
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePlatinum–(phosphinito–phosphinous acid) complexes as bi-talented catalysts for oxidative fragmentation of piperidinols: an entry to primary amines†
Romain Membrata,
Alexandre Vasseur b,
Delphine Moraledaa,
Sabine Michaud-Chevalliera,
Alexandre Martinez*a,
Laurent Giordano*a and
Didier Nuel*a
b,
Delphine Moraledaa,
Sabine Michaud-Chevalliera,
Alexandre Martinez*a,
Laurent Giordano*a and
Didier Nuel*a
aAix-Marseille Univ, CNRS, Centrale Marseille, iSm2, Marseille, France. E-mail: didier.nuel@centrale-marseille.fr
bUniversité de Lorraine, CNRS, L2CM, F-54000 Nancy, France
First published on 20th November 2019
Abstract
Platinum–(phosphinito–phosphinous acid) complex catalyzes the oxidative fragmentation of hindered piperidinols according to a hydrogen transfer induced methodology. This catalyst acts successively as both a hydrogen carrier and soft Lewis acid in a one pot – two steps process. This method can be applied to the synthesis of a wide variety of primary amines in a pure form by a simple acid–base extraction without further purification.
Introduction
The terminology “metal-catalyzed hydrogen transfer methodology” refers to any chemical process relying on the generation of a metal hydride from a hydrogen source and its transfer to an unsaturated hydrogen acceptor.1,2 A substantial number of consistent versions of this concept have been reported and implemented with palladium catalysts for alcohols oxidation,3–5 hydrogenation,6,7 C–C,8,9 C–N,10 C-halogen bond formation,11 double bond isomerization,12 cross aldolisation13 or else amination14 with a view to increasing the molecular complexity. The critical feature of metal-catalyzed hydrogen transfer methodology lays in the design of suitable ligands, able to induce the formation of a reactive metal hydride intermediate.4,7,15 In this context, we recently reported that palladium and platinum – phosphinous acid (PA) complexes can generate active metal hydrides.16 This property is due to the self-assembled hydrogen bond negatively charged assisted structure of two PA ligands (M/PAP complexes, Scheme 1).17 These catalysts allowed therefore the anaerobic oxidation of notably reluctant alcohols such as N-alkyl-(2,2,6,6)-tetramethylpiperidin-4-ols 1.18 In earlier years, the M/PAP structure proved to be also active as a Lewis acid toward sterically congested substrates in tandem [2 + 1] cycloaddition-ring expansion of norbornene derivatives with propargylic acetates.19 In order to benefit from this noteworthy M/PAP's multiskilling20 we focused our effort on a new hydrogen transfer induced transformation where the catalyst acts as both a hydrogen carrier and a Lewis acid in a single chemical system (Scheme 1(a)).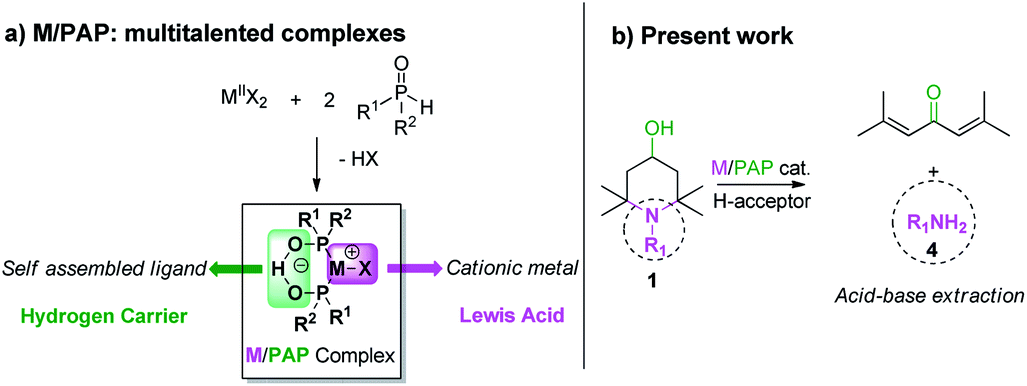 | ||
| Scheme 1 M/PAP complexes as bitalented catalyst. | ||
On one hand, we recently reported the Pt(II)-SPO complex-catalyzed oxidation the N-alkyl-2,2,6,6-tetramethylpiperidin-4-ols into their corresponding piperidinone derivatives.18 On the other hand N-alkyl piperidinone motif have been reported earlier as a source of N-alkyl primary amines.21–26 We hypothesized that the electrophilic features of bisphosphinite metallacycle would also promote the double C(sp3)–N bond cleavage. Combining these two steps with a single catalyst the whole process would emerge as a readily access to primary amines (Scheme 1b). Herein we report our results in this regard.
Results and discussion
During our studies on the palladium catalyzed anaerobic oxidation of N-alkyl-2,2,6,6-tetramethylpiperidin-4-ols18 derivatives, we observed, in the case of 1a, that product 2a was found only in trace amount along with phorone (2,6-dimethylhepta-2,5-dien-4-one) 3, which was isolated in 90% yield (Scheme 2). | ||
| Scheme 2 M/PAP complex catalyzed oxidative fragmentation of piperidinols. | ||
We hypothesise that formation of 3 arose through the cleavage of C–N bonds in the piperidinone 2a leading to methylamine 4a (not isolated). According to this result, the development of a one pot oxidative fragmentation of piperidinols 1 to deliver primary amines 4 should be possible (Scheme 1(b)). Thus, a preliminary study was carried out using N-benzyl-2,2,6,6-tetramethylpiperidin-4-ol 1b in order to isolate benzylamine 4b (Scheme 2).
The reaction of 1b and electron-poor alkene 5a in the presence of 5 mol% of Pd/PAP or Pt/PAP complexes 6 in toluene/water (9/1) at 105 °C was examined (Table 1).

The experiment conducted in 4 hours with the catalyst 6a proved disappointing as the conversion was incomplete and only 14% of phorone 3 were detected (Table 1, entry 1). Increasing the reaction time led to complete conversion but the ring opening remained incomplete affording a mixture of 2b![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 in a 70:30 ratio (Table 1, entry 2). Catalyst 6b also led to complete conversion with a decreased yield of phorone 3 (Table 1, entry 3). The best ratio of 2b:3 (64:36) was observed with the platinum complex 6c and improved to 40:60 with a longer reaction time (Table 1, entries 4 and 5) suggesting a sequential oxidation-opening process. In a previous work, we showed that treatment of 6c with NaOH afforded a hydroxy-platinum catalyst 6d as active species (see Table 2) for the chemoselective anaerobic oxidation of 1b with 5a as H-acceptor and allowed the oxidation to occur at room temperature. We decided then to try the reaction in these conditions to obtain the ketone prior to optimize the ring opening step (Table 2).
:3 in a 70:30 ratio (Table 1, entry 2). Catalyst 6b also led to complete conversion with a decreased yield of phorone 3 (Table 1, entry 3). The best ratio of 2b:3 (64:36) was observed with the platinum complex 6c and improved to 40:60 with a longer reaction time (Table 1, entries 4 and 5) suggesting a sequential oxidation-opening process. In a previous work, we showed that treatment of 6c with NaOH afforded a hydroxy-platinum catalyst 6d as active species (see Table 2) for the chemoselective anaerobic oxidation of 1b with 5a as H-acceptor and allowed the oxidation to occur at room temperature. We decided then to try the reaction in these conditions to obtain the ketone prior to optimize the ring opening step (Table 2).
|
||||
|---|---|---|---|---|
| Entry | H-acceptor | Acid (equiv.) | t (h) | 2b:3:4b ratiob (%) |
| a All reactions were carried out with 1 mmol of 1b in toluene/H2O 9/1 at 105 °C, 4 h. 1/5b/6c = 1/2/0.05 and then addition of acid (5 equiv.), 105 °C.b Determined by 1H NMR spectroscopy.c 4b is detected in trace amounts. | ||||
| 1 | 5a | — | 16 | 100:0:0 |
| 2 | 5a | HCl (1) | 16 | 82:18:NDc |
| 3 | 5a | HCl (5) | 16 | 100:0:0 |
| 4 | 5a | AcOH (1) | 16 | 72:28:NDc |
| 5 | 5a | AcOH (5) | 16 | 0:100:NDc |
| 6 | 5b | AcOH (5) | 16 | 0:100:100 |
| 7 | 5b | AcOH (5) | 5 | 0:100:100 |

Under these conditions, the oxidation of 1b to 2b was complete in 4 hours and the amine 4b was not detected even under prolonged reaction time like 16 hours at 105 °C (Table 2, entry 1). In this case, we supposed that the relative basicity of the medium precludes the cleavage process of 2b. Indeed, in the reaction conditions described in Table 1 (with complexes 6a, 6b and 6c) no base was added, and catalytic traces of HCl or AcOH are probably released during the formation of the [M]–OH active species, which probably promotes the cleavage of 2b. Thus, once the oxidation reaction under basic conditions is over (typically after 4 hours), 1 equivalent of HCl or AcOH (Table 2, entries 2 and 4) was added directly to the mixture.
Results comparable to the reactions done in neutral medium using 6a, 6b or 6c as catalyst (Table 1, entries 2–4) were obtained. However, an excess of HCl (5 equiv.) resulted in a complete deactivation of the catalyst (Table 2, entry 3). In contrast, the use of an excess of acetic acid (5 equiv.) (Table 2, entry 5) was shown to be very beneficial as quantitative deprotection of piperidinone 2b was observed after 16 h at 105 °C. Unfortunately only traces of free amine 4b were detected probably due to the possible 1,4-addition of the amine to the MVK 5a (Table 2, entries 2, 4 and 5).27 We then seek for a less reactive hydrogen acceptor to the addition of amine. Trans-phenyl-but-3-en-2-one 5b was finally selected as the H-acceptor as it gives a clean total conversion and 4b was obtained in a pure form in 88% isolated yield by simple acid–base extraction without further purification (Table 2, entry 6). The optimal conditions for the reaction were refined to 105 °C during 4 hours for the first step and 5 hours for the second (Table 2, entry 7).
Having established the feasibility of the oxidation-opening sequence, we synthesized various N-alkyl-2,2,6,6-tetramethylpiperidin-4-ols 1b–q to extend its applicability.28,29 Under optimized conditions, the conversion of 1 proceeded cleanly and the amines 4 were obtained in good to excellent yields without chromatography (Scheme 3). All benzylic amines were obtained in very good yields whatever is the substitution on the phenyl ring (Scheme 3, 4b–4j). The electronic features of the functional groups within the phenyl moiety have no major influence on the outcome of the reaction (compare for instance 4d with 4i). Allylic amines are also efficiently produced (compounds 4k and 4l) as well as aliphatic amines (4m, 4n). The obtention of product 4p stressed the already presumed necessity of the activation of the protective group by a ketone function. Even a congested piperidinol (1q) was able to deliver amine 4q although in lower yield (38%). Interestingly, the method could be utilized on a large scale (4 grams) as 4b was obtained in 86% yield through simple acid–base extraction. In addition, treatment of the organic phase with ammonia (14 M in MeOH) allowed the recovery of 2,2,6,6-tetramethylpiperidin-4-one 2 in quantitative yield.30
 | ||
| Scheme 3 Scope of the process. | ||
It should be stressed that amino-ester (see 1r) could be directly involved in a three steps one pot process oxidation–fragmentation–cyclization reaction to give the corresponding pure lactam 7 in 98% isolated yield without further purification (Scheme 4).
 | ||
| Scheme 4 Application to three steps-one pot oxidation–fragmentation–cyclization. | ||
Complementary experiments were performed in order to elucidate the oxidative fragmentation mechanism of piperidinone 2 into primary amines 4. A E1CB type mechanism involving the enolate form of product 2 can be ruled out because this ring opening process was unsuccessful under basic conditions (Table 2, entry 1) and required the addition of acetic acid to occur (Table 2, entries 5–7). In order to get insights into this mechanism, we decided to compare the reactivity of two different substrates N-benzyl-2,6-diphenylpiperidin-4-ol 1s and N-benzyl-2,6-dimethyl-3,5-diphenylpiperidin-4-ol 1t. After reactions under our optimized conditions 1s gave a mixture of 2s:4b in a 38:62 ratio while the reaction involving 1t stopped at the oxidized product 2t (Scheme 5).31 Since 2t is present as a single diastereomer, in which the methyl and phenyl substituents are all in equatorial position the absence of fragmentation suggests the requirement of an anti-relationship between the H at C2 and the nitrogen atom (Scheme 5).32
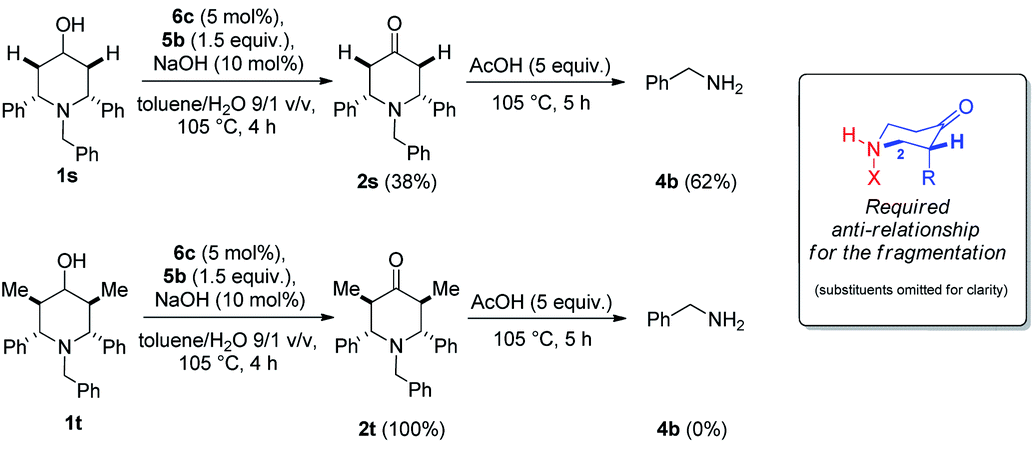 | ||
| Scheme 5 Complementary experiments. | ||
For these reasons, we suggest that an Hofmann type degradation could proceed via the anti-deprotonation of an amino–platinum complex.33,34 At this stage of the study, elucidating the role of the metal in the fragmentation has been found to be crucial. M/PAP moiety has been chosen because of its supposed Lewis acid activity.19 To support this assumption, reactions with 2b were performed in an acidic medium adding either Brønsted or Lewis acid (Table 3).

AcOH or HCl (1 equiv.) were inefficient for the aminone C–N bond cleavage process (Table 3, entries 1 and 2). A stoichiometric amount of BF3· Et2O allowed a complete conversion (Table 3, entry 4). However, a catalytic amount of BF3· Et2O led to a low conversion (Table 3, entry 3). The produced primary amine probably easily coordinates BF3 thus rendering it ineffective for further ring opening. As a result, the yield of amine matches the amount of BF3 used (5%). This also maintains the amine in the organic phase and we were unable to isolate it in pure form. Other Lewis acids such as scandium triflate (Table 3, entry 5) gave poorer results even when used in stoichiometric quantities. PtCl2 also known as a soft Lewis35 acid did not gave a better result (3 was not detected) (Table 3, entry 6). A cationic platinum complex [PtCl(dppp)][PF6], in which the covalent dppp ligand mimic the supramolecular bisphosphinite chelate structure, was also inefficient (Table 3, entry 7). All these results tend to show that the PAP ligand provides the right Lewis acid character to our platinum catalyst 6c (Table 3, entry 8) which is consistent with a Hofmann type fragmentation.
According to these results, a possible pathway for the sequence is proposed in Scheme 6. First, in basic conditions (NaOH, Scheme 6, 1), the Pt/PAP catalyst acted as a hydrogen carrier to promote the oxidation of 1 to 2 according to our previously reported mechanism.18 Thereafter, a one pot minor modification of the experimental conditions (AcOH, Scheme 6, 2) switched Pt/PAP's properties and triggered the ring opening process. A mechanism involving a Pt(II) cationic species 6f playing the role of Lewis acid is suggested and can be compared to a Hofmann degradation.34 It appeared that the combined effect of both Lewis and Brønsted acid is crucial for the reaction outcome. The pathway can be described as follows: (i) the nitrogen atom coordinates to the Lewis acid (complex 8), thus promoting the ring opening (complex 9), (ii) the Brønsted acid allows the Pt–N cleavage and the resulting protonated amine 4 is carried away in the aqueous layer. In this phase transfer mechanism, the bi-talented nature of the Pt/PAP structure was clearly highlighted.
 | ||
| Scheme 6 Plausible pathway for cascade oxidation fragmentation by M/PAP catalysts. | ||
Conclusions
M/PAP complexes can be used as unique catalyst to carry out oxidation of N-alkyl-2,2,6,6-tetramethylpiperidin-4-ols 1 through hydrogen transfer methodology followed by C(sp3)–N bond cleavage in a single operation. The self-assembled negatively charged structure of the ligand may ease the abstracting hydrogen step from 1 while the metal cationic centre may promote the formation of amino–platinum complex species from 2 responsible fora Hofmann-type degradation so as to afford a primary amine 4. As N-alkyl-2,2,6,6-tetramethylpiperidin-4-ols are easily accessible from the cheap 2,2,6,6-tetramethylpiperidin-4-ol,28 this cascade reaction sequence provides an interesting entry to aliphatic primary amines merely isolated after simple acid–base extraction. Finally, the generated phorone 3 could readily be recycled into 2,2,6,6-tetramethylpiperidin-4-ol by reacting with ammonia and the method is practicable on a large scale (see ESI†). The proposed mechanism for this sequence clearly shows the multiskilling aspect of M/PAP catalysts.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Ministère de l'Enseignement Supérieur et de la Recherche (R. Membrat PhD grant). The authors want to thank Dr. Valérie Monnier and Dr Christophe Chendo for ESI-MS experiments and Dr Alphonse Tenaglia for fruitful discussions. Umicore AG & Co. KG is acknowledged for the generous gift of platinum complexes.Notes and references
- J. Muzart, Eur. J. Org. Chem., 2015, 2015, 5693–5707 CrossRef CAS.
- D. Wang and D. Astruc, Chem. Rev., 2015, 115, 6621–6686 CrossRef CAS PubMed.
- B. Karimi and A. Zamani, J. Iran. Chem. Soc., 2008, 5, S1–S20 CrossRef CAS.
- D. Wang, A. B. Weinstein, P. B. White and S. S. Stahl, Chem. Rev., 2018, 118, 2636–2679 CrossRef CAS PubMed.
- J. Muzart, Tetrahedron, 2003, 59, 5789–5816 CrossRef CAS.
- B. Ding, Z. Zhang, Y. Liu, M. Sugiya, T. Imamoto and W. Zhang, Org. Lett., 2013, 15, 3690–3693 CrossRef CAS PubMed.
- Y. Tsuchiya, Y. Hamashima and M. Sodeoka, Org. Lett., 2006, 8, 4851–4854 CrossRef CAS PubMed.
- K. M. Gligorich, S. A. Cummings and M. S. Sigman, J. Am. Chem. Soc., 2007, 129, 14193–14195 CrossRef CAS PubMed.
- S. M. Podhajsky, Y. Iwai, A. Cook-Sneathen and M. S. Sigman, Tetrahedron, 2011, 67, 4435–4441 CrossRef CAS PubMed.
- H. Hikawa, T. Koike, K. Izumi, S. Kikkawa and I. Azumaya, Adv. Synth. Catal., 2016, 358, 784–791 CrossRef CAS.
- S. M. Podhajsky and M. S. Sigman, Organometallics, 2007, 26, 5680–5686 CrossRef CAS PubMed.
- M. J. Spallek, S. Stockinger, R. Goddard and O. Trapp, Adv. Synth. Catal., 2012, 354, 1466–1480 CrossRef CAS.
- O. Kose and S. Saito, Org. Biomol. Chem., 2010, 8, 896–900 RSC.
- A. Martínez-Asencio, M. Yus and D. J. Ramón, Synthesis, 2011, 2011, 3730–3740 CrossRef.
- G.-J. ten Brink, I. W. C. E. Arends and R. A. Sheldon, Adv. Synth. Catal., 2002, 344, 355–369 CrossRef CAS.
- A. Vasseur, R. Membrat, D. Gatineau, A. Tenaglia, D. Nuel and L. Giordano, ChemCatChem, 2017, 9, 728–732 CrossRef CAS.
- P. M. Castro, H. Gulyás, J. Benet-Buchholz, C. Bo, Z. Freixa and P. W. N. M. van Leeuwen, Catal. Sci. Technol., 2011, 1, 401 RSC.
- R. Membrat, A. Vasseur, A. Martinez, L. Giordano and D. Nuel, Eur. J. Org. Chem., 2018, 2018, 5427–5434 CrossRef CAS.
- J. Bigeault, I. de Riggi, Y. Gimbert, L. Giordano and G. Buono, Synlett, 2008, 2008, 1071–1075 CrossRef.
- T. Achard, CHIMIA International Journal for Chemistry, 2016, 70, 8–19 CrossRef CAS PubMed.
- J.-Y. Laronze, J. Sapi and J. Lévy, Synthesis, 1988, 1988, 619–621 CrossRef.
- J. Lévy, J.-Y. Laronze and J. Sapi, Tetrahedron Lett., 1988, 29, 3303–3306 CrossRef.
- P. Aschwanden, C. R. J. Stephenson and E. M. Carreira, Org. Lett., 2006, 8, 2437–2440 CrossRef CAS PubMed.
- M. Shimano and A. I. Meyers, J. Org. Chem., 1995, 60, 7445–7455 CrossRef CAS.
- H. Shi, D. J. Babinski and T. Ritter, J. Am. Chem. Soc., 2015, 137, 3775–3778 CrossRef CAS PubMed.
- H. Seo, M. H. Katcher and T. F. Jamison, Nat. Chem., 2017, 9, 453–456 CrossRef CAS PubMed.
- 1,4 Addition products and polymers were detected by 1H NMR spectroscopy and low resolution ESIMS experiments.
- R. Membrat, A. Vasseur, L. Giordano, A. Martinez and D. Nuel, Tetrahedron Lett., 2019, 60, 240–243 CrossRef CAS.
- For aliphatic electrophiles, see the procedure in the ESI (Section 2.3†).
- For procedure and yield see ESI (Section 2.5†).
- The fact that 1s is less reactive than the other 1 substrates clearly stress that the presence of the two methyl groups in α position due to the nitrogen atom is crucial for the ring opening to occur (see ref. 34).
- T. Ravindran, R. Jeyaraman, R. W. Murray and M. Singh, J. Org. Chem., 1991, 56, 4833–4840 CrossRef CAS.
- A. M. Belostotskii and A. B. Shapiro, Chem. Heterocycl. Compd., 1984, 20, 761–766 CrossRef.
- A. M. Belostotskii and A. B. Shapiro, Chem. Heterocycl. Compd., 1987, 23, 665–669 CrossRef.
- S. Kobayashi, T. Busujima and S. Nagayama, Chem.–Eur. J., 2000, 6, 3491–3494 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra08709e |
| This journal is © The Royal Society of Chemistry 2019 |