
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTowards the complete synthetic O-antigen of Vibrio cholerae O1, serotype inaba: improved synthesis of the conjugation-ready upstream terminal hexasaccharide determinant†
Mana Mohan Mukherjee *a,
Peng Xua,
Edwin D. Stevensb and
Pavol Kováč*a
*a,
Peng Xua,
Edwin D. Stevensb and
Pavol Kováč*a
aLBC, NIDDK, National Institutes of Health, Bethesda, MD 20892-0815, USA. E-mail: mana.mukherjee@nih.gov; kpn@helix.nih.gov
bDepartment of Chemistry, Western Kentucky University, Bowling Green, KY 42101, USA
First published on 8th November 2019
Abstract
Synthesis of the upstream terminal hexasaccharide part of the lipopolysaccharides (LPS) of Vibrio cholerae O1, serotype Inaba has been improved. The key improvements include but are not limited to optimized conditions for the stereoselectivity of glycosylation reactions involved and fewer number of synthetic steps, compared to previous approaches. Particularly noteworthy is conducting the glycosylation of the very reactive glycosyl acceptor 8-azido-3,6-dioxaoctanol with the fully assembled hexasaccharide trichloroacetimidate under thermodynamic control. It produced the desired α glycoside with an α![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :β ratio of 7:1, compared with the ratio of 1.1:1, observed when the coupling was conducted conventionally. Several substances, which were previously obtained in purity acceptable only for synthetic intermediates, were now obtained in the analytically pure state and were fully characterized. The structure of the key trisaccharide glycosyl acceptor was confirmed by single-crystal X-ray structure determination.
:β ratio of 7:1, compared with the ratio of 1.1:1, observed when the coupling was conducted conventionally. Several substances, which were previously obtained in purity acceptable only for synthetic intermediates, were now obtained in the analytically pure state and were fully characterized. The structure of the key trisaccharide glycosyl acceptor was confirmed by single-crystal X-ray structure determination.
Introduction
The upstream, terminal part of the lipopolysaccharides (LPS) of most Gram-negative bacteria consist of several copies of oligosaccharide, sometimes monosaccharide repeating units. The said macromolecules are termed O-specific polysaccharides (O-SP) and are recognized as protective antigens of these pathogens. O-SPs confer specificity to their homologous antibodies and they, or fragments thereof, are paramount in the development of synthetic/conjugate vaccines. We have been involved in work towards vaccines from synthetic fragments of O-SPs for a number of years. One of our major targets has been a conjugate vaccine for cholera from synthetic fragments of O-SPs of Vibrio cholerae.1–3The O-SP of Vibrio cholerae O1, serotype Inaba consists of 12–18 repeats4 of (1→2)-α-linked perosamine (4-amino-4,6-dideoxy-D-mannose) whose amino group is acylated with 3-deoxy-L-glycero-tetronic acid. We have previously synthesized hexasaccharide fragments of the O-SP and have determined essential immune responses of conjugates therefrom in mice.5 Chemical structures of O-SPs of the two strains (Inaba and Ogawa) of Vibrio cholerae O1 are the same, except that the terminal, upstream perosamine residue in the Ogawa strain carries a methyl group at O-2. To be able to compare immune responses of the conjugates from the hexasaccharide fragments of the O-SP of both serotypes of Vibrio cholerae O1 with those of similar conjugates from synthetic polymers representing the complete O-SPs, we intend to synthesize glycoconjugates from the analogous octadecasaccharides. Syntheses of such structures are much more involved undertakings. Previous syntheses of the hexasaccharide antigens comprised up to more than 40 linear steps, depending on the individual approach.1,2 The key intermediates within our strategy towards the octadecasaccharides will be synthons derived from the related hexasaccharides. With the aim to increase the feasibility of a large-scale synthesis required by future immunization studies and decrease the number of synthetic steps involved in making such substances, the objective of this work was to test the practicability and scalability of the current, new synthetic scheme. Thus, we synthesized on large scale the trisaccharide glycosyl donor and acceptor 5 and 4, respectively, and used these to prepare, also on large scale, the related hexasaccharide 3 (Fig. 1). These substances are versatile intermediates, which we intend to use to make considerably larger fragments of the O-SP, up to the complete bacterial O-specific antigen, octadecasaccharide. Using hexasaccharide 3, we proceeded to complete the synthesis of the title hexasaccharide 1. The present pathway is the shortest published to date (33 linear steps). An additional advantage of the approach described here is that the stereoselectivity of the critical glycosylation reaction, which converted the terminal determinant to a conjugation-ready form, was substantially increased by controlling it thermodynamically.2,6
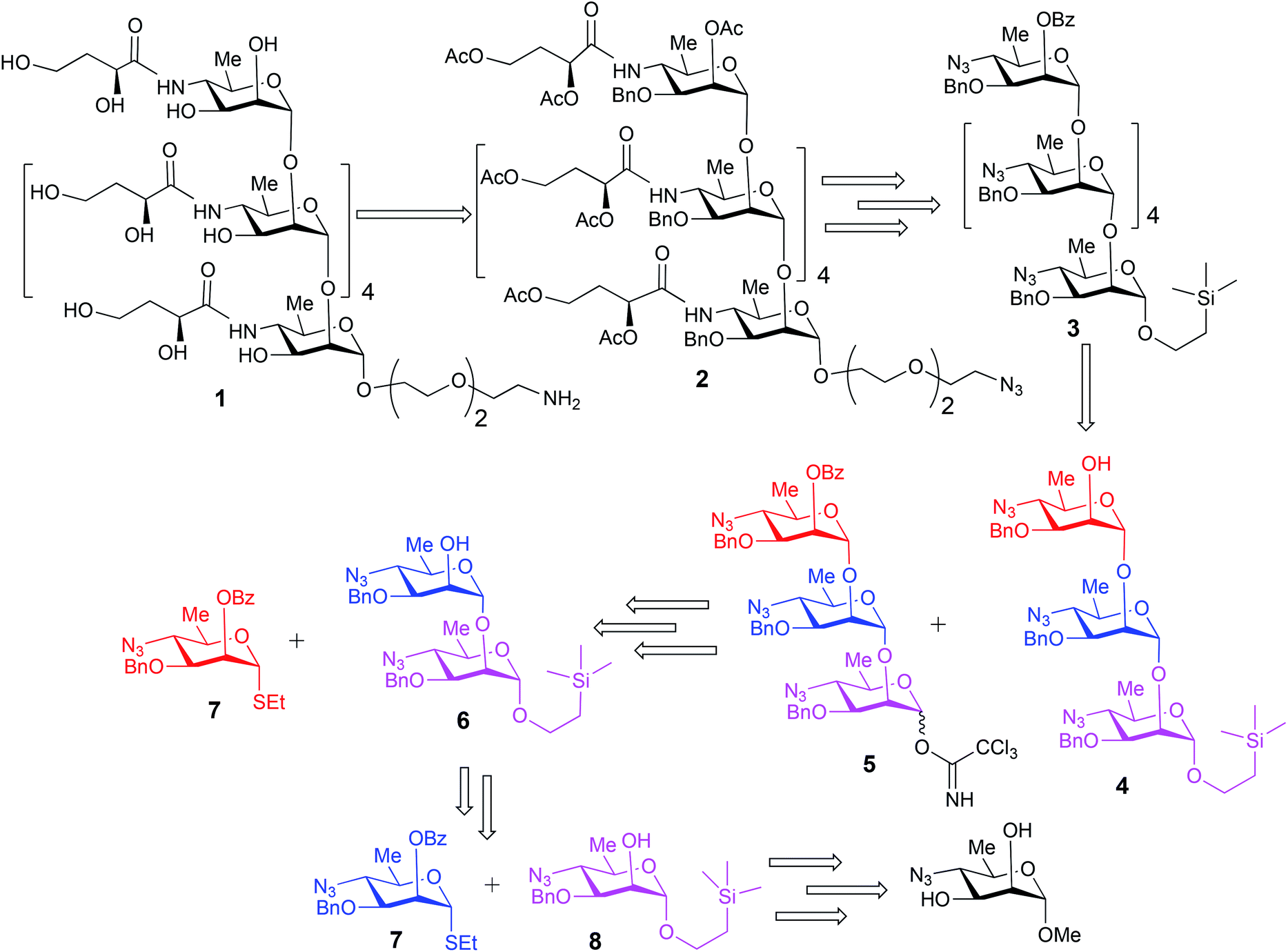 | ||
| Fig. 1 Retrosynthetic analysis of the hexasaccharide 1. | ||
Results and discussion
Synthesis of the title hexasaccharide started with the known7 methyl 4-azido-2-O-benzoyl-3-O-benzyl-4,6-dideoxy-α-D-mannopyranoside (10). It was treated with Ac2O in presence of H2SO4, and the formed (Scheme 1) 1-O-acetyl-4-azido-2-O-benzoyl-3-O-benzyl-4,6-dideoxy-α,β-D-mannopyranose (11)7 where the α-anomer largely predominated was resolved by chromatography. Unlike previously, the α-acetate 11a was now obtained crystalline for the first time. We planned to use the latter as a glycosyl donor under BF3·Et2O or TMSOTf catalysis8,9 to make the corresponding 2-trimethylsilylethyl (SE) glycoside 13a, and perhaps also higher oligosaccharides in this series, thereby improving the economy of the overall synthesis. Unfortunately, this approach was unsuccessful: only the corresponding 1-OH compound was formed. Similarly unsuccessful was the reaction of the corresponding 2-O-acetyl derivative 12 (→13b), prepared conventionally10 (these reactions are not described in the Experimental). Explanation for these failures was not sought, but in their extensive synthetic study towards this class of substances, Saksena et al.11 observed that formation of 2-trimethylsilylethyl mannopyranosides is a complex process whose outcome is largely unpredictable. In contrast with the unsuccessful Lewis acid mediated reaction of 11 or 12 with 2-trimethylsilylethanol, the BF3·Et2O-catalyzed reaction of 11 with ethanethiol readily produced ethyl 4-azido-2-O-benzoyl-3-O-benzyl-4,6-dideoxy-1-thio-α,β-D-mannopyranoside (7a and hitherto unknown 7b),11 which was fully characterized. The latter mixture of anomers was converted to its corresponding SE glycoside 13a as described,11 and the pure α-anomer 7a was used as glycosyl donor towards oligosaccharide synthesis (Schemes 2 and 3). The initial glycosyl acceptor 87 (Scheme 1) was prepared by conventional debenzoylation (Zemplén12) from the foregoing 2-O-benzoate 13a.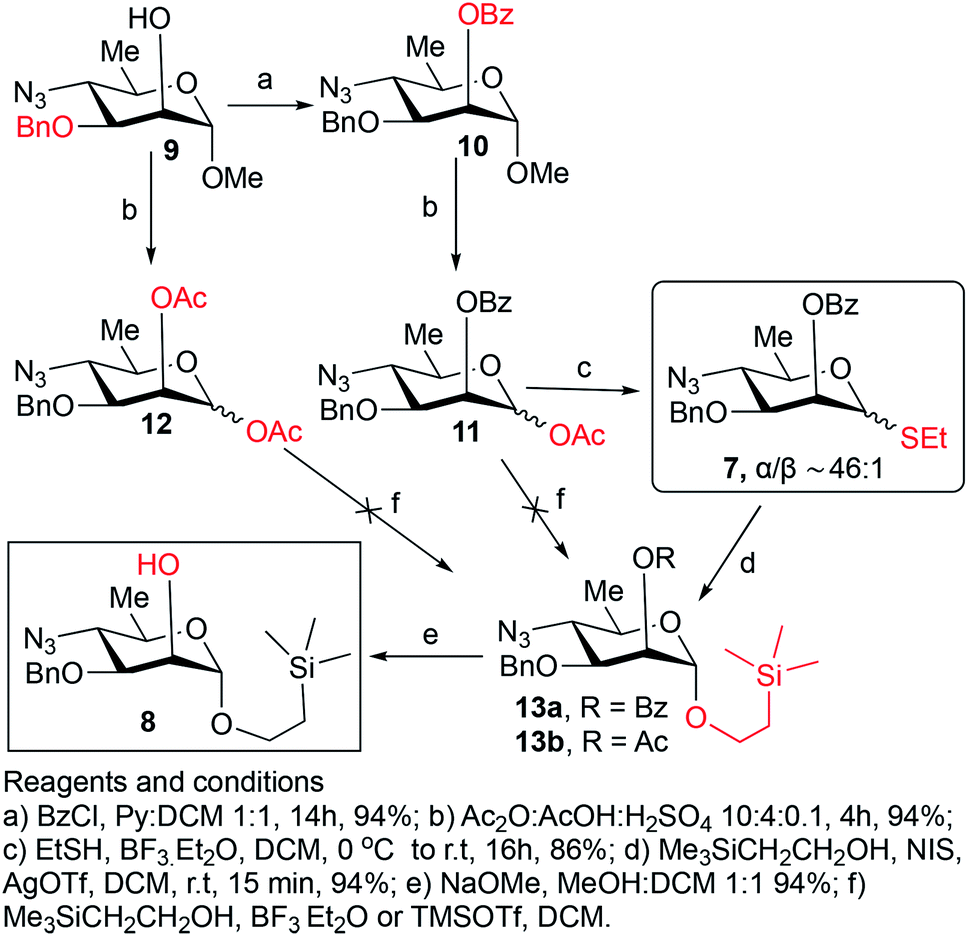 | ||
| Scheme 1 Synthesis of the monosaccharide building blocks. | ||
 | ||
| Scheme 2 Synthesis of the disaccharide acceptor 6. | ||
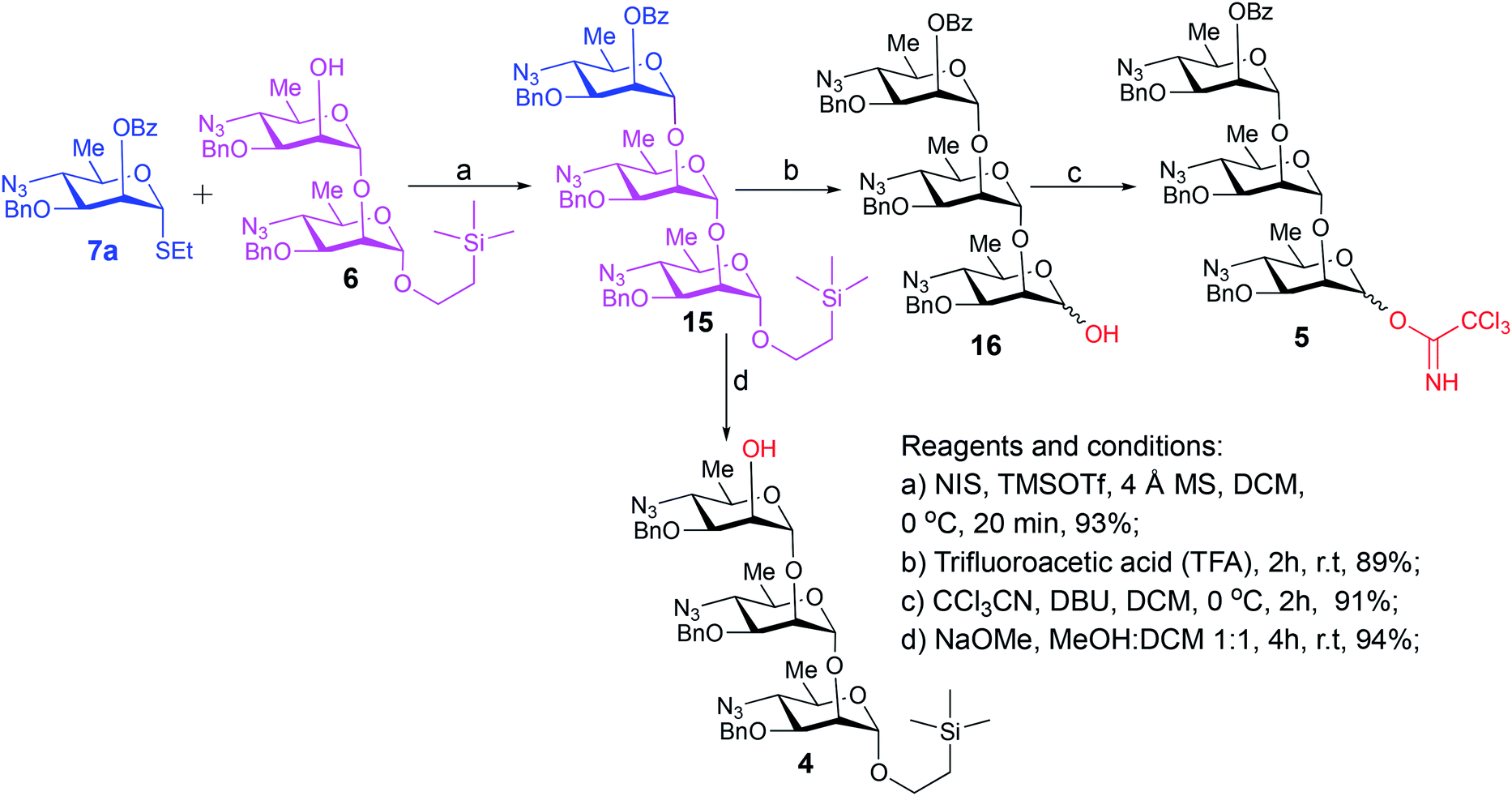 | ||
| Scheme 3 Synthesis of trisaccharide donor 5 and acceptor 4. | ||
With suitably equipped glycosyl donor and acceptor at hand, we set out for oligosaccharide synthesis on the multigram scale (see Experimental). Accordingly, glycosyl acceptor 8 and thioglycoside donor 7a were coupled using NIS/TMSOTf as promoter, affording disaccharide 14 (94%, Scheme 2). The NMR spectra of the disaccharide showed signals characteristic of the presence of both donor and acceptor moieties (165.3 ppm for the benzoyl carbonyl carbon from the donor and −1.32 ppm for SiMe3 carbon from the acceptor), while the α-configuration of the interglycosidic linkage was confirmed from the corresponding NMR spectra, mainly JC,H at the newly formed glycosidic linkage (173.3 Hz).13 Zemplén debenzoylation of 14 furnished disaccharide acceptor 6,7 which was subsequently used for chain elongation (Scheme 3). Coupling of disaccharide acceptor 6 and thioglycoside donor 7a in presence of NIS-TMSOTf at 0 °C produced trisaccharide 15 (93%, Scheme 3). Formation of the desired trisaccharide 15 was confirmed by HRMS and the stereochemistry of the newly formed interglycosidic linkage followed from the 13C–1H coupling constant for the anomeric carbon center at 99.2 ppm (C-1III, JC–H = 173.3 Hz). Zemplén debenzoylation of 15 furnished trisaccharide acceptor 4, which was obtained crystalline (CCDC no. 1939745†). Compound 4 crystallizes with 2 independent molecules in the asymmetric unit of the unit cell. Only one free –OH group is available on each molecule for hydrogen bonding, and in each case, a hydrogen bond is formed to an O5 acceptor on a mannopyranoside ring in an adjacent molecule. A modest number of weaker C–H⋯O and C–H⋯N interactions are also observed. Given the limited number of free hydroxyl groups available for hydrogen bonding, the majority of the intermolecular interactions will be weaker non-polar van der Waals type interactions. In this case, the conformations of the polysaccharide chains of the two independent molecules are more likely to reflect a conformational energy minimum for the chain, then would be the case if extensive hydrogen bonding were present. Significantly, the relative conformations of the trisaccharide chains of both independent molecules are very similar, while the orientations of the substituent groups (especially the benzyl and trimethylsilylethyl groups) show greater variation (Fig. 2). Further analysis of the conformation of X-ray data for compound 4 will be reported in a subsequent communication.
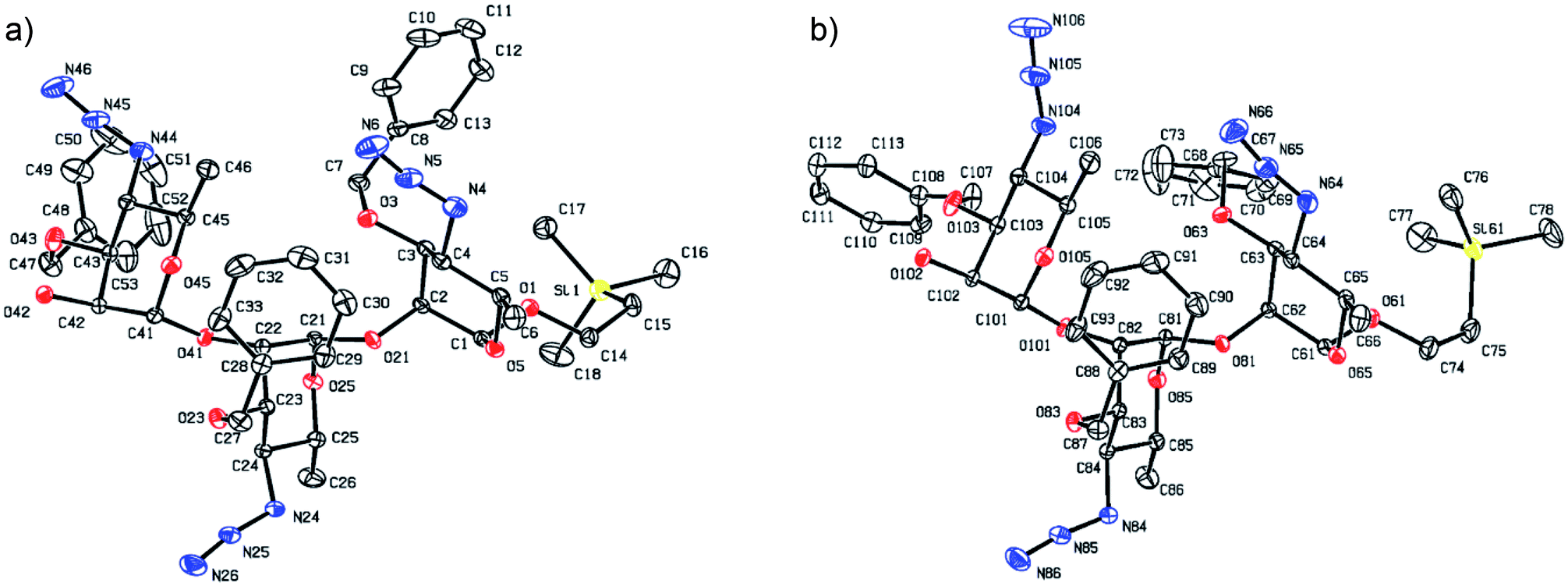 | ||
| Fig. 2 Stereo projections of the two independent molecules of compound 4 at 120 K. Thermal ellipsoids are plotted at the 50% level, and hydrogen atoms have been omitted for clarity. The projection directions have been chosen to highlight the similarity of the core conformations of the two trisaccharide chains. | ||
Compared to the NMR spectra of parent compound 15, disappearance of the carbonyl carbon signal at 165.3 ppm in the 13C NMR spectrum of 4 and upfield shift of H-2III (from δ 5.59 ppm, dd, J2–3 = 2.9 Hz, J2–1 = 2.0 Hz to δ 3.98 ppm, ddd, J2–3 = 2.8 Hz, J2-OH-2 = 1.7 Hz, J2–1 = 1.3 Hz) in the 1H NMR spectrum confirmed the removal of the benzoyl group and formation of the corresponding 2-hydroxy product 4. Treatment of the foregoing 2-trimethylsilyl ethyl glycoside 15 with trifluoroacetic acid (TFA) produced the corresponding trisaccharide hemiacetal 16 (89%). Absence of signals for the 2-(trimethylsilylethyl) group in the 1H NMR spectrum of 16, together with presence of two anomeric C-1I signals in the 13C NMR spectrum (δ 93.5 and 93.2 ppm) confirmed the successful hydrolysis and formation of the desired hemiacetal 16. Subsequent base-catalyzed reaction of 16 with trichloroacetonitrile and DBU produced the corresponding trichloroacetimidate donor 5 (Scheme 3).
Large-scale glycosylation (24.3 g of trichloroacetimidate donor 5 with 19 g of trisaccharide acceptor 4) produced hexasaccharide 3 (92%, Scheme 4) with excellent stereoselectivity (α:β ∼ 34:1, as shown from integration of signals at δ 5.32 ppm and 5.59 ppm for H-1V of 3a and H-2VI of 3b, respectively, in the NMR spectrum of the crude reaction mixture). Strong contours for anomeric carbons at 101.2 ppm (C-1II), 101.06 ppm (C-1III), 101.04 ppm (C-1IV), 101.03 ppm (C-1V), 100.3 ppm (C-1VI), 99.16 ppm (C-1I) in the HSQC spectrum confirmed the formation of the hexasaccharide. That the predominant isomer contained the desired α-configuration followed from the 13C–1H coupling constant for the 13C carbon involved in the newly formed interglycosidic linkage. For the major isomer, 13C signal of C-1IV at δ 101.04 ppm showed coupling constant JC-1–H-1 = 174.4 Hz whereas the same for the minor isomer was at δ 96.3 with JC-1–H-1 = 155.5 Hz (Fig. 3).
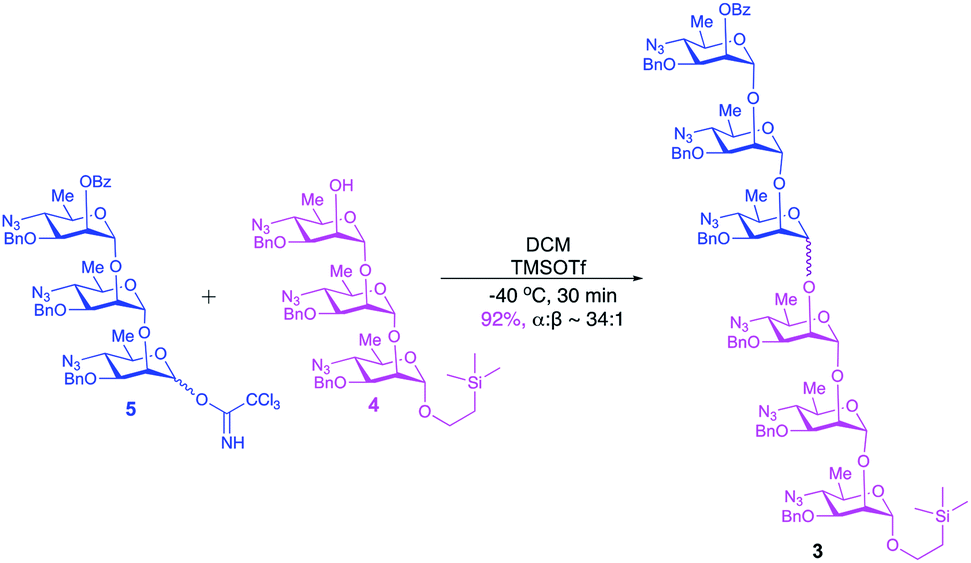 | ||
| Scheme 4 Synthesis of the hexasaccharide derivative 3. | ||
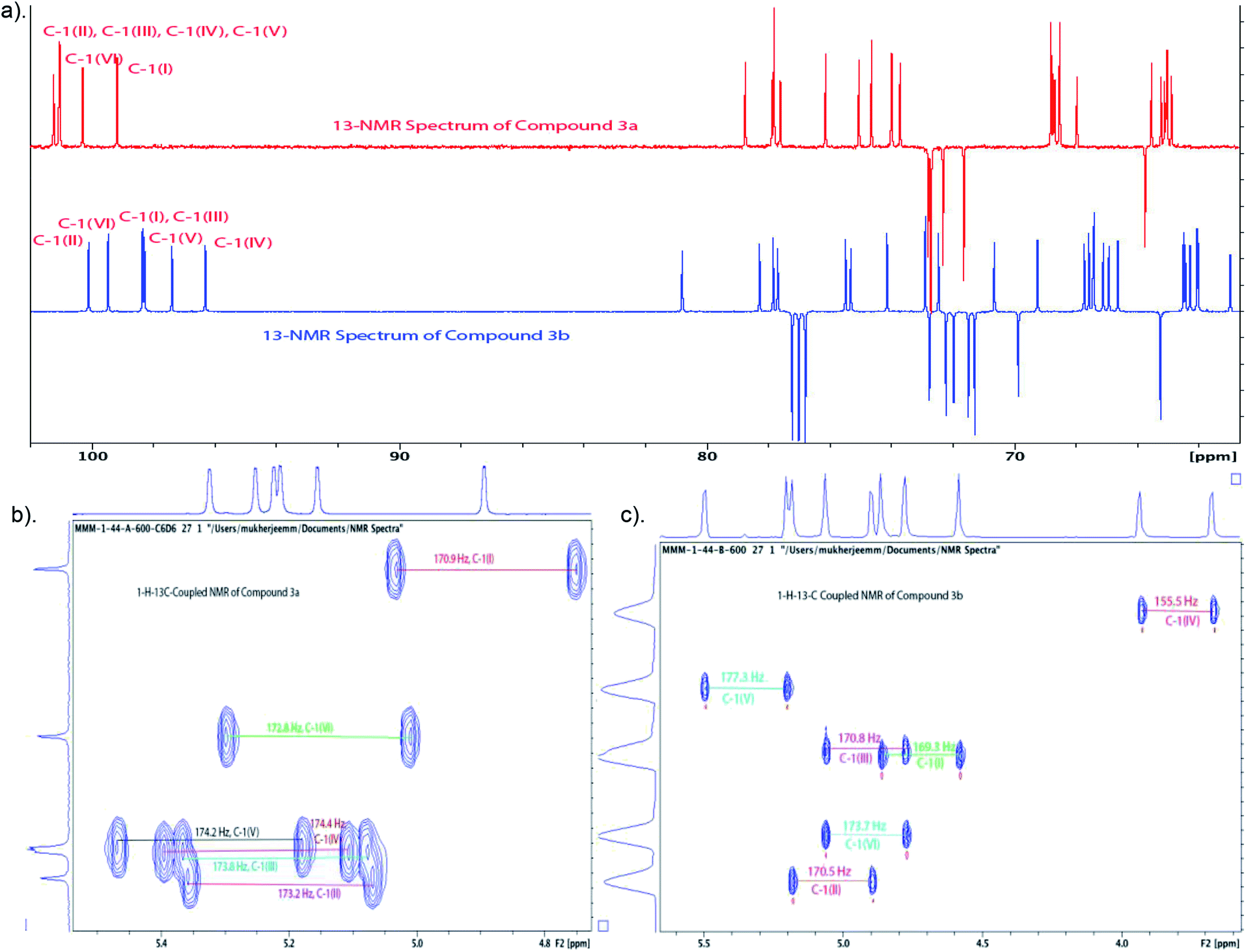 | ||
| Fig. 3 (a) Comparison of 13C-NMR spectra of compounds 3a (150 MHz, C6D6) and 3b (150 MHz, CDCl3); (b) 13C–1H coupled 2D-NMR spectrum of compound 3a (C6D6); (c) 13C–1H coupled 2D-NMR spectrum of compound 3b (CDCl3). | ||
Debenzoylation of hexasaccharide 3a under Zemplén conditions produced the 2-hydroxyl group-free intermediate 17.14 The six azido groups in the foregoing hexasaccharide were transformed, into amines by H2S reduction (→18,14 92%, Scheme 5). The next task was to introduce the N-3-deoxy-L-glycero-tetronoyl groups into the foregoing hexaamine 18. This was performed conventionally2,6 with 2,4-di-O-acetyl-3-deoxy-L-glycero-tetronic acid15 and 1-[3-(dimethylamino)propyl]-3-ethylcarbodiimide hydrochloride (EDAC). For easier isolation and next reaction, the crude product was directly acetylated, to give the fully protected hexasaccharide amide 19 (84% over two steps, Scheme 5).
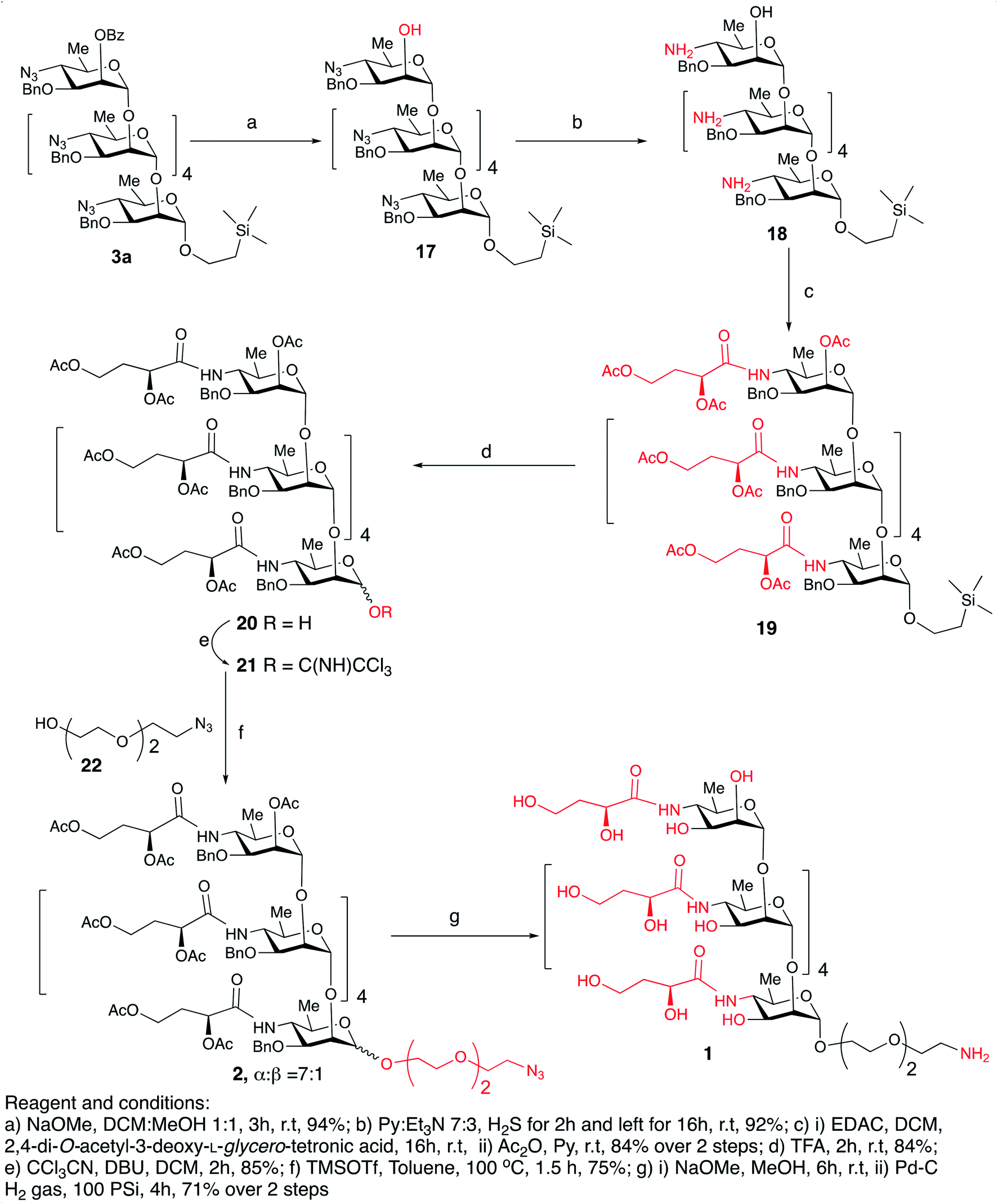 | ||
| Scheme 5 Synthesis of the title hexasaccharide. | ||
To convert compound 19 into a conjugation-ready form, aglycone in the silyl ethyl glycoside had to be replaced with a suitably equipped linker molecule. Cleavage of 19 with trifluoroacetic acid (TFA), and subsequent conversion of the reducing sugar 20, thus obtained (Scheme 5), to the corresponding trichloroacetimidate donor 21 brought the synthesis of the title antigen to a synthetic step that proved difficult in the past. In most situations across carbohydrate chemistry, synthesis of the 1,2-trans-glycosidic linkage is not problematic because a participating group can be introduced into 2-positions of the glycosyl donors. However, in our case, the 2-O-position of the donor is glycosylated and, therefore, no participating group could be introduced. Furthermore, as we found during our previous syntheses of oligosaccharides within the Vibrio cholerae O1 series, the selectivity of α-mannosylation is impaired when the glycosylation is conducted with donors having the 3-deoxy-L-glycero-tetronic acid side chain already in place.2,6,16 It is especially noteworthy that when we7,14 previously glycosylated a fully assembled hexasaccharide Vibrio cholerae O1 donor with a primary hydroxyl group-containing linker molecule, the reaction showed almost no stereoselectivity. This was the reason why more recent syntheses of similar oligosaccharides utilized glycosyl donors containing 4-azido groups,1,17 unlike in the earlier works where oligosaccharides related to the O-SP of Vibrio cholerae O1 were synthesized using glycosyl donors where the tetronamido side chain was already in place.18,19 Similarly, in the initial reaction of 21 with 8-azido-3,6-dioxaoctanol (22) under Ogawa's14 conditions, the α and β glycosides were formed in a ratio of ∼1.1:1. When we took advantage of the glycosylation under thermodynamic control developed in this laboratory,2,6 the stereoselectivity of the same glycosylation increased by many folds (α:β ∼ 7:1). Conversion of oligosaccharides into conjugation-ready forms often involves α-mannosylation of very reactive aglycons. Such reactions are characterized by poor stereo selectivity. Performing such reactions under thermodynamic control remarkably increases the stereo selectivity of such conversions, and constitutes a considerable improvement of the synthesis of this and similar conjugation-ready oligosaccharides over existing protocols. Conventional deacetylation (Zemplén12) of 2a, followed by hydrogenation/hydrogenolysis (Pd/C) yielded the final glycoside 1 (71%, over two steps). The structure of compound 1 was confirmed by HRMS and NMR spectra.
Conclusions
We have modified existing approaches leading to antigenic determinants of Vibrio cholerae O1 and verified scalability of reactions involved. Intermediates to oligosaccharides in this series, up to and including the hexasaccharide have been successfully prepared on multigram scales. Using such hexasaccharide, the synthesis of the terminal hexasaccharide fragment of the O-specific polysaccharide of Vibrio cholerae O1, serotype Inaba was improved. The number of linear synthetic steps toward the compound was reduced, and the stereoselectivity of the critical 1,2-trans-glycosylation of the very reactive 8-azido-3,6-dioxaoctanol with the fully assembled hexasaccharide trichloroacetimidate was markedly increased, from α:β = 1.1:1 to 7:1 thereby increasing considerably the yield of the conjugation-ready title compound manifold.
Experimental
Materials and methods, crystallography
Unless specified otherwise, all reagents and solvents were purchased from Sigma-Aldrich Chemical Company and used as supplied. Reactions were monitored by thin-layer chromatography (TLC) on silica gel 60 glass slides. Spots were visualized by charring with H2SO4 in EtOH (5% v/v) and/or UV light. Melting points were determined with a Kofler hot stage. Optical rotations were measured at ambient temperature with a Jasco P-2000 digital polarimeter. NMR spectra were measured at 25 °C for solutions in benzene-d6, methanol-d4 or CDCl3, at 400 MHz, 500 MHz or 600 MHz for 1H, and at 100 MHz, 125 MHz or 150 MHz for 13C with Bruker Avance Spectrometers. Assignments of NMR signals were aided by 1D and 2D experiments (1H–1H homonuclear decoupling, APT, COSY, HSQC, TOCSY and HMBC) run with the software supplied with the spectrometer. Chemical shifts were referenced to that of tetramethylsilane (0 ppm) or signals of residual non-deuterated solvents. Crystals of 4 suitable for X-ray data collection were obtained by slow evaporation of ethanol from ethanolic solution. X-ray intensity measurements were collected at low temperature from a colorless needle-shaped crystal using a Bruker Kappa APEX II 4K CCD diffractometer with MoKα radiation. An Oxford Cryosystems 700 low temperature system was used to generate a stream of cold N2 gas that cooled the sample crystal to 120(2) K during data collection. Data were collected using both ω and ϕ scans with a scan width of 0.50° per frame and a rate of 30 s per frame, with the detector center located 40.0 mm from the crystal at 2θ = 30.00° or 60.00°. The data were processed using the Bruker APEX III software package. The crystal structure was solved and refined using the SHELXL20 software package. The absolute configuration (Flack) parameter, determined from the X-ray data during the refinement, correctly identified the absolute configuration of the structure, which was also established by the known configuration of the α-D-mannopyranoside rings. The positions of H atoms attached to C and O atoms were calculated using idealized sp2 or sp3 geometry and included as riding atoms in the least-squares refinement. For methyl and –OH hydrogens, the torsion angles about the X-Me or C–OH bond were also optimized during the refinement. Details of the crystal data and structure refinement are given in ESI.† The 7 N solution of NH3 in MeOH was purchased from Sigma-Aldrich. Solutions in organic solvents were dried with anhydrous MgSO4 and concentrated at reduced pressure at <40 °C.1-O-Acetyl-4-azido-2-O-benzoyl-3-O-benzyl-4,6-dideoxy-α-D-mannopyranose (11a) and 1-O-acetyl-4-azido-2-O-benzoyl-3-O-benzyl-4,6-dideoxy-β-D-mannopyranose (11b)
This compound was prepared from 115 g (289.4 mmol) of 10 as described,7 yielding 115.7 g (94%) of anomeric mixture of 11a and 11b (α/β ∼ 9:1) as white solid. Rf = 0.45 (hexane/ethyl acetate, 4/1). Chromatography gave first 11a (104.4 g, 85%). m.p. 107–108 °C (EtOAc–hexane). [α]D +1.5 (c 1.0, CHCl3), lit17 [α]D +1.9 (c 1.9, CHCl3) for amorphous material. 1H NMR (400 MHz, CDCl3): δ 8.07–8.05 (d, 2H, J = 7.9 Hz, Ar–H), 7.59 (t, 1H, J = 7.3 Hz, Ar–H), 7.49–7.43 (t, 2H, J = 7.4 Hz, Ar–H), 7.36–7.24 (m, 5H, Ar–H), 6.15 (d, 1H, J1–2 = 1.4 Hz, H-1), 5.56 (2d, 1H, J2–3 = 2.8, J2–1 = 1.9 Hz, H-2), 4.80 (d, 1H, J = 11.2 Hz, CH2Ph), 4.59 (d, 1H, J = 11.2 Hz, CH2Ph), 3.91 (dd, 1H, J3–4 = 9.7 Hz, J3–2 = 3.2 Hz, H-3), 3.67 (dq, 1H, J5–4 = 10.1 Hz, J5–6 = 6.0 Hz, H-5), 3.57 (2d, 1H, J4–3 = 9.8 Hz, J4–5 = 10.0 Hz, H-4), 2.12 (s, 3H, COCCH3), 1.39 (d, 3H, J6–5 = 6.0 Hz, CH3). 13C{1H} NMR (100 MHz, CDCl3): δ 168.4 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 165.4 (CO), 136.9, 133.4, 129.9 (2C), 129.3, 128.5 (2C), 128.4 (2C), 128.2 (2C), 127.9, 91.1 (C-1), 75.8 (C-3), 71.6 (CH2Ph), 69.3 (C-2), 66.7 (C-5), 63.8 (C-4), 20.9 (COCH3), 18.7 (C-6). HRMS (ESI-TOF): m/z [M + Na]+ calcd for C22H23O6N3Na 448.1485, found 448.1488. Anal. calcd for C22H23O6N3: C, 62.11; H, 5.45; N, 9.88. Found C, 62.20; H, 5.40; N, 10.06.
O), 165.4 (CO), 136.9, 133.4, 129.9 (2C), 129.3, 128.5 (2C), 128.4 (2C), 128.2 (2C), 127.9, 91.1 (C-1), 75.8 (C-3), 71.6 (CH2Ph), 69.3 (C-2), 66.7 (C-5), 63.8 (C-4), 20.9 (COCH3), 18.7 (C-6). HRMS (ESI-TOF): m/z [M + Na]+ calcd for C22H23O6N3Na 448.1485, found 448.1488. Anal. calcd for C22H23O6N3: C, 62.11; H, 5.45; N, 9.88. Found C, 62.20; H, 5.40; N, 10.06.
Continued elution gave the β-linked derivative 11b as colorless syrup 11.3 g, 9.2%. Rf = 0.43 (hexane/ethyl acetate, 4/1). [α]D −51.1 (c 1.0, CHCl3), lit17 [α]D −50.2 (c 1.2, CHCl3). 1H NMR (400 MHz, CDCl3): δ 8.11–8.09 (d, 2H, J = 7.9 Hz, Ar–H), 7.59 (t, 1H, J = 7.3 Hz, Ar–H), 7.49–7.43 (t, 2H, J = 7.7 Hz, Ar–H), 7.36–7.24 (m, 5H, Ar–H), 5.82 (d, 1H, J1–2 = 3.1 Hz, H-2), 5.77 (s, 1H, H-1), 4.80 (d, 1H, J = 11.2 Hz, CH2Ph), 4.56 (d, 1H, J = 11.2 Hz, CH2Ph), 3.68 (dd, 1H, J3–4 = 9.6 Hz, J3–2 = 3.0 Hz, H-3), 3.53 (t, 1H, J = 9.7 Hz, H-4), 3.39 (m, 1H, H-5), 2.04 (s, 3H, COCCH3), 1.45 (d, 3H, J6–5 = 6.1 Hz, CH3). 13C{1H} NMR (100 MHz, CDCl3): δ 168.8 (CO), 165.8 (CO), 136.6, 133.4, 129.9 (2C), 129.5, 128.5 (2C), 128.4 (2C), 128.3 (2C), 128.1, 91.1 (C-1), 78.0 (C-3), 72.1 (C-2), 71.4 (CH2Ph), 66.8 (C-5), 63.6 (C-4), 20.7 (COCH3), 18.6 (C-6). HRMS (ESI-TOF): m/z [M + Na]+ calcd for C22H23O6N3Na 448.1485, found 448.1481. Anal. calcd for C22H23O6N3: C, 62.11; H, 5.45; N, 9.88. Found C, 62.21; H, 5.47; N, 10.01.
Ethyl 4-azido-2-O-benzoyl-3-O-benzyl-4,6-dideoxy-1-thio-α-D-mannopyranoside (7a) and ethyl 4-azido-2-O-benzoyl-3-O-benzyl-4,6-dideoxy-1-thio-β-D-mannopyranoside (7b)
To a stirred suspension of anomeric mixture of compounds 11a and 11b (115 g, 270.3 mmol) and powdered 4 Å molecular sieves (10.0 g) in dichloromethane (800 mL), ethanethiol (39 mL, 540.6 mmol) followed by boron trifluoride etherate (100 mL, 811 mmol) were added dropwise at 0 °C and the mixture was stirred at room temperature overnight, when TLC (4:1 hexane–EtOAc) showed that the reaction was complete. The mixture was neutralized with NEt3 (113 mL, 811 mmol), filtered through a Celite pad and the filtrate was concentrated with Chlorox in the receiving flask, to give crude product. A solution of the residue in DCM (300 mL) was washed with aq NaHCO3, and the aqueous layer was backwashed with DCM (3× 100 mL). Concentration of the organic phase and chromatography (10:1 hexane–EtOAc) gave first the α-linked glycoside (7a, 97.3 g, 84%) as colorless syrup. Rf = 0.70 (hexane/ethyl acetate, 4/1). [α]D +63.7 (c 1.3, CHCl3), lit11 [α]D +65 (c 1.45, CHCl3), 1H NMR (400 MHz, CDCl3): δ 8.07–8.05 (d, 2H, J = 7.8 Hz, Ar–H), 7.59 (app t, 1H, J = 7.5, 7.3 Hz, Ar–H), 7.49–7.43 (t, 2H, J = 7.7 Hz, Ar–H), 7.36–7.24 (m, 5H, Ar–H), 5.62 (t, 1H, J = 1.5 Hz, H-2), 5.34 (s, 1H, H-1), 4.74 (d, 1H, J = 11.3 Hz, CH2Ph), 4.56 (d, 1H, J = 11.2 Hz, CH2Ph), 3.96 (dq, 1H, J5–4 = 10.0 Hz, J5–6 = 6.2 Hz, H-5), 3.84 (dd, 1H, J3–4 = 10.1 Hz, J3–2 = 3.2 Hz, H-3), 3.55 (t, 1H, J = 9.9 Hz, H-4), 2.71–2.56 (m, 2H, SCH2CH3), 1.38 (d, 3H, J6–5 = 6.3 Hz, CH3), 1.29 (t, 3H, J = 7.4 Hz, SCH2CH3). 13C{1H} NMR (100 MHz, CDCl3): δ 165.6 (CO), 137.0, 133.3, 129.9 (2C), 129.7, 128.43 (2C), 128.4 (2C), 128.2 (2C), 127.9, 82.4 (C-1), 76.4 (C-3), 71.5 (CH2Ph), 69.8 (C-2), 67.5 (C-5), 64.6 (C-4), 25.7 (SCH2CH3), 18.6 (C-6), 14.9 (SCH2CH3). HRMS (ESI-TOF): m/z [M + Na]+ calcd for C22H25O4N3SNa 450.1463, found 450.1459.
Continued elution gave the β-linked glycoside 7b as colorless syrup (2.1 g, 1.8%, Total 99.4 g, 86% overall, α/β ∼ 46:1). Rf = 0.69 (hexane/ethyl acetate, 4/1). Data for 7b, [α]D −97.2 (c 1.0, CHCl3). 1H NMR (500 MHz, CDCl3): δ 8.13–8.09 (d, 2H, J = 7.8 Hz, Ar–H), 7.57 (app t, 1H, J = 7.5, 7.4 Hz, Ar–H), 7.47–7.42 (t, 2H, J = 7.7 Hz, Ar–H), 7.38–7.25 (m, 5H, Ar–H), 5.86 (d, 1H, J2–3 = 3.2 Hz, H-2), 4.86 (d, 1H, J = 11.2 Hz, CH2Ph), 4.70 (s, 1H, H-1), 4.56 (d, 1H, J = 11.8 Hz, CH2Ph), 3.61 (dd, 1H, J3–4 = 9.9 Hz, J3–2 = 3.3 Hz, H-3), 3.49 (t, 1H, J = 9.5 Hz, H-4), 3.61 (dq, 1H, J5–4 = 9.7 Hz, J5–6 = 6.2 Hz, H-5), 2.77–2.68 (m, 2H, SCH2CH3), 1.45 (d, 3H, J6–5 = 6.3 Hz, CH3), 1.30–1.24 (t, 3H, J = 7.3 Hz, SCH2CH3). 13C{1H} NMR (125 MHz, CDCl3): δ 165.8 (CO), 136.9, 133.3 (2C), 130.1, 129.4 (2C), 128.42 (2C), 128.39 (2C), 128.37, 127.9, 82.2 (C-1), 79.3 (C-3), 75.4 (C-5), 71.4 (CH2Ph), 69.2 (C-2), 63.9 (C-4), 25.6 (SCH2CH3), 18.9 (C-6), 14.8 (SCH2CH3). HRMS (ESI-TOF): m/z [M + Na]+ calcd for C22H25O4N3SNa 450.1463, found 450.1461. Anal. calcd for C22H25O4N3S: C, 61.81; H, 5.89; N, 9.83. Found C, 61.93; H, 5.89; N, 9.58.
2-(Trimethylsilyl)ethyl 4-azido-2-O-benzoyl-3-O-benzyl-4,6-dideoxy-α-D-mannopyranoside (13a)11
:1), prepared as described above, as glycosyl donors.When compounds 11a and 11b (0.25 g, 0.6 mmol) were treated with 2-(trimethylsilyl) ethanol (0.25 mL, 1.8 mmol) in presence of either boron trifluoride etherate (0.12 mL, 0.9 mmol) or TMSOTf (0.1 mL, 0.6 mmol) as described above for the synthesis of thioglycosides 7a and 7b, TLC showed that a complex mixture was formed where the product of hydrolysis of the anomeric OAc group largely predominated. Optimization of reaction conditions for this approach was not attempted.
This compound was prepared as described11 from 36 g (84.2 mmol) of anomeric mixture of 7a and 7b resulting in 38.3 g (94%) of pure compound 13a as white solid. Rf = 0.45 (hexane/ethyl acetate, 9/1). Mp. 75–76 °C (hexane), lit11 mp. 73–75 °C. [α]D −0.6 (c 1.0, CHCl3), lit11 [α]D −1 (c 1, CHCl3). 1H NMR (400 MHz, CDCl3): δ 8.06–8.05 (d, 2H, J = 7.8 Hz, Ar–H), 7.57 (app t, 1H, J = 7.5, 7.2 Hz, Ar–H), 7.47–7.43 (m, 2H, Ar–H), 7.34–7.24 (m, 5H, Ar–H), 5.52 (s, 1H, H-2), 4.89 (s, 1H, H-1), 4.76 (d, 1H, J = 11.7 Hz, CH2Ph), 4.56 (d, 1H, J = 11.3 Hz, CH2Ph), 3.93 (dd, 1H, J3–4 = 9.8 Hz, J3–2 = 3.0 Hz, H-3), 3.78 (ddd, 1H, J = 6.8, 6.6, 6.4 Hz, OCH2CH2Si), 3.63 (ddd, 1H, J = 6.0, 6.2, 4.1 Hz, OCH2CH2Si), 3.56–3.47 (m, 2H, H-4,5), 1.37 (d, 3H, J6–5 = 6.5 Hz, CH3), 1.01–0.87 (m, 2H, OCH2CH2Si), 0.28 (s, 9H, (CH3)3Si). 13C{1H} NMR (100 MHz, CDCl3): δ 165.9 (CO), 137.5, 133.3, 129.9 (2C), 129.7, 128.4 (2C), 128.3 (2C), 128.2 (2C), 127.8, 97.3 (C-1), 76.2 (C-3), 71.4 (CH2Ph), 68.1 (C-2), 66.9 (C-5), 65.5 (OCH2Si), 64.4 (C-4), 18.7 (CH3), 17.9 (CH2Si), −1.33 [3C, (CH3)3Si]. HRMS (ESI-TOF): m/z [M + Na]+ calcd for C25H33O5N3SiNa 506.2087, found 506.2089.
2-(Trimethylsilyl)ethyl 4-azido-3-O-benzyl-4,6-dideoxy-α-D-mannopyranoside (8)7
This compound was prepared as described7 from 38 g (78.6 mmol) of 13a giving pure compound 8 as colorless syrup (28 g, 94%). Rf = 0.47 (hexane/ethyl acetate, 4/1). [α]D +122.1 (c 1.6, CHCl3), lit7 [α]D +121 (c 1.0, CHCl3). 1H NMR (400 MHz, CDCl3): δ 7.41–7.30 (m, 5H, Ar–H), 4.82 (d, 1H, J1–2 = 1.5 Hz, H-1), 4.71, 4.66 (2d, 1H each, 2J = 11.3 Hz, CH2Ph), 3.95 (t, 1H, J = 1.5 Hz, H-2), 3.76 (dd, 1H, J = 9.9, 6.2 Hz, OCH2CH2Si), 3.73 (dd, 1H, J3–4 = 9.7 Hz, J3–2 = 3.5 Hz, H-3), 3.58–3.37 (m, 3H, OCH2CH2Si, H-4,5), 2.39 (d, 1H, J = 1.6 Hz, OH), 1.32 (d, 3H, J6–5 = 6.2 Hz, CH3), 0.98–0.82 (m, 2H, OCH2CH2Si), 0.01 [s, 9H, (CH3)3Si]. 13C{1H} NMR (100 MHz, CDCl3): δ 137.3, 128.6, 128.2 (2C), 128.1 (2C), 98.5 (C-1), 78.4 (C-3), 72.0 (CH2Ph), 67.4 (C-2), 66.5 (C-5), 65.2 (OCH2Si), 64.1 (C-4), 18.4 (CH3), 17.9 (CH2Si), −1.34 [3C, (CH3)3Si]. HRMS (ESI-TOF): m/z [M + NH4+] calcd for C18H33O4N4Si 397.2271, found 397.2275.2-(Trimethylsilyl)ethyl 4-azido-2-O-benzoyl-3-O-benzyl-4,6-dideoxy-α-D-mannopyranosyl-(1→2)-4-azido-3-O-benzyl-4,6-dideoxy-α-D-mannopyranoside (14)
To a solution of the glycosyl acceptor 8 (26 g, 68.5 mmol) and thioglycoside donor 7a (32.2 g, 75.3 mmol) in dry DCM (500 mL) was added 10 g of 4 Å powdered molecular sieves, and the mixture was stirred for 15 min. N-Iodosuccinimide (22.1 g, 90.4 mmol) was added, which resulted in slight pink color development. The stirring was continued for another 5 min, the reaction mixture was cooled at 0 °C, and TMSOTf (5.3 mL, 29.4 mmol) was added dropwise. Red color developed immediately and, after 20 min, when TLC (solvent 1, Rf = 0.72 at 4:1 hexane–EtOAc. Solvent 2, Rf = 0.26 at neat toluene) in both solvents showed complet conversion, the reaction was quenched by addition of triethylamine (4 mL, 29.4 mmol). The precipitate formed was filtered off (a pad of Celite) directly into a separating funnel containing excess of 2:1 (v/v) sodium thiosulfate (10%)-sodium bicarbonate (saturated) solution. The organic layer was extracted with DCM (3× 50 mL), dried and concentrated. The residue was chromatographed (10:1 hexane–EtOAc) and title compound 14 was obtained as colorless syrup (47.9 g, 94%), [α]D +23.8 (c 1.0, CHCl3). 1H NMR (600 MHz, CDCl3): δ 8.05–8.03 (d, 2H, J = 7.5 Hz, Ar–H), 7.58 (t, 1H, J = 7.5 Hz, Ar–H), 7.48–7.45 (t, 2H, J = 7.5 Hz, Ar–H), 7.38–7.33 (m, 4H, ArH), 7.31–7.24 (m, 5H, ArH), 7.17 (t, 1H, J = 7.5 Hz, Ar–H), 5.62 (t, 1H, J = 2.5 Hz, H-2II), 4.94 (d, 1H, J1–2 = 1.6 Hz, H-1II), 4.78 (d, 1H, J = 11.2 Hz, CH2Ph), 4.71 (d, 1H, J1–2 = 1.4 Hz, H-1I), 4.69 (d, 1H, J = 11.5 Hz, CH2Ph), 4.61 (d, 1H, J = 11.5 Hz, CH2Ph), 4.57 (d, 1H, J = 11.5 Hz, CH2Ph), 3.90 (dd, 1H, J3–4 = 9.8 Hz, J3–2 = 3.1 Hz, H-3II), 3.83 (t, 1H, J = 2.2 Hz, H-2I), 3.75 (dd, 1H, J3–4 = 9.8 Hz, J3–2 = 2.9 Hz, H-3I), 3.71 (m, 1H, H-5I), 3.66 (m, 1H, OCHaCH2Si), 3.52–3.47 (m, 2H, H-4II, H-5II), 3.43 (m, 1H, OCHbCH2Si), 3.33 (t, 1H, J = 9.9 Hz, H-4I), 1.34 (d, 3H, J6–5 = 6.2 Hz, CH3, H-6I), 1.29 (d, 3H, J6–5 = 6.3 Hz, CH3, H-6II), 0.94–0.82 (m, 2H, CH2CH2Si), 0.01 [s, 9H, (CH3)3Si]. 13C{1H} NMR (150 MHz, CDCl3): δ 165.3 (CO), 137.6, 137.1, 133.2, 129.9 (2C), 129.7, 128.5 (2C), 128.4 (2C), 128.3 (2C), 127.94 (2C), 127.90 (2C), 127.8 (2C), 99.5 (C-1II, JC-1,H-1 = 173.3 Hz), 98.2 (C-1I, JC-1,H-1 = 167.7 Hz), 77.8 (C-3I), 75.3 (C-3II), 74.4 (C-2I), 72.1 (CH2Ph), 71.4 (CH2Ph), 67.7 (C-2II), 67.6 (C-5I), 66.9 (C-5II), 65.2 (OCH2CH2Si), 64.2 (C-4I), 64.1 (C-4II), 18.7 (CH3, C-6I), 18.6 (CH3, C-6II), 17.5 (CH2Si), −1.32 [3C, (CH3)3Si]. HRMS (ESI-TOF): m/z [M + NH4+] calcd for C38H52O8N7Si 762.3647, found 762.3637. Anal. calcd for C38H48O8N6Si: C, 61.27; H, 6.5; N, 11.28. Found C, 61.54; H, 6.36; N, 11.09.
2-(Trimethylsilyl)ethyl 4-azido-3-O-benzyl-4,6-dideoxy-α-D-mannopyranosyl-(1→2)-4-azido-3-O-benzyl-4,6-dideoxy-α-D-mannopyranoside (6)7
This compound was prepared as described7 from 47.5 g (63.8 mmol) of 14, to give 38.4 g (94%) of pure compound 6 as colorless syrup. Rf = 0.41 at hexane/ethyl acetate, 4/1. [α]D +101.7 (c 1.1, CHCl3), lit7 [α]D +102 (c 1.0, CHCl3). 1H NMR (500 MHz, CDCl3): δ 7.41–7.29 (m, 10H, Ar–H), 4.94 (d, 1H, J1–2 = 1.4 Hz, H-1II), 4.72, 4.68, 4.64, 4.61 (partially overlapped 4d, 4H, 2J ∼ 11.5 Hz, 2 CH2Ph), 4.66 (partially overlapped d, 1H, J1–2 = 4.4 Hz, H-1I), 3.99 (m, 1H, H-2II), 3.88 (t, 1H, J = 2.5 Hz, H-2I), 3.75–3.68 (m, 3H, H-3I,II, OCHaCH2Si), 3.61 (m, 1H, H-5I), 3.51–3.40 (m, 3H, H-4I, H-5II, OCHbCH2Si), 3.29 (t, 1H, J = 10.0 Hz, H-4II), 2.29 (d, 1H, J = 1.7 Hz, OH), 1.30, 1.29 (overlapped 2d, 6H, J6–5 = 6.2 Hz, CH3, H-6I,II), 0.94–0.82 (m, 2H, CH2CH2Si), 0.01 [s, 9H, (CH3)3Si]. 13C{1H} NMR (125 MHz, CDCl3): δ 137.4, 137.1, 128.6 (2C), 128.5 (2C), 128.3 (2C), 128.2 (2C), 127.9 (2C), 100.7 (C-1II), 98.3 (C-1I), 77.9 (C-3I), 77.6 (C-3II), 73.8 (C-2I), 72.1 (2C, CH2Ph), 67.2 (C-2II), 67.1 (C-5I), 66.9 (C-5II), 65.2 (OCH2Si), 64.3 (C-4II), 63.8 (C-4I), 18.6 (CH3), 18.4 (CH3), 17.7 (CH2Si), −1.32 [3C, (CH3)3Si]. HRMS (ESI-TOF): m/z [M + NH4+] calcd for C31H48O7N7Si 658.3384, found 658.3389.2-(Trimethylsilyl)ethyl 4-azido-2-O-benzoyl-3-O-benzyl-4,6-dideoxy-α-D-mannopyranosyl-(1→2)-4-azido-3-O-benzyl-4,6-dideoxy-α-D-mannopyranosyl-(1→2)-4-azido-3-O-benzyl-4,6-dideoxy-α-D-mannopyranoside (15)
To a solution of disaccharide acceptor 6 (38 g, 59.3 mmol) and thioglycoside donor 7a (28 g, 65.2 mmol) in dry DCM (500 mL) was added 10 g of 4 Å powdered molecular sieves, and the mixture was stirred for 15 min. N-Iodosuccinimide (19.2 g, 78.2 mmol) was added, which resulted in slight pink color development. The stirring was continued for another 5 min, and the reaction mixture was cooled at 0 °C. TMSOTf (4.7 mL, 26.1 mmol) was added, whereupon red color developed immediately. After 20 min at 0 °C, when TLC in 2 solvents (1. Rf = 0.72 at 4:1 hexane–EtOAc; 2. Rf = 0.32 at neat toluene) showed that the reaction was complete, the reaction was terminated by addition of triethylamine (3.7 mL). The precipitate formed was filtered through a pad of Celite directly into a separating funnel containing excess of 2:1 (v/v) sodium thiosulfate (10%)–sodium bicarbonate (saturated) solution. The mixture was extracted with DCM (3× 50 mL), the combined organic layers were dried, concentrated, and chromatography (12:1 hexane:EtOAc) gave product 15 as colorless syrup (55.8 g, 93%). [α]D +34.0 (c 1.0, CHCl3). 1H NMR (600 MHz, CDCl3): δ 8.07–8.05 (d, 2H, J = 7.3 Hz, Ar–H), 7.61 (t, 1H, J = 7.4 Hz, Ar–H), 7.50–7.47 (t, 2H, J = 7.7 Hz, Ar–H), 7.38–7.37 (m, 2H, ArH), 7.34–7.25 (m, 11H, ArH), 7.20–7.15 (m, 2H, Ar–H), 5.59 (dd, 1H, J2–3 = 2.9 Hz, J2–1 = 2.0 Hz, H-2III), 4.97 (d, 1H, J1–2 = 1.6 Hz, H-1II), 4.89 (d, 1H, J1–2 = 1.6 Hz, H-1III), 4.76 (d, 1H, J = 11.0 Hz, CH2Ph), 4.71 (d, 1H, J = 11.7 Hz, CH2Ph), 4.65 (d, 1H, J1–2 = 1.6 Hz, H-1I), 4.63 (d, 1H, J = 11.6 Hz, CH2Ph), 4.60–4.55 (3d, 3H, J = 11.5 Hz, 11.4 Hz, 11.3 Hz, 3CH2Ph), 3.88–3.85 (m, 2H, H-2II, H-3III), 3.82 (t, 1H, J = 2.3 Hz, H-2I), 3.73 (dd, 1H, J3–4 = 9.9 Hz, J3–2 = 2.9 Hz, H-3II), 3.73–3.68 (m, 2H, H-3I, OCHaCH2Si), 3.60–3.51 (m, 2H, H-5III, H-5II), 3.49–3.40 (m, 3H, H-4III, H-5I, OCHbCH2Si), 3.35 (t, 1H, J = 10.0 Hz, H-4II), 3.22 (t, 1H, J = 9.9 Hz, H-4I), 1.28, 1.27 (overlapped 2d, 3H each, J6–5 = 6.1 Hz, CH3, H-6II, H-6III), 1.23 (d, 3H, J6–5 = 6.1 Hz, CH3, H-6I), 0.93–0.82 (m, 2H, CH2CH2Si), 0.01 [s, 9H, (CH3)3Si]. 13C{1H} NMR (150 MHz, CDCl3): δ 165.3 (CO), 137.4, 137.3, 137.1, 133.3, 129.9, 129.8 (2C), 128.6, 128.5 (2C), 128.46 (2C), 128.45 (2C), 128.4 (2C), 128.3 (2C), 128.1 (2C), 128.0 (2C), 127.9 (2C), 100.4 (C-1II, JC–H = 173.7 Hz), 99.2 (C-1III, JC–H = 173.3 Hz), 98.3 (C-1I, JC–H = 168.6 Hz), 77.7 (C-3I), 76.7 (C-3II), 75.3 (C-3III), 74.1 (C-2II), 74.0 (C-2I), 72.1 (2C, 2 CH2Ph), 71.4 (CH2Ph), 67.7 (C-5II), 67.64 (C-5III), 67.6 (C-2III), 67.0 (C-5I), 65.2 (OCH2CH2Si), 64.5 (C-4I), 64.1 (C-4III), 64.0 (C-4II), 18.7 (CH3, C-6I), 18.6 (CH3, C-6II), 18.58 (CH3, C-6III), 17.8 (CH2Si), −1.32 [3C, (CH3)3Si]. HRMS (ESI-TOF): m/z [M + NH4+] calcd for C51H67O11N10Si 1023.4760, found 1023.4742. Anal. calcd for C51H63O11N9Si: C, 60.88; H, 6.31; N, 12.53. Found C, 61.17; H, 6.05; N, 12.32.
2-(Trimethylsilyl)ethyl 4-azido-3-O-benzyl-4,6-dideoxy-α-D-mannopyranosyl-(1→2)-4-azido-3-O-benzyl-4,6-dideoxy-α-D-mannopyranosyl-(1→2)-4-azido-3-O-benzyl-4,6-dideoxy-α-D-mannopyranoside (4)
To the solution of trisaccharide 15 (23 g, 22.8 mmol) in a mixture of dry DCM (50 mL) and dry MeOH (50 mL), methanolic NaOMe (1 M, 10 mL) was added under Ar, and the mixture was stirred at room temperature overnight, when TLC (Rf = 0.54 at 4:1 hexane:EtOAc) showed that the reaction was complete and that a much slower moving product was formed. The mixture was neutralized with Dowex 50W resin, filtered, and the solvent was removed. The crude product was passed through a short pad of silica and elution with 5:1 hexane–EA afforded pure 4 as white solid. Crystallization from hot hexane gave needles (19.4 g, 94%), mp. 72–73 °C, [α]D +102.86 (c 1.0, CHCl3). 1H NMR (600 MHz, CDCl3): δ 7.40–7.29 (m, 15H, Ar–H), 4.96 (d, 1H, J1–2 = 1.3 Hz, H-1III), 4.95 (d, 1H, J1–2 = 1.6 Hz, H-1II), 4.72 (d, 1H, J = 11.4 Hz, CH2Ph), 4.69–4.64 (m, 3H, 2 CH2Ph, H-1I), 4.62 (d, 1H, J = 11.5 Hz, CH2Ph), 4.61 (d, 1H, J = 11.6 Hz, CH2Ph), 4.57 (d, 1H, J = 11.6 Hz, CH2Ph), 3.98 (ddd, 1H, J2–3 = 2.8 Hz, J2-OH-2 = 1.7 Hz, J2–1 = 1.3 Hz, H-2III), 3.93 (t, 1H, J = 2.2 Hz, H-2II), 3.81 (t, 1H, J = 2.3 Hz, H-2I), 3.74–3.71 (m, 1H, H-3II), 3.71–3.67 (m, 3H, H-3III, H-3I, OCHaCH2Si), 3.56–3.51 (m, 1H, H-5II), 3.51–3.47 (m, 1H, H-5III), 3.47–3.38 (m, 3H, H-5I, OCHbCH2Si, H-4III), 3.32 (t, 1H, J = 10.0 Hz, H-4II), 3.22 (t, 1H, J = 9.9 Hz, H-4I), 2.27 (d, 1H, J2-OH-2 = 1.8 Hz, OH), 1.28 (2d, 3H each, J6–5 = 6.1 Hz, CH3, H-6II, H-6I), 1.18 (d, 3H, J6–5 = 6.1 Hz, CH3, H-6III), 0.92–0.83 (m, 2H, CH2CH2Si), 0.01 [s, 9H, (CH3)3Si]. 13C{1H} NMR (150 MHz, CDCl3): δ 137.31, 137.26, 137.1, 128.6 (2C), 128.57, 128.3 (2C), 128.26 (2C), 128.21 (2C), 128.17 (2C), 128.1 (2C), 128.0 (2C), 100.42 (C-1II), 100.4 (C-1III), 98.3 (C-1I), 77.6 (C-3II), 77.5 (C-3I), 76.9 (C-3III), 73.9 (C-2II), 73.2 (C-2I), 72.2 (2C, 2 CH2Ph), 72.1 (CH2Ph), 67.7 (C-5II), 67.3 (C-5III), 67.1 (C-2III), 67.0 (C-5I), 65.2 (OCH2CH2Si), 64.4 (C-4I), 64.2 (C-4II), 63.8 (C-4III), 18.64 (CH3, C-6I), 18.56 (CH3, C-6III), 18.3 (CH3, C-6II), 17.7 (CH2Si), −1.32 [3C, (CH3)3Si]. HRMS (ESI-TOF): m/z [M + NH4+] calcd for C44H63O10N10Si 919.4498; found 919.4483. Anal. calcd for C44H59O10N9Si: C, 58.58; H, 6.59; N, 13.97. Found C, 58.77; H, 6.44; N, 13.77.
4-Azido-2-O-benzoyl-3-O-benzyl-4,6-dideoxy-α-D-mannopyranosyl-(1→2)-4-azido-3-O-benzyl-4,6-dideoxy-α-D-mannopyranosyl-(1→2)-4-azido-3-O-benzyl-4,6-dideoxy-D-mannopyranose (16)
A solution of glycoside 16 (30 g, 30 mmol) in trifluoroacetic acid (TFA, 120 mL) was stirred at room temperature for 2 hours, when TLC (4:1 hexane–EtOAc) showed that the reaction was complete and that a much slower moving product was formed. The mixture was concentrated and a solution of the residue in DCM was washed with saturated Na2CO3 solution. The aqueous layer was extracted with DCM (3× 50 mL), and the combined organic phase was dried and concentrated. The residue was chromatographed (Rf = 0.27 at 4:1 hexane–EtOAc) to give pure product 16 as white foam (23.9 g, 89%). 1H NMR for the major α anomer (600 MHz, CDCl3): δ 8.07–8.06 (d, 2H, J = 7.3 Hz, Ar–H), 7.61 (t, 1H, J = 7.5 Hz, Ar–H), 7.50–7.47 (t, 2H, J = 7.7 Hz, Ar–H), 7.39–7.27 (m, 13H, Ar–H), 7.20–7.16 (m, 2H, Ar–H), 5.59 (dd, 1H, J2–3 = 2.9 Hz, J2–1 = 2.0 Hz, H-2III), 5.08 (dd, 1H, J1-1-OH = 3.2 Hz, J1–2 = 1.8 Hz, H-1I), 4.98 (d, 1H, J1–2 = 1.6 Hz, H-1II), 4.89 (d, 1H, J1–2 = 1.6 Hz, H-1III), 4.77 (d, 1H, J = 11.6 Hz, CH2Ph), 4.71 (d, 1H, J = 11.7 Hz, CH2Ph), 4.65–4.55 (m, 4H, 4CH2Ph), 3.89–3.84 (m, 3H, H-2I, H-2II, H-3III), 3.75 (dd, 1H, J3–4 = 10.0 Hz, J3–2 = 2.8 Hz, H-3I), 3.73 (dd, 1H, J3–4 = 10.0 Hz, J3–2 = 2.9 Hz, H-3II), 3.68 (m, 1H, H-5I), 3.58 (m, 1H, H-5III), 3.52 (m, 1H, H-5II), 3.47 (t, 1H, J = 10.0 Hz, H-4III), 3.35 (t, 1H, J = 10.0 Hz, H-4II), 3.22 (t, 1H, J = 10.0 Hz, H-4I), 2.54 (d, 1H, J1-OH-1 = 3.5 Hz, OH), 1.28, 1.27 (overlapped 2d, 3H each, J6–5 = 6.1 Hz each, CH3, H-6II, H-6I), 1.23 (d, 3H, J6–5 = 6.1 Hz, CH3, H-6III). 13C{1H} NMR (150 MHz, CDCl3): δ 165.3 (CO), 137.4, 137.3, 137.1, 133.3, 129.9, 129.7, 128.52, 128.5 (2C), 128.44 (2C), 128.43, 128.4 (2C), 128.3 (2C), 128.2 (2C), 128.1 (2C), 128.04 (2C), 128.0, 127.9, 100.3 (C-1II), 99.2 (C-1III), 93.5 (C-1I), 77.0 (C-3I), 76.6 (C-3II), 75.3 (C-3III), 74.0 (C-2II), 73.9 (C-2I), 72.2 (CH2Ph), 72.0 (CH2Ph), 71.3 (CH2Ph), 67.7 (C-5II), 67.63 (C-5III), 67.6 (C-2III), 67.2 (C-5I), 64.4 (C-4I), 64.1 (C-4III), 64.0 (C-4II), 18.7 (CH3, C-6I), 18.6 (CH3, C-6II), 18.5 (CH3, C-6III). HRMS (ESI-TOF): m/z [M + NH4+] calcd for C46H55O11N10 923.4052; found 923.4054.
4-Azido-2-O-benzoyl-3-O-benzyl-4,6-dideoxy-α-D-mannopyranosyl-(1→2)-4-azido-3-O-benzyl-4,6-dideoxy-α-D-mannopyranosyl-(1→2)-4-azido-3-O-benzyl-4,6-dideoxy-D-mannopyranosyl trichloroacetimidate (5)
1,8-Diazabicyclo[5,4,0]undec-7-ene (DBU, 1.95 mL, 13 mmol) was added at 0 °C to a solution of hemiacetal 16 (23.5 g, 26 mmol) and trichloroacetonitrile (3.9 mL, 39 mmol) in dry DCM (100 mL). The mixture was stirred for 2 h, when TLC (Rf = 0.57 at 4:1 hexane–EtOAc) showed complete consumption of starting material and formation of a faster moving product. The mixture was concentrated to a small volume and applied onto a short column of silica gel. Elution with 10:1 hexane–EtOAc gave trichloroacetimidate donor 5 as colorless syrup (24.8 g, 91%). 1H NMR for the major isomer (600 MHz, CDCl3): δ 8.59 (s, 1H, NH), 8.07–8.06 (d, 2H, J = 7.8 Hz, Ar–H), 7.61 (t, 1H, J = 7.5 Hz, Ar–H), 7.50–7.47 (t, 2H, J = 7.6 Hz, Ar–H), 7.41–7.39 (m, 2H, Ar–H), 7.36–7.21 (m, 13H, Ar–H), 6.07 (s, 1H, H-1I), 5.61 (s, 1H, H-2III), 4.99 (s, 1H, H-1II), 4.98 (s, 1H, H-1III), 4.78 (d, 1H, J = 11.5 Hz, CH2Ph), 4.75 (d, 1H, J = 11.5 Hz, CH2Ph), 4.67–4.56 (m, 4H, 4CH2Ph), 3.93–3.86 (m, 3H, H-2I, H-2II, H-3III), 3.75–3.71 (m, 2H, H-3I, H-3II), 3.66–3.55 (m, 3H, H-5I, H-5II, H-5III), 3.49 (t, 1H, J = 9.9 Hz, H-4III), 3.38 (t, 1H, J = 10.0 Hz, H-4II), 3.33 (t, 1H, J = 10.0 Hz, H-4I), 1.32 (d, 3H, J6–5 = 6.3 Hz, CH3, H-6II), 1.30 (d, 3H, J6–5 = 6.1 Hz, CH3, H-6I), 1.28 (d, 3H, J6–5 = 6.2 Hz, CH3, H-6III). 13C{1H} NMR (150 MHz, CDCl3): δ 165.3 (CO), 159.9 (CO), 137.3, 137.1, 136.8, 133.3, 129.9 (2C), 129.7, 128.6 (2C), 128.5 (2C), 128.44 (2C), 128.42 (2C), 128.4 (2C), 128.3 (2C), 128.2 (2C), 128.1 (2C), 127.9, 100.4 (C-1II), 99.2 (C-1III), 96.2 (C-1I), 76.7 (C-3I), 76.5 (C-3II), 75.3 (C-3III), 73.8 (C-2I), 72.5 (CH2Ph), 72.2 (CH2Ph), 71.9 (C-2II), 71.4 (CH2Ph), 70.1 (C-5II), 68.1 (C-5III), 67.7 (C-2III), 67.6 (C-5I), 64.0 (C-4I), 63.9 (C-4III), 63.7 (C-4II), 18.64 (CH3, C-6I), 18.62 (CH3, C-6II), 18.5 (CH3, C-6III). HRMS (ESI-TOF): m/z [M + 18]+ calcd for C48H51O11N10Cl3 1048.2804; found 1048.2810.
2-(Trimethylsilyl)ethyl 4-azido-2-O-benzoyl-3-O-benzyl-4,6-dideoxy-α-D-mannopyranosyl-(1→2)-[4-azido-3-O-benzyl-4,6-dideoxy-α-D-mannopyranosyl-(1→2)]4-4-azido-3-O-benzyl-4,6-dideoxy-α-D-mannopyranoside (3a) and 2-(Trimethylsilyl)ethyl 4-azido-2-O-benzoyl-3-O-benzyl-4,6-dideoxy-α-D-mannopyranosyl-(1→2)-4-azido-3-O-benzyl-4,6-dideoxy-α-D-mannopyranosyl-(1→2)-4-azido-3-O-benzyl-4,6-dideoxy-β-D-mannopyranosyl-(1→2)-4-azido-3-O-benzyl-4,6-dideoxy-α-D-mannopyranosyl-(1→2)-4-azido-3-O-benzyl-4,6-dideoxy-α-D-mannopyranosyl-(1→2)-4-azido-3-O-benzyl-4,6-dideoxy-α-D-mannopyranoside (3b)
To a solution of the trisaccharide acceptor 4 (19 g, 21 mmol) and trisaccharide trichloroacetimidate donor 5 (24.3 g, 23.2 mmol) in dry DCM (300 mL) was added 5 g of 4 Å powdered molecular sieves, and the mixture was stirred for 15 min. The mixture was cooled to −40 °C, TMSOTf (1.7 mL, 9.3 mmol) was added dropwise at the rate of 0.1 mL min−1 over a period of 17 min using a syringe pump. After additional 30 min at −40 °C TLC in two solvents (1. Rf = 0.74 at 4:1 hexane–EtOAc and 2. Rf = 0.40 at neat toluene) showed that the glycosyl acceptor was completely consumed. Two faster moving spots were formed, one of which strongly predominated, indicating formation of anomeric hexasaccharides. The mixture was neutralized with NEt3 (1.3 mL, 9.3 mmol), filtered through Celite pad and filtrate combined with the washings were concentrated. The residue was chromatographed (8:1 hexane–EtOAc) to give first the α-linked hexasaccharide (3a, 33.72 g, 90%) as colorless syrup, [α]D +73.0 (c 2.0, CHCl3). 1H NMR (600 MHz, C6D6): δ 8.20–8.19 (d, 2H, J = 7.3 Hz, Ar–H), 7.43–7.38 (d, 2H, J = 7.4 Hz, Ar–H), 7.38–7.31 (m, 10H, Ar–H), 7.31–7.19 (m, 11H, Ar–H), 7.19–7.08 (m, 5H, Ar–H), 7.06–7.01 (m, 5H, Ar–H), 5.93 (dd, 1H, J2–3 = 2.9 Hz, J2–1 = 1.9 Hz, H-2VI), 5.32 (d, 1H, J = 1.3 Hz, H-1V), 5.25 (d, 1H, J = 1.1 Hz, H-1IV), 2.23 (d, 1H, J = 1.2 Hz, H-1III), 5.21 (d, 1H, J = 1.2 Hz, H-1II), 5.16 (d, 1H, J = 1.3 Hz, H-1VI), 4.89 (d, 1H, J = 1.3 Hz, H-1I), 4.59 (d, 1H, J = 11.5 Hz, CH2Ph), 4.49–4.43 (dd, 2H, I = 16.6, 11.5 Hz, CH2Ph), 4.42–4.33 (m, 7H, CH2Ph), 4.31 (d, 1H, J = 11.8 Hz, CH2Ph), 4.30 (d, 1H, J = 11.3 Hz, CH2Ph), 4.15 (t, 1H, J = 2.4 Hz, H-2IV), 4.12 (dd, 1H, J3–4 = 10.0 Hz, J3–2 = 3.2 Hz, H-3VI), 4.10 (t, 1H, J = 2.2 Hz, H-2III), 4.09 (t, 1H, J = 2.4 Hz, H-2II), 4.05 (t, 1H, J = 2.3 Hz, H-2V), 3.98 (t, 1H, J = 2.4 Hz, H-2I), 3.96–3.88 (m, 6H, H-5VI, H-3V, H-3IV, H-3III H-3II, H-3I), 3.80 (m, 1H, H-5V), 3.77–3.68 (m, 5H, OCHbCH2Si, H-4VI, H-5IV, H-5III, H-5I), 3.67–3.61 (m, 2H, H-5II, H-4I), 3.60–3.51 (m, 4H, H4V, H-4IV, H-4III, H-4II), 3.34 (m, 1H, OCHbCH2Si), 1.33 (d, 3H, J6–5 = 6.4 Hz, CH3, H-6II), 1.32 (d, 3H, J6–5 = 6.5 Hz, CH3, H-6V), 1.30 (2d, 6H, J = 6.1 Hz, H-6VI, H-6I), 1.28 (d, 3H, J6–5 = 6.5 Hz, CH3, H-6IV), 1.26 (d, 3H, J6–5 = 6.0 Hz, CH3, H-6III), 0.85–0.76 (m, 2H, CH2CH2Si), 0.06 (s, 9H, (CH3)3Si). 13C{1H} NMR (150 MHz, C6D6): δ 165.9 (CO), 138.3, 138.1, 137.9, 137.89, 137.88 (2C), 133.7, 130.7, 130.5 (2C), 129.3 (2C), 129.28 (2C), 129.24 (2C), 129.22 (2C), 129.2 (2C), 129.17 (2C), 129.15 (2C), 129.1 (2C), 129.07 (2C), 129.05 (3C), 129.0 (2C), 128.9 (3C), 128.8 (3C), 128.7 (2C), 128.6, 101.2 (C-1II, JC-1,H-1 = 173.2 Hz), 101.06 (C-1III, JC-1,H-1 = 173.8 Hz), 101.04 (C-1IV, JC-1,H-1 = 174.4 Hz), 101.03 (C-1V, JC-1,H-1 = 174.2 Hz), 100.3 (C-1VI, JC-1,H-1 = 172.8 Hz), 99.16 (C-1I, JC-1,H-1 = 170.9 Hz), 78.7 (C-3I), 77.9 (C-3IV), 77.8 (2C, C-3II, C-3III), 77.6 (C-3V), 76.1 (C-3VI), 75.0 (C-2V), 74.6 (C-2I), 73.99 (C-2II), 73.97 (C-2IV), 73.7 (C-2III), 72.8 (CH2Ph), 72.7 (3C, 3× CH2Ph), 72.3 (CH2Ph), 71.6 (CH2Ph), 68.8 (2C, C-5I, C-5IV), 68.7 (C-5V), 68.6 (C-5III), 68.5 (2C, C-5VI, C-2VI), 67.9 (C-5II), 65.7 (OCH2CH2Si), 65.5 (C-4V), 65.2 (C-4IV), 65.1 (C-4III), 65.05 (C-4VI), 65.02 (C-4II), 64.9 (C-4I), 19.14 (3× CH3, C-6VI, C-6III, C-6II), 19.11 (CH3, C-6V), 19.09 (CH3, C-6IV), 19.08 (CH3, C-6I), 18.2 (CH2Si), −0.98 [3C, (CH3)3Si]. HRMS (ESI-TOF): m/z [M + NH4+] calcd for C90H112O20N19Si 1806.8100; found 1806.8083. Anal. calcd for C90H108O20N18Si: C, 60.39; H, 6.08; N, 14.08. Found C, 60.22; H, 5.92; N, 13.89.
Continued elution gave the β-linked hexasaccharide 3b as colorless syrup (0.98 g, 2.5%, Total yield of the glycosylation, 34.7 g, 92%, α/β ∼ 34:1). Data for 3b, [α]D +17.6 (c 2.0, CHCl3). 1H NMR (600 MHz, CDCl3): δ 8.02–7.98 (d, 2H, J = 7.1 Hz, Ar–H), 7.56 (t, 1H, J = 7.5 Hz, Ar–H), 7.50–7.47 (d, 2H, J = 7.5 Hz, Ar–H), 7.46–7.43 (t, 2H, J = 7.8 Hz, Ar–H), 7.43–7.36 (m, 8H, Ar–H), 7.36–7.31 (m, 6H, Ar–H), 7.31–7.22 (m, 5H, Ar–H), 7.22–7.17 (m, 3H, Ar–H), 7.17–7.09 (m, 5H, Ar–H), 7.04 (m, 1H, Ar–H), 5.59 (dd, 1H, J2–3 = 2.8 Hz, J2–1 = 2.0 Hz, H-2VI), 5.31 (d, 1H, J = 0.9 Hz, H-1V), 4.99 (d, 1H, J = 1.0 Hz, H-1II), 4.87 (s, 1H, H-1VI), 4.87 (s, 1H, H-1III), 4.79 (d, 1H, J = 10.8 Hz, CH2Ph), 4.76 (d, 1H, J = 11.0 Hz, CH2Ph), 4.71 (d, 1H, J = 11.8 Hz, CH2Ph), 4.70 (d, 1H, J = 10.8 Hz, CH2Ph), 4.68 (d, 1H, J = 1.5 Hz, H-1I), 4.66 (d, 1H, J = 10.8 Hz, CH2Ph), 4.60 (d, 1H, J = 11.6 Hz, CH2Ph), 4.57 (d, 1H, J = 11.0 Hz, CH2Ph), 4.54 (d, 1H, J = 11.6 Hz, CH2Ph), 4.52 (d, 1H, J = 11.60 Hz, CH2Ph), 4.46 (d, 1H, J = 10.8 Hz, CH2Ph), 4.24 (d, 1H, J = 11.3 Hz, CH2Ph), 4.15 (dd, 1H, J = 3.3, 1.5 Hz, H-2III), 4.10 (t, 1H, J = 2.2 Hz, H-2II), 4.03 (d, 1H, J = 1.9 Hz, H-2IV), 3.99 (d, 1H, J = 11.5 Hz, CH2Ph), 3.92 (m, 1H, H-5V), 3.89 (t, 1H, J = 2.2 Hz, H-2I), 3.86 (t, 1H, J = 2.0 Hz, H-2V), 3.83 (dd, 1H, J3–4 = 9.5 Hz, J3–2 = 3.0 Hz, H-3VI), 3.81 (dd, 1H, J3–4 = 11.3 Hz, J3–2 = 3.0 Hz, H-3V), 3.77–3.69 (m, 4H, OCHaCH2Si, H-1IV, H-3II, H-3I), 3.68 (dd, 1H, J3–4 = 10.1 Hz, J3–2 = 3.4 Hz, H-3III), 3.63 (t, 1H, J = 10.0 Hz, H-4III), 3.54 (m, 1H, H-5II), 3.51–3.36 (m, 6H, OCHbCH2Si, H-4VI, H-4V, H-5VI, H-5III, H-5I), 3.34 (t, 1H, J = 10.0 Hz, H-4II), 3.33 (t, 1H, J = 10.0 Hz, H-4I), 3.29 (t, 1H, J = 9.9 Hz, H-4IV), 3.00 (dd, 1H, J3–4 = 9.8 Hz, J3–2 = 2.3 Hz, H-3IV), 2.46 (dq, 1H, J5–4 = 3.8 Hz, J5–6 = 6.1 Hz, H-5IV), 1.34 (d, 3H, J6–5 = 6.1 Hz, CH3, H-6V), 1.31 (d, 3H, J6–5 = 6.5 Hz, CH3, H-6I), 1.29 (d, 3H, J6–5 = 6.5 Hz, CH3, H-6II), 1.22 (d, 3H, J6–5 = 6.1 Hz, CH3, H-6IV), 1.17 (d, 3H, J6–5 = 6.2 Hz, CH3, H-6III), 1.12 (d, 3H, J6–5 = 6.0 Hz, CH3, H-6VI), 0.95–0.83 (m, 2H, CH2CH2Si), 0.01 (s, 9H, (CH3)3Si). 13C{1H} NMR (150 MHz, CDCl3): δ 165.4 (CO), 138.2, 137.9, 137.4, 137.2, 137.1, 136.9, 133.1, 129.9 (2C), 129.8, 128.7 (2C), 128.6 (2C), 128.5 (2C), 128.45 (2C), 124.43 (3C), 128.1 (3C), 128.06 (3C), 128.03 (3C), 128.0 (3C), 127.8 (3C), 127.7 (3C), 127.6 (2C), 127.3, 100.1 (C-1II, JC-1,H-1 = 170.5 Hz), 99.4 (C-1VI, JC-1,H-1 = 173.7 Hz), 98.33 (C-1I, JC-1,H-1 = 169.3 Hz), 98.26 (C-1III, JC-1,H-1 = 170.8 Hz), 97.4 (C-1V, JC-1,H-1 = 177.3 Hz), 96.3 (C-1IV, JC-1,H-1 = 155.5 Hz), 80.8 (C-3IV), 78.3 (C-3I), 77.8 (C-3II), 77.7 (C-3V), 75.5 (C-3VI), 75.3 (C-3III), 74.1 (C-2V), 72.9 (C-2I), 72.7 (CH2Ph), 72.5 (C-2II), 72.2 (CH2Ph), 71.9 (CH2Ph), 71.5 (CH2Ph), 71.3 (CH2Ph), 70.6 (C-5IV), 69.9 (CH2Ph), 69.2 (C-2III), 67.7 (C-5II), 67.6 (C-5VI), 67.43 (C-5III), 67.40 (C-2VI), 67.10 (C-5I), 66.9 (C-5V), 66.6 (C-2IV), 65.2 (OCH2CH2Si), 64.5 (C-4I), 64.4 (C-4II), 64.3 (C-4V), 64.05 (C-4VI), 64.01 (C-4IV), 62.9 (C-4III), 18.9 (CH3, C-6V), 18.6 (CH3, C-6I), 18.5 (CH3, C-6II), 18.3 (CH3, C-6IV), 18.23 (CH3, C-6III), 18.21 (CH3, C-6VI), 17.7 (CH2Si), −1.33 [3C, (CH3)3Si]. HRMS (ESI-TOF): m/z [M + NH4+] calcd for C90H112O20N19Si 1806.8100; found 1806.8089. Anal. calcd for C90H108O20N18Si: C, 60.39; H, 6.08; N, 14.08. Found C, 60.46; H, 6.15; N, 14.06.
2-(Trimethylsilyl)ethyl [4-azido-3-O-benzyl-4,6-dideoxy-α-d-mannopyranosyl-(1→2)]5-4-azido-3-O-benzyl-4,6-dideoxy-α-D-mannopyranoside (17)14
This compound was prepared as described14 from 2 g (1.1 mmol) of 3a giving 1.8 g (94%) of pure compound 17 as colorless syrup. Rf = 0.55 at 4:1 hexane–EtOAc. [α]D +115.3 (c 1.0, CHCl3), lit14 [α]D +112. 1H NMR (600 MHz, C6D6): δ 7.38–7.34 (m, 4H, Ar–H), 7.34–7.29 (m, 6H, Ar–H), 7.29–7.24 (m, 7H, Ar–H), 7.24–7.18 (m, 7H, Ar–H), 7.18–7.12 (m, 4H, Ar–H), 7.12–7.08 (m, 2H, Ar–H), 5.29 (d, 1H, J = 1.3 Hz, H-1VI), 5.25 (brs, 1H, H-1V), 5.21 (d, 1H, J = 1.3 Hz, H-1II), 5.19 (brs, 2H, H-1III, H-1IV), 4.88 (d, 1H, J = 1.4 Hz, H-1I), 4.47–4.31 (m, 8H, CH2Ph), 4.31–4.27 (m, 2H, CH2Ph), 4.19–4.13 (m, 3H, 2 CH2Ph, H-2V), 4.01 (t, 1H, J = 2.2 Hz, H-2II), 4.09–4.06 (m, 2H, H-2III, H-2IV), 4.01 (brs, 1H, H-2VI), 3.97 (t, 1H, J = 2.3 Hz, H-2I), 3.96–3.86 (m, 5H, H-3I–V), 3.80–3.60 (m, 9H, H-5I–VI, H-4II, H-3VI, OCHaCH2Si), 3.59–3.49 (m, 4H, H-4I–V), 3.46 (t, 1H, J = 10.0 Hz, H-4VI), 3.33 (m, 1H, OCHbCH2Si), 2.08 (d, 1H, J = 1.8 Hz, H-2VI-OH), 1.32 (d, 3H, J6–5 = 6.3 Hz, CH3, H-6), 1.31–1.23 (m, 15H, 5 CH3, H-6), 0.83–0.76 (m, 2H, CH2CH2Si), 0.06 (s, 9H, (CH3)3Si). 13C{1H} NMR (150 MHz, C6D6): δ 138.1 (2C), 138.0 (2C), 137.9 (2C), 129.33 (3C), 129.3, 129.28 (3C), 129.23 (3C), 129.2 (3C), 129.13 (3C), 129.1 (2C), 128.98 (2C), 128.96 (2C), 128.8 (3C), 128.75 (2C), 128.7 (2C), 128.6 (2C), 101.5 (C-1VI), 101.2 (C-1IV), 101.1 (C-1V), 100.06 (C-1II), 100.04 (C-1III), 99.2 (C-1I), 78.7 (C-3I), 78.5 (C-3VI), 77.82 (C-3IV), 77.8 (2C, C-3II,II), 77.7 (C-3V), 74.6 (C-2I), 74.0 (C-2IV), 73.8 (C-2III), 73.7 (C-2II), 73.3 (C-2V), 72.8 (CH2Ph), 72.7 (CH2Ph), 72.69 (CH2Ph), 72.6 (CH2Ph), 72.3 (CH2Ph), 71.8 (CH2Ph), 68.83 (C-5V), 68.78 (C-5VI), 68.72 (2C, C-5I,III), 68.3 (C-5II), 67.9 (C-5IV), 67.6 (C-2VI), 65.7 (OCH2CH2Si), 65.5 (C-4II), 65.14, 65.12, 65.1 and 65.07, (4C, C-4I, C-4III, C-4IV and C-4V), 64.5 (C-4VI), 19.14 (2× CH3, 2× C-6), 19.11 (CH3, C-6), 19.1 (CH3, C-6), 19.08 (CH3, C-6), 18.9 (CH3, C-6), 18.2 (CH2Si), −0.98 [3C, (CH3)3Si]. HRMS (ESI-TOF): m/z [M + NH4+] calcd for C83H108O19N19Si 1702.7838; found 1702.7826.
2-(Trimethylsilyl)ethyl [4-amino-3-O-benzyl-4,6-dideoxy-α-D-mannopyranosyl-(1→2)]5-4-amino-3-O-benzyl-4,6-dideoxy-α-D-mannopyranoside (18)14
Compound 17 (0.75 g, 0.45 mmol) was dissolved in pyridine/triethylamine 7:3 (20 mL). Hydrogen sulfide (H2S) was passed through the above solution for 2 h and the resulting dark solution was stirred at room temperature, in the same flask equipped with an empty balloon, to ensure exclusion of atmospheric oxygen, overnight. TLC (Rf = 0.51 at DCM–MeOH 10:1) showed that the reaction was complete and that a slower moving product was formed. The reaction mixture was concentrated to dryness and coevaporated with toluene. The crude product was chromatographed and eluted with 1–3% ammonia (7 N solution in MeOH) in DCM (v/v). Compound 18 was obtained as a white foam (0.62 g, 92%), [α]D −5.0 (c 2.1, CHCl3), 1H NMR (600 MHz, CDCl3): δ 7.37–7.27 (m, 30H, Ar–H), 5.09 (s, 1H, H-1), 5.08–5.06 (s, 3H, 3× H-1), 5.01 (s, 1H, H-1VI), 4.78 (s, 1H, H-1I), 4.70–4.65 (m, 6H, CH2Ph), 4.51 (d, 1H, J = 11.5 Hz, CH2Ph), 4.47 (d, 1H, J = 12.0 Hz, CH2Ph), 4.44–4.38 (m, 4H, CH2Ph), 4.08–4.06 (m, 2H, 2× H-2), 4.03 (brs, 3H, 3× H-2), 3.92 (t, 1H, J = 2.3 Hz, H-2I), 3.77–3.72 (m, 1H, OCHaCH2Si), 3.65–3.46 (m, 12H, 6× H-3, 6× H-5), 3.46–3.42 (m, 1H, OCHbCH2Si), 2.88–2.79 (m, 6H, 6× H-4), 1.38–1.21 (m, 18H, 12× NH2, 2× CH3, 2× H-6), 1.18–1.14 (m, 12H, 4× CH3, 4× H-6), 0.96–0.90 (m, 1H, CH2CHaSi), 0.89–0.84 (m, 1H, CH2CHbSi), 0.02 (s, 9H, (CH3)3Si). 13C{1H} NMR (150 MHz, CDCl3): δ 137.9 (2C), 137.8 (2C), 137.7, 137.6, 128.6 (2C), 128.56 (3C), 128.53 (2C), 128.5 (3C), 128.4 (2C), 128.36 (3C), 128.32 (3C), 128.2 (2C), 128.1 (2C), 128.0 (3C), 127.98 (3C), 127.9 (2C), 101.1 (C-1), 100.98 (C-1), 100.96 (C-1), 100.94 (2C, 2× C-1), 98.8 (C-1), 79.7 (C-3), 79.2 (3C, 3× C-3), 78.8 (C-3), 78.7 (C-3), 73.2 (C-2), 73.1 (C-2), 73.0 (C-2), 72.9 (C-2), 72.8 (C-2), 71.4 (CH2Ph), 71.2 (CH2Ph), 71.15 (CH2Ph), 71.1 (CH2Ph), 71.0 (2C, 2× CH2Ph), 70.3 (4C, 4 C-5), 69.6 (C-5), 69.5 (C-5), 66.5 (C-2), 64.7 (OCH2CH2Si), 53.7 (2C, 2× C-4), 53.6 (3C, 3× C-4), 53.3 (C-4), 18.3 (CH3, C-6), 18.2 (3C, CH3, 3× C-6), 18.1 (CH3, C-6), 17.9 (CH3, C-6), 17.8 (CH2Si), −1.3 [3C, (CH3)3Si]. HRMS (ESI-TOF): m/z [M + H+] calcd for C83H117O19N6Si 1529.8143; found 1529.8136.
2-(Trimethylsilyl)ethyl 2-O-acetyl-3-O-benzyl-4-(2,4-di-O-acetyl-3-deoxy-L-glycero-tetronamido)-4,6-dideoxy-α-D-mannopyranosyl-(1→2)-[3-O-benzyl-4-(2,4-di-O-acetyl-3-deoxy-L-glycero-tetronamido)-4,6-dideoxy-α-D-mannopyranosyl-(1→2)]4-3-O-benzyl-4-(2,4-di-O-acetyl-3-deoxy-L-glycero-tetronamido)-4,6-dideoxy-α-D-mannopyranoside (19)
EDAC (0.45 g, 2.4 mmol) was added portion wise at room temperature to a stirred solution of amino sugar 18 (0.5 g, 0.33 mmol) and 2,4-di-O-acetyl-3-deoxy-L-glycero-tetronic acid15 (0.5 g, 2.5 mmol) in dry dichloromethane (20 mL). Stirring was continued overnight, when TLC showed complete consumption of starting material and formation of a much faster moving product (15:1 DCM–MeOH). The mixture was diluted with DCM (25 mL), washed with aq NaHCO3 solution (3× 10 mL), brine (3× 10 mL), dried and concentrated. HRMS analysis of the crude product confirmed formation of the desired coupling product only (HRMS (ESI-TOF): m/z [M + H+] calcd for C131H177O49N6Si 2646.1312; found 2646.1313). The crude product was dissolved in dry pyridine (2 mL) and treated with acetic anhydride (0.1 mL) overnight at room temperature. TLC showed complete conversion of the starting material and formation of a faster moving product (Rf = 0.54 at 15:1 DCM–MeOH). After concentration, a solution of the residue in DCM (10 mL) was washed with cold 4 N HCl (3× 10 mL), aq NaHCO3 solution (3× 10 mL), the phases were separated, and the aqueous phase was backwashed with DCM. The organic phase was dried, concentrated, and chromatography (DCM–MeOH–Py 10:1:0.1) gave pure 19 as colorless syrup (738 mg, 84% over 2 steps), [α]D −11.6 (c 1.0, CHCl3), 1H NMR (600 MHz, CD3OD): δ 7.45–7.41 (m, 4H, Ar–H), 7.39–7.37 (m, 6H, Ar–H), 7.36–7.32 (m, 6H, Ar–H), 7.31–7.25 (m, 10H, Ar–H), 7.25–7.18 (m, 4H, Ar–H), 5.40 (t, 1H, J = 2.0 Hz, H-2VI), 5.10–5.00 (m, 10H, 6× H-2′, H-1II, H-1III, H-1IV, H-1V), 4.77 (s, 1H, H-1I), 4.65–4.55 (m, 12H, 11× CH2Ph and H-1VI at 4.56), 4.41 (d, 1H, J = 11.3 Hz, CH2Ph), 4.21–4.03 (m, 22H, 12× H-4′, 6× H-4I–VI, 4× H-2II–V), 3.98–3.91 (m, 6H, 6× H-3I–VI), 3.88–3.76 (m, 7H, H-2I, 5× H-5, OCHaCH2Si), 3.71 (m, 1H, H-5), 4.48 (m, 1H, OCHbCH2Si), 2.15–1.98 (m, 51H, 6× Ha,b-3′ incl. 12 s at 2.14, 2.11, 2.08, 2.07, 2.06, 2.05, 2.04, 2.03, 2.02, 2.00, 1.998, 1.99 for 13× COCH3), 1.14–1.11 (m, 6H, 2× H-6), 1.06–1.02 (m, 12H, 4× H-6), 0.96–0.92 (m, 1H, CH2CHaSi), 0.90–0.86 (m, 1H, CH2CHbSi), 0.02 (s, 9H, (CH3)3Si). 13C{1H} NMR (150 MHz, CD3OD): δ 172.9 (CH3CO), 172.87 (CH3CO), 172.8 (CH3CO), 172.79 (CH3CO), 172.77 (CH3CO), 172.7 (CH3CO), 172.61 (3C, 3× CH3CO), 172.60 (CH3CO), 172.5 (2C, 2× CH3CO), 171.84 (NHCO), 171.8 (NHCO), 171.78 (NHCO), 171.76 (NHCO), 171.73 (NHCO), 171.69 (NHCO), 171.5 (CH3CO), 139.8, 139.7, 139.68, 139.63 (2C), 139.6, 129.7, 129.67 (2C), 129.6 (2C), 129.5 (2C), 129.4 (3C), 129.23 (3C), 129.2 (3C), 129.1 (2C), 129.0 (2C), 128.9 (2C), 128.8 (2C), 128.7 (2C), 128.6 (2C), 128.5, 102.5 and 102.3 (4C, 4× C-1II–V), 100.9 (C-1VI), 99.8 (C-1I), 77.0, 76.9, 76.6, 76.5, 76.4 (10C, 5× C-3, 5× C-2), 75.6 (C-3VI), 73.0 (CH2Ph), 72.8 (3C, 3× CH2Ph), 72.6 (CH2Ph), 72.56 (CH2Ph), 72.5 (C-2′), 72.43 (2C, 2× C-2′), 72.4 (3C, 3× C-2′), 69.8 (C-5), 69.7 (C-5), 69.6 (C-5), 69.5 (C-5), 68.9 (2C, C-5, C-2VI), 68.8 (C-5), 66.1 (OCH2CH2Si), 61.3 (4C, 4× C-4′), 61.25 (C-4′), 61.2 (C-4′), 53.6 (C-4), 53.4 (C-4), 53.3 (C-4), 53.2 (3C, 3× C-4), 32.4 (C-3′), 32.3 (5C, 5× C-3′), 20.9 (COCH3), 20.83 (5C, 5× COCH3), 20.81 (COCH3), 20.78 (COCH3), 20.73 (COCH3), 20.71 (3C, 3× COCH3), 20.70 (COCH3), 18.7 (CH3, C-6), 18.57 (CH3, C-6), 18.54 (CH2Si), 18.48 (2C, CH3, 2× C-6), 18.4 (CH3, C-6), 18.3 (CH3, C-6), −1.2 [3C, (CH3)3Si]. HRMS (ESI-TOF): m/z [M + H+] calcd for C133H179O50N6Si 2688.1418; found 2688.1423. Anal. calcd for C133H178O50N6Si: C, 59.41; H, 6.67; N, 3.13. Found C, 59.51; H, 6.66; N, 3.06.
2-O-Acetyl-3-O-benzyl-4-(2,4-di-O-acetyl-3-deoxy-L-glycero-tetronamido)-4,6-dideoxy-α-D-mannopyranosyl-(1→2)-[3-O-benzyl-4-(2,4-di-O-acetyl-3-deoxy-L-glycero-tetronamido)-4,6-dideoxy-α-D-mannopyranosyl-(1→2)]4-3-O-benzyl-4-(2,4-di-O-acetyl-3-deoxy-L-glycero-tetronamido)-4,6-dideoxy-α-D-mannopyranose (20)
A solution of 19 (700 mg, 0.26 mmol) in TFA (12 mL) was kept at room temperature for 2 hours, when TLC (Rf = 0.31 at 15:1 DCM–MeOH) showed that the reaction was complete and that a much slower moving product was formed. The mixture was processed as described above for a similar reaction, and chromatography (19:1 DCM–MeOH) gave 20 as foam (565 mg, 84%). 1H NMR (600 MHz, CD3OD) for the major anomer: δ 8.14–7.92 (m, 6H, NH), 7.43–7.17 (m, 30H, Ar–H), 5.39 (brs, 1H, H-2VI), 5.09–5.00 (11H, 6× H-2′, 5× H-1I–V at 5.083, 5.076, 5.056, 5.056, 5.012), 4.69–4.55 (m, 12H, 11× CH2Ph and H-1VI at 4.56), 4.41 (d, 1H, J = 11.4 Hz, CH2Ph), 4.20–4.03 (m, 21H, 12× H-4′, 4× H-2, 4× H-4 and H-5VI), 3.97–3.78 (m, 14H, H-2I, 6× H-3, 2× H-4, 5× H-5), 2.17–1.95 (m, 51H, 6× Ha,b-3′ incl. 12 s at 2.136, 2.13, 2.10, 2.09, 2.08, 2.07, 2.06, 2.05, 2.03, 2.02, 2.01, 2.00, 1.99 for 13× COCH3), 1.16–0.99 (m, 18H, 6× H-6). 13C{1H} NMR (150 MHz, CD3OD): δ 172.98 (CH3CO), 172.95 (CH3CO), 172.9 (CH3CO), 172.87 (CH3CO), 172.84 (CH3CO), 172.80 (CH3CO), 172.78 (CH3CO), 172.76 (CH3CO), 172.7 (CH3CO), 172.6 (2C, 2× CH3CO), 172.5 (CH3CO), 171.85 (NHCO), 171.8 (NHCO), 171.78 (NHCO), 171.76 (NHCO), 171.73 (NHCO), 171.7 (NHCO), 171.6 (CH3CO), 139.8, 139.76, 139.72, 139.64, 139.64, 139.6, 129.7, 129.6 (2C), 129.55 (2C), 129.5 (2C), 129.4 (2C), 129.2 (2C), 129.19 (2C), 129.1 (2C), 129.0 (3C), 128.86 (3C), 128.84 (2C), 128.80 (2C), 128.7 (2C), 128.6 (2C), 128.5, 102.4 and 102.2 (4C, 4× C-1II–V), 100.9 (C-1VI), 94.5 (C-1I), 76.9, 76.8, 76.6, 76.56, 76.5, 75.6, 75.5 (11C, 6× C-3, 5 C-2), 72.9 (2C, 2× CH2Ph), 72.8 (3C, 3× CH2Ph), 72.6 (CH2Ph), 72.5 (C-2′), 72.47 (C-2′), 72.45 (2C, 2× C-2′), 72.4 (2C, 2× C-2′), 69.76 (C-5), 69.7 (C-5), 69.5 (2 C, C-5), 68.9 (2C, C-5, C-2VI), 68.3 (C-5), 61.3 (4C, 4× C-4′), 61.25 (C-4′), 61.2 (C-4′), 53.9 (C-4), 53.8 (C-4), 53.5 (C-4), 53.4 (C-4), 53.3 (C-4), 53.2 (C-4), 32.4 (6C, 6× C-3′), 20.9 (COCH3), 20.8 (6C, 6× COCH3), 20.78 (COCH3), 20.74 (COCH3), 20.72 (2C, 2× COCH3), 20.70 (2C, 2× COCH3), 18.7 (CH3, C-6), 18.6 (CH3, C-6), 18.5 (3C, CH3, 3× C-6), 18.3 (CH3, C-6). HRMS (ESI-TOF): m/z [M + H+] calcd for C128H167O50N6 2588.0710; found 2588.0725.
8-Azido-3,6-dioxaoctyl 2-O-acetyl-3-O-benzyl-[4-(2,4-di-O-acetyl-3-deoxy-L-glycero-tetronamido)-4,6-dideoxy-α-D-mannopyranosyl]-(1→2)-[3-O-benzyl-[4-(2,4-di-O-acetyl-3-deoxy-L-glycero-tetronamido)-4,6-dideoxy-α-D-mannopyranosyl-(1→2)]4-3-O-benzyl-4-(2,4-di-O-acetyl-3-deoxy-L-glycero-tetronamido)-4,6-dideoxy-α-D-mannopyranoside (2)
1,8-Diazabicyclo[5,4,0]undec-7-ene (DBU, 15 μL, 0.1 mmol) was added at 0 °C to a stirred solution of hemiacetal 21 (500 mg, 0.19 mmol) and trichloroacetonitrile (30 μL, 0.29 mmol) in dry DCM (10 mL). The stirring was continued for 2 hours when TLC (15:1 DCM–MeOH) showed complete formation of a faster moving product. The mixture was concentrated, and passed through a small pad of silica gel which was eluted with (DCM:MeOH = 19:1) containing a few drops of triethylamine, to obtain pure compound 21 as light brown syrup (448 mg, 85%). HRMS: m/z [M + NH4+] calcd for C130H170O50N8Cl3 2748.0071; found 2748.0081. The material was sufficiently pure for the next step.
:2 toluene–acetone) showed complete consumption of the glycosyl donor, the reaction was terminated by addition of triethylamine (1 drop). The mixture was filtered through a pad of Celite directly into a separating funnel containing excess of sodium bicarbonate (saturated) solution. The mixture was extracted with DCM (3× 5 mL), the combined organic layers were dried and concentrated, to give crude product (65.3 mg, 65%). NMR showed that the reaction produced the two anomers with poor selectivity (α:β = 1.1:1).:2 toluene–acetone) showed complete consumption of the glycosyl donor, the mixture was processed as described above. 1H NMR of the crude mixture showed the ratio of the α and β anomers formed was 7:1. Chromatography (3:2 toluene–acetone) gave first the α-linked hexasaccharide (2a, 132 mg, 66%) as colorless syrup. [α]D −10.8 (c 2.0, CHCl3). 1H NMR (600 MHz, CD3OD): δ 7.74–7.71 (m, 2H, Ar–H), 7.40–7.38 (m, 5H, Ar–H), 7.36–7.33 (m, 5H, Ar–H), 7.30–7.26 (m, 6H, Ar–H), 7.23–7.19 (m, 6H, Ar–H), 7.16–7.14 (m, 4H, Ar–H), 7.13–7.10 (m, 2H, Ar–H), 5.40 (brs, 1H, H-2VI), 5.09–5.00 (m, 10H, 6× H-2′, 4× H-1II–V at 5.09, 5.065, 5.06 and 5.02), 4.83 (d, 1H, J1–2 = 1.5 Hz, H-1I), 4.66–4.55 (m, 12H, 11× CH2Ph and H-1VI at 4.57), 4.41 (d, 1H, J = 11.3 Hz, CH2Ph), 4.20–4.05 (m, 22H, 12× H-4′, 4× H-2II–V, 6× H-4I–VI), 3.99–3.92 (m, 6H, 5× H-3II–V, H-2I), 3.91–3.88 (m, 2H, H-3, H-5), 3.86–3.79 (m, 4H, 4× H-5), 3.78–3.73 (m, 2H, OCHa, H-5), 3.69–3.63 (m, 8H, 8× OCH2), 3.59 (m, 1H, OCHb), 3.37 (t, 2H, J = 5.0 Hz, CH2N3), 2.15–1.98 (m, 51H, 6× Ha,b-3′ incl. 12 s at 2.145, 2.143, 2.12, 2.08, 2.07, 2.06, 2.04, 2.03, 2.02, 2.00, 1.999 and 1.99 for 13× COCH3), 1.15–1.11 (m, 6H, 2× CH3, 2× H-6), 1.09–1.02 (m, 12H, 4× CH3, 4× H-6). 13C{1H} NMR (150 MHz, CD3OD): δ 172.88 (CH3CO), 172.84 (CH3CO), 172.82 (CH3CO), 172.77 (CH3CO), 172.75 (CH3CO), 172.68 (CH3CO), 172.59 (2C, 2× CH3CO), 172.58 (2C, 2× CH3CO), 172.54 (2C, 2× CH3CO), 171.81 (2C, 2× NHCO), 171.76 (NHCO), 171.74 (NHCO), 171.71 (NHCO), 171.67 (NHCO), 171.5 (CH3CO), 139.74, 139.72, 139.70 (2C), 139.6 (2C), 129.9, 129.7 (2C), 129.6 (2C), 129.56 (2C), 129.5 (2C), 129.4 (2C), 129.22 (2C), 129.21 (2C), 129.18 (2C), 129.1 (2C), 129.0 (2C), 128.9 (2C), 128.8 (2C), 128.7 (2C), 128.6, 128.5, 126.3, 102.4 and 102.3 (4C, 4× C-1II–V), 100.9 (C-1VI), 100.5 (C-1I), 76.98, 76.9, 76.6, 76.5, 76.4, 75.6 (11C, 6× C-3, 5 C-2), 72.86 (2C, 2× CH2Ph), 72.8 (2C, 2× CH2Ph), 72.6 (CH2Ph), 72.54 (CH2Ph), 72.5 (C-2′), 72.4 (3C, 3× C-2′), 72.3 (2C, 2× C-2′), 71.7 (OCH2), 71.5 (OCH2), 71.4 (OCH2), 71.2 (OCH2), 69.8 (C-5), 69.7 (2C, 2× C-5), 69.6 (C-5), 69.5 (C-5), 68.9 (C-2VI), 68.8 (C-5), 68.1 (OCH2), 61.3 (4C, 4× C-4′), 61.24 (C-4′), 61.2 (C-4′), 53.5 (C-4), 53.4 (C-4), 53.3 (2C, 2× C-4), 53.2 (2C, 2× C-4), 51.7 (CH2N3), 32.42 (C-3′), 32.4 (2C, 2× C-3′), 32.36 (3C, 3× C-3′), 21.5 (COCH3), 20.9 (COCH3), 20.84 (5C, 5× COCH3), 20.82 (COCH3), 20.8 (COCH3), 20.73 (3C, 3× COCH3), 20.7 (COCH3), 18.7 (CH3, C-6), 18.6 (CH3, C-6), 18.5 (CH3, C-6), 18.47 (CH3, C-6), 18.4 (CH3, C-6), 18.3 (CH3, C-6). HRMS (ESI-TOF): m/z [M + NH4+] calcd for C134H181O52N10 2762.1826; found 2762.1836. Anal. calcd for C134H177O52N9: C, 59.61; H, 6.50; N, 4.59. Found C, 59.48; H, 6.60; N, 4.47.Continued elution gave the β-linked hexasaccharide 2b as colorless syrup (19 mg, 9%, total yield of glycosylation, 151 mg, 75%, α/β ∼ 6.9:1). Data for 2b: [α]D −18.3 (c 0.9, CHCl3). 1H NMR (600 MHz, CD3OD): δ 7.52–7.17 (m, 30H, Ar–H), 5.41 (t, 1H, J = 2.1 Hz, H-2VI), 5.21 (d, 1H, J1–2 = 1.9 Hz, H-1I), 5.15–4.98 (m, 9H, 6× H-2′, 3× H-1II–V at 5.10, 5.08 and 5.04), 4.73–4.55 (m, 12H, 11× CH2Ph and H-1VI at 4.58), 4.49 (s, 1H, H-1I), 4.43 (d, 1H, J = 11.6 Hz, CH2Ph), 4.24–4.03 (m, 22H, 12× H-4′, 4× H-2II–V, 6× H-4I–VI), 3.99–3.75 (m, 13H, H-2I, 6× H-3II–V, 6× H-5), 3.69–3.63 (m, 8H, 8× OCH2), 3.59 (m, 2H, OCH2), 3.38 (2d, 2H, J = 4.5 Hz, CH2N3), 2.15–1.98 (m, 51H, 6× Ha,b-3′ incl. 11 s at 2.11, 2.09, 2.08, 2.06, 2.057, 2.05, 2.04, 2.03, 2.02, 2.01 and 2.00 for 13× COCH3), 1.15–1.11 (m, 6H, 2× CH3, 2× H-6), 1.09–1.02 (m, 12H, 4× CH3, 4× H-6). 13C{1H} NMR (150 MHz, CD3OD): δ 172.9 (CH3CO), 172.88 (CH3CO), 172.84 (CH3CO), 172.78 (4C, 4× CH3CO), 172.6 (2C, 2× CH3CO), 172.59 (3C, 3× CH3CO), 171.82 (NHCO), 171.81 (NHCO), 171.77 (NHCO), 171.75 (NHCO), 171.72 (NHCO), 171.7 (NHCO), 171.6 (CH3CO), 139.8, 139.73, 139.71, 139.65, 139.6, 139.5, 129.7 (2C), 129.6 (2C), 129.5 (2C), 129.4 (2C), 129.3 (2C), 129.24 (2C), 129.2 (2C), 129.1 (2C), 129.0 (2C), 128.96 (2C), 128.9 (2C), 128.8 (2C), 128.7 (2C), 128.6 (2C), 128.5 (2C), 101.5 (3C, 3× C-1II–V), 100.8 (3C, 3× C-1II–V), 76.9, 76.6, 76.2, 75.6 (11C, 6× C-3, 5 C-2), 73.2 (2C, 2× CH2Ph), 72.8 (3C, 3× CH2Ph), 72.6 (CH2Ph), 72.54, 72.5, 72.4, 72.40, 72.39 (6C, 6× C-2′), 71.6 (OCH2), 71.5 (OCH2), 71.4 (OCH2), 71.1 (OCH2), 69.8 (OCH2), 69.8 (C-5), 69.7 (3C, 3× C-5), 69.6 (C-5), 69.5 (C-5), 68.9 (C-2VI), 61.3 (C-4′), 61.28 (2C, 2× C-4′), 61.26 (2C, 2× C-4′), 61.23 (C-4′), 53.7 (C-4), 53.4 (2C, 2× C-4), 53.3 (3C, 3× C-4), 51.7 (CH2N3), 32.5 (C-3′), 32.4 (2C, 2× C-3′), 32.38 (3C, 3× C-3′), 20.98 (COCH3), 20.85 (2C, 2× COCH3), 20.83 (3C, 3× COCH3), 20.8 (COCH3), 20.78 (COCH3), 20.75 (COCH3), 20.7 (4C, 4× COCH3), 18.7 (CH3, C-6), 18.6 (CH3, C-6), 18.5 (3CH3, 3× C-6), 18.4 (CH3, C-6). HRMS (ESI-TOF): m/z [M + NH4+] calcd for C134H181O52N10 2762.1826; found 2762.1838. Anal. calcd for C134H177O52N9: C, 59.61; H, 6.50; N, 4.59. Found C, 58.99; H, 6.50; N, 4.49.
8-Amino-3,6-dioxaoctyl [4-(3-deoxy-L-glycero-tetronamido)-4,6-dideoxy-α-D-mannopyranosyl-(1→2)]5-4-(3-deoxy-L-glycero-tetronamido)-4,6-dideoxy-α-D-mannopyranopyranoside (1)
To a solution of the linker-equipped hexasaccharide 2a (100 mg, 0.04 mmol) in dry MeOH (10 mL), methanolic NaOMe (1 M, 1 mL) was added with exclusion of moisture and atmospheric CO2, and the mixture was kept at room temperature for 6 hours, when TLC (3:2 toluene–acetone) showed that the starting material was consumed and that a much slower moving product was formed (Rf = 0.34 at 4:1 DCM–methanol with 1 drop of acetic acid). After neutralization (Dowex 50W H+ resin) and filtration, the solvent was removed, and the residue, showing correct HRMS (ESI-TOF: m/z [M + H+] calcd for C108H152O39N9 2199.0187; found 2199.0188), was used for the next step without further purification.
To a solution of the above product in MeOH (2 mL), 10 mg of Pd–C was added, and the mixture was stirred at room temperature under 100 Psi pressure of hydrogen gas for 4 hours. TLC showed complete consumption of starting material and presence of a much more polar product. HRMS analysis confirmed the completion of global reduction. The mixture was filtered over a Celite pad, the solids were washed several times with methanol and the solvent was removed. A solution of the product in MeOH:H2O (2:1) was filtered through a 0.2 μm porosity syringe filter and lyophilized to collect pure product as white foam (42 mg, 71% over 2 steps), [α]D +1.8 (c 1.0, CH3OH). 1H NMR (600 MHz, CD3OD): δ 5.16–5.11 (m, 4H, 4× H-1II–V), 4.98 (s, 1H, H-1VI), 4.87 (s, 1H, H-1I), 4.22–4.14 (m, 6H, 6× H-2′), 4.13–4.08 (m, 4H, 4× H-2II–V), 4.05–3.98 (m, 5H, 4× H-3II–V, H-2VI at 4.02), 3.98–3.83 (m, 13H, 2× H-3I,VI, 6× H-4I–VI, 5× H-5II–VI), 3.83–3.78 (m, 3H, H-2I, H-5I, OCHa), 3.77–3.71 (m, 12H, 12× H-4′), 3.71–3.52 (m, 10H, 10× OCH2), 3.13 (t, 1H, J = 4.8 Hz, OCH2CHaNH2), 2.06–1.97 (m, 6H, 6× H-3′a), 1.87–1.78 (m, 6H, 6× H-3′b), 1.22–1.10 (m, 18H, 6× H-6). 13C{1H} NMR (150 MHz, CD3OD): δ 178.0 (5C, 5× NHCO), 177.9 (NHCO), 103.8 (C-1VI), 102.7, 102.5, 105.46, 102.4 (4C, 4× C-1II–V), 100.4 (C-1I), 79.6, 79.23, 79.2 (5C, 5× C-2), 71.6, 71.4, 71.30 (3C, 3× OCH2), 70.9 (C-2), 70.7 (6C, 6× C-2′), 70.0, 69.5, 69.4, 69.3 (11C, 6× C-3, 5× C-5), 68.7 (C-5), 68.1 (OCH2), 67.8 (OCH2), 59.4 (6C, 6× C-4′), 54.8–54.2 (6C, C-4I–VI), 40.7 (CH2NH2), 38.3 (6C, 6× C-3′), 18.4 (2C, CH3, 2× C-6), 18.3 (3C, CH3, 3× C-6), 18.2 (CH3, C-6). HRMS (ESI-TOF): m/z [M + H+] calcd for C66H118O39N7 1632.7465; found 1632.7467.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Intramural Research Program of the National Institutes of Health, NIDDK.Notes and references
- P. Kováč, Studies toward a rationally designed conjugate vaccine for cholera using synthetic carbohydrate antigens, in Protein-carbohydrate interactions in infectious diseases. Studies toward a rationally designed conjugate vaccine for cholera using synthetic carbohydrate antigens, ed. C. Bewley, Royal Society of Chemistry London, 2006, vol. 10, pp. 175–220 Search PubMed.
- S. J. Hou and P. Kovac, Carbohydr. Res., 2010, 345, 999–1007 CrossRef CAS.
- X. Lu, H. B. Pfister, S. E. Soliman and P. Kováč, Eur. J. Org. Chem., 2018, 2944–2957 CrossRef CAS.
- S. N. Chatterjee and K. Chaudhuri, Biochim. Biophys. Acta, Mol. Basis Dis., 2003, 1639, 65–79 CrossRef CAS.
- M. D. Meeks, R. Saksena, X. Ma, T. K. Wade, R. K. Taylor, P. Kováč and W. F. Wade, Infect. Immun., 2004, 72, 4090 CrossRef CAS PubMed.
- R. Adamo and P. Kováč, Eur. J. Org. Chem., 2007, 988–1000 CrossRef CAS.
- Y. Ogawa, P.-s. Lei and P. Kováč, Carbohydr. Res., 1996, 288, 85–98 CrossRef CAS.
- H. Paulsen and M. Brenken, Liebigs Ann. Chem., 1988, 649–654 CrossRef CAS.
- H. Paulsen and M. Paal, Carbohydr. Res., 1984, 135, 53–69 CrossRef CAS.
- D. R. Bundle, M. Gerken and T. Peters, Carbohydr. Res., 1988, 174, 239–251 CrossRef CAS.
- R. Saksena, J. Zhang and P. Kováč, J. Carbohydr. Chem., 2002, 21, 453–470 CrossRef CAS.
- G. Zemplén, Berichte der deutschen chemischen Gesellschaft (A and B Series), 1926, 59, 1254–1266 CrossRef.
- K. Bock and C. Pedersen, J. Chem. Soc., Perkin Trans. 2, 1974, 293–297 RSC.
- Y. Ogawa, P.-s. Lei and P. Kováč, Carbohydr. Res., 1996, 293, 173–194 CrossRef CAS PubMed.
- A. Arencibia-Mohar, A. Ariosa-Alvarez, O. Madrazo-Alonso, G. E. Abreu, L. García -Imia, G. Sierra-González and V. Verez-Bencomo, Carbohydr. Res., 1998, 306, 163–170 CrossRef CAS.
- R. Adamo and P. Kováč, Eur. J. Org. Chem., 2006, 2803–2809 CrossRef CAS.
- N. V. Ganesh, J. M. Sadowska, S. Sarkar, L. Howells, J. McGiven and D. R. Bundle, J. Am. Chem. Soc., 2014, 136, 16260–16269 CrossRef CAS.
- M. Gotoh and P. Kovác, J. Carbohydr. Chem., 1993, 12, 981–983 CrossRef CAS.
- X. Ma, R. Saksena, A. Chernyak and P. Kováč, Org. Biomol. Chem., 2003, 1, 775–784 RSC.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Copy of 1H, 13C NMR of all compounds, 1H, 13C, COSY, and HSQC NMR spectra of new compounds and table for crystallographic and structural refinement parameters (pdf). CCDC 1939745. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra08232h |
| This journal is © The Royal Society of Chemistry 2019 |