
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChemoradiotherapy screening in a novel biomimetic polymer based pancreatic cancer model†
Priyanka Guptaa,
Stella Tottia,
Pedro A. Pérez-Mancerab,
Eleanor Dykec,
Andrew Nisbetcd,
Giuseppe Schettinode,
Roger Webbf and
Eirini G. Velliou *a
*a
aBioprocess and Biochemical Engineering Group (BioProChem), Department of Chemical and Process Engineering, University of Surrey, Guildford, GU2 7XH, UK. E-mail: e.velliou@surrey.ac.uk
bDepartment of Molecular and Clinical Cancer Medicine, University of Liverpool, Ashton Street, Liverpool, L69 3GE, UK
cDepartment of Medical Physics, The Royal Surrey County Hospital, NHS Foundation Trust, Guildford, GU2 7XX, UK
dDepartment of Physics, University of Surrey, Guildford GU2 7XH, UK
eMedical Radiation Science Group, The National Physical Laboratory, Teddington, TW11 0LW, UK
fThe Ion Beam Centre, University of Surrey, Guildford, GU2 7XH, UK
First published on 17th December 2019
Abstract
Pancreatic Ductal Adenocarcinoma (PDAC) is a deadly and aggressive disease with a very low survival rate. This is partly due to the resistance of the disease to currently available treatment options. Herein, we report for the first time the use of a novel polyurethane scaffold based PDAC model for screening the short and relatively long term (1 and 17 days post-treatment) responses of chemotherapy, radiotherapy and their combination. We show a dose dependent cell viability reduction and apoptosis induction for both chemotherapy and radiotherapy. Furthermore, we observe a change in the impact of the treatment depending on the time-frame, especially for radiation for which the PDAC scaffolds showed resistance after 1 day but responded more 17 days post-treatment. This is the first study to report a viable PDAC culture in a scaffold for more than 2 months and the first to perform long-term (17 days) post-treatment observations in vitro. This is particularly important as a longer time-frame is much closer to animal studies and to patient treatment regimes, highlighting that our scaffold system has great potential to be used as an animal free model for screening of PDAC.
1. Introduction
Pancreatic Ductal Adenocarcinoma (PDAC) is a deadly disease with very low prognosis. The 5 year survival rate is only 5–7% and it has barely improved over the last decades making it the 5th leading cause of cancer related death in the UK (https://www.cancerresearchuk.org/health-professional/cancer-statistics/statistics-by-cancer-type/pancreatic-cancer; accessed August 2019), 4th in the USA1 and the 7th leading cause of cancer related death worldwide.2–5 These disheartening statistics have been partly attributed to the disease's high resistance to current therapeutic regimes (radiotherapy & chemotherapy). PDAC's high resistance to treatment is linked to the tumour's complexity.6 More specifically, the so-called tumour microenvironment (TME), which is a cocktail of biochemical, cellular, biomechanical and structural components has been shown to affect the efficiency of treatment delivery to the tumour.4,7–9Traditionally, PDAC research including treatment screening, is conducted in (a) 2D in vitro systems10–13 (tissue culture flasks or Petri-plates) or in (b) animals, mainly mice.10,11,14–18 Generally, 2D in vitro cultures are responsive to radiotherapy and chemotherapy, reproducible and very easy to use.4,13,19 However, the system is unable to capture key features of the TME such as structure, stiffness, spatial orientation, cell–cell cross talk, cell-extracellular matrix (ECM) interactions and environmental gradients.4,20–22 Animal models (mostly murine) are more accurate/realistic than the 2D in vitro models in capturing a patient's situation and are currently the most widely used model for drug and radiotherapy testing at a pre-clinical level.2,10,14,15,23–27 However, they are expensive, difficult to reproduce and complex to use.4,7,21,23,26,28 Additionally, there is evidence that animal models undergo significant genetic changes that diverge from the evolutionary course observed in human disease, raising concerns about the models' translatability and application for personalised therapies.2
More recently, 3D in vitro models have emerged and are being developed as low cost, promising alternatives to animals.7,29–48 However, most studies focus on the development of the actual 3D model and, to the best of our knowledge, there are very limited 3D studies on PDAC treatment screening (chemotherapy and radiotherapy) with most studies conducted in 3D spheroids (cell aggregates).14,32,34–36,49–52 Generally, these studies report a higher resistance of PDAC to treatment in 3D when compared to 2D cultures, a trend which is in closer alignment with in vivo studies.34,49,52 For example, Longati et al. (2013) studied the impact of the chemotherapeutic drug Gemcitabine (GEM, 0–1 μM) on the viability of PDAC spheroids and observed a higher resistance of the spheroids to the drug, as compared to a 2D culture. The same study also reported that chemo-resistance differed amongst different cell lines with PANC-1 showing a higher resistance to GEM in comparison to BxPC-3 and Capan-1, 7 days after drug administration.34 Ki et al., (2014) also observed increased resistance of the PDAC cell line COLO-357 to GEM within hydrogel based 3D structures as compared to a 2D culture, 4 days post-treatment.52 More specifically, there was a 6-fold increase in apoptosis in the 2D culture which was treated with GEM (1 μM) in comparison to the 3D hydrogels for which there was less than 3-fold apoptosis induction. Al-Ramadan et al. (2018) also used a spheroid model for assessment of radiation (0–6 Gy) on the PDAC cell line BON1 and showed a dose dependent increase in cell apoptosis within the 3D system over a period of 7 days.49
However, even though spheroid type models are easy to fabricate and responsive to drugs, they cannot mimic robustly the TME, mainly due to their nature as well as their structural and spatial organisation.4,20,53,54 More specifically: (i) they lack mechanical stability and mechanical tunability (ii) they cannot maintain specific spatial cellular orientation (iii) simulation of mass transfer limitations which realistically occur in a dense PDAC tissue in vivo are not accurate (iv) the ECM production cannot be efficiently replicated in a spheroid structure, mainly due to the high variability of the aggregates and the non-uniform secretion of endogenous ECM (v) due to lack of porosity and perfusion, they form unrealistically high environmental gradients which do not necessarily occur in vivo, limiting their accuracy as well as the culture duration, i.e., to a few days without requiring re-suspension, however the latter would destroy the formed TME and the formed cell–matrix interactions. The short culture duration is a key bottleneck for treatment related studies. More specifically, as described above, most treatment studies in spheroids have been monitored for a maximum of 7 days,32,34,35,52 which is significantly shorter in comparison to patients' treatment time-frame and to animal models which are usually assessed over a time frame of several weeks.2,11,14–17,55–57 Furthermore, in vitro models that allow long term post-treatment monitoring would enable the conduction of fractionated radiation screening wherein radiotherapeutic treatment is provided to the patients in serial smaller dosages over a specific time interval to minimise radiation related side effects.58–61
We have recently developed a robust polyurethane (PU) scaffold for PDAC (re-)modelling, which overcomes some of the challenges faced in spheroid systems.7 More specifically, due to the porosity and interconnectivity of our scaffolds, pancreatic cancer cells remain live for more than 4 weeks without need of resuspension, which to our knowledge is the longest in vitro culturing period. Furthermore, with appropriate surface modification of our scaffolds with ECM proteins such as fibronectin we observed enhancement of cell proliferation, ECM secretion by the cells as well as the presence of HIF 1α, in our previously reported work. In this study, we investigate radio-chemo treatment screening in our 3D bioinspired polymer based PDAC model. It should be stated that this is the first reported PDAC treatment screening in a scaffold-based system. We performed chemotherapy and radiotherapy treatment on our scaffolds at week 4 of culture, i.e., the time-point at which we have previously observed physiological behaviour such as significant ECM (collagen-I) secretion, environmental gradients and dense cell masses. More specifically, chemotherapy (with Gemcitabine) as well as radiotherapy (X-rays) treatments were conducted in our scaffolds and the cell evolution, i.e., viability, apoptosis, cell mass formation was monitored for 17 days post-treatment and was spatially visualised with appropriate scaffold sectioning, staining and imaging.
2. Materials and methods
2.1 Polymer scaffold preparation & surface modification
The scaffolds were fabricated via the thermal induced phase separation method prepared as reported previously.7,62–64 Briefly, PU beads (Noveon, Belgium) were dissolved in dioxane (5% w/v) (99.8% anhydrous pure, Sigma-Aldrich, UK) followed by quenching at −80 °C for 3 h. The solvent was then removed by freeze drying in a poly-ethylene glycol (PEG) bath at −15 °C under 0.01 mbar vacuum pressure. The scaffolds were then cut into 5 × 5 × 5 mm3 cubes and sterilised by exposing them to 70% ethanol (3 h) and UV ray (1 h). As previously reported, the average pore size of the scaffolds was 100–150 μm, the porosity was 85–90% and the elastic modulus 20 ± 2 kPa (ESI Fig. 1†). It should be stated that the stiffness of the scaffolds was similar to that of PDAC ex vivo tissue.65–67Thereafter, as previously described, the generated scaffolds were surface modified (adsorption) with fibronectin for ECM mimicry.7 Briefly, the scaffolds were dipped in Phosphate Buffered Saline (PBS, Sigma-Aldrich, UK) and centrifuged (in PBS) for 10 min at 2500 rpm. Then, they were transferred to fibronectin solution (25 μg ml−1) and centrifuged for 20 min at 2000 rpm, followed by a final centrifugation step in PBS for 10 min at 1500 rpm to unblock the surface pores of the scaffolds.
2.2 3D cell culture
The 3D cell culture was conducted as previously described.7 Briefly, the human pancreatic adenocarcinoma cell line PANC-1 (Sigma-Aldrich, UK) was expanded in Dulbecco's modified Eagle's medium (DMEM) with high glucose (Lonza, UK) supplemented with 10% fetal bovine serum (Fisher Scientific, UK), 1% penicillin/streptomycin (Fisher Scientific, UK) and 2 mM L-glutamine (Sigma-Aldrich, UK) in a humidified incubator at 37 °C with 5% CO2. Cells were passaged regularly on reaching 90% confluency till the required cell density was obtained. For all experimental conditions, 0.5 × 106 cells (re-suspended in 30 μl of cell culture media) were seeded in each scaffold and the scaffolds were placed in 24 well plates. Immediately after seeding, the scaffolds were placed in the incubator to facilitate cell attachment for 1 hour. Thereafter, 1.5 ml of cell culture media was added to each well and incubation took place in a humidified incubator at 37 °C with 5% CO2 for the entire duration of the experiment (2 months). The cell culture medium was changed every two days. Furthermore, to avoid cell confluency at the bottom of the wells due to cell egress from the scaffolds, all scaffolds were placed in a new well plate once a week.2.3 Treatment protocol in the 3D scaffolds
At week 4 of culture the scaffolds were exposed to different treatment regimes, i.e., chemotherapy, radiotherapy or a combination. This time point was selected as we have observed and previously reported that, on week 4 of culture, the pancreatic cancer cells form high cell masses within the scaffolds (ESI Fig. 2†) along with secretion of collagen-I and presence of HIF 1α in some areas within our scaffold. Furthermore, at this stage of culture the cells within the scaffold are highly proliferative. Overall, at this point of the culture the cells exhibit physiological features, therefore, that time point is ideal for the conduction of treatment screening.7For the conduction of chemotherapy, GEM at concentrations of 10, 50 and 100 μM was added to the culture for 1 feeding cycle (48 h) and removed thereafter.76 These concentrations were selected based on dosages used in previously published PDAC in vitro studies.10,33,35,68,72 Thereafter, the scaffolds were characterised 24 h and 17 days after treatment with sectioning, staining and advanced imaging. These time points were selected to study the immediate responses to GEM33,34,36 and to mimic in vivo treatment regimes.14,77
2.4 Spatial evaluation of live and dead cells in the scaffolds via confocal imaging
To visualise the spatial distribution of live and dead cells pre- and post-treatment, scaffolds were collected, sectioned, stained and further imaged. More specifically, scaffolds were collected at appropriate time points, snap frozen in liquid nitrogen for 15 min and then preserved at −80 °C. The method of scaffold preservation has been widely used in the field of tissue engineering and is known to aid in preservation without harming the cells.83–85 For live/dead cell analysis a Live/Dead Viability/Cytotoxicity kit was used7 (Molecular Probes, Thermo Scientific, UK). Prior to analysis scaffolds were sectioned and washed twice with PBS, stained with 2 μM of Calcein-AM (4 mM stock) and 4 μM of Ethidium Homodimer (2 mM stock) and were incubated at 37 °C for 1 h. The solution was then removed, and the samples were washed twice in PBS followed by imaging using a Nikon Ti-Eclipse inverted confocal microscope (Nikon Instruments, Europe).2.5 Spatial evaluation of apoptotic cells (caspase 3/7 activity) in the scaffolds via confocal imaging
The caspase 3/7 activity was visualised and quantified in situ to assess the induction of cellular apoptosis after different treatments. Scaffolds were collected at different time points and processed as described above (Section 2.4). Thereafter, the scaffolds were incubated in culture medium containing (i) the Cell Event Caspase-3/7 green detection reagent (Fisher Scientific, UK) and (ii) DAPI (Fisher Scientific, UK) for 1 h at 37 °C. The presence of caspase 3/7 positive cells (green) was immediately evaluated with a Nikon Ti-Eclipse inverted confocal microscope (Nikon Instruments, Europe).2.6 Confocal laser scanning microscopy (CLSM) imaging
Immunofluorescent samples (prepared as described in Sections 2.4 & 2.5 above) were imaged on a Nikon Ti-Eclipse inverted confocal microscope (Nikon Instruments, Europe) and processed with the NIS-Elements software, using 405, 488 and 561 nm lasers for DAPI (blue), green fluorescence (calcein and caspase 3/7) and Ethidium Homodimer (red) staining respectively. Confocal images were captured using a 10× objective and a 5–10 μm Z-stack distance. The same acquisition conditions were used for the positive controls. Multiple scaffolds as well as multiple areas and sections per scaffold were imaged to ensure reproducibility. Representative images are presented in this manuscript.2.7 Image analysis
For the quantitative evaluation of (i) the live (green) and dead (red) population as well as (ii) the caspase positive/apoptotic (green) and non-apoptotic (blue) population of each image, the percentage of green vs. red (live/dead) or green vs. blue (caspase positive/caspase negative) areas of each image were calculated using Image J® software (Wayne Rasband, NIH, Bethesda, MD, USA). The particle analyser macro (Image J®, Wayne Rasband, NIH, Bethesda, MD, USA) was used in each individual channel (green or read for live/dead and green or blue for the caspase 3/7 respectively).This approach is hugely beneficial in 3D scaffolds as it provides a quantitative indication of the live/dead and apoptotic/non apoptotic cell distribution within the various scaffold areas for different treatment conditions.41,86 The latter is particularly important as we have seen that metabolic activity dependent assays like the MTT assay and Alamar Blue assay are affected by various parameters including cytotoxic agents and are not sensitive enough to identify differences in cell population within the scaffolds, especially for very high cell numbers.87–89 In contrast, imaging is much more representative and reproducible. As described above, multiple scaffold sections (at least 3) from at least 3 replicate scaffolds were analysed for each condition to ensure reproducibility of the results.
2.8 Statistical analysis
Statistical analysis was performed for at least 3 independent experiments with at least 3 replicates per time-point (N ≥ 3, n ≥ 3). Analysis of Variance (one-way ANOVA) followed by the Tukey's multiple comparison test using the Graph Pad Prism® software (version 6.00 for Windows) in order to find statistically significant differences between data (p < 0.05) was carried out. Untreated samples were considered as control in all cases. The error bars in the graphs represent standard error of mean.3. Results
3.1 Chemotherapy assessment in the 3D scaffolds
As mentioned previously, Gemcitabine (GEM), a standard chemotherapeutic agent against PDAC73–75,90,91 was introduced to the culture medium surrounding the scaffolds at week 4 of culture and at different concentrations, i.e. 10, 50 and 100 μM, for 1 feeding cycle, i.e. 48 h. Thereafter, the medium containing the drug was removed and replaced with fresh medium. The scaffold culture was further maintained and monitored for 17 days post-treatment. In order to assess the short and long term responses to GEM, the scaffolds were assessed 24 h and 17 days post-treatment. Staining, sectioning and imaging of the scaffolds at those different time points (as described in Sections 2.4–2.7) with CLSM enabled the spatial assessment and quantification of the impact of GEM on the cell viability and apoptosis induction in the scaffolds (Fig. 1 and 2). More specifically, Fig. 1A shows representative live areas on scaffold sections for various concentrations of GEM while Fig. 1B shows the distribution of apoptotic areas on representative scaffold sections, 24 h post treatment. Fig. 1C and D show the equivalent quantifications (performed as described in Section 2.7). A day after treatment, a drug dose dependent decrease in cell viability was observed within the scaffolds (Fig. 1A and C). From a quantitative point of view this reduction was statistically significant only for 50 μM and 100 μM GEM, i.e., the higher doses used. It should be stated that, for the higher doses of GEM, there was a significantly lower number of cells in the scaffolds, due to the detachment of severely damaged/dead cells92 from the polymeric matrix. In terms of apoptosis, an increase of apoptosis with higher GEM concentrations was observed, as evaluated with caspase 3/7 activity (Fig. 1B and D). Furthermore, a high number of apoptotic cells was observed even for low concentrations of GEM (Fig. 1D), indicating that the drug had an immediate action and had impacted/stressed the cells even at low concentrations. More specifically, for 10 and 50 μM GEM a mixed cell population with both apoptotic and non-apoptotic cells was observed while for 100 μM GEM almost all the population was apoptotic. Furthermore, as can be seen in Fig. 1D, the quantitative difference of the apoptotic induction was statistically significant for all drug concentrations.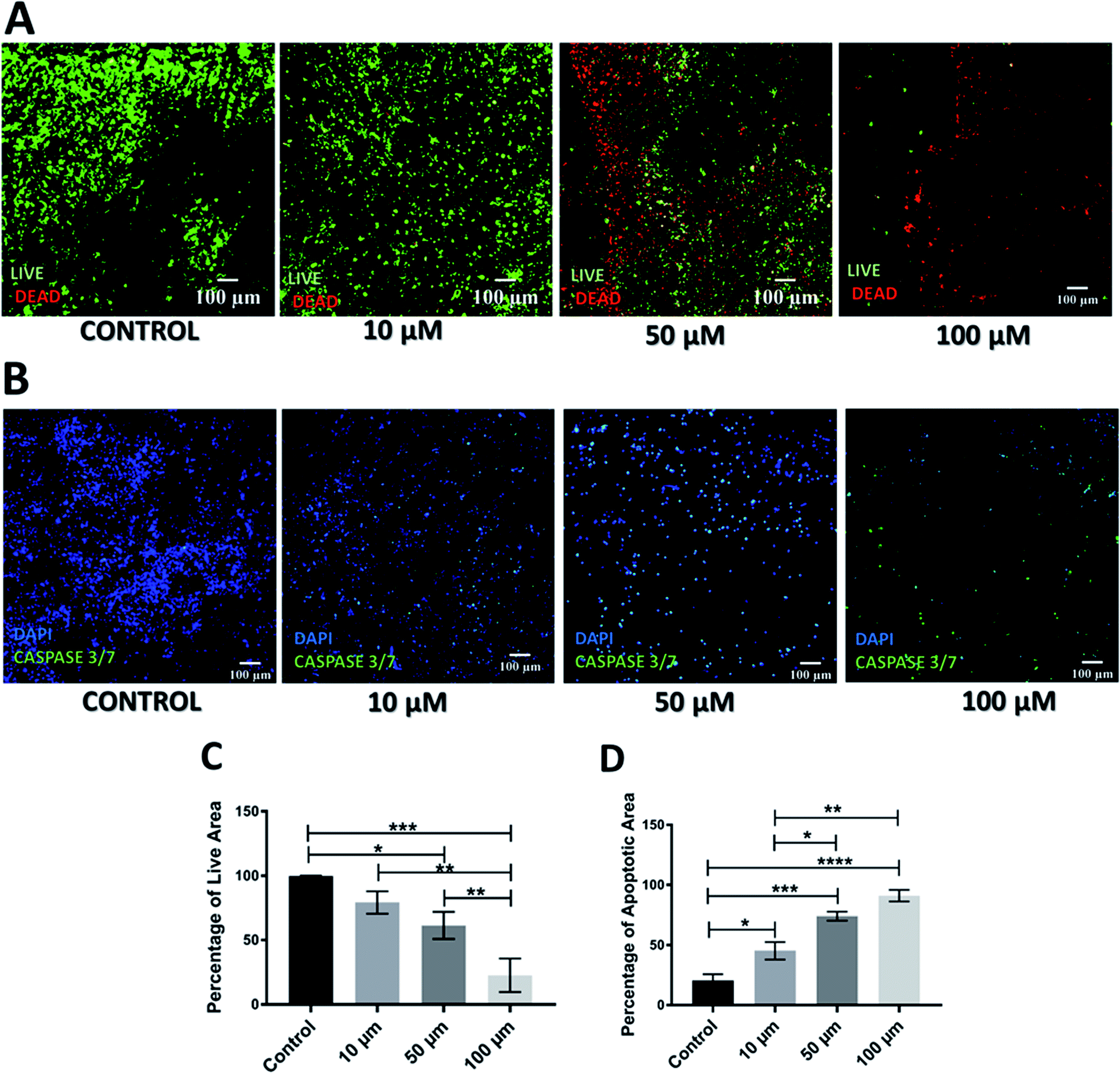 | ||
| Fig. 1 Effect of GEM in the scaffolds 24 h post-treatment. (A) Representative images for live-dead staining. (B) Representative image for apoptosis (caspase 3/7) staining. (C) Image analysis based quantification of live (green) image areas for different GEM concentrations. (D) Image analysis based quantification of apoptotic areas on scaffold sections for various GEM concentrations. Multiple images (≥3) and multiple scaffolds (≥3) were analysed the mean values were used here. | ||
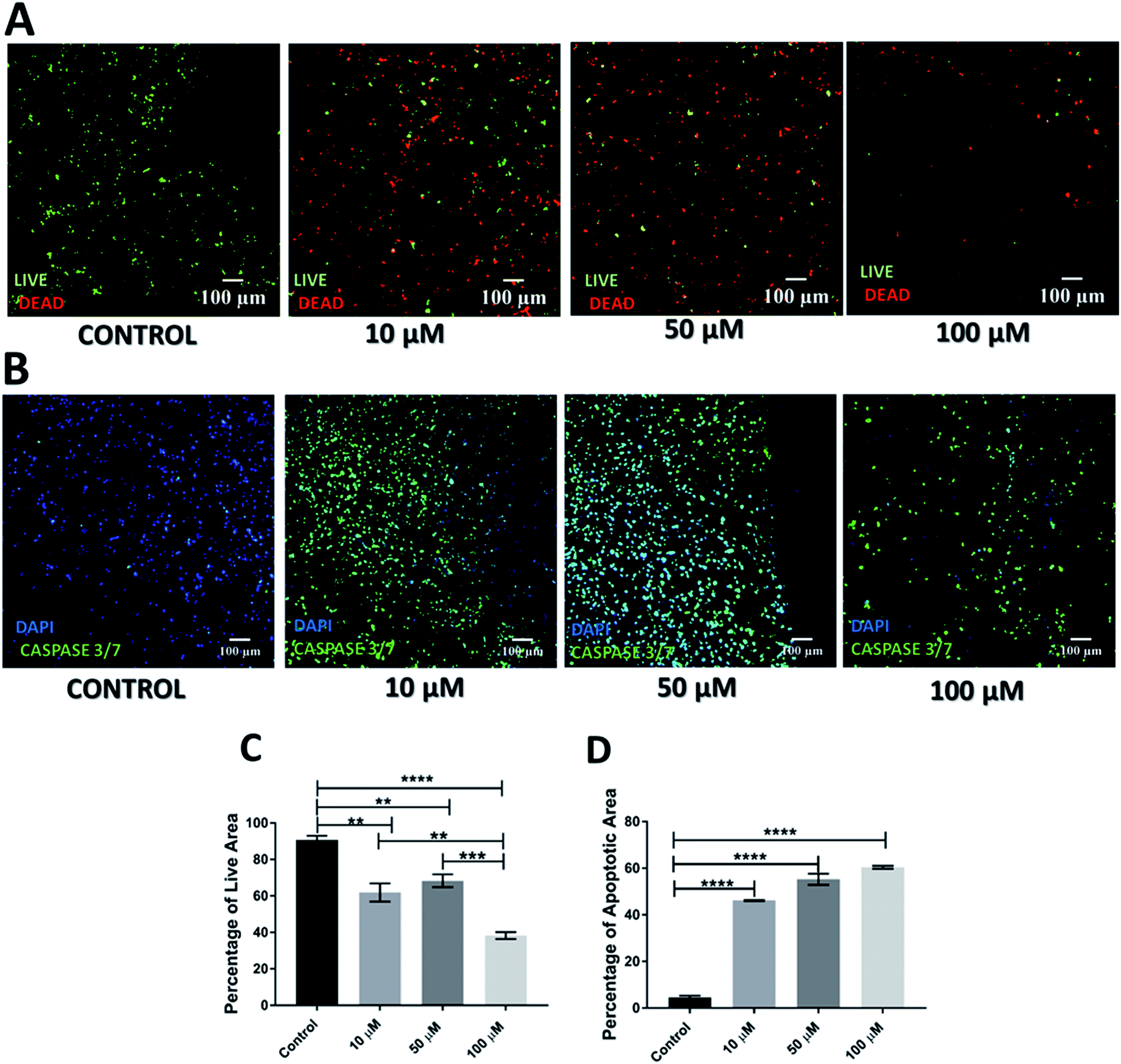 | ||
| Fig. 2 Effect of GEM in the scaffolds 17 days post-treatment. (A) Representative images for live-dead staining. (B) Representative image for apoptosis (caspase 3/7) staining. (C) Image analysis based quantification of live (green) image areas for different GEM concentrations. (D) Image analysis based quantification of apoptotic areas on scaffold sections for various GEM concentrations. Multiple images (≥3) and multiple scaffolds (≥3) were analysed the mean values were used here. | ||
The cell death and apoptosis levels were also assessed 17 days post treatment to evaluate the long-term response to the drug in the scaffolds (Fig. 2). It should be stated that this is the first time that such an extended culturing period has been reported in vitro. Interestingly the control remained very high in viability (Fig. 2A) and low in apoptotic levels (Fig. 2C) after almost 2 months in culture. The live-dead staining (Fig. 2A) and quantification (Fig. 2C) showed a significant decrease in green/live image areas of the scaffold sections for all tested concentration of GEM as compared to untreated scaffolds in contrast to the 24 h post-treatment response which was significant only for the higher drug doses (50 and 100 μM GEM). The apoptosis induction was significantly higher as compared to the untreated scaffolds for all drug concentrations under study with no significant differences for the two higher drug doses, for which the majority of the population was apoptotic (Fig. 2B and D). Overall, these results indicate the importance of having a system that enables long term post-treatment drug response monitoring.
3.2 Radiotherapy assessment in the 3D scaffolds
Application of radiotherapy (X-rays) in the 3D scaffolds was carried out at the Royal Surrey County Hospital, NHS Foundation Trust (see Section 2.3.2). More specifically, the scaffolds were exposed to one-off radiation treatment for doses of 2 Gy, 6 Gy and 8 Gy. As described for the chemotherapy experiments, to assess the short and relatively long term responses to radiation, the scaffolds were analysed 24 h and 17 days post-treatment. More specifically, sectioning, staining and imaging of the scaffolds (as described in Sections 2.4–2.7) with CLSM enabled the spatial assessment and quantification of the radiation responses on the cell viability and apoptosis induction 24 h (Fig. 3) and 17 days (Fig. 4) post-treatment.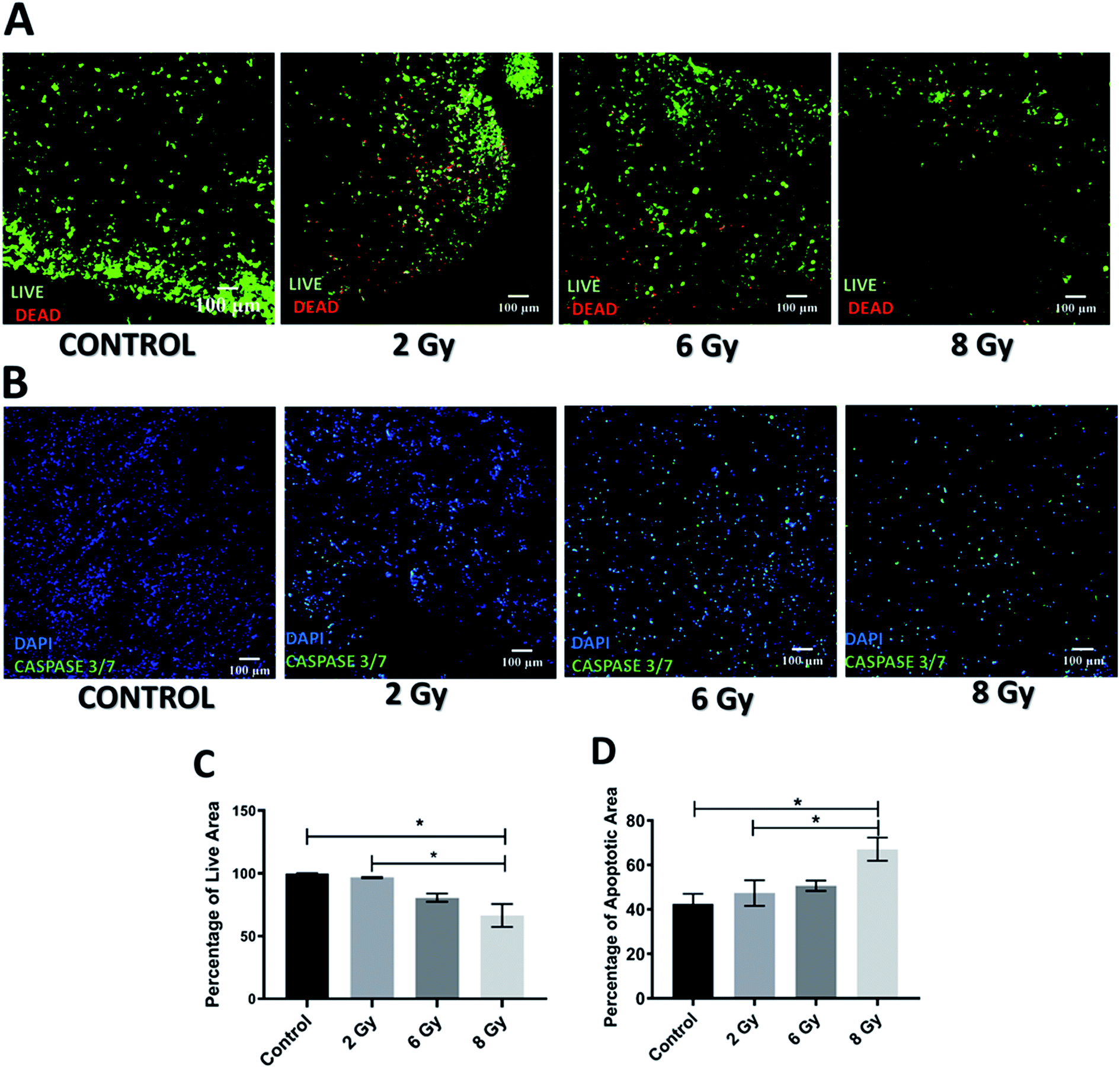 | ||
| Fig. 3 Effect of radiotherapy in the scaffolds 24 h post-treatment. (A) Representative images for live-dead staining. (B) Representative image for apoptosis (caspase 3/7) staining. (C) Image analysis based quantification of live (green) image areas for different dosage of radiation. (D) Image analysis based quantification of apoptotic areas cells on scaffold sections for various dosages of radiation. Multiple images (≥3) and multiple scaffolds (≥3) were analysed the mean values were used here. | ||
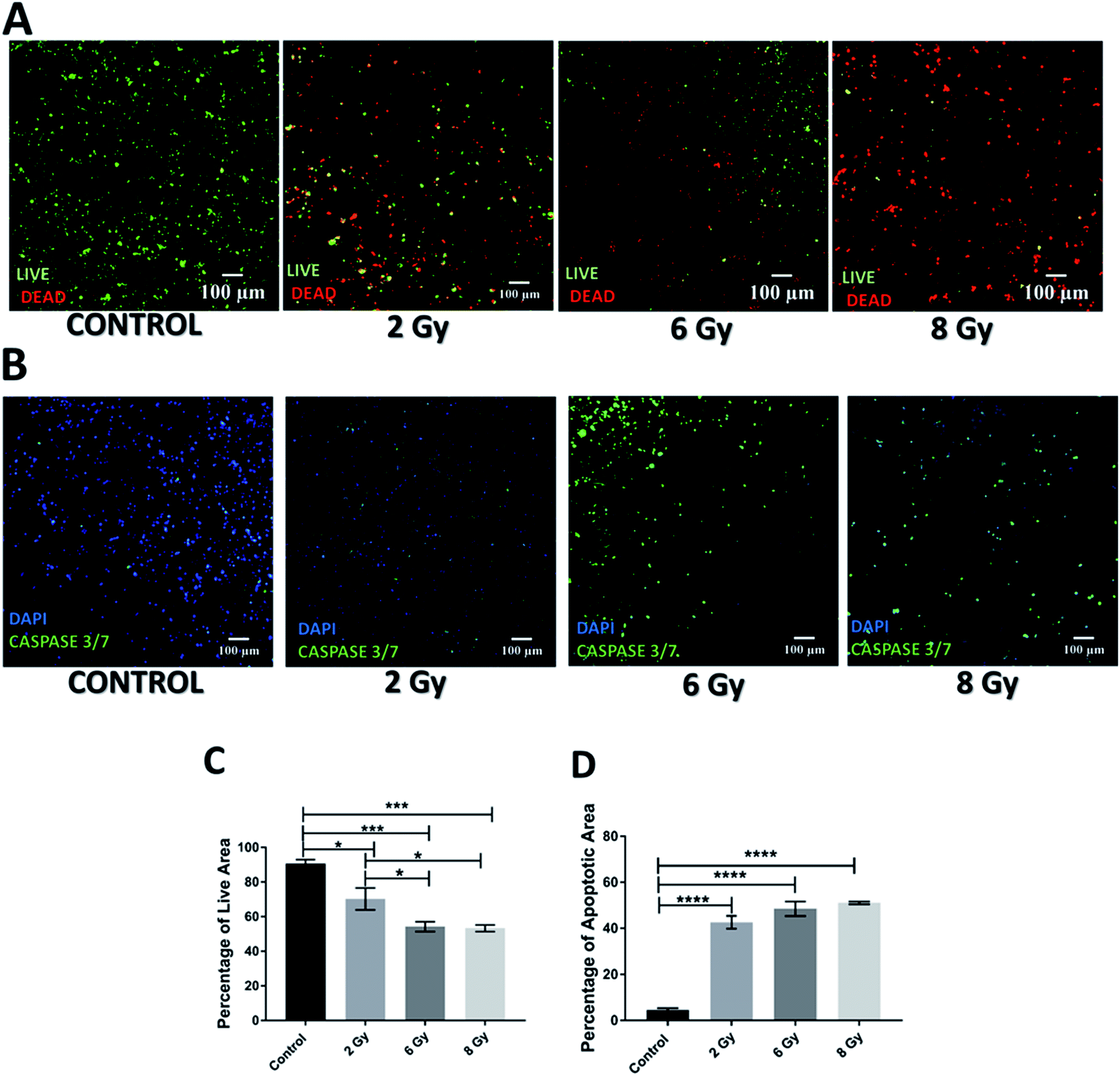 | ||
| Fig. 4 Effect of radiotherapy in the scaffolds 17 days post-treatment. (A) Representative images for live-dead staining. (B) Representative image for apoptosis (caspase 3/7) staining. (C) Image analysis based quantification of live (green) image areas for different dosage of radiation. (D) Image analysis based quantification of apoptotic areas cells on scaffold sections for various dosages of radiation. Multiple images (≥3) and multiple scaffolds (≥3) were analysed the mean values were used here. | ||
As can be seen on Fig. 3A and C, 24 h after radiation treatment, there was no significant impact from low dosages of radiation, i.e., 2 Gy and 6 Gy, on the viability while a significant decrease in viability was observed for the highest radiation dose, i.e., 8 Gy. Additionally, much more live cells were retained in the scaffolds after radiation treatment (Fig. 3A) as compared to the GEM treated samples (Fig. 1A). Similarly, the apoptosis (caspase 3/7 activity) assessment showed that only exposure to 8 Gy resulted in a significant increase in cell apoptosis within the scaffolds (Fig. 3B and D), in contrast to the effect of GEM, which led to high cell apoptosis even for the lowest drug concentration (Fig. 1A). Overall, these findings indicate that the immediate response (24 h post-treatment) to 10 μM GEM on the scaffolds was much higher than that of 2 Gy radiotherapy treatment, resulting in higher death and higher apoptosis.
However, the impact of radiation on the viability and apoptosis induction in the scaffolds was much more significant 17 days post-treatment (Fig. 4A and C). More specifically, scaffolds treated with radiation showed a dose dependent viability reduction. Moreover, scaffolds treated with 8 Gy radiation, were virtually empty suggesting extreme lethal dosage, the impact of which was realistically captured 17 days after treatment. In terms of apoptosis, a significant increase was observed for all treatment doses as compared to untreated scaffolds (Fig. 4B and D). This data suggests that unlike chemotherapy, the response to radiation on the PDAC scaffolds is not immediate but can be seen more clearly long-term; therefore, having a platform that allows long-term treatment monitoring can be very informative.
3.3 Combined chemotherapy and radiotherapy treatment in the scaffolds
Further to independent chemotherapy and radiotherapy treatments, a chemo-radiotherapy combinatorial treatment regime was tested in our 3D scaffolds. More specifically, a combination of 10 μM GEM and 6 Gy radiation was applied. These levels of treatment were selected based on our findings from the individual treatments, i.e., they were found to be independently efficient enough to induce cell death but not total death, therefore, their combination would allow the evaluation of potential synergies (Fig. 1–4). Indeed, as observed in Fig. 5 and 6, both 24 h and 17 days post treatment, the use of a chemo-radiotherapy combination showed a significantly higher cell death and apoptosis induction as compared to the independent treatments. This highlights the potential synergy of the different treatment methods in enhancing cell death and apoptosis induction both short and long term.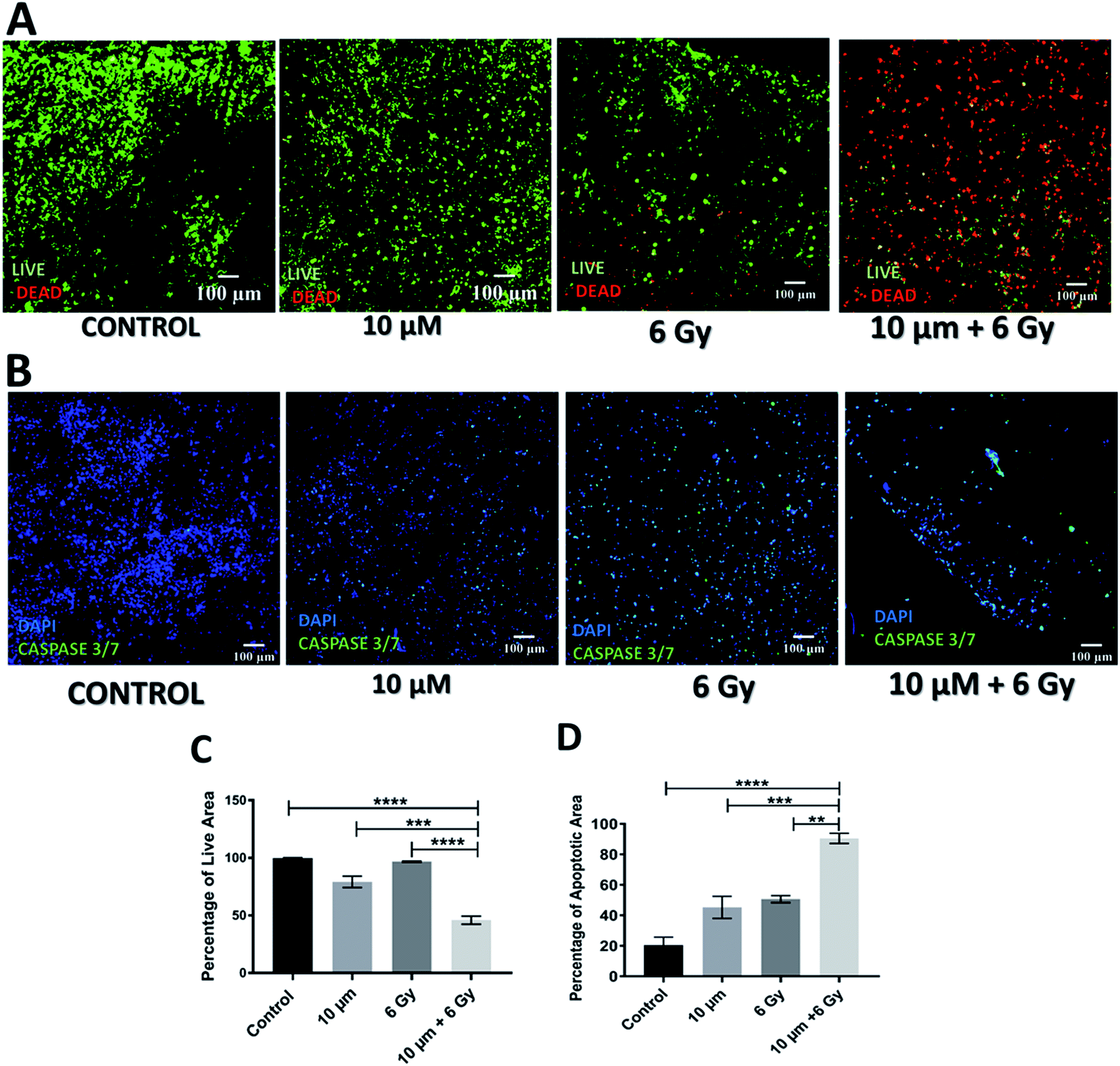 | ||
| Fig. 5 Effect of combinatorial treatment (GEM + radiation) approach 24 h post treatment. (A and B) Representative image of live-dead staining and caspase 3/7 staining on scaffold sections respectively. (C) Image analysis based quantification of percentage of live/green image area. (D) Image analysis based quantification of apoptotic area. Multiple images (≥3) and multiple scaffolds (≥3) were analysed the mean values were used here. | ||
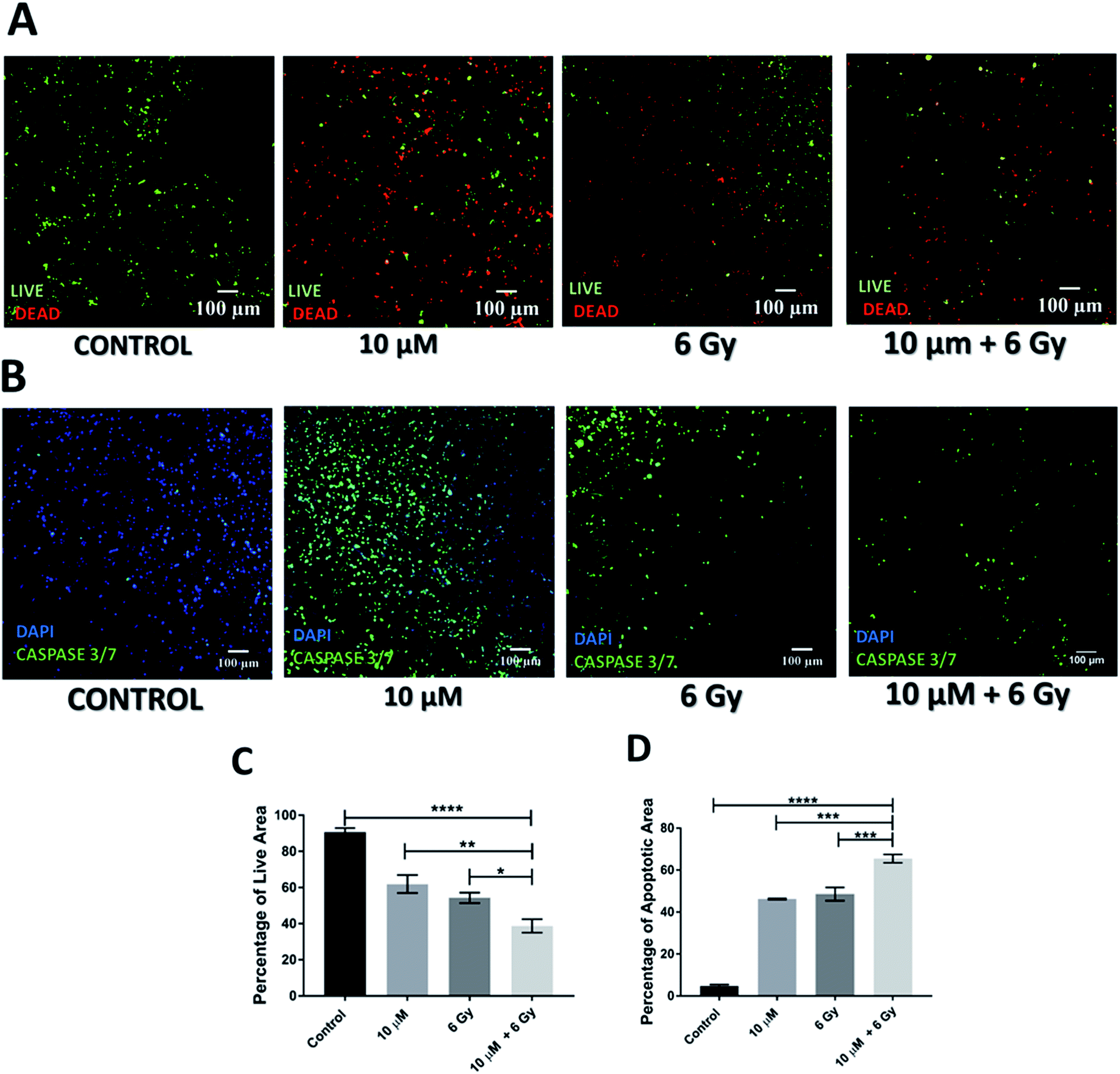 | ||
| Fig. 6 Effect of combinatorial treatment (GEM + radiation) approach 17 days post treatment. (A and B) Representative image of live-dead staining and caspase 3/7 staining on scaffold sections respectively. (C) Image analysis based quantification of percentage of live/green image area. (D) Image analysis based quantification of apoptotic area. Multiple images (≥3) and multiple scaffolds (≥3) were analysed the mean values were used here. | ||
4. Discussion
In the current work, treatment screening, i.e., chemotherapy, radiotherapy and a combination of the two were performed, for the first time, in a novel polymer scaffold based PDAC model. We have developed and recently published a PU based highly porous polymeric scaffold, decorated with ECM matrix features, i.e., fibronectin, for PDAC re-modelling. Due to the porosity and pore interconnectivity within the scaffolds, we have recently reported that pancreatic cancer cell lines remain alive for more than a month in culture, without the requirement of cell re-suspension, which is the longest reported culturing period in vitro. On week 4 of culture, the cells in the scaffold developed some in vivo like features such as the formation of dense cell masses, beginning of the secretion of ECM protein (collagen I), and some degree of oxidative stress. Furthermore, majority of the population was proliferative throughout the whole scaffold after a month in culture. It should be stated that a similar proliferation distribution within the tumour has been recently reported in a PDAC orthotopic mice model (Fig. 7A) Therefore, in this work we performed treatment screening on week 4 culture of PANC-1 cells in the scaffolds, as at this time point the scaffold had significant in vivo like features. More specifically, the scaffolds were treated with chemotherapy (GEM), radiotherapy (X-rays) and a combination of both. Sectioning, staining and image analysis of multiple scaffolds pre and post-treatment enabled us to spatially map the viable as well as the apoptotic population for various conditions both short-term, i.e., 24 h post-treatment (Fig. 1, 3, and 5), and relatively long term, i.e., 17 days post-treatment (Fig. 2, 4, and 6). To the best of our knowledge, we are the first to report in vitro such long post-treatment observations/monitoring, similar to the time-frame employed in animal studies2,12,14,15,17,23,55 and in patients.56,69,81 In contrast, most treatment screening studies in in vitro models to date have post-treatment assessment time-frame of a maximum of one week and they are mainly conducted for chemotherapy.33–36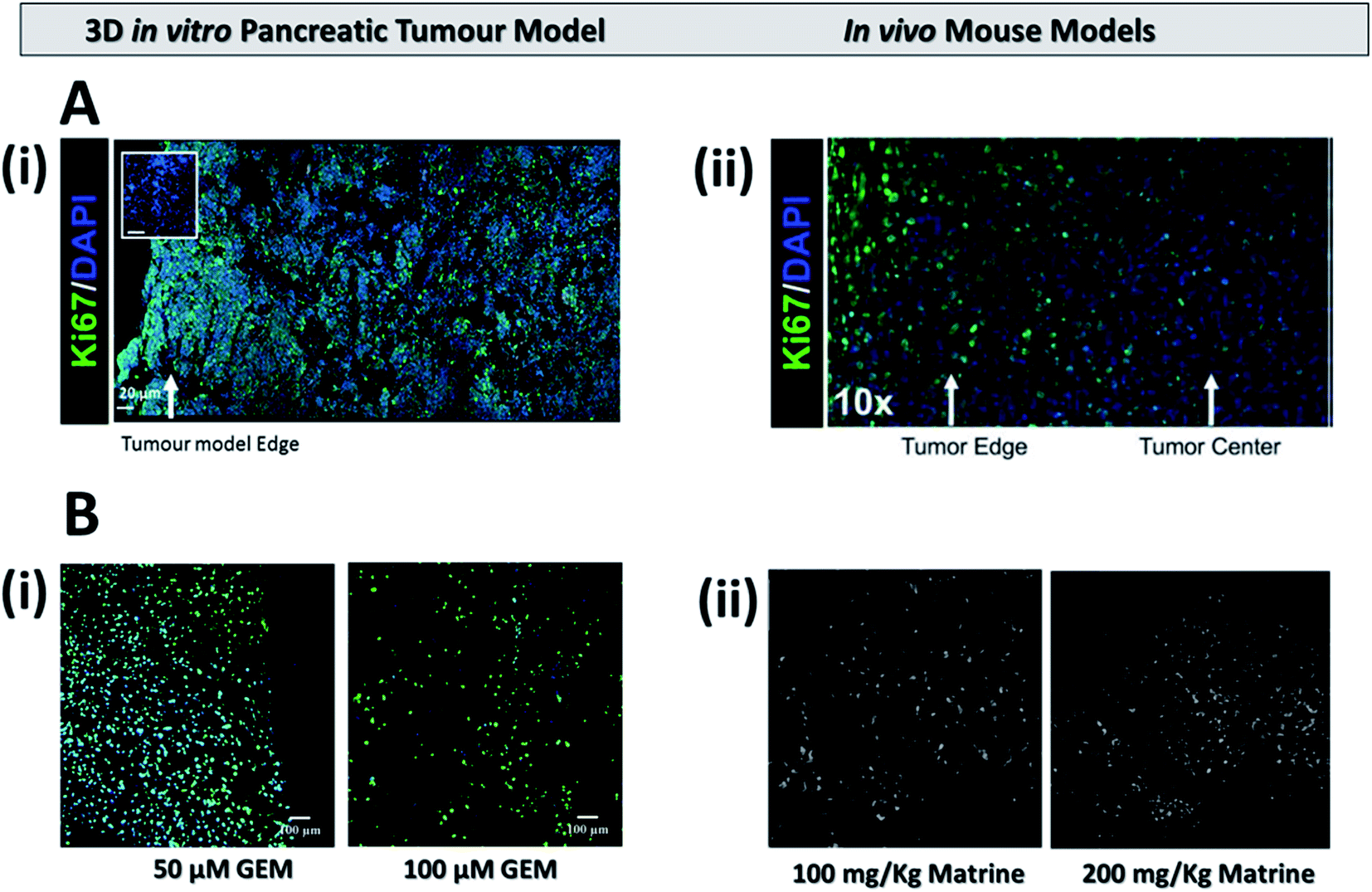 | ||
| Fig. 7 (A) Representative immunofluorescent staining for cell distribution (DAPI) and proliferation (Ki-67) (i) on our 3D scaffold based pancreatic cancer model (adapted with permission from Totti et al., 2018)7 and (ii) on an mouse model (adapted with permission from Erstad et al., 2018).2 (B) Apoptosis expression (i) on the 3D pancreatic cancer model 17 days post treatment for 50 μM (left) and 100 μM (right) of GEM and (ii) on a mouse model 18 days post treatment for similar doses of the drug Matrine (adapted with permission from Liu et al., 2010).17 | ||
4.1 Chemotherapy treatment in the scaffolds
As previously mentioned, (see Sections 2.3.1 and 3.1), the chemotherapeutic agent Gemcitabine (GEM) was used for conducting chemotherapy treatment on our scaffolds. GEM is a drug that is used for PDAC treatment clinically56,69 as well as in various in vivo12,14,15,18 and in vitro studies.12,19,33,34,72,75,91Most in vitro studies are in 2D systems and very few in 3D, mostly spheroid type models. Furthermore, most in vitro studies (2D and 3D) are relatively short term and hence do not capture the long-term response to various drugs, unlike in vivo studies that are much longer, i.e., several weeks, both for animals and patients. For example, Lee et al.33 (2018), studied the impact of several drugs (gemcitabine, paclitaxel and oxaliplatin) on the viability of PANC-1 cells in a 3D spheroid system and showed a dose dependent decrease in cell viability for most of the drugs, 72 h post treatment. Longati et al., (2013) treated PANC-1 and BxPC-3 cells in 2D as well as 3D spheroids with GEM and performed post treatment analysis after 7 days. Cells in 3D were more chemo-resistant than in the 2D culture.34 Lazzari et al., (2018) monitored the viability of GEM treated pancreatic cancer spheroids for 72 hours and reported that at 10 and 50 μM of GEM the tumour spheroid viability loss was 20% and 30% respectively, highlighting the dose dependent nature of GEM's action on PDAC.32 Ki et al., investigated the efficiency of 1 μM of GEM through monitoring of apoptosis (caspase 3/7 expression) in a hydrogel based pancreatic cancer model. The caspase 3/7 activation was 6-fold higher as compared to the untreated hydrogels 4 days after treatment.52 However, to the best of our knowledge scaffold based in vitro models of PDAC have not been used for treatment studies to date.
In contrast to reported 3D in vitro studies which last a maximum of one week post-treatment, animal studies and clinical trials are carried out for a more extended time period, i.e., several weeks highlighting a key drawback for most in vitro 3D models. For example, Awasthi et al., (2019) studied orthotopic tumours of PDAC cell lines in mice for a total of 4 weeks which included 2 weeks post treatment observation.14 Liu et al., (2010) studied the effect (growth inhibition and gene expression of apoptotic related pathways) of the drug Matrine on PDAC xenografts in a mouse model for about 3 weeks post-treatment.17 The longest reported mouse study was carried out by Krzykawska-Serda et al., in 2018 wherein they carried out analysis 4 weeks post treatment with the total experiment running for 10–12 weeks.77 In a similar fashion, clinical trials are also carried out for a longer time frame with drug doses distributed over months to avoid excessive toxicity.16,56,58,69
In our work, we studied the effect of 10, 50 and 100 μM of GEM on the viability and apoptosis induction of pancreatic cancer cells in polymer based scaffolds short term (24 hours post treatment) and relatively long term (17 days post treatment), the latter being similar to the post-treatment pattern followed in animal studies.2,12,14,15,17,23,55 Due to the high cell death at 100 μM GEM, i.e., less than 50% viability was observed 24 h post-treatment (Fig. 1), we did not test higher drug concentrations. Similar to other reported in vitro studies for spheroids/hydrogels, the effect of GEM on the viability and apoptosis induction (caspase 3/7 activation) was generally dose and/or day dependent. More specifically, 24 h post-treatment, the viability of the scaffolds did not significantly decrease as compared to the control for 10 μM of GEM but higher death (including emptier scaffold sections due to detachment of dead cells from the scaffolds) were observed for 50 and 100 μM (Fig. 1A and C) while the induction of apoptosis followed a more clear drug dose-dependent trend (Fig. 1B and D). In contrast, 17 days post treatment, an impact of the drug on the viability was observed for all concentrations under study in a clear dose dependent manner (Fig. 2). We also observed significantly higher loss of cell viability for 100 μM GEM in comparison to 10 μM and 50 μM GEM highlighting further dose dependence for the drug (Fig. 2A and C). In terms of apoptosis induction, there was a significant induction of apoptosis for all drug concentrations under study as compared to the untreated scaffold (the apoptosis level of which remained very low even after 2 months in culture). The apoptosis induction was significantly higher for higher drug doses (50 μM and 100 μM GEM) as compared to the low drug dose used (10 μM) (Fig. 2B and C). As mentioned previously, we are the first to perform long-term post-treatment monitoring in vitro, therefore, there is no existing literature to compare our results in that length of time-frame. However, the cell death and apoptotic patterns we observe are similar to PDAC mouse studies (Fig. 7B). More specifically, as can be seen in Fig. 7B(ii), approximately 3 weeks post-treatment monitoring of apoptosis in mice resulted in a similar dose dependent increase in apoptotic induction17 to the one observed in our system, highlighting the great potential of our scaffold for the performance of chemotherapy screening in a more realistic time regime which is closer to the animal23,77,93 and human56–58,69 treatment situation.
4.2 Radiotherapy treatment in the scaffolds
Generally, there are limited radiotherapy studies in PDAC, even though it is a common treatment option, especially for patients with advanced disease.58,69 Tuli et al., studied the cell viability reduction and the DNA damage of PDAC in response to various doses of radiation (0–10 Gy) in a 2D mono-layer culture system and reported a radiation dose dependent decrease in cell viability along with instant DNA damage for the highest radiation dosage.55 Giagkousiklidis et al., (2007) also reported in a 2D system that XIAP inhibition increased the sensitivity of PDAC cell lines to radiation (10 Gy, 20 Gy) 96 h post treatment.94 To the best of our knowledge, there are only two studies that explore the radiation efficacy utilizing 3D pancreatic cancer models in the form of spheroids and for a relatively short time duration, i.e. a maximum of 7 days.49,50 Hehlgans et al. (2009) used a 3D spheroid model to study the response of Caveolin-1 (membrane protein) knockdown on the PDAC MiaPaCa2 cell line response to radiation (0–6 Gy)50 and reported an increased sensitivity to radiation of the Caveolin-1 knockdown in 3D as compared to the wildtype. Furthermore, a 12 days study on tumour spheroids by Al-Ramadan et al. (2018), showed that spheroids of the PDAC cell line BON1 exhibited a dose dependent (0–6 Gy) increase in the apoptotic maker caspase 7 days post treatment.49 Small animal models (mice) have also been used by different groups to study the effects of radiation on PDAC.55,95,96 For example, Tuli et al., (2014) reported that a mouse model developed with orthotopic implant of PDAC cells, was able to survive for up to 39 days post treatment (5 Gy, single fraction radiation).Based on the radiation dosages reported,49,50,55 we performed radiation with X-rays for doses of 2, 6 and 8 Gy (see Sections 2.3.2 and 3.2). Similarly to published data for 2D,55 3D in vitro50 and in vivo55 systems, we generally observed a dose/time dependent trend on cell death and apoptotic induction (caspase 3/7 expression) throughout the assessment period (24 h and 17 days post-treatment) (Fig. 3 and 4). More specifically, a significant increase in apoptosis and death 24 h post-treatment was only observed at 8 Gy (Fig. 3), in contrast to GEM, which resulted in a significant increase in apoptosis 24 h post-treatment for all drug doses under study (Fig. 1). The enhanced efficiency of chemotherapy compared to radiotherapy for pancreatic cancer has also been reported by European clinical trials for pancreatic cancer. Namely, the European Study Group for Pancreatic Cancer (ESPAC) assessed the efficiency of systemic chemotherapy and radiation for pancreatic cancer patients and reported the benefit of chemotherapy in comparison to radiotherapy.82,97
In our work, 17 days post-treatment, a significantly higher impact on the viability reduction and apoptosis induction was observed for all radiation doses under study, as compared to the untreated scaffolds (Fig. 4). The increased radio-resistance we observe 24 h post-treatment is in accordance with in vitro studies in other systems for similar post-treatment time frames.49,50 However, our long post-treatment observations clearly show that radiation does influence the PDAC scaffolds, but not immediately.
Overall, we are the first to report long-term radiation responses in a novel polymer based scaffolding system. Having a low-cost robust screening system for PDAC radiotherapy screening is particularly important not only because radiation on PDAC has been understudied, but furthermore, due to the limited availability of preclinical (animal) facilities for standard radiotherapy screening and the complete lack of animal facilities to screen novel radiotherapy regimes. More specifically, novel radiotherapy regimes, e.g., with protons or in the presence of high magnetic fields as with MRI-linear accelerators,98–100 are generally understudied and, considering the output of the ESPAC trial and others for PDAC (see above), novel radiation approaches could be more efficient. Indeed, there are some clinical trials on proton therapy which show promising results for PDAC.101–104 Our platform could help accelerate those studies in a reproducible in vitro environment which mimics closer the in vivo situation, as shown in Fig. 7.
4.3 Assessment of combination of chemotherapy and radiotherapy in the scaffolds
As previously mentioned, in recent years, clinical trials are focusing on the potential synergistic effect of radiotherapy and chemotherapy but with contradictory results.82,97,105,106 Eladawei et al., (2017) have shown in their clinical trial that a combined chemo-radiotherapy is more effective than chemotherapy alone on patients who have undergone surgery, although some level of toxicity was observed.105 In contrast, Neoptolemos et al., (2001) reported in their clinical trial that chemo-radiotherapy showed no added benefit for patients in comparison to chemotherapy alone.82 To the best of our knowledge, very limited studies have been carried out in vitro to understand the response of chemoradiotherapy on PDAC.78,79 Weiss et al., (2003) reported that combinatory treatment resulted in significantly increased cell death and DNA damage for BxPC-3 PDAC cell lines, 24 h post treatment in a 2D system.79 Similarly, Mukubou et al., (2010) carried out both in vitro (2D) and in vivo (mouse model) combinatorial chemo-radiotherapy treatment involving GEM and radiation.78 A dose dependent increase in apoptosis for the combinatorial treatment regime along with increased cell death was observed in vitro, 7 day post-treatment (GEM concentrations of 0, 65 and 135 nM with radiation between 0–8 Gy). In vivo, they observed a decreased tumour volume for a combinatorial treatment (GEM = 300 mg per kg per week and single dose of 5 Gy radiation) in comparison to chemotherapy or radiotherapy alone, 40 days post treatment.In this work, as described in Sections 2.3.3 and 3.3 above, we performed a chemo-radiotherapy regime with 10 μM GEM and 6 Gy radiation and performed short (24 h, Fig. 5) and relatively long term (17 days, Fig. 6) analysis of the treatment response. Similarly to reported animal studies,78 in vitro studies78,79 and clinical studies,82,105,106 for both short and long post-treatment time points of analysis, our results show a significantly lower viability and higher apoptosis in our scaffolds as compared to the individual/independent treatment regimes.
It should be stated that all reported in vitro studies have a different time-regime, i.e., our long-term study is more aligned with the time-frame of animal experiments2,11,14,17,23,55,78,79,95 (Fig. 7) and clinical trials,56,58,69,73,105,106 pointing to the great potential of our scaffold in providing a reproducible, low cost powerful tool for replacing animals in long-term realistic treatment screening for PDAC.
5. Conclusions
Overall, in this work we performed treatment screening, i.e., chemotherapy, radiotherapy and a combination of the two, on our recently published scaffold based PDAC model.7 We were able to capture short (24 h) and long-term (17 days) post-treatment responses of (i) the chemotherapeutic drug Gemcitabine and (ii) radiation (X-rays) on the viability reduction and apoptosis induction in the scaffolds. We observed a change of the impact of the treatment depending on the time-frame, especially for radiation for which the PDAC scaffolds were resistant 24 h post-treatment but responded much more after 17 days post-treatment. It should be stated that, to the best of our knowledge, we are the first to report a viable PDAC culture in a scaffold for more than 2 months (without the need of resuspension) and the first to perform relatively long-term (17 days) post-treatment observations in vitro. The latter is particularly important as this time-frame is much closer to animal studies and to the patient time-frame treatment regimes, highlighting that our scaffold system has great potential to be used as a good animal free model for screening of PDAC treatments including treatment personalisation. Current work of our group focuses on the incorporation of (i) biological complexity (co-culture of multiple cell types) and (ii) perfusion/interstitial flow mimicry in the PDAC model for a better recapitulation of the in vivo niche. Future work will focus on treatment screening in PDAC models of higher complexity, to capture more accurately the impact of multiple compartments of the tumour tissue on the response and potential resistance to treatment.Conflicts of interest
The authors declare that they have no conflict of interest.Acknowledgements
This work was supported by the Chemical & Process Engineering Department of the University of Surrey, Impact Acceleration Grant (IAA-KN9149C) from the University of Surrey, IAA–EPSRC Grant (RN0281J) and the Royal Society. P.G. is supported by Commonwealth Rutherford Post-Doctoral Fellowship.References
- American Cancer Society, Cancer Facts & Figures 2018, Atlanta, 2018 Search PubMed.
- D. J. Erstad, M. Sojoodi, M. S. Taylor, S. Ghoshal, A. A. Razavi, K. A. Graham-O'Regan, N. Bardeesy, C. R. Ferrone, M. Lanuti and P. Caravan, Dis. Models Mech., 2018, 11, dmm034793 CrossRef PubMed.
- P. Rawla, T. Sunkara and V. Gaduputi, World J. Oncol., 2019, 10, 10–27 CrossRef PubMed.
- S. Totti, S. I. Vernardis, L. Meira, P. A. Pérez-Mancera, E. Costello, W. Greenhalf, D. Palmer, J. Neoptolemos, A. Mantalaris and E. G. Velliou, Drug discovery today, 2017, 22, 690–701 CrossRef CAS PubMed.
- R. L. Siegel, K. D. Miller and A. Jemal, Ca-Cancer J. Clin., 2018, 68, 7–30 CrossRef PubMed.
- J. Kleeff, M. Korc, M. Apte, C. La Vecchia, C. D. Johnson, A. V. Biankin, R. E. Neale, M. Tempero, D. A. Tuveson and R. H. Hruban, Nat. Rev. Dis. Primers, 2016, 2, 16022 CrossRef PubMed.
- S. Totti, M. C. Allenby, S. B. Dos Santos, A. Mantalaris and E. G. Velliou, RSC Adv., 2018, 8, 20928–20940 RSC.
- D. Ansari, H. Friess, M. Bauden, J. Samnegård and R. Andersson, Oncotarget, 2018, 9, 6644 Search PubMed.
- S. Chand, K. O'Hayer, F. F. Blanco, J. M. Winter and J. R. Brody, Int. J. Biol. Sci., 2016, 12, 273 CrossRef CAS PubMed.
- N. Awasthi, M. A. Schwarz and R. E. Schwarz, Cancer Chemother. Pharmacol., 2011, 68, 571–582 CrossRef CAS PubMed.
- D. I. Dovzhanskiy, S. M. Arnold, T. Hackert, I. Oehme, O. Witt, K. Felix, N. Giese and J. Werner, BMC Cancer, 2012, 12, 226 CrossRef CAS PubMed.
- K. B. Harikumar, A. B. Kunnumakkara, G. Sethi, P. Diagaradjane, P. Anand, M. K. Pandey, J. Gelovani, S. Krishnan, S. Guha and B. B. Aggarwal, Int. J. Cancer, 2010, 127, 257–268 CAS.
- H. Onishi, Y. Morifuji, M. Kai, K. Suyama, H. Iwasaki and M. Katano, Cancer Sci., 2012, 103, 1272–1279 CrossRef CAS PubMed.
- N. Awasthi, D. Kronenberger, A. Stefaniak, M. S. Hassan, U. von Holzen, M. A. Schwarz and R. E. Schwarz, Cancer Lett., 2019, 459, 41–49 CrossRef CAS PubMed.
- A. Courtin, F. M. Richards, T. E. Bapiro, J. L. Bramhall, A. Neesse, N. Cook, B.-F. Krippendorff, D. A. Tuveson and D. I. Jodrell, PLoS One, 2013, 8, e67330 CrossRef CAS PubMed.
- V. Heinemann, M. Haas and S. Boeck, Ann. Oncol., 2013, 24, 2484–2492 CrossRef CAS PubMed.
- T. Liu, Y. Song, H. Chen, S. Pan and X. Sun, Biol. Pharm. Bull., 2010, 33, 1740–1745 CrossRef CAS PubMed.
- W. W. Tseng, D. Winer, J. A. Kenkel, O. Choi, A. H. Shain, J. R. Pollack, R. French, A. M. Lowy and E. G. Engleman, Clin. Cancer Res., 2010, 16, 3684–3695 CrossRef CAS PubMed.
- L. R. Jaidev, U. M. Krishnan and S. Sethuraman, Mater. Sci. Eng. C, 2015, 47, 40–47 CrossRef CAS PubMed.
- L. K. Chim and A. G. Mikos, Curr. Opin. Biomed. Eng., 2018, 6, 42–50 CrossRef PubMed.
- A. F. Adcock, G. Trivedi, R. Edmondson, C. Spearman and L. Yang, J. Anal. Bioanal. Tech., 2015, 6, 2 Search PubMed.
- G. Feldmann, S. Rauenzahn and A. Maitra, Expert Opin. Drug Discovery, 2009, 4, 429–443 CrossRef CAS PubMed.
- P. A. Pérez-Mancera, C. Guerra, M. Barbacid and D. A. Tuveson, Gastroenterology, 2012, 142, 1079–1092 CrossRef PubMed.
- T. J. Humpton, B. Alagesan, G. M. DeNicola, D. Lu, G. N. Yordanov, C. S. Leonhardt, M. A. Yao, P. Alagesan, M. N. Zaatari and Y. Park, Cancer Discov., 2019, 9, 1268–1287 CrossRef PubMed.
- E. J. Poulin, A. K. Bera, J. Lu, Y.-J. Lin, S. D. Strasser, J. A. Paulo, T. Q. Huang, C. Morales, W. Yan and J. Cook, Cancer Discov., 2019, 9, 738–755 CrossRef PubMed.
- L. Ireland, A. Santos, M. S. Ahmed, C. Rainer, S. R. Nielsen, V. Quaranta, U. Weyer-Czernilofsky, D. D. Engle, P. A. Perez-Mancera and S. E. Coupland, Cancer Res., 2016, 76, 6851–6863 CrossRef CAS PubMed.
- C. Bermejo-Rodríguez and P. A. Pérez-Mancera, Curr. Opin. Biotechnol., 2015, 35, 103–110 CrossRef PubMed.
- X. Yan, L. Zhou, Z. Wu, X. Wang, X. Chen, F. Yang, Y. Guo, M. Wu, Y. Chen, W. Li, J. Wang and Y. Du, Biomaterials, 2019, 198, 167–179 CrossRef CAS PubMed.
- S. J. Coleman, J. Watt, P. Arumugam, L. Solaini, E. Carapuca, M. Ghallab, R. P. Grose and H. M. Kocher, World J. Gastroenterol., 2014, 20, 8471 CrossRef PubMed.
- F. Di Maggio, P. Arumugam, F. R. Delvecchio, S. Batista, T. Lechertier, K. Hodivala-Dilke and H. M. Kocher, Pancreatology, 2016, 16, 995–1004 CrossRef CAS PubMed.
- F. E. Froeling, T. A. Mirza, R. M. Feakins, A. Seedhar, G. Elia, I. R. Hart and H. M. Kocher, Am. J. Pathol., 2009, 175, 636–648 CrossRef CAS PubMed.
- G. Lazzari, V. Nicolas, M. Matsusaki, M. Akashi, P. Couvreur and S. Mura, Acta Biomater., 2018, 78, 296–307 CrossRef CAS PubMed.
- J.-H. Lee, S.-K. Kim, I. A. Khawar, S.-Y. Jeong, S. Chung and H.-J. Kuh, J. Exp. Clin. Cancer Res., 2018, 37, 4 CrossRef PubMed.
- P. Longati, X. Jia, J. Eimer, A. Wagman, M.-R. Witt, S. Rehnmark, C. Verbeke, R. Toftgård, M. Löhr and R. L. Heuchel, BMC Cancer, 2013, 13, 95 CrossRef CAS PubMed.
- Z. Wen, Q. Liao, Y. Hu, L. You, L. Zhou and Y. Zhao, Braz. J. Med. Biol. Res., 2013, 46, 634–642 CrossRef CAS PubMed.
- S.-E. Yeon, S.-H. Lee, S. W. Nam, I.-H. Oh, J. Lee and H.-J. Kuh, PLoS One, 2013, 8, e73345 CrossRef CAS PubMed.
- N. Mauro, F. Chiellini, C. Bartoli, M. Gazzarri, M. Laus, D. Antonioli, P. Griffiths, A. Manfredi, E. Ranucci and P. Ferruti, J. Tissue Eng. Regener. Med., 2017, 11, 2164–2175 CrossRef CAS PubMed.
- M. G. Gandolfi, F. Zamparini, M. Degli Esposti, F. Chiellini, F. Fava, P. Fabbri, P. Taddei and C. Prati, Mater. Sci. Eng. C, 2019, 102, 341–361 CrossRef CAS PubMed.
- C. Ricci, C. Mota, S. Moscato, D. D'Alessandro, S. Ugel, S. Sartoris, V. Bronte, U. Boggi, D. Campani, N. Funel, L. Moroni and S. Danti, Biomatter, 2014, 4, e955386 CrossRef PubMed.
- C. Ricci, S. Danti, N. Funel, S. Moscato, E. Pugliesi, D. D'alessandro, R. Pini, D. Campani and S. Berrettini, Front. Bioeng. Biotechnol., 2016 DOI:10.3389/conf.FBIOE.2016.01.01674.
- S. Danti, L. P. Serino, D. D'Alessandro, S. Moscato, S. Danti, L. Trombi, D. Dinucci, F. Chiellini, A. Pietrabissa and M. Lisanti, Tissue Eng., Part C, 2013, 19, 911–924 CrossRef CAS PubMed.
- S. Totti, K. W. Ng, L. Dale, G. Lian, T. Chen and E. G. Velliou, Sens. Actuators, B, 2019, 296, 126652 CrossRef CAS.
- B. Matta-Domjan, A. King, S. Totti, C. Matta, G. Dover, P. Martinez, A. Zakhidov, R. La Ragione, H. Macedo, I. Jurewicz, A. Dalton and E. G. Velliou, J. Biomed. Mater. Res., Part B, 2018, 106, 1637–1644 CrossRef CAS PubMed.
- S. F. Boj, C.-I. Hwang, L. A. Baker, I. I. C. Chio, D. D. Engle, V. Corbo, M. Jager, M. Ponz-Sarvise, H. Tiriac and M. S. Spector, Cell, 2015, 160, 324–338 CrossRef CAS PubMed.
- S. F. Boj, C.-I. Hwang, L. A. Baker, D. D. Engle, D. A. Tuveson and H. Clevers, Mol. Cell. Oncol., 2016, 3, e1014757 CrossRef.
- F. Chiellini, D. Puppi, A. M. Piras, A. Morelli, C. Bartoli and C. Migone, RSC Adv., 2016, 6, 54226–54235 RSC.
- V. Angeloni, N. Contessi, C. De Marco, S. Bertoldi, M. C. Tanzi, M. G. Daidone and S. Farè, Acta Biomater., 2017, 63, 306–316 CrossRef CAS PubMed.
- S. S. Suner, S. Demirci, B. Yetiskin, R. Fakhrullin, E. Naumenko, O. Okay, R. S. Ayyala and N. Sahiner, Int. J. Biol. Macromol., 2019, 130, 627–635 CrossRef CAS PubMed.
- A. Al-Ramadan, A. C. Mortensen, J. Carlsson and M. V. Nestor, Oncol. Lett., 2018, 15, 3008–3016 Search PubMed.
- S. Hehlgans, I. Eke, K. Storch, M. Haase, G. B. Baretton and N. Cordes, Radiother. Oncol., 2009, 92, 362–370 CrossRef CAS.
- L. Huang, A. Holtzinger, I. Jagan, M. BeGora, I. Lohse, N. Ngai, C. Nostro, R. Wang, L. B. Muthuswamy and H. C. Crawford, Nat. Med., 2015, 21, 1364 CrossRef CAS.
- C. S. Ki, T.-Y. Lin, M. Korc and C.-C. Lin, Biomaterials, 2014, 35, 9668–9677 CrossRef CAS PubMed.
- S. Pradhan, J. M. Clary, D. Seliktar and E. A. Lipke, Biomaterials, 2017, 115, 141–154 CrossRef CAS PubMed.
- L. Moreira, B. Bakir, P. Chatterji, Z. Dantes, M. Reichert and A. K. Rustgi, Cell. Mol. Gastroenterol. Hepatol., 2018, 5, 289–298 CrossRef PubMed.
- R. Tuli, A. J. Surmak, J. Reyes, M. Armour, A. Hacker-Prietz, J. Wong, T. L. DeWeese and J. M. Herman, Transl. Oncol., 2014, 7, 439–445 CrossRef PubMed.
- V. Heinemann, D. Quietzsch, F. Gieseler, M. Gonnermann, H. Schönekäs, A. Rost, H. Neuhaus, C. Haag, M. Clemens and B. Heinrich, J. Clin. Oncol., 2006, 24, 3946–3952 CrossRef CAS.
- J. R. Infante, B. G. Somer, J. O. Park, C.-P. Li, M. E. Scheulen, S. M. Kasubhai, D.-Y. Oh, Y. Liu, S. Redhu and K. Steplewski, Eur. J. Cancer, 2014, 50, 2072–2081 CrossRef CAS PubMed.
- M. Hoyer, H. Roed, L. Sengelov, A. Traberg, L. Ohlhuis, J. Pedersen, H. Nellemann, A. K. Berthelsen, F. Eberholst and S. A. Engelholm, Radiother. Oncol., 2005, 76, 48–53 CrossRef.
- P. J. Loehrer Sr, Y. Feng, H. Cardenes, L. Wagner, J. M. Brell, D. Cella, P. Flynn, R. K. Ramanathan, C. H. Crane and S. R. Alberts, J. Clin. Oncol., 2011, 29, 4105 CrossRef PubMed.
- A. Mahadevan, S. Jain, M. Goldstein, R. Miksad, D. Pleskow, M. Sawhney, D. Brennan, M. Callery and C. Vollmer, Int. J. Radiat. Oncol., Biol., Phys., 2010, 78, 735–742 CrossRef PubMed.
- D. Schellenberg, K. A. Goodman, F. Lee, S. Chang, T. Kuo, J. M. Ford, G. A. Fisher, A. Quon, T. S. Desser and J. Norton, Int. J. Radiat. Oncol., Biol., Phys., 2008, 72, 678–686 CrossRef CAS.
- T. M. Blanco, A. Mantalaris, A. Bismarck and N. Panoskaltsis, Biomaterials, 2010, 31, 2243–2251 CrossRef CAS.
- L. Safinia, A. Mantalaris and A. Bismarck, Langmuir, 2006, 22, 3235–3242 CrossRef CAS PubMed.
- E. G. Velliou, S. B. Dos Santos, M. M. Papathanasiou, M. Fuentes-Gari, R. Misener, N. Panoskaltsis, E. N. Pistikopoulos and A. Mantalaris, Bioprocess Biosyst. Eng., 2015, 38, 1589–1600 CrossRef CAS PubMed.
- T. Chantarojanasiri and P. Kongkam, World J. Gastrointest. Endosc., 2017, 9, 506 CrossRef PubMed.
- A. Rice, E. Cortes, D. Lachowski, B. Cheung, S. Karim, J. Morton and A. Del Rio Hernandez, Oncogenesis, 2017, 6, e352 CrossRef CAS PubMed.
- R. Pozzi, I. Parzanese, A. Baccarin, M. Giunta, C. B. Conti, P. Cantù, G. Casazza, A. Tenca, R. Rosa and D. Gridavilla, Pancreatology, 2017, 17, 905–910 CrossRef PubMed.
- R. J. Bold, S. Virudachalam and D. J. McConkey, J. Surg. Res., 2001, 100, 11–17 CrossRef CAS.
- H. R. Cardenes, A. M. Moore, C. S. Johnson, M. Yu, P. Helft, E. G. Chiorean, J. Vinson, T. J. Howard, A. W. Stephens and D. F. Tai, Am. J. Clin. Oncol., 2011, 34, 460–465 CrossRef CAS.
- T. Conroy, F. Desseigne, M. Ychou, O. Bouché, R. Guimbaud, Y. Bécouarn, A. Adenis, J.-L. Raoul, S. Gourgou-Bourgade and C. de la Fouchardière, N. Engl. J. Med., 2011, 364, 1817–1825 CrossRef CAS.
- P. C. Hermann, S. L. Huber, T. Herrler, A. Aicher, J. W. Ellwart, M. Guba, C. J. Bruns and C. Heeschen, Cell Stem Cell, 2007, 1, 313–323 CrossRef CAS PubMed.
- S. H. Lee, J. K. Ryu, K.-Y. Lee, S. M. Woo, J. K. Park, J. W. Yoo, Y.-T. Kim and Y. B. Yoon, Cancer Lett., 2008, 259, 39–49 CrossRef CAS PubMed.
- E. Van Cutsem, H. Van De Velde, P. Karasek, H. Oettle, W. Vervenne, A. Szawlowski, P. Schoffski, S. Post, C. Verslype and H. Neumann, J. Clin. Oncol., 2004, 22, 1430–1438 CrossRef CAS PubMed.
- D. D. Von Hoff, T. Ervin, F. P. Arena, E. G. Chiorean, J. Infante, M. Moore, T. Seay, S. A. Tjulandin, W. W. Ma and M. N. Saleh, N. Engl. J. Med., 2013, 369, 1691–1703 CrossRef CAS.
- Z. Zhang, Q. Duan, H. Zhao, T. Liu, H. Wu, Q. Shen, C. Wang and T. Yin, Cancer Lett., 2016, 382, 53–63 CrossRef CAS PubMed.
- M. Prescott, J. Mitchell, S. Totti, J. Lee, E. Velliou and M. Bussemaker, Ultrason. Sonochem., 2018, 40, 72–80 CrossRef CAS PubMed.
- M. Krzykawska-Serda, M. S. Agha, J. C.-S. Ho, M. J. Ware, J. J. Law, J. M. Newton, L. Nguyen, S. A. Curley and S. J. Corr, Transl. Oncol., 2018, 11, 664–671 CrossRef PubMed.
- H. Mukubou, T. Tsujimura, R. Sasaki and Y. Ku, Int. J. Oncol., 2010, 37, 821–828 CAS.
- C. Weiss, G. G. Grabenbauer, R. Sauer and L. Distel, Strahlenther. Onkol., 2003, 179, 93–98 CrossRef PubMed.
- D. C. Lauffer, P. A. Kuhn, M. Kueng, S. U. Thalmann, G. Risse, P.-A. Tercier, B. Egger and A. S. Allal, Cureus, 2018, 10 Search PubMed.
- S. Lewis, S. C. Sastri, S. Arya, S. Mehta, P. Patil, S. Shrivastava, R. Phurailatpam, S. V. Shrikhande and R. Engineer, J. Gastrointest. Oncol., 2019, 10, 474 CrossRef.
- J. Neoptolemos, J. Dunn, D. Stocken, J. Almond, K. Link, H. Beger, C. Bassi, M. Falconi, P. Pederzoli and C. Dervenis, Lancet, 2001, 358, 1576–1585 CrossRef CAS.
- M. C. Allenby, R. Misener, N. Panoskaltsis and A. Mantalaris, Tissue Eng., Part C, 2017, 23, 108–117 CrossRef PubMed.
- M. C. Allenby, N. Panoskaltsis, A. Tahlawi, S. B. Dos Santos and A. Mantalaris, Biomaterials, 2019, 188, 24–37 CrossRef CAS PubMed.
- A. Tahlawi, M. E. Klontzas, M. C. Allenby, J. C. Morais, N. Panoskaltsis and A. Mantalaris, J. Tissue Eng. Regener. Med., 2019, 13, 232–243 CAS.
- O. Sirenko, T. Mitlo, J. Hesley, S. Luke, W. Owens and E. F. Cromwell, Assay Drug Dev. Technol., 2015, 13, 402–414 CrossRef CAS PubMed.
- E. Ulukaya, M. Colakogullari and E. J. Wood, Chemotherapy, 2004, 50, 43–50 CrossRef CAS.
- D. T. Vistica, P. Skehan, D. Scudiero, A. Monks, A. Pittman and M. R. Boyd, Cancer Res., 1991, 51, 2515–2520 CAS.
- T. L. Riss, R. A. Moravec, A. L. Niles, S. Duellman, H. A. Benink, T. J. Worzella and L. Minor, in Assay Guidance Manual [Internet], Eli Lilly & Company and the National Center for Advancing Translational Sciences, 2016 Search PubMed.
- E. G. Velliou, S. B. Dos Santos, M. M. Papathanasiou, M. Fuentes-Gari, R. Misener, N. Panoskaltsis, E. N. Pistikopoulos and A. Mantalaris, Bioprocess Biosyst. Eng., 2015, 38, 1589–1600 CrossRef CAS.
- L. Zhou, L. Qi, L. Jiang, P. Zhou, J. Ma, X. Xu and P. Li, AAPS J., 2014, 16, 246–257 CrossRef CAS PubMed.
- E. Brauchle, S. Thude, S. Y. Brucker and K. Schenke-Layland, Sci. Rep., 2014, 4, 4698 CrossRef PubMed.
- L. Evers, P. A. Perez-Mancera, E. Lenkiewicz, N. Tang, D. Aust, T. Knösel, P. Rümmele, T. Holley, M. Kassner and M. Aziz, Genome Med., 2014, 6, 9 CrossRef.
- S. Giagkousiklidis, S. H. Vellanki, K. M. Debatin and S. Fulda, Oncogene, 2007, 26, 7006 CrossRef CAS PubMed.
- Z. A. Cao, D. Daniel and D. Hanahan, BMC Cancer, 2002, 2, 11 CrossRef PubMed.
- L. Seifert, G. Werba, S. Tiwari, N. N. Giao Ly, S. Nguy, S. Alothman, D. Alqunaibit, A. Avanzi, D. Daley, R. Barilla, D. Tippens, A. Torres-Hernandez, M. Hundeyin, V. R. Mani, C. Hajdu, I. Pellicciotta, P. Oh, K. Du and G. Miller, Gastroenterology, 2016, 150, 1659–1672 CrossRef PubMed.
- J. P. Neoptolemos, D. H. Palmer, P. Ghaneh, E. E. Psarelli, J. W. Valle, C. M. Halloran, O. Faluyi, D. A. O'Reilly, D. Cunningham and J. Wadsley, Lancet, 2017, 389, 1011–1024 CrossRef CAS.
- J. K. Mohajer, A. Nisbet, E. Velliou, M. Ajaz and G. Schettino, Br. J. Radiol., 2019, 92, 20180484 CrossRef PubMed.
- H. R. Withers, Lancet, 1992, 339, 156 CrossRef CAS.
- A. Vincent, J. Herman, R. Schulick, R. H. Hruban and M. Goggins, Lancet, 2011, 378, 607 CrossRef.
- T. S. Hong, D. P. Ryan, D. R. Borger, L. S. Blaszkowsky, B. Y. Yeap, M. Ancukiewicz, V. Deshpande, S. Shinagare, J. Y. Wo and Y. Boucher, Int. J. Radiat. Oncol., Biol., Phys., 2014, 89, 830–838 CrossRef CAS PubMed.
- T. S. Hong, D. P. Ryan, L. S. Blaszkowsky, H. J. Mamon, E. L. Kwak, M. Mino-Kenudson, J. Adams, B. Yeap, B. Winrich and T. F. DeLaney, Int. J. Radiat. Oncol., Biol., Phys., 2011, 79, 151–157 CrossRef CAS PubMed.
- M. Bouchard, R. A. Amos, T. M. Briere, S. Beddar and C. H. Crane, Radiother. Oncol., 2009, 92, 238–243 CrossRef PubMed.
- K. Terashima, Y. Demizu, N. Hashimoto, D. Jin, M. Mima, O. Fujii, Y. Niwa, K. Takatori, N. Kitajima and S. Sirakawa, Radiother. Oncol., 2012, 103, 25–31 CrossRef PubMed.
- S Attia, et al., Research in Oncology, 2017, 13.1, 2–7 CrossRef.
- V. Damarla, D. Dobrosotskaya, F. Siddiqui, I. Wollner, M. Raoufi, J. Li, D. Kwon and G. Khan, Eur. J. Cancer, 2018, 92, S8–S9 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra09123h |
| This journal is © The Royal Society of Chemistry 2019 |