DOI:
10.1039/C9RA08489D
(Paper)
RSC Adv., 2019,
9, 41591-41602
Construction of novel Ag/HKUST-1/g-C3N4 towards enhanced photocatalytic activity for the degradation of pollutants under visible light†
Received
17th October 2019
, Accepted 20th November 2019
First published on 16th December 2019
Abstract
A novel Ag/metal–organic framework/graphitic carbon nitride (Ag/HKUST-1/g-C3N4, AHC) photocatalyst was prepared via an in situ growth strategy and photo-deposition technique for environmental remediation. The as-obtained samples were characterized by X-ray diffraction (XRD), transmission electron microscopy (TEM), scanning electron microscopy (SEM), X-ray photoelectron spectroscopy (XPS), N2 adsorption–desorption isotherm measurement, UV-vis diffuse reflection spectroscopy (UV-vis DRS), and photoluminescence (PL) spectroscopy. The results indicated that the hybrids have large surface area, mesoporous structure and enhanced visible-light absorption. The as-prepared hybrid samples exhibited considerable improvement in photocatalytic activity and stability for rhodamine B (RhB) degradation under visible light irradiation (λ > 420 nm). In addition, they also have good adsorption properties. Compared to the pure g-C3N4 and Ag/g-C3N4, the 5% AHC photocatalyst showed superior photocatalytic activity. Moreover, 5% AHC exhibits good photocatalytic activity even after four cycles. Additionally, the active species trapping and electron spin resonance (ESR) experiments indicated that h+ and ·OH were the main active species.
Introduction
With environmental degradation becoming increasingly prominent throughout the world, the effective control of environmental pollutants and government intervention have become urgent to address this issue. During the past decade, a variety of strategies have been employed to solve this problem.1–3 The semiconductor photocatalytic technique using the renewable solar energy to degrade organic pollutants to non-toxic products has received word-wide attention.4 Simultaneously, visible-light-driven photocatalysts have attracted considerable attention because sunlight consists of about 50% visible light and only about 4% ultraviolet (UV) light. Additionally, nanomaterials with adsorptive properties can also increase the rate of removal of pollutants in water. For this purpose, materials with good adsorptive properties are combined with the semiconductor photocatalysts, and the respective advantages are brought into play. The low-concentration and highly toxic pollutants in wastewater are adsorbed on the surface of the photocatalysts, and then decomposed into H2O, CO2 and non-toxic inorganic acids.
Graphitic carbon nitride (g-C3N4), a metal-free polymeric semiconductor, has attracted extensive attention due to its good thermochemical stability, electronic properties, optical characteristics and suitable band gap (around 2.70 eV).5–7 To date, the recent improvements with regards to g-C3N4 have been published in a large number of articles. For example, the different morphologies of g-C3N4, such as g-C3N4 quantum dots (QDs) and two-dimensionally ultrathin g-C3N4 nanosheets, were prepared successfully. In addition, g-C3N4 as a novel visible-light-driven photocatalyst has been reported for hydrogen evolution and pollutant degradation.8–12 However, as a single-phase catalyst, g-C3N4 is restricted by its high recombination rate of photoinduced electron–hole pairs, leading to its poor quantum efficiency and low photocatalytic activity. In order to improve the photocatalytic performance of g-C3N4, numerous methods have been employed, such as doping of metal/nonmetal elements13,14 and coupling with other semiconductors.15,16 Hong et al. reported a novel direct solid-state Z-scheme V2O5/g-C3N4 heterojunction obtained by a facile in situ growth strategy for the first time. The photocatalytic performance was evaluated by the degradation of RhB and TC under visible light irradiation (λ > 420 nm).17 Zhu et al. synthesized a highly dispersed Ag/Fe3O4/g-C3N4 composite photocatalyst by means of the selective photo-deposition technique. It exhibited a significantly improved photocatalytic activity and stability for degrading TC, besides retaining the recyclable magnetic property.18 Recent studies have shown that noble metal loading proved to be an effective way of increasing the photocatalytic activity. This is because the localized surface plasmon resonance (SPR) of a noble metal can enhance the localized electric fields by several distinct orders of magnitude at the noble metal/g-C3N4 photocatalyst interface. Noble metal nanoparticles can serve as antennae to capture photoinduced electrons from the conduction band (CB) of g-C3N4, thus decreasing the recombination of the electron–hole pairs and further improving the photocatalytic activity. Ag, as a low-cost and widely used noble metal, has been employed to synthesize composite photocatalysts, such as Ag@AgCl, Ag/AgX/GO and Ag/g-C3N4. However, there are still some disadvantages that restrict the application of Ag/g-C3N4 in environmental remediation,19 particularly its low surface area.20 Therefore, in order to further enlarge the adsorption capacity and improve the stability, it is necessary to modify Ag/g-C3N4 using porous materials.
As important porous materials, metal–organic frameworks (MOFs) have the advantages of adjustable pore size and pore surface, ultra-low density, ultra-high specific surface area, as well as insolubility in common solvents among other characteristics, compared with traditional pore materials, such as zeolites, mesoporous silica and activated carbon.21–23 Consequently, much attention has been paid to the potential applications of MOFs in gas capture and storage, chemical separation, sensor devices, drug delivery and catalysis.24–29 Wang et al. reported the synthesis of g-C3N4/Ti-benzenedicarboxylate (MIL-125(Ti)) heterostructure photocatalysts. By optimizing the compositions, an environment-friendly composite was found to show excellent photocatalytic activity towards RhB dye degradation under visible-light irradiation. The significantly reduced electron–hole recombination rate of photocatalysts was not only caused by the sufficient surface contact of g-C3N4 and MIL-125(Ti), but also induced by the synergic effect between g-C3N4 and MIL-125(Ti).30 He et al. manifested the photocatalytic hydrogen production efficiency of CdS nanoparticles embedded on MIL-101 under visible-light irradiation, which was superior to that of bare CdS due to the large specific surface area, more active adsorption sites, photocatalytic reaction centers and good visible-light harvesting capability.31 Liang et al. used a simple self-assembly strategy for increasing the interfacial contact to prepare MIL-53(Fe)-reduced graphene oxide nanocomposites.32 Therefore, combining MOFs with semiconductor photocatalysts can improve the photocatalytic efficiency.
Herein, we report a facile process for the preparation of plasmonic AHC photocatalysts. First, the metal-free bulk g-C3N4 was synthesized by the thermal treatment of melamine. Second, we prepared HKUST-1/g-C3N4 composites by a facile in situ growth strategy. Finally, the AHC composite photocatalysts were successfully synthesized through a photo-deposition technique. In addition, the possible photocatalytic mechanism is tentatively proposed based on the active species trapping and ESR experiments. Furthermore, the stability of AHC composite photocatalyst was tested by the circulation experiments.
Experimental section
Materials and methods
AgNO3, Cu(NO3)2, trimesinic acid (H3BTC), melamine, ethanol, acetic acid, polyethylene glycol 800 (PEG 800), triethanolamine (TEOA), isopropanol (IPA), and 5,5-dimethyl-1-pyrroline N-oxide (DMPO) were analytical grade agents and purchased from Aladdin (China). RhB was analytically pure and used without further purification. All of the phase compositions and crystal structures of the prepared samples were determined by the powder X-ray diffraction (XRD) method using Cu Kα radiation (λ = 1.54178 Å) (D/MAX-2500 diffractometer, Rigaku, Japan) with a Cu Kα radiation source (k = 1.54056) over the 2θ range of 5–90° at a scanning rate of 10° min−1. The morphologies and structure details of the as-prepared samples were obtained using scanning electron microscopy (SEM, JSM-6510) and transmission electron microscopy (TEM, JEM-2100F). X-ray photoelectron spectroscopy (XPS) maps were obtained by a Thermo ESCALAB 250X (America) electron spectrometer using a 150 W Al Kα X-ray source. The specific surface area of these samples was determined from the nitrogen absorption–desorption data and Brunauer–Emmett–Teller (BET) measurements (ASAP2020, Micromeritics, USA). The UV-vis diffused reflectance spectra (DRS) of the samples were obtained from a UV-vis spectrophotometer (UV-2450, Shimadzu, Japan); BaSO4 was used as a reflectance standard. The photoluminescence (PL) spectra for the solid samples were obtained on a F4500 (Hitachi, Japan) photoluminescence detector. The photocurrent and electrochemical impedance spectroscopy (EIS) measurements were conducted using a CHI660C (Chenhua Instruments Co. Shanghai China) electrochemical workstation. The electron spin resonance (ESR) analysis was conducted with a Bruker EPR A 300-10/12 spectrometer.
Photocatalysts preparation
Synthesis of g-C3N4 sheets. The metal-free bulk g-C3N4 was synthesized by the thermal treatment of melamine, according to a previous report.33 Typically, 10 g melamine was put into a 50 mL alumina crucible with a cover, and then heated in a muffle furnace at a rate of 2.3 °C min−1 and kept for 4.0 h at 550 °C. After being cooled to room temperature, the resulting products were collected and milled into powder in an agate mortar for further use.
Synthesis of AHC. The HKUST-1/g-C3N4 composites were synthesized according to a modified method.34 First, a certain amount of g-C3N4 was dispersed into ethanol with the aid of ultrasonication. Then, H3BTC (0.1 mmol) was added into a mixture of g-C3N4/ethanol (30 mL) and sonicated for 30 min to afford a homogeneous solution. Second, copper acetate (Cu(OAc)2·H2O; 0.15 mmol) was dissolved in acetic acid solution (HAc: 3.0 mL; H2O: 30 mL) to form another solution. Subsequently, the solutions obtained in the first step and the second step were mixed and stirred for an hour at high speed. The products were collected by centrifugation at 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 3.0 min, washed with ethanol several times, and finally dried in an oven at 60 °C for 10 h.
000 rpm for 3.0 min, washed with ethanol several times, and finally dried in an oven at 60 °C for 10 h.A certain amount of the as-prepared HKUST-1/g-C3N4 was mixed with 200 mL of deionized water by ultrasonication for 30 min. Then, 1.0 mL of 5% polyethylene glycol (PEG) 800 solution was added and the dispersion was stirred for another 10 min.35,36 For deposition of silver on the surface of HKUST-1/g-C3N4, a photo-deposition technique was applied as follows. Initially, 5.0 mL of AgNO3 solution (0.015 mol L−1) was added to the dispersion. Then, the suspension was transferred to a water-cooled reactor (250 mL) and irradiated under a PLS-SXE300 Xe lamp with 250 W illumination intensity for 60 min. The theoretical value of the Ag loading amount was 4.0 wt%. Subsequently, 5% AHC, 6% AHC, and 7% AHC were synthesized.
Photocatalytic experiments
The photocatalytic activities of the as-prepared samples were investigated by the degradation of RhB under visible light irradiation. The typical photocatalytic experiment was performed in a Pyrex reactor with refluxed water at room temperature and illuminated using a 250 W xenon lamp with a 420 nm cut off filter. In detail, 50 mg of the powered samples was dispersed in the 100 mL of 0.5 mg L−1 pollutant aqueous solutions. In order to ensure the adsorption equilibrium, the suspension was continuously stirred for 45 min in the dark before irradiation. The concentration changes of the pollutant aqueous solutions were monitored by measuring the UV-vis absorption of the suspensions at 15 min intervals. During irradiation, 5.0 mL of the suspension was taken out and centrifuged (10000 rpm, 2.0 min) to remove the photocatalyst before measurements. Then, the supernatant was monitored by a UV-vis spectrophotometer at the characteristic absorption of RhB at 553 nm.
Photoelectrochemical measurements
The photocurrent and electrochemical impedance spectra tests were performed using a CHI660C electrochemical workstation in a standard three-electrode configuration, with the as-prepared samples as the working electrodes, a Pt foil as the counter electrode and an Ag/AgCl electrode as a reference electrode. A 0.5 mol L−1 Na2SO4 aqueous solution was used as the electrolyte. The working electrodes were prepared as the follows: 0.3 g of each sample, 0.03 mL oleic acid and 0.01 g of polyvinyl pyrrolidone (PVP) were dissolved in 3.0 mL of ethanol to form a suspension, which was then spin coated onto a 15 mm × 20 mm indium tin oxide (ITO) conducting glass electrode by using a drop-casting method and annealed at 500 °C in air for 2.0 h. The visible light source was provided by a 300 W xenon lamp with a 420 nm cut off filter was used to filter out UV light (λ < 420 nm).
Active species trapping and ESR experiments
It is well known that the holes (h+), superoxide radical (·O2−) and hydroxyl radicals (·OH) were the major reactive species for the photocatalytic oxidation. In the active species trapping experiments, 1.0 mM TEOA and 1.0 mM IPA were respectively employed as the scavengers for h+ and ·OH. The method is similar to the former photocatalytic activity test. Furthermore, the ESR technique was further used to discover the existence of ·O2− and ·OH radicals in the photocatalytic reaction system under visible light (λ > 420 nm). The samples for ESR measurements were prepared as follows: 10 mg of each of the samples was dissolved in 0.5 mL methanol (DMPO–·O2−) or 0.5 mL deionized water (DMPO–·OH) and then, 45 μL DMPO was added to the solution, followed by ultrasonic dispersion for 5.0 min.
Results and discussion
Structure and morphology analysis
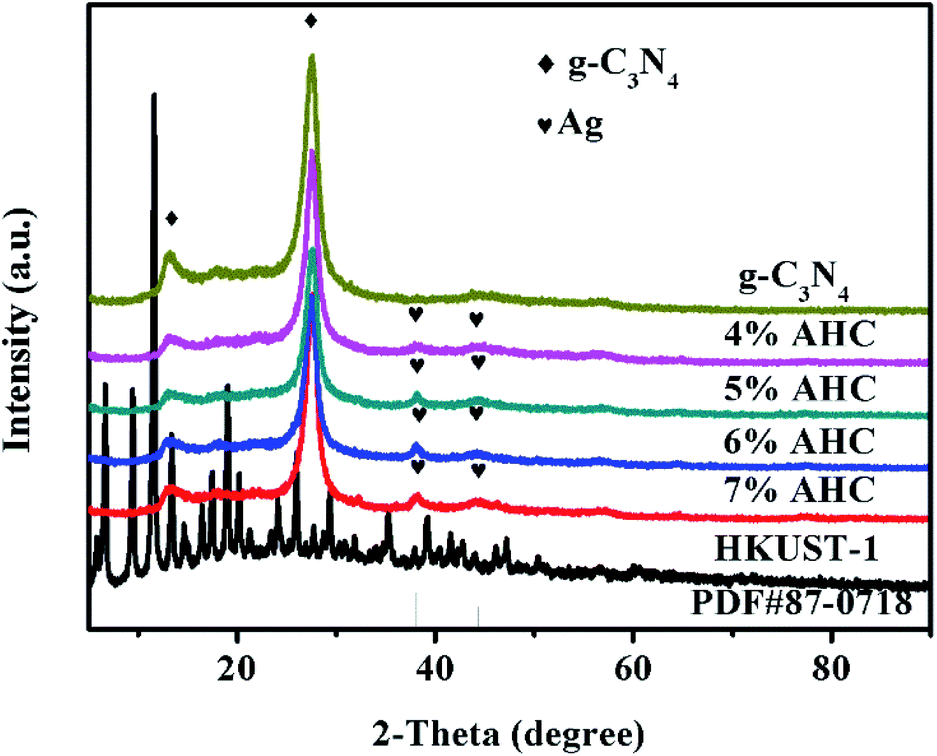
The XRD patterns of the as-prepared g-C3N4, 4% AHC, 5% AHC, 6% AHC, 7% AHC, HKUST-1 and Ag (PDF#87-0718) are shown in Fig. 1. As can be seen, pure g-C3N4 shows one diffraction peak ascribed to the (100) plane at about 2θ = 13.1°, corresponding to the characteristic interlayer structural packing. Another diffraction peak ascribed to the (002) plane at 27.2° corresponds to the interplanar stacking peaks of the aromatic systems.37,38 Compared with pure g-C3N4, the 4% AHC, 5% AHC, 6% AHC and 7% AHC samples exhibit two additional peaks at 38.2° and 44.4°, which are indexed to the (111) and (200) planes of Ag, respectively. Such observations indicate that the Ag NPs were successfully loaded on g-C3N4 after irradiation.39 Nevertheless, the diffraction peaks of HKUST-1 were not detected in the samples of AHC, which may be due to its low loading content and high dispersity.
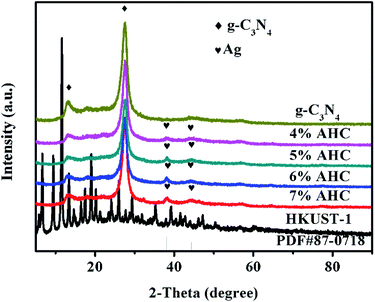 |
| | Fig. 1 XRD patterns of the as-prepared g-C3N4, 4% AHC, 5% AHC, 6% AHC, 7% AHC and HKUST-1 composites. | |
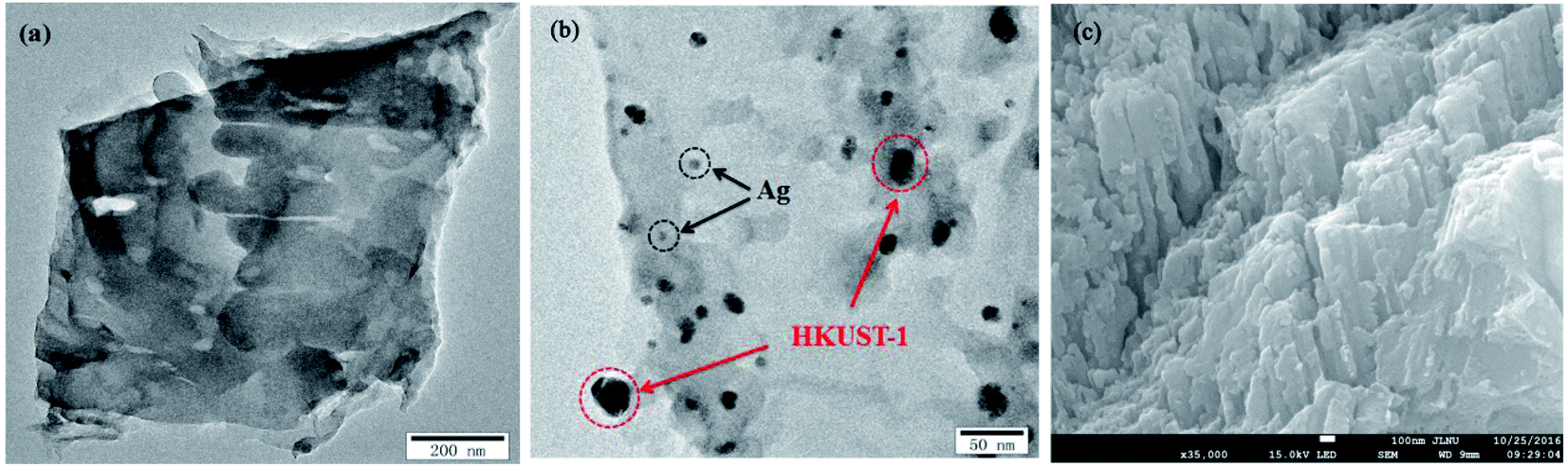
The morphologies and microstructures of g-C3N4 and 5% AHC composite photocatalyst were further investigated by TEM and SEM (Fig. 2). As shown in Fig. 2(a), it could be observed that g-C3N4 displays two dimensional and overlapped lamellar structures, whereas as shown in Fig. 2(b), for 5% AHC, irregular Ag NPs and HKUST-1 particles, which covered with bits of g-C3N4 nanosheets, were found to be embedded in the wrinkling g-C3N4 lamellar structure. The SEM image of 5% AHC (Fig. 2(c)) and its EDS elemental mappings (Fig. S1†) further confirm that the 5% AHC composite photocatalyst was composed of Ag, Cu, C, N and O elements. Thus, the composite obtained by combining Ag, HKUST-1 and g-C3N4 was successfully fabricated.
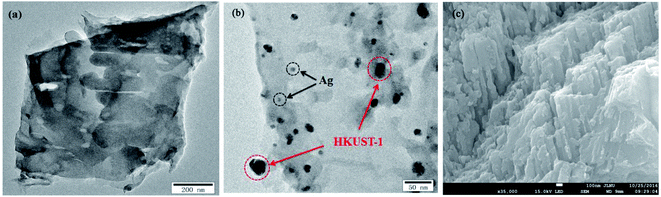 |
| | Fig. 2 TEM images of (a) pure g-C3N4; (b) 5% AHC; (c) SEM image of 5% AHC. | |
Herein, based on the above experimental results, it could be demonstrated that the AHC composite photocatalysts were successfully synthesized, which were expected to facilitate the transfer of photo-generated charge carriers.
Chemical states analysis
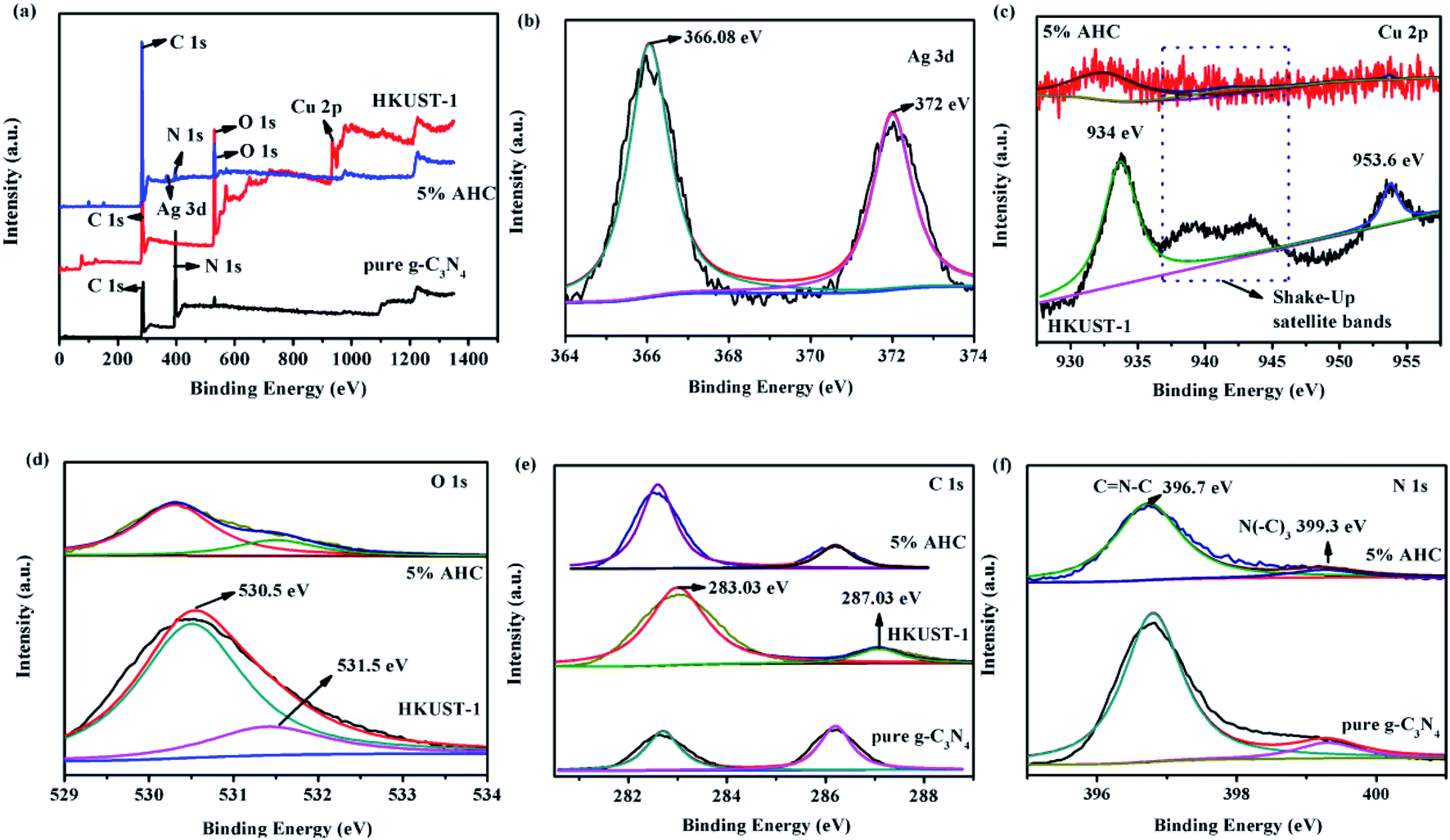
XPS spectra were employed to analyze the detailed chemical status of the samples. The spectra were recorded to analyze the surface chemical compositions of g-C3N4, HKUST-1 and 5% AHC composite photocatalyst. The XPS survey spectra of g-C3N4, HKUST-1 and 5% AHC samples are shown in Fig. 3(a). The spectra indicated that C and N exist on the surface of g-C3N4, Cu, C and O exist on the surface of HKUST-1, and Ag, C, N and O exist in 5% AHC. The doublet peaks of Ag 3d5/2 and Ag 3d3/2 were located at about 366.08 eV and 372 eV, respectively, which were very close to the binding energy values of Ag (Fig. 3(b)).40 The Cu 2p XPS spectra for HKUST-1 and 5% AHC are shown in Fig. 3(c). For HKUST-1, the peaks of Cu 2p3/2 and Cu 2p1/2 located at 934 eV and 953.6 eV, respectively. However, when HKUST-1 combined with g-C3N4, the Cu 2p3/2 peak of 5% AHC displayed a slight shift. Furthermore, the well-known ‘‘shake-up satellite bands’’ (at the 937–946 eV regions) were clearly observed, which was indicative of the paramagnetic chemical state of Cu2+.41 Correspondingly, for HKUST-1, the peaks of O 1s are located at 530.5 eV and 531.5 eV, respectively. However, one of the O 1s peaks of 5% AHC also exhibited a slight shift to the lower binding energy at about 530.4 eV (Fig. 3(d)). Fig. 3(e) displays two strong peaks at 283.03 eV and 287.03 eV, which were assigned to C 1s, which should originate from HKUST-1. The binding energy of C 1s in the 5% AHC composite photocatalyst also exhibited a slight shift to the low energy region compared to that of HKUST-1. These binding energy shifts in the spectra could be attributed to the presence of strong interactions between g-C3N4 and HKUST-1. In the N 1s spectrum (Fig. 3(f)), two peaks at 396.7 eV and 399.3 eV are separately ascribed to sp2 N bonded to two carbon atoms (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–C) and tertiary nitrogen (N–(C)3).42,43 The XPS results validate the coexistence of Ag, g-C3N4 and HKUST-1 in the 5% AHC composite photocatalyst. In addition, it is also concluded that there are strong interfacial interactions between g-C3N4 and HKUST-1, which could facilitate the rapid transfer of photoinduced charge and improve the photocatalytic performance of 5% AHC.
N–C) and tertiary nitrogen (N–(C)3).42,43 The XPS results validate the coexistence of Ag, g-C3N4 and HKUST-1 in the 5% AHC composite photocatalyst. In addition, it is also concluded that there are strong interfacial interactions between g-C3N4 and HKUST-1, which could facilitate the rapid transfer of photoinduced charge and improve the photocatalytic performance of 5% AHC.
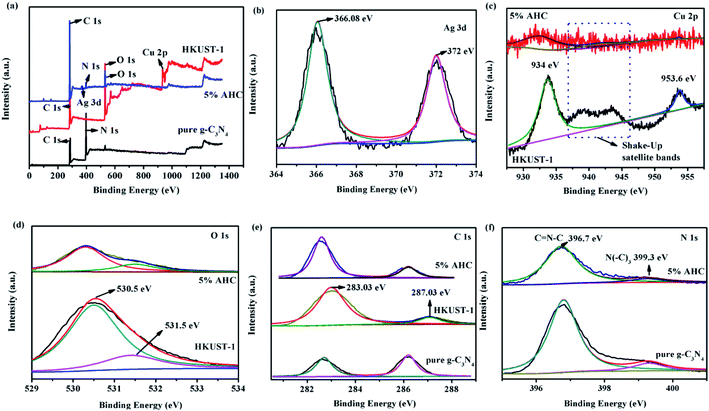 |
| | Fig. 3 XPS spectra of pure g-C3N4, HKUST-1 and 5% AHC samples: (a) survey spectrum; (b) Ag 3d; (c) Cu 2p; (d) O 1s; (e) C 1s and (f) N 1s. | |
Specific surface area and optical properties
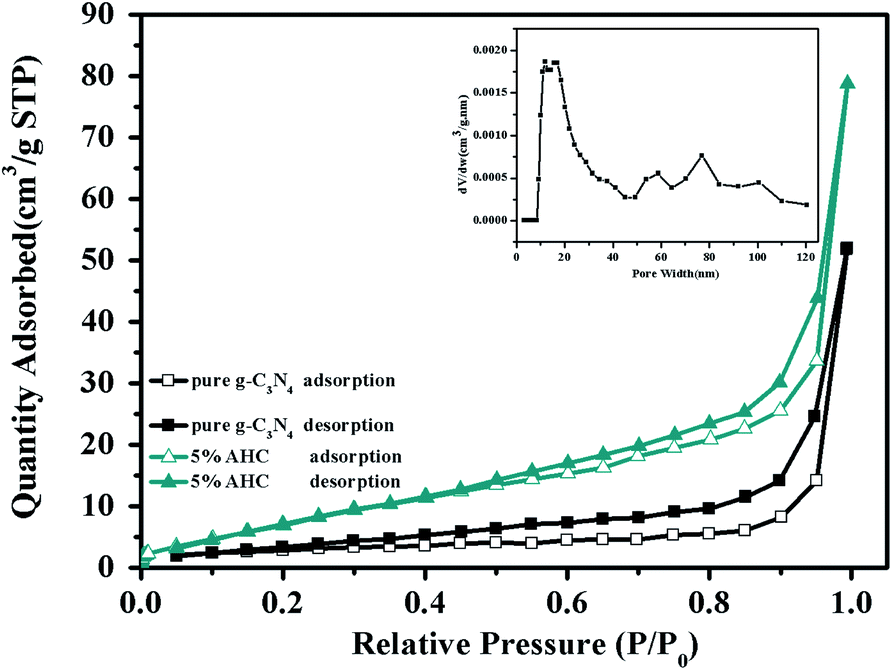
The N2 adsorption–desorption isotherms of pure g-C3N4 and 5% AHC are shown in Fig. 4. It can be seen that the isotherms are similar and all of them are classical type IV, suggesting the presence of abundant mesopores and macropores. The BET surface area of pure g-C3N4 was found to be 10.4 m2 g−1, while that of 5% AHC was 27.4 m2 g−1, which was higher than that of pure g-C3N4. Correspondingly, the total pore volume of 5% AHC was 0.12 cm3 g−1, which was higher than that of pure g-C3N4 (0.08 cm3 g−1). The pore size distribution (PSD) of 5% AHC was calculated via nonlocal density functional theory (NLDFT) (the inset in Fig. 4). The 5% AHC composite photocatalyst exhibits a dominant pore width of about 20 nm and several smaller pores in the pore-size range of 60–100 nm. The PSD curves agree with the shape of the nitrogen isotherms (Fig. 4) and suggest that the polymers are predominantly mesoporous and macropores.
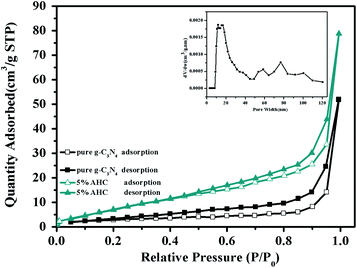 |
| | Fig. 4 N2 adsorption–desorption isotherms of the as-prepared pure g-C3N4 and 5% AHC samples (closed symbols: desorption; open symbols: adsorption; inset: pore size of 5% AHC). | |
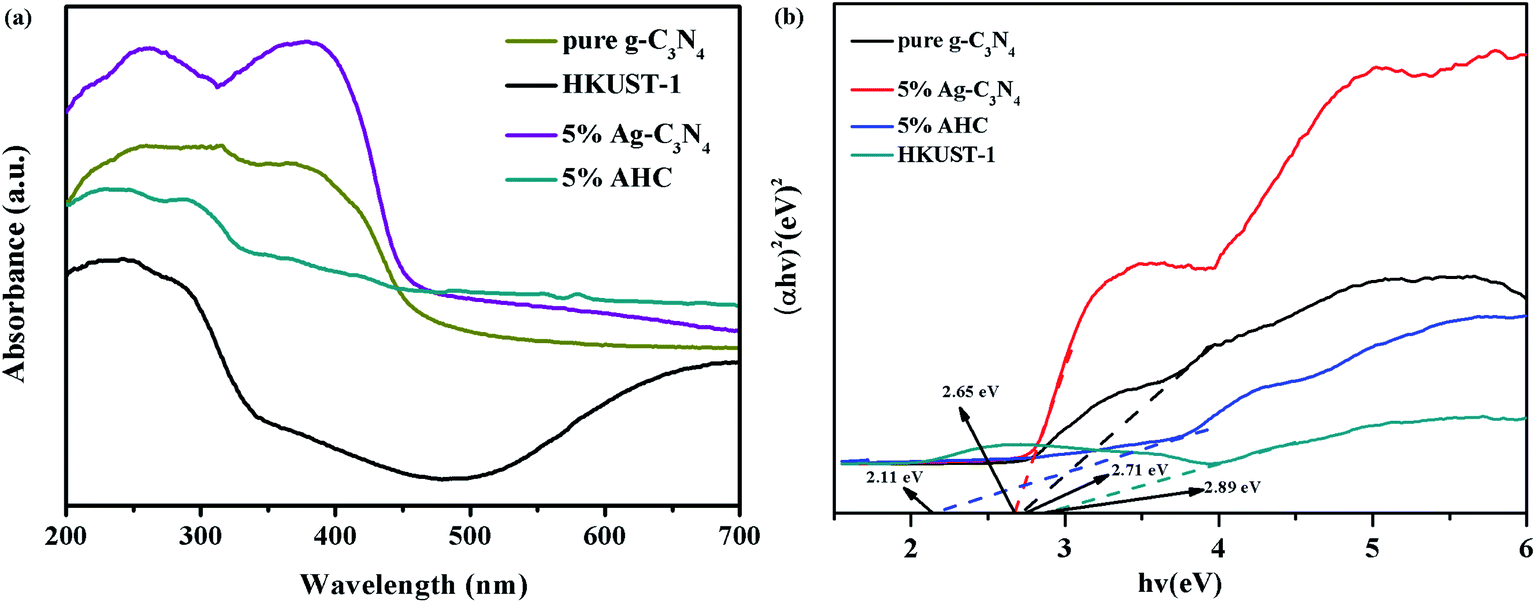
The optical properties of the as-prepared g-C3N4, HKUST-1, 5% Ag/g-C3N4 (the method of synthesis was in the ESI), and 5% AHC composite photocatalyst were measured via the UV-vis DRS technique. As shown in Fig. 5(a), it is clear that pure g-C3N4 shows absorption wavelengths from UV to visible range up to 460 nm, and its band gap is around 2.70 eV, which agrees well with the previous reports.44,45 Compared with that of pure g-C3N4, the absorption edge of the 5% Ag/g-C3N4 composite photocatalyst shows a systematic slight red shift. This may be attributed to the surface plasmon resonance (SPR) effect of Ag species.46 Furthermore, HKUST-1 displays an absorption band lower than about 460 nm, resulting in a low visible light utilization efficiency. However, as the wavelength increases, the response of HKUST-1 to the visible light becomes stronger. Therefore, the 5% AHC composite photocatalyst has a strong absorption under visible light irradiation, suggesting that it can efficiently utilize the visible light for the degradation of organic dyes pollutants. Furthermore, in order to confirm the relative band gap position of the samples, the band gap energy (Eg) of sample was calculated by the following formula based on the DRS results (1):
where
α is the absorption coefficient,
ν is the optical frequency,
Eg is the band gap energy, and
A is a constant. As shown in
Fig. 5(b), the band gap energy of g-C
3N
4, HKUST-1, 5% Ag/g-C
3N
4 and 5% AHC can be estimated to be 2.71 eV, 2.89 eV, 2.63 eV and 2.11 eV, respectively. Similarly, the band gap energy of g-C
3N
4, HKUST-1, 5% Ag/g-C
3N
4 and 5% AHC can also be inferred from a plot of (
αhv)
2 versus energy (
hv).
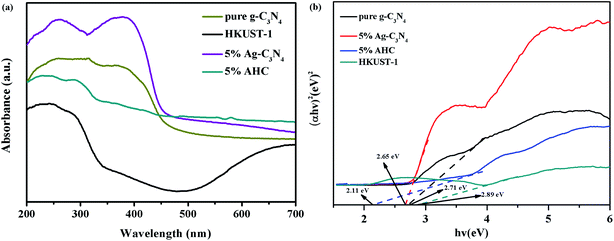 |
| | Fig. 5 (a) UV-vis diffuse reflectance spectra of pure g-C3N4, HKUST-1 and 5% AHC; (b) the plots of (αhv)2 versus hv for the band gap energies of pure g-C3N4, HKUST-1 and 5% AHC. | |
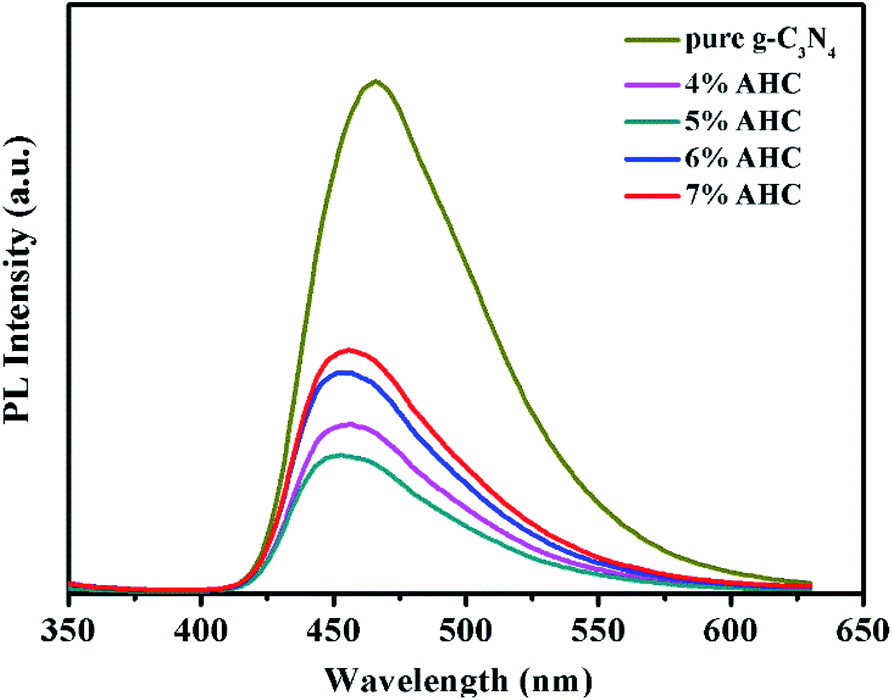
The transfer and recombination processes of electron–hole pairs in the interfaces of the composite photocatalysts are one of most important factors that affect the photocatalysis activity. PL analysis was used to estimate the photoinduced charge carrier separation and transfer ability of the as-prepared samples. It is well acknowledged that the higher PL intensity indicates the fast recombination of the charge carriers, resulting in lower photocatalytic activity. Fig. 6 presents the PL spectra of g-C3N4 and AHC with an excitation wavelength of 325 nm.47 Pure g-C3N4 has a strong peak at around 466 nm in the PL spectrum, which can be attributed to the band gap recombination of electron–hole pairs.48 However, the quenching phenomenon exists in the PL spectrum of AHC, which is related to good electrical conductivity and induced SPR effect of the Ag species. Therefore, AHC inhibits a more effective recombination of photogenerated charge carriers to improve photocatalytic activity. Moreover, the PL intensity of the 5% AHC composite photocatalyst possessed the lowest PL intensity, suggesting its high photocatalytic activity.
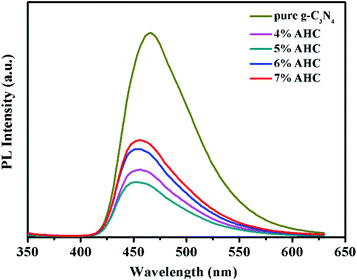 |
| | Fig. 6 Photoluminescence (PL) spectra of pure g-C3N4 and AHC. | |
Photocatalytic activity
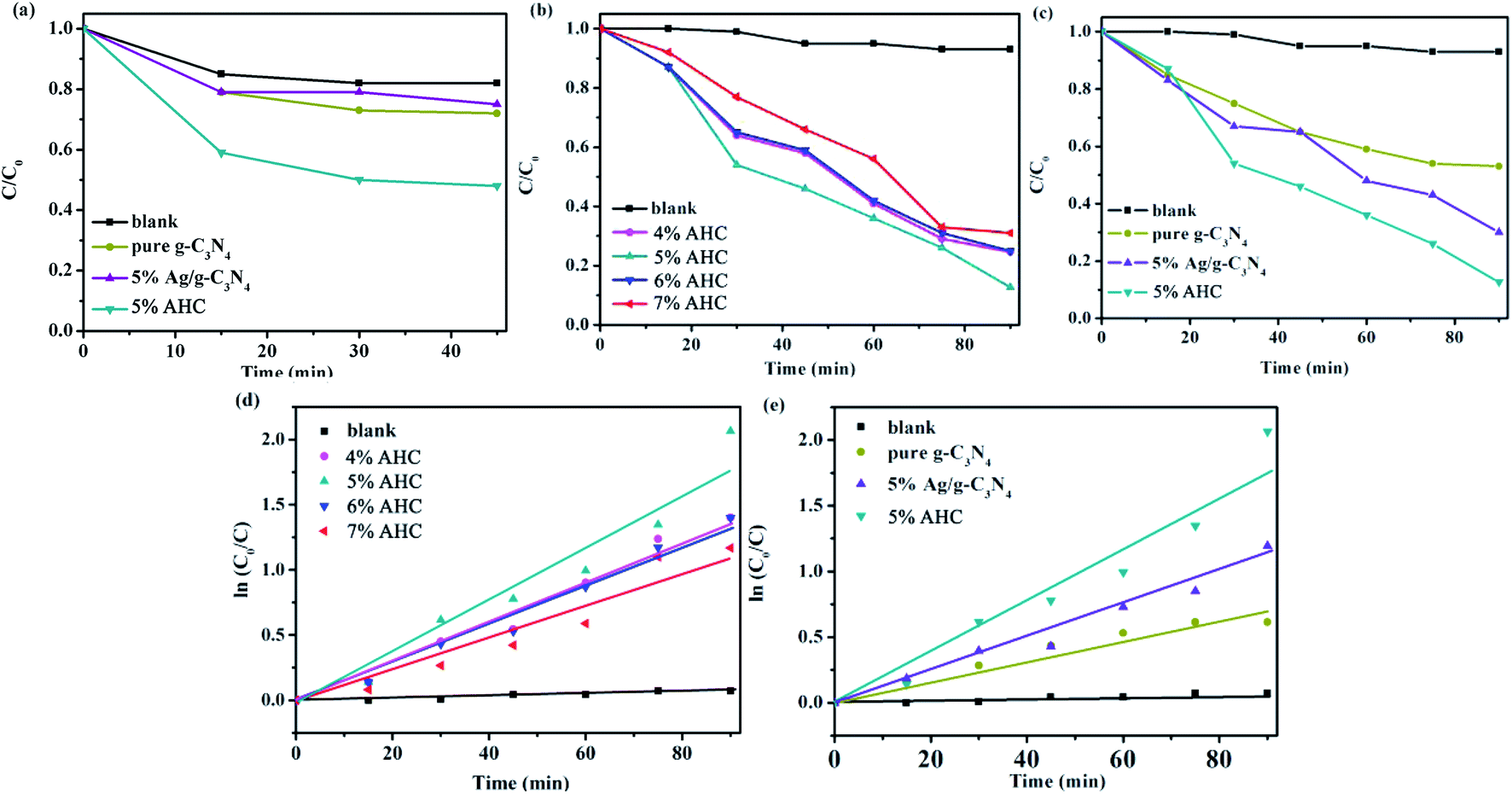
The photocatalytic activities of the as-prepared samples were evaluated by the degradation of RhB under visible light irradiation (λ > 420 nm). As shown in Fig. 7(a), the concentration of RhB could reach adsorption–desorption equilibrium after reaction under dark for 45 min. It can be seen that the adsorption of 5% AHC was significantly better than that of pure g-C3N4 and 5% Ag/g-C3N4. Hence, more RhB molecules can be adsorbed, which was beneficial for improving their visible light activities.
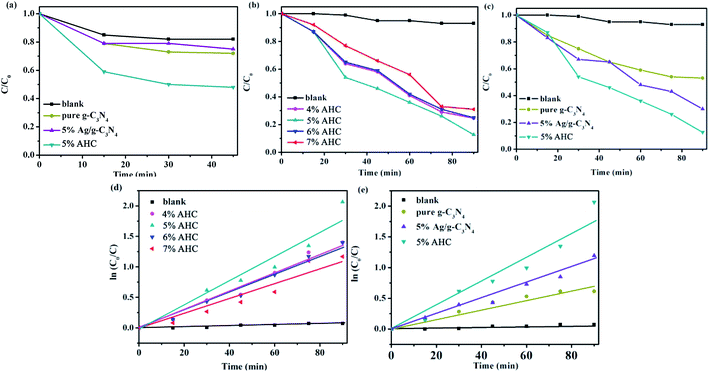 |
| | Fig. 7 (a) The dark adsorption–desorption equilibrium of RhB in catalysts; (b and c) the photocatalytic activities of as-prepared samples for RhB degradation under visible-light (λ > 420 nm); (d and e) the first-kinetic of the photocatalytic degradation of RhB. | |
The photocatalytic activities of the AHC composite photocatalysts with different Ag contents as well as the blank, pure g-C3N4 and 5% Ag/g-C3N4 were tested under visible light irradiation. As shown in Fig. 7(b) and (c), the blank test without a catalyst reveals that the photolysis of RhB occurs very slowly, which can be neglected. As shown in Fig. 7(b), when the content of Ag species increases to 5.0 wt%, the photocatalytic activity of AHC is optimal and the degradation rate of RhB reaches 87.4% while undergoing a photocatalytic reaction for 90 min. In particular, with respect to the introduction of Ag species, Ag NPs deposited on the surface of HKUST-1/g-C3N4 not only induce a plasmon resonance effect to widen light absorption ability, but also export photogenerated electrons on the HKUST-1/g-C3N4, which more efficiently improve the separation ability of the electron–hole pairs, thus resulting in the higher photocatalytic activity. However, the excess Ag species will lead to the reduction of photocatalytic activity owing to the shielding effect that weakens light harvesting ability of AHC. Fig. 7(c) indicates that the pure g-C3N4 sample exhibits low photocatalytic performance for the degradation of RhB under visible light. Furthermore, using 5% Ag/g-C3N4, 70% RhB was degraded. However, the as-synthesized 5% AHC composite photocatalyst can efficiently enhance the photocatalytic activity compared with the g-C3N4 and 5% Ag/g-C3N4. This is mainly because HKUST-1 not only has good adsorption performance, but can also be used as a medium for the rapid transfer of photogenerated charges.
At the same time, the kinetic behavior of photocatalytic degradation using the composite photocatalysts was further investigated, and the results are shown in Fig. 7(d) and (e); all of the plots fit well with the pseudo-first-order correlation:
where
C is the concentration of RhB remaining in the solution at irradiation time
t,
C0 is the initial concentration at
t = 0, and
k is the apparent rate constant for degradation. The order of the RhB degradation rate for the as-prepared photocatalysts was 5% AHC (0.021 min
−1) > 4% AHC (0.016 min
−1) > 6% AHC (0.015 min
−1) > 5% Ag/g-C
3N
4 (0.014 min
−1) > 7% AHC (0.012 min
−1) > pure g-C
3N
4 (0.007 min
−1) > blank (0.00095 min
−1).
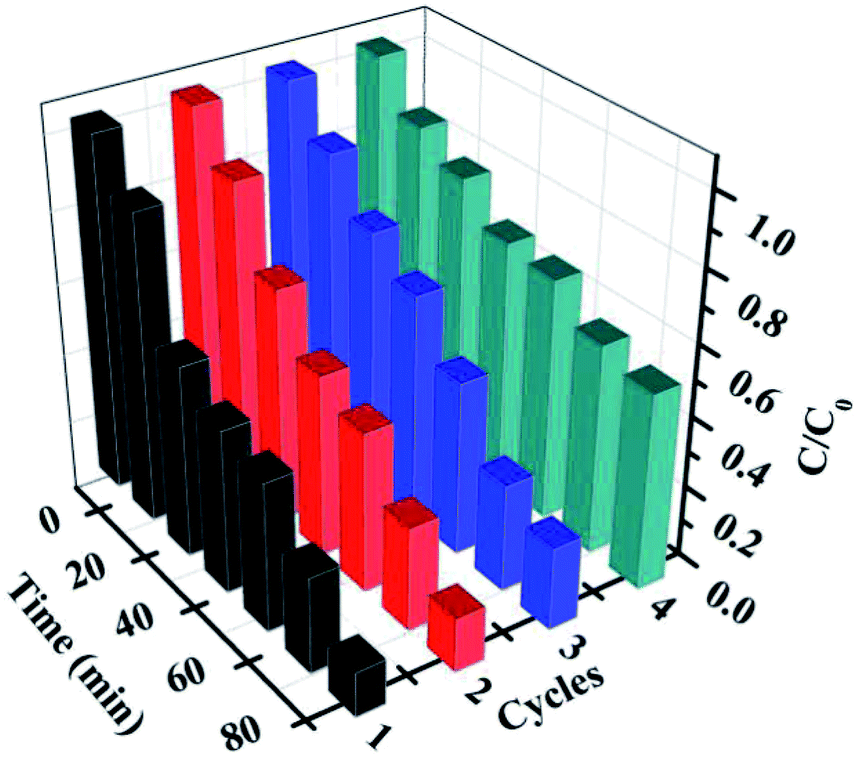
Furthermore, for the photocatalysts, the durability and stability are also crucially important for their practical applications. After each cycling test, the samples were collected and washed with distilled water and absolute ethanol three times and then, the as-obtained samples were dried in vacuum at 80 °C for 12 h for further cycling. The recycling capability of 5% AHC was verified by performing a four-run test for the degradation of RhB. As shown in Fig. 8, no significant inactivation of the photocatalysts was observed, revealing that the 5% AHC composite photocatalyst possesses high stability for its practical application. Moreover, in order to further testify the reusability and stability of the catalysts, the XRD pattern of the 5% AHC composite photocatalyst after 4th run was obtained (Fig. S2†). There was no apparent change in intensity after the photocatalytic reaction, which once again proved the stability of the catalyst.
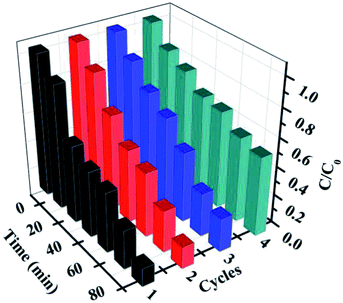 |
| | Fig. 8 The four reaction cycles of 5% AHC photocatalyst for the degradation of RhB. | |
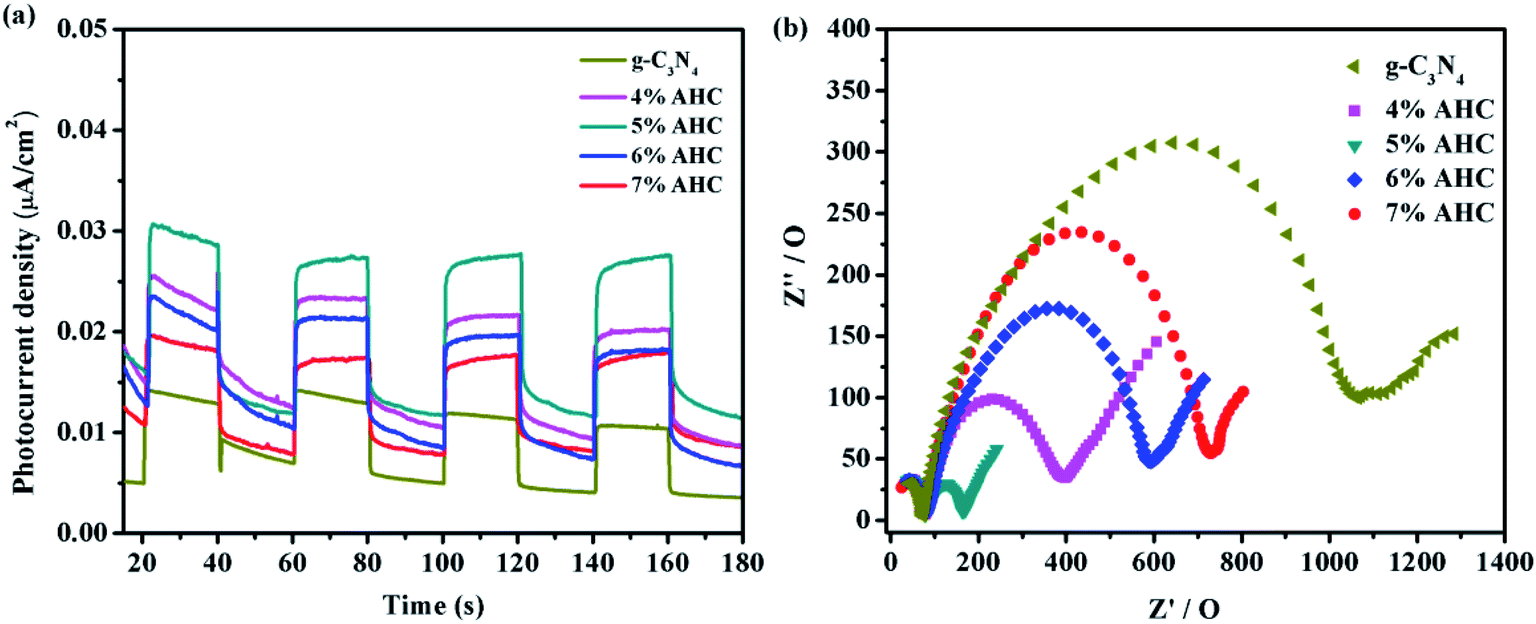
Photocatalytic activity photocurrents and EIS analyses
To further understand the separation and recombination of electron–hole pairs in the composite photocatalysts, the photocurrents over all the as-prepared samples have been investigated. Generally, the corresponding relationship was that the higher photocurrent implies the higher electron–hole separation efficiency, thus leading to higher photocatalytic activity.49 It can be clearly observed from Fig. 9(a) that rapid and stable photocurrent responses are detected in all the as-prepared samples as electrodes under visible light illumination. Clearly, the 5% AHC composite photocatalyst presents the higher photocurrent response intensity than other composites, which indicates that it has the lowest electron–hole recombination rate. This conclusion can be confirmed by PL measurements. In addition, the separation of photogenerated charge carriers can be illustrated by the EIS.50,51 EIS was performed to explore the process of their charge transfer resistance. The radius of the arc in the EIS Nyquist plot represented the charge transfer rate occurring at the contact interface between the working electrode and electrolyte solution. The smaller radius of the Nyquist arc indicates lower charge transfer resistance.52 As shown in Fig. 9(b), all the AHC samples feature significantly smaller radius than pure g-C3N4. All the above results indicated that introducing Ag had a positive effect on the separation and transfer of photogenerated charge carriers. Moreover, the arc radius of the 5% AHC composite photocatalyst was the smallest, demonstrating the fast interfacial charge transfer property of 5% AHC.
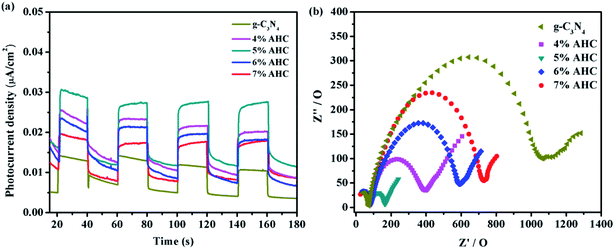 |
| | Fig. 9 (a) Transient photocurrent response of the as-prepared samples; (b) electrochemical impedance spectroscopy of as-prepared samples. | |
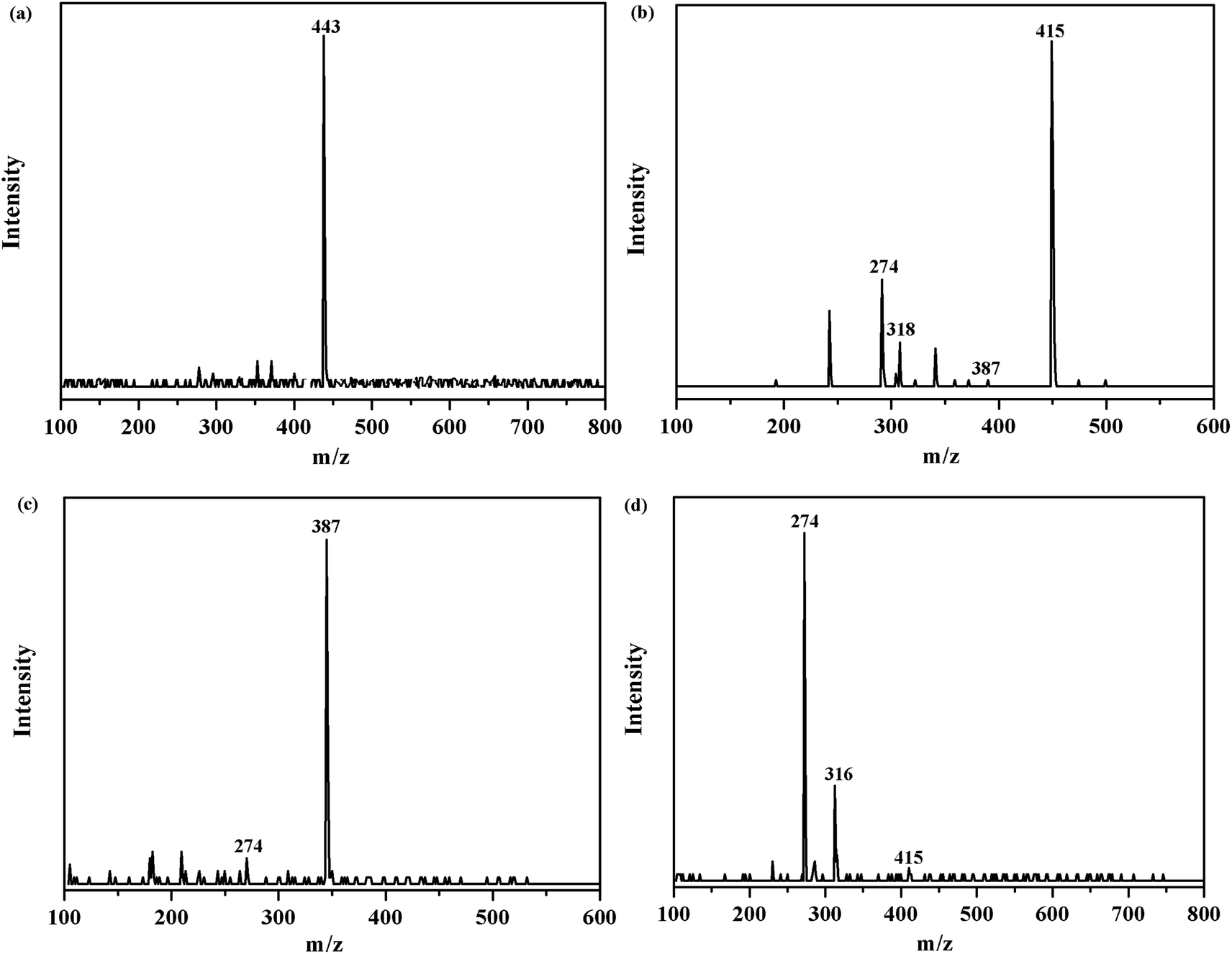
HPLC-MS spectra analysis
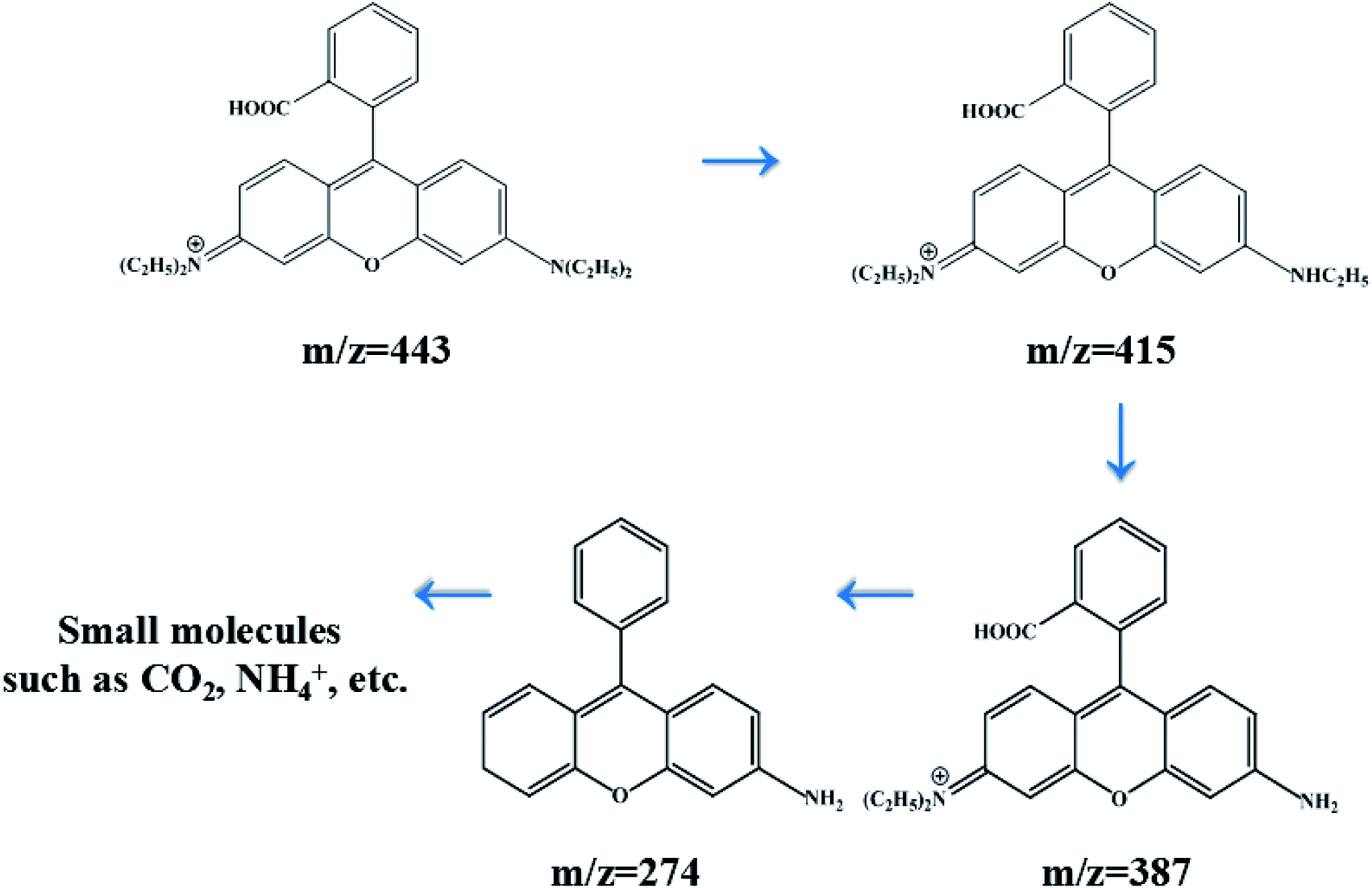
In order to investigate the intermediates and pathways in depth during the degradation of RhB, HPLC-MS experiments were performed. As shown in Fig. 10, RhB was depicted with a fragment ion at m/z 443, which is in good consistency with deprotonated RhB. As shown in Fig. 11, on progression of the reaction, RhB is resolved into small molecules with m/z = 443, m/z = 415, m/z = 387, and m/z = 274. The rest of the fragments may be derived from other complex intermediate products. Finally, with the progress of photocatalytic reaction, the intermediate products would be degraded to CO2, H2O, and NH4+ and some other small inorganic molecular materials.
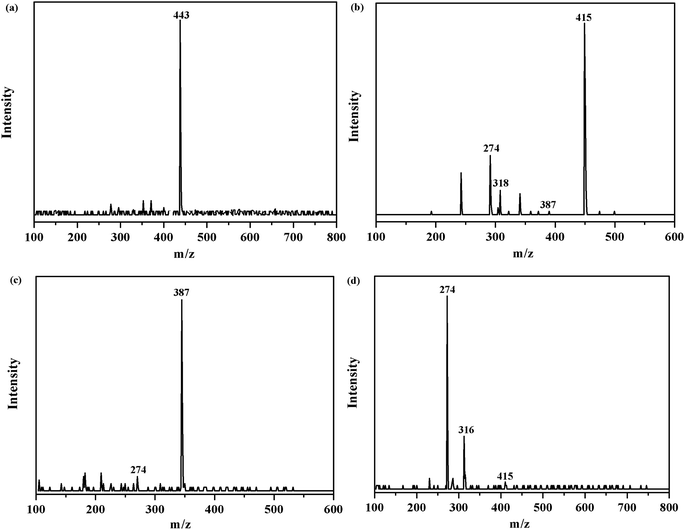 |
| | Fig. 10 m/z of degraded RhB over the 5% AHC composite photocatalyst: (a) the initial RhB solution; (b) degradation of RhB after 30 min; (c) degradation of RhB after 60 min; (d) degradation of RhB after 90 min. | |
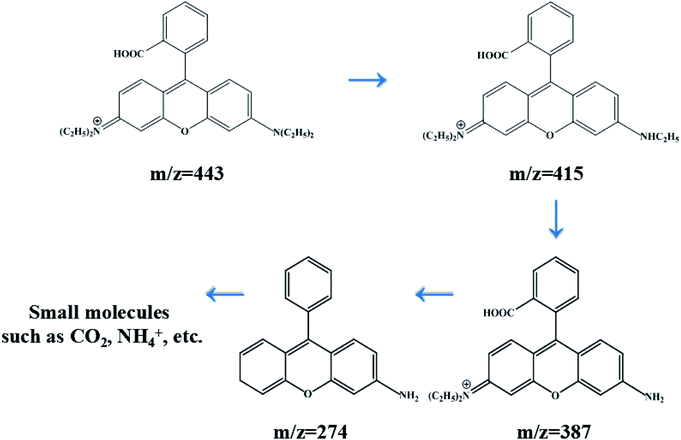 |
| | Fig. 11 Degradation process of RhB with its intermediate products over 5% AHC. | |
Photocatalytic mechanism
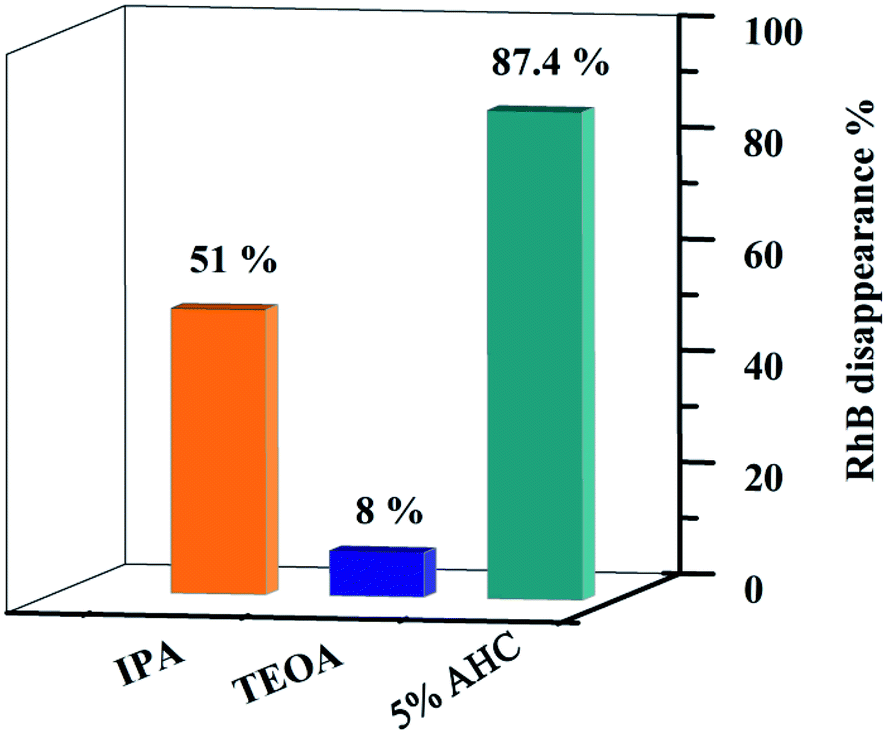
In order to explore the photocatalytic mechanism of RhB, different active species trapping experiments were conducted. The interaction between the capturer and photocatalyst played a dominant role in impacting the degradation efficiency of organic molecules.18 Therefore, a series of radical trapping experiments were performed using TEOA (a quencher of h+) and IPA (a quencher of ·OH) over the 5% AHC composite photocatalyst.53–55 As shown in Fig. 12 and S3,† for the 5% AHC sample (87.4%), it can be seen that when TEOA was added, the photocatalytic degradation rate significantly decreased (8%), indicating that h+ was the predominant active species. When IPA was added into reaction solution, the degradation rate of RhB was also inhibited (51%), suggesting that ·OH also played an important role in the photocatalytic degradation process.
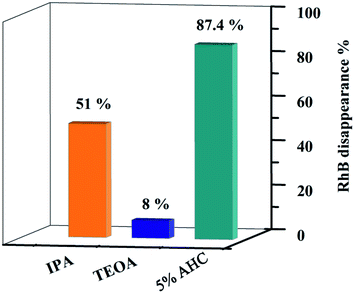 |
| | Fig. 12 Photodegradation rates of RhB over 5% AHC in the present of different scavengers. | |
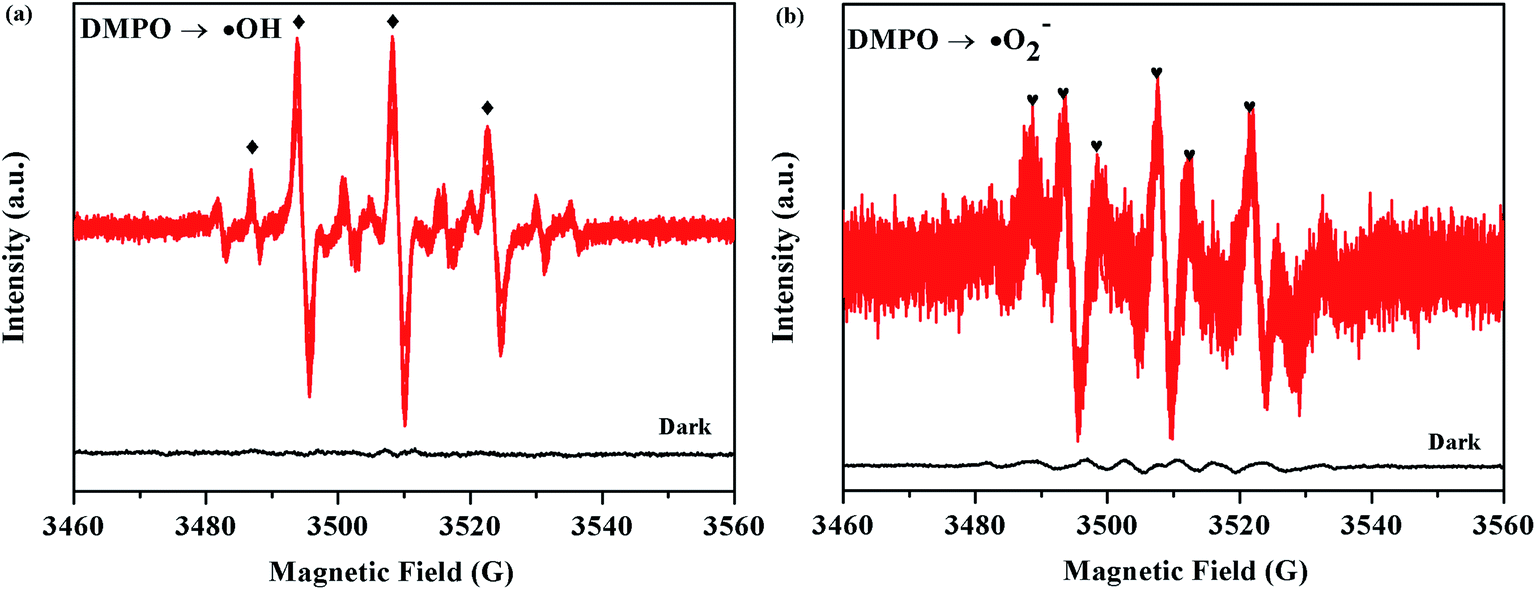
The ESR technique was further used to detect the presence of ·OH and ·O2− radicals in the 5% AHC photocatalytic reaction systems under visible light. As can be seen from Fig. 11a and b, no ESR signals were observed when the reaction was performed in the dark. As expected, for the 5% AHC sample, the four characteristic peaks of the DMPO–·OH adducts were observed (Fig. 13(a)), indicating that the ·OH radicals were produced in 5% AHC reaction systems. In addition, weak characteristic peaks of DMPO–·O2− adducts were observed (Fig. 13(b)), indicating that ·O2− radicals were produced in the 5% AHC reaction system. However, the number of ·O2− radicals were less than the number of ·OH radicals.
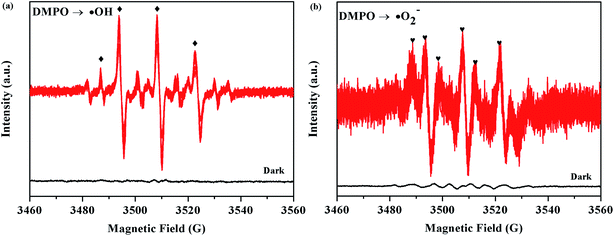 |
| | Fig. 13 ESR spectra of the 5% AHC sample for (a) DMPO–·OH and (b) DMPO–·O2−. | |
Thus, the influence order of the activated species in the process of RhB photodegradation was h+ > ·OH > ·O2−. Therefore, according to the above results of the active species trapping experiments and ESR analysis, we can make a conclusion that the h+ and ·OH play major roles and ·O2− plays a secondary role in the 5% AHC photocatalytic degradation reaction.
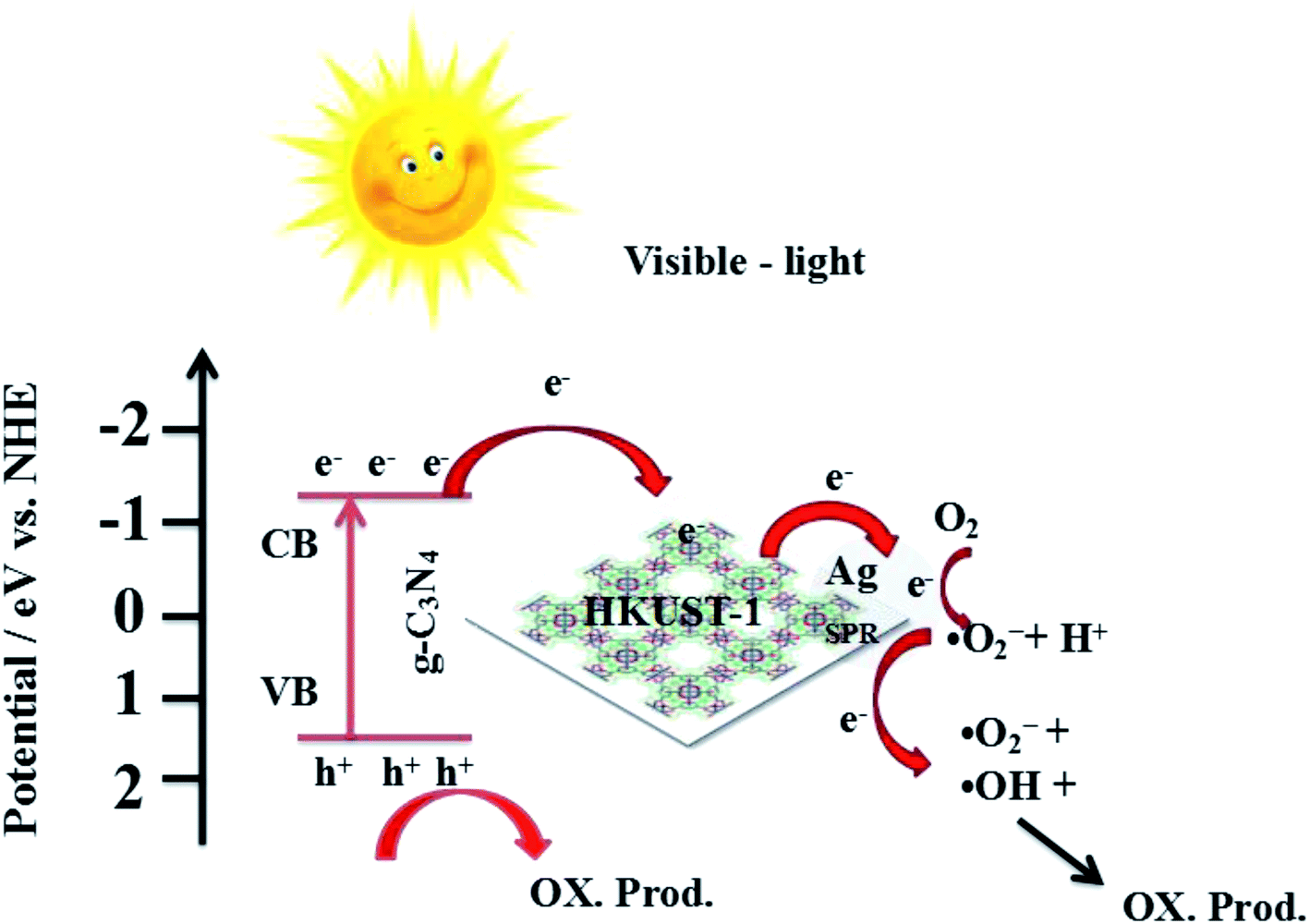
Combined with the above discussion, the possible photocatalytic mechanism for the degradation of RhB with 5% AHC composite photocatalyst is proposed, as shown in Fig. 14. First, under visible-light irradiation, g-C3N4 can be excited and photo-induced electrons and holes are generated (eqn (2)). Electrons are rapidly transferred from the g-C3N4 conduction band to the HKUST-1 surface. Due to this, the organic bridging ligands of HKUST-1 can serve as antennae to harvest visible light and then effectively transfer the energy to the inorganic copper-oxygen cluster parts. Subsequently, the photogenerated electrons on HKUST-1 rapidly migrate to the Ag surface. Some of the photoelectrons on the g-C3N4 conduction band indirectly transfer to Ag NPs via HKUST-1 and serve as terminal electron acceptors, thus prolonging the photogenerated electron lifetime and facilitating the charge separation in the entire photocatalytic system. This result has been confirmed by the PL spectra, photocurrent test and EIS analyses. Second, the photogenerated electrons react with O2 absorbed on the surface of Ag NPs to produce the active species ·O2− (eqn (3)) because the ECB value of g-C3N4 (−1.13 eV) was more negative than the potential of O2/·O2− (−0.33 eV). Then, ·O2− may also react with H+ to further produce ·OH (eqn (4)–(7)) based on the results of active species trapping and ESR analysis. Thus, both ·O2− and ·OH can oxidize RhB (eqn (8) and (9)). Moreover, the EVB value of g-C3N4 (+1.57 eV) was lower than the standard redox potential of ·OH/OH− (+1.99 eV), and photogenerated holes (h+) left on the VB of g-C3N4 could not react with H2O or OH− to generate the active oxidative species ·OH. Hence, h+ on the VB of g-C3N4 would be consumed by directly decomposing RhB (eqn (10)).56,57 The major routes of the photocatalytic degradation of RhB under visible-light irradiation are proposed as follows:
| | |
g-C3N4 + hν → g-C3N4 (e− + h+)
| (2) |
| | |
·O2− + 2H+ + e− → H2O2
| (4) |
| | |
H2O2 + e− → ·OH + OH−
| (5) |
| | |
·O2− + RhB → OX. prod.
| (8) |
| | |
·OH + RhB → OX. prod.
| (9) |
| | |
h+ + RhB → OX. prod.
| (10) |
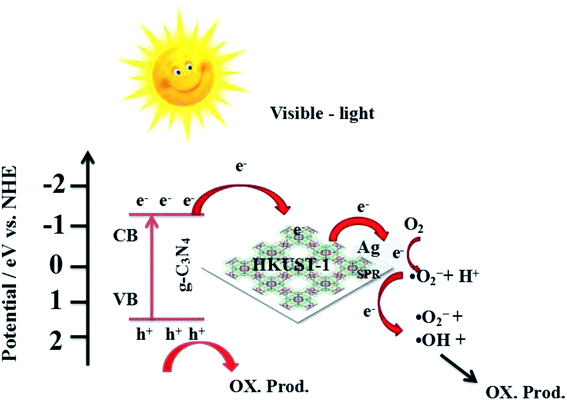 |
| | Fig. 14 Possible mechanism for the photo-induced charge transfer route of 5% AHC photocatalyst. | |
Conclusions
In summary, in situ growth strategy and photo-deposition technique were utilized to fabricate the 5% AHC composite photocatalyst. The 5% AHC composite photocatalyst exhibited superior photocatalytic performance in the degradation of RhB compared with pure g-C3N4 and other percentages of AHC. The enhanced photocatalytic activity under visible light irradiation can be ascribed to the adsorption and superior electron transport property of HKUST-1 and surface SPR of Ag, which greatly promoted the efficient separation of the photoexcited electron–hole pairs as well as depressed the recombination of the charge carriers. Finally, the present study provides the feasibility of designing new composite photocatalysts via a facile synthetic method and gives a new direction to design other high-efficiency composite semiconductor photocatalysts for practical applications.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This study was supported by the National Natural Science Foundation (No. 21905107, 21902060, 21576112, 21607051), Natural Science Foundation Project of Jilin Province (20180623042TC, 20180101181JC, 20150623024TC-19, 20170520147JH), the Project of Human Resources and Social Security Department of Jilin Province (2017956), and the Project of Education Department of Jilin Province (JJKH20180775KJ, JJKH20191009KJ).
References
- S. Debén, J. A. Fernández, A. Carballeira and J. R. Aboal, Environ. Pollut., 2016, 210, 315–322 CrossRef PubMed.
- H. K. Yan, N. Wang, T. L. Yu, Q. Fu and C. Liang, Mar. Pollut. Bull., 2013, 71, 29–40 CrossRef CAS PubMed.
- A. Álvarez-Martín, M. S. Rodríguez-Cruz, M. S. Andrades and M. J. Sánchez-Martín, Environ. Sci. Pollut. Res., 2016, 23, 9192–9230 CrossRef PubMed.
- H. Li, Y. Jing, X. L. Ma, T. Y. Liu, L. F. Yang, F. Liu, S. Yin, Y. Z. Wei and Y. H. Wang, RSC Adv., 2017, 7, 8688–8693 RSC.
- S. Bai, X. Wang, X. Hu, M. Xie, J. Jiang and Y. Xiong, Chem. Commun., 2014, 50, 6094–6097 RSC.
- C. M. Li, Y. Xu, W. G. Tu, G. Chen and R. Xu, Green Chem., 2017, 19, 882–899 RSC.
- Z. Tong, D. Yang, Z. Li, Y. Nan, F. Ding and Y. Shen, ACS Nano, 2017, 11, 1103–1112 CrossRef CAS PubMed.
- D. D. Zheng, C. J. Huang and X. C. Wang, Nanoscale, 2015, 7, 465–470 RSC.
- H. N. Che, C. B. Liu, W. Hu, H. Hu, J. Q. Li, J. Y. Dou, W. D. Shi, C. M. Li and H. J. Dong, Catal. Sci. Technol., 2018, 8, 622–631 RSC.
- Y. Y. Kang, Y. Q. Yang, L. C. Yin, X. D. Kang, L. Z. Wang, G. Liu and H. M. Cheng, Adv. Mater., 2016, 28, 6471–6477 CrossRef CAS PubMed.
- L. J. Fang, Y. H. Li, P. F. Liu, D. P. Wang, H. D. Zeng, X. L. Wang and H. G. Yang, ACS Sustainable Chem. Eng., 2017, 5, 2039–2043 CrossRef CAS.
- Z. C. Sun, M. S. Zhu, M. Fujitsuka, A. J. Wang, C. Shi and T. Majima, ACS Appl. Mater. Interfaces, 2017, 9, 30583–30590 CrossRef CAS PubMed.
- Z. Zhou, Y. Zhang, Y. Shen, S. Liu and Y. Zhang, Chem. Soc. Rev., 2018, 47, 2298–2321 RSC.
- M. Wang, P. Y. Guo, Y. Zhang, C. M. Lv, T. Y. Chai, Y. H. Xie, Y. Z. Wang and T. Zhu, J. Hazard. Mater., 2018, 349, 224–233 CrossRef CAS PubMed.
- Z. Xie, Y. Feng, F. Wang, D. Chen, Q. Zhang, Y. Zeng, W. Lv and G. Liu, Appl. Catal., B, 2018, 229, 96–104 CrossRef CAS.
- Z. Mo, H. Xu, Z. G. Chen, X. J. She, Y. H. Song, J. J. Wu, P. G. Yan, L. Xu, Y. C. Lei, S. Q. Yuan and H. M. Li, Appl. Catal., B, 2018, 225, 154–161 CrossRef CAS.
- Y. Z. Hong, Y. H. Jiang, C. S. Li, W. Q. Fan, X. Yan, M. Yan and W. D. Shi, Appl. Catal., B, 2016, 180, 663–673 CrossRef CAS.
- Z. Zhu, Z. Y. Lu, D. D. Wang, X. Tang, Y. S. Yan, W. D. Shi, Y. S. Wang, N. L. Gao, X. Yao and H. J. Dong, Appl. Catal., B, 2016, 182, 115–122 CrossRef CAS.
- X. J. Bai, S. C. Yan, J. J. Wang, L. Wang, W. J. Jiang, S. L. Wu, C. P. Sun and Y. F. Zhu, J. Mater. Chem. A, 2014, 2, 17521–17529 RSC.
- Y. Wang, J. S. Zhang, X. C. Wang, M. Antonietti and H. R. Li, Angew. Chem., Int. Ed., 2010, 49, 3356–3359 CrossRef CAS PubMed.
- A. Dailly and E. Poirier, Energy Environ. Sci., 2011, 4, 3527–3543 RSC.
- F. J. Ma, S. X. Liu, C. Y. Sun, D. D. Liang, G. J. Ren, F. Wei, Y. G. Chen and Z. M. Su, J. Am. Chem. Soc., 2011, 133, 4178–4181 CrossRef CAS PubMed.
- H. K. Chae, D. Y. Siberio-Perez, J. Kim, Y. Go, M. Eddaoudi, A. J. Matzger, M. O'Keeffe and O. M. Yaghi, Nature, 2004, 427, 523–527 CrossRef CAS PubMed.
- Y. Qiao, Y. F. Ma, W. Jiang, X. Y. Wang, W. S. Guan, G. B. Che, W. K. Li and F. Qin, CrystEngComm, 2018, 20, 7782–7794 RSC.
- J. D. Xiao, Q. C. Shang, Y. J. Xiong, Q. Zhang, Y. Luo and S. H. Yu, Angew. Chem., Int. Ed., 2016, 128, 9389–9393 CrossRef PubMed.
- W. Xu, Z. X. Si, M. Xie, L. X. Zhou and Y. Q. Zheng, Cryst. Growth Des., 2017, 17, 2147–2157 CrossRef CAS.
- P. Li, N. A. Vermeulen, C. D. Malliakas, D. A. G. Gualdrón, A. J. Howarth, B. L. Mehdi, A. Dohnalkova, N. D. Browning, M. O'Keeffe and O. K. Farha, Science, 2017, 356, 624–627 CrossRef CAS PubMed.
- Z. M. Li, Y. Qiao, C. B. Liu, Y. F. Zhou, X. Y. Wang, P. A. Charpentier, G. B. Che, W. Z. Xu, L. H. Liu and E. W. Zhu, Dalton Trans., 2018, 47, 7761–7775 RSC.
- E. Barea, C. Montoro and J. A. R. Navarro, Chem. Soc. Rev., 2014, 43, 5419–5430 RSC.
- H. Wang, X. Z. Yuan, Y. Wu, G. M. Zeng, X. H. Chen, L. J. Leng, Z. B. Wu, L. B. Jiang and H. Li, Appl. Catal., B, 2015, 174, 445–454 CrossRef.
- J. He, Z. Y. Yan, J. Q. Wang, J. Xie, L. Jiang, Y. M. Shi, F. G. Yuan, F. Yu and Y. J. Sun, Chem. Commun., 2013, 49, 6761–6763 RSC.
- R. Liang, L. J. Shen, F. F. Jing, N. Qin and L. Wu, ACS Appl. Mater. Interfaces, 2015, 7, 9507–9515 CrossRef CAS PubMed.
- S. C. Yan, Z. S. Li and Z. G. Zou, Langmuir, 2009, 25, 10397–10401 CrossRef CAS PubMed.
- H. Wang, X. Z. Yuan, Y. Wu, G. M. Zeng, X. H. Chen, L. J. Leng, Z. B. Wu, L. B. Jiang and H. Li, J. Hazard. Mater., 2015, 286, 187–194 CrossRef CAS PubMed.
- G. Cheng, M. S. Akhtar, O. B. Yang and F. J. Stadler, ACS Appl. Mater. Interfaces, 2013, 5, 6635–6642 CrossRef CAS PubMed.
- Q. J. Xiang, J. G. Yu and M. Jaroniec, J. Phys. Chem. C, 2011, 115, 7355–7363 CrossRef CAS.
- J. H. Liu, T. K. Zhang, Z. C. Wang, G. Dawson and W. Chen, J. Mater. Chem., 2011, 21, 14398–14401 RSC.
- J. X. Sun, Y. P. Yuan, L. G. Qiu, X. Jiang, A. J. Xie, Y. H. Shen and J. F. Zhu, Dalton Trans., 2012, 41, 6756–6763 RSC.
- X. P. Sun, S. J. Dong and E. Wang, Angew. Chem., Int. Ed., 2004, 43, 6360–6363 CrossRef CAS PubMed.
- S. W. Zhang, J. X. Li, X. K. Wang, Y. S. Huang, M. Y. Zeng and J. Z. Xu, ACS Appl. Mater. Interfaces, 2014, 6, 22116–22125 CrossRef CAS PubMed.
- Q. X. Luo, B. W. An, M. Ji, S. E. Park, C. Hao and Y. Q. Li, J. Porous Mater., 2015, 22, 247–259 CrossRef CAS.
- J. H. Liu, T. K. Zhang, Z. C. Wang, G. Dawson and W. Chen, J. Mater. Chem., 2011, 21, 14398–14401 RSC.
- F. Dong, L. W. Wu, Y. J. Sun, M. Fu, Z. B. Wu and S. C. Lee, J. Mater. Chem., 2011, 221, 15171–15174 RSC.
- J. H. Liu, Y. W. Zhang, L. H. Lu, G. Wu and W. Chen, Chem. Commun., 2012, 48, 8826–8828 RSC.
- Y. J. Wang, X. J. Bai, C. S. Pan, J. He and Y. F. Zhu, J. Mater. Chem., 2012, 22, 11568–11573 RSC.
- H. H. Yin, K. Yu, C. Q. Song, R. Huang and Z. Q. Zhu, ACS Appl. Mater. Interfaces, 2014, 6, 14851–14860 CrossRef CAS PubMed.
- Z. W. Tong, D. Yang, T. X. Xiao, Y. Tian and Z. Y. Jiang, Chem. Eng. J., 2015, 260, 117–125 CrossRef CAS.
- D. Li, Z. D. Wu, C. S. Xing, D. L. Jiang, M. Chen, W. D. Shi and S. Q. Yuan, J. Mol. Catal. A: Chem., 2014, 395, 261–268 CrossRef CAS.
- Y. J. Lin, S. Zhou, X. H. Liu, S. Sheehan and D. W. Wang, J. Am. Chem. Soc., 2009, 131, 2772–2773 CrossRef CAS PubMed.
- W. D. Zhang, L. C. Jiang and J. S. Ye, J. Phys. Chem. C, 2009, 113, 16247–16253 CrossRef CAS.
- W. R. Zhao, Y. Wang, Y. Yang, J. Tang and Y. Yang, Appl. Catal., B, 2012, 115, 90–99 CrossRef.
- J. Di, J. X. Xia, S. Yin, H. Xu, L. Xu, Y. G. Xu, M. Q. He and H. M. Li, J. Mater. Chem. A, 2014, 2, 5340–5351 RSC.
- Z. A. Huang, Q. Sun, K. L. Lv, Z. H. Zhang, M. Li and B. Li, Appl. Catal., B, 2015, 164, 420–427 CrossRef CAS.
- D. S. Wang, H. T. Sun, Q. Z. Luo, X. L. Yang and R. Yin, Appl. Catal., B, 2014, 156, 323–330 CrossRef.
- W. J. Li, D. Z. Li, Y. M. Lin, P. X. Wang, W. Chen, X. Z. Fu and Y. Shao, J. Phys. Chem. C, 2012, 116, 3552–3560 CrossRef CAS.
- H. T. Ren, S. Y. Jia, Y. Wu, S. H. Wu, T. H. Zhang and X. Han, Ind. Eng. Chem. Res., 2014, 53, 17645–17653 CrossRef CAS.
- M. Xu, L. Han and S. J. Dong, ACS Appl. Mater. Interfaces, 2013, 5, 12533–12540 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra08489d |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab,
Qian Hana,
Dong Lid,
Hongji Lia,
Bing Weic,
Guangbo Che
ab,
Qian Hana,
Dong Lid,
Hongji Lia,
Bing Weic,
Guangbo Che