.
- (a) M. T. Schumperli, C. Hammond and I. Hermans, ACS Catal., 2012, 2, 1108–1117 CrossRef CAS; (b) Y. Zhang, K. Xu, X. Chen, T. Hu, Y. Yu, J. Zhang and J. Huang, Catal. Commun., 2010, 11, 951–954 CrossRef CAS; (c) K. Yamaguchi and N. Mizuno, Angew. Chem., Int. Ed., 2003, 42, 1480–1483 CrossRef CAS PubMed.
- (a) S. Enthaler, Chem. – Eur. J., 2011, 17, 9316–9319 CrossRef CAS PubMed; (b) S. Sueoka, T. Mitsudome, T. Mizugaki, K. Jitsukawa and K. Kaneda, Chem. Commun., 2010, 46, 8243–8245 RSC; (c) S. Zhou, D. Addis, S. Das, K. Junge and M. Beller, Chem. Commun., 2009, 4883–4885 RSC; (d) K. Yamaguchi, H. Fujiwara, Y. Ogasawara, M. Kotani and N. Mizuno, Angew. Chem., Int. Ed., 2007, 46, 3922–3975 CrossRef CAS PubMed; (e) K. Ishihara, Y. Furuya and H. Yamamoto, Angew. Chem., Int. Ed., 2002, 41, 2983–2986 CrossRef CAS.
- (a) P. Anbarasan, T. Schareina and M. Beller, Chem. Soc. Rev., 2011, 40, 5049–5067 RSC; (b) G. Yan, J. Yu and L. Zhang, Chin. J. Org. Chem., 2012, 32, 294–303 CrossRef CAS.
- (a) D. J. C. Constable, A. D. Curzons and V. L. Cunningham, Green Chem., 2002, 4, 521–527 RSC; (b) R. A. Sheldon, ACS Sustainable Chem. Eng., 2018, 6, 32–48 CrossRef CAS.
- (a) S. Iida and H. Togo, Synlett, 2007, 407–410 CAS; (b) C. Zhu, C. Sun and Y. Wei, Synthesis, 2010, 4235–4241 CAS; (c) H. Shimojo, K. Moriyama and H. Togo, Synthesis, 2013, 2155–2164 CAS; (d) W. Yin, C. Wang and Y. Huang, Org. Lett., 2013, 15, 1850–1853 CrossRef CAS PubMed; (e) D. K. T. Yadav and B. M. Bhanage, Eur. J. Org. Chem., 2013, 5106–5110 CrossRef CAS.
- H.-R. Bjørsvik, L. Liguori, F. Costantino and F. Minisci, Org. Process Res. Dev., 2002, 6, 197–200 CrossRef.
- M. Ferreri, A. Drageset, C. Gambarotti and H.-R. Bjørsvik, React. Chem. Eng., 2016, 1, 379–386 RSC.
- A. H. Sandtorv and H.-R. Bjørsvik, Adv. Synth. Catal., 2013, 355, 499–507 CrossRef CAS.
- A. Drageset and H.-R. Bjørsvik, Eur. J. Org. Chem., 2018, 4436–4445 CrossRef CAS.
- Montanari-type oxidation is a TEMPO-mediated oxidation that originally involved sodium hypochlorite as the terminal oxidant. This oxidation method can be used to transform alcohols to their corresponding carbonyl compounds, see: (a) P. L. Anelli, C. Biffi and F. Montanari, J. Org. Chem., 1987, 52, 2559–2562 CrossRef; (b) P. L. Anelli, S. Banfi, F. Montanari and S. Quici, J. Org. Chem., 1989, 54, 2970–2972 CrossRef CAS; (c) P. L. Anelli, S. Banfi, F. Montanari and S. Quici, Org. Synth., 1990, 69, 212–219 CrossRef CAS.
- Comparison of the cost of DIH and DCH as research chemicals. A cost ratio DCH mole : DIH mole gives a mass ratio of 1 kg : 1.924 kg that corresponds to a cost ratio of 1 : 220. The cost data was retrieved from Sigma-Aldrich (Merck) web page: https://www.sigmaaldrich.com/norway.html, (November 2017) .
- (a) A. Drageset and H.-R. Bjørsvik, React. Chem. Eng., 2016, 1, 436–444 RSC; (b) M. Ferreri, A. Drageset, C. Gambarotti and H.-R. Bjørsvik, React. Chem. Eng., 2016, 1, 379–386 RSC; (c) A. Drageset, V. Elumalai and H.-R. Bjørsvik, React. Chem. Eng., 2018, 3, 550–558 RSC.
- (a) L. Liguori and H.-R. Bjørsvik, Org. Process Res. Dev., 2011, 15, 997–1009 CrossRef CAS; (b) H.-R. Bjørsvik and L. Liguori, Org. Process Res. Dev., 2014, 18, 1509–1515 CrossRef.
- (a) R. Ohmura, M. Takahata and H. Togo, Tetrahedron Lett., 2010, 51, 4378–4381 CrossRef CAS; (b) H. Shimojo, K. Moriyama and H. Togo, Synthesis, 2013, 45, 2155–2164 CrossRef CAS.
- (a) R. Mundil, Bayer Ag. US3077383A, 1963; (b) J.-P. Schirmann and P. Bourdauducq, Hydrazine, Ullmann's Encyclopedia of Industrial Chemistry, 2001 Search PubMed.
- (a) E. Schmitz, Angew. Chem., 1961, 73, 23–25 CrossRef CAS; (b) A. T. Nielsen, D. W. Moore, R. L. Atkins, D. Mallory, J. DiPol and J. M. LaBerge, J. Org. Chem., 1976, 41, 3221–3229 CrossRef CAS.
- D. O. Cicero, G. Barbato and R. Bazzo, J. Magn. Reson., 2001, 148, 209–213 CrossRef CAS PubMed.
- A. T. Nielsen, D. W. Moore, R. L. Atkins, D. Mallory, J. DiPol and J. M. LaBerge, J. Org. Chem., 1976, 41, 3221–3229 CrossRef CAS.
- A. Drageset and H.-R. Bjørsvik, React. Chem. Eng., 2016, 1, 436–444 RSC.
- APEX2, 2014.2011–2010, Bruker-AXS, Madison, Wisconsin, USA, 2014 Search PubMed.
- SAINT, 7.68A, Bruker-AXS, Madison, Wisconsin, USA, 2010 Search PubMed.
- SADABS 2008/1, see L. Krause, R. Herbst-Irmer, G. M. Sheldrick and D. Stalke, J. Appl. Crystallogr., 2015, 48, 3–10 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- V. Dyadkin, P. Pattison, V. Dmitriev and D. Chernyshov, J. Synchrotron Radiat., 2016, 23, 825–829 CrossRef CAS PubMed.
- Rigaku 2014, Rigaku Oxford Diffraction, CrysalisPro Software System, Version 1.171.37.34. http://www.rigaku.com/en Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8re00129d |
| This journal is © The Royal Society of Chemistry 2019 |