
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCD44-targeted hyaluronic acid–curcumin reverses chemotherapeutics resistance by inhibiting P-gp and anti-apoptotic pathways†
Lu Diao ab,
Ao Shenc,
Yunxu Yangb,
Jin Taob and
Ying Hu*ab
ab,
Ao Shenc,
Yunxu Yangb,
Jin Taob and
Ying Hu*ab
aSchool of Pharmaceutical Sciences, Wenzhou Medical University, Wenzhou, Zhejiang 325035, China
bZhejiang Pharmaceutical College, No. 888, East Section, YInXian Main Road, The Zone of Higher Education, Ningbo, Zhejiang 315100, China. E-mail: pharmhawk@126.com; Fax: +86 5748822 3023; Tel: +86 5748822 7845
cThe University of Queensland, Brisbane 4072, Queensland, Australia
First published on 11th December 2019
Abstract
Chemotherapeutic drug resistance poses a great challenge in cancer therapy. Drug efflux and anti-apoptotic processes are the two most common mechanisms leading to chemotherapy resistance. In this study, we focused on the applicability of curcumin (CUR) as a sensitizer for chemotherapeutics (doxorubicin [DOX] as the model drug) modified with hyaluronic acid (HA) as an effective therapeutic strategy against multidrug resistance (MDR) in cancer cells. We constructed an HA–CUR/DOX delivery system measuring approximately 180 nm with superior encapsulation efficacy and serum stabilities. In vitro, we found that HA modification could facilitate the efficient delivery of chemotherapeutics through CD44 receptor-mediated targeted delivery. MTT assay results confirmed that the combination of CUR and DOX/paclitaxel (PTX) had a significant synergistic effect and significantly reversed MDR. Further experiments including real-time polymerase chain reaction and western blotting proved that the main mechanisms by which CUR reversed MDR in tumor cells were inhibiting the expression and activity of P-glycoprotein (P-gp) and inducing apoptosis through mitochondrial pathway. Taken together, our new engineered tumor-targeting nanoparticle delivery system may have the potential for overcoming MDR in cancer.
Introduction
Chemotherapeutics play important roles in cancer treatment. Multidrug resistance (MDR) is a major obstacle to the use of many chemotherapeutic agents in clinical oncology.1,2 Doxorubicin (DOX) is widely used to treat various tumors because of its broad spectrum of antitumor activity. However, the clinical application of the drug is severely limited by its severe cardiotoxicity and genotoxicity, as well as the development of MDR.3,4The mechanisms of tumor cell MDR are complicated, primarily including increased cellular drug efflux and anti-apoptotic activity.5 P-glycoprotein (P-gp) has an energy-dependent “drug pump” function through which intracellular drugs are pumped out of the cell, reducing the drug concentration inside the cells and making the cells resistant.6 P-gp is often highly expressed on the surface of tumor cells, reflecting a major hindrance to achieving maximum therapeutic efficacy.7 Studies have illustrated that effective inhibition of P-gp expression can reverse MDR in tumors and prolong the survival of patients.8,9 To date, several drugs that inhibit P-gp have been developed, such as verapamil (first-generation P-gp inhibitor), dexniguldipine (second-generation P-gp inhibitor), and tetrandrine (third-generation P-gp inhibitor). However, they still have side effects such as cardiotoxicity, immunosuppression, and neutropenia.10
Curcumin (CUR) is a polyphenol derived from the underground rhizome of the traditional Chinese medicine turmeric of the family Zingiberaceae. The substance exhibits various pharmacological activities, including anti-oxidation, anti-infection, anti-atherosclerosis, and anti-tumor activities.11 Furthermore, CUR displays massive potential as an effective MDR-reversing agent by downregulating P-gp, MRP-1, and mitoxantrone resistance protein (encoded by ABCG2).12,13 However, some shortcomings of CUR have limited its clinical application, such as low solubility, poor stability, and low absorption rates.14 To solve these problems, hyaluronic acid (HA), a negatively charged water-soluble natural chain mucopolysaccharide, was used to modify CUR via covalent bonding. Prior research found that the HA-specific receptor CD44 can be expressed on the surface of various cancer cells, such as breast cancer, ovarian cancer, and lung adenocarcinoma cells.15,16 The amphiphilic polymer obtained by combining HA and CUR can self-assemble into nanoparticles (NPs) in water. These NPs can target CUR and anticancer drugs to cancer cells and enhance the ability of CUR to reverse MDR.
Recently, many studies have revealed that CUR and DOX have synergistic effects and that these agents can reverse MDR in tumor cells.17–19 In the present study, we aimed to construct a CD44-targeted self-assembled NP system (HA–CUR/DOX-NPs) that can selectively co-deliver CUR and DOX to tumor cells in an effort to investigate the potential of CUR to reverse cancer drug resistance and explore its underlying mechanisms of action.
Experimental
Materials
Doxorubicin hydrochloride (DOX·HCl) and CUR were purchased from Dalian Meilun Biotechnology Co., Ltd (Dalian, China). Hyaluronic acid was purchased from Bloomage Freda Biopharm Co., Ltd (Jinan, Shandong, China). 4-Dimethylaminopyridine (DMAP), N,N-dimethylformamide (DMF) was purchased from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China). N-Hydroxysuccinimide was purchased from Alfa Aesar (Ward Hill, MA, USA). 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) was purchased from Aladdin Reagent Database Inc. (Shanghai, China). All reagents were of HPLC or analytical grade and were used without further purification. Anti-P-gp antibody and an MDR assay kit (fluorometric) were purchased from Abcam (Cambridge, UK). Anti-Bcl-2 and anti-Bax antibodies were purchased from Cell Signaling Technology (Boston, MA, USA).Cell lines
The human hepatocellular carcinoma cell line HepG2, human non-small lung cancer cell line A549, DOX-resistant non-small lung cancer cell line A549/ADR, human ileocecal cancer cell line HCT-8, and DOX-resistant ileocecal cancer cell line HCT-8/ADR cell line were all kindly provided by Professor Yongzhuo Huang (Chinese Academy of Sciences, China). These cells were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS), penicillin, and streptomycin at 37 °C with 5% CO2. The cell suspension was centrifuged at 1000 rpm for 3 min and then re-suspended in growth medium for further studies. The receipt of gifted cells was approved by the institutional review board of Chinese Academy of Sciences.Synthesis of HA–CUR conjugates
HA–CUR was synthesized as described previously.10,20 In total, HA powder (80 mg) were dissolved in 4 ml of water to obtain the HA solution. NHS (2.41 mg), EDC (2.87 mg), and DMAP (4.35 mg) were added to the solution followed by stirring at 4 °C overnight to activate the carboxyl group on HA. Then, 11 mg of CUR were dissolved in 1 ml of DMF to obtain CUR solution, which was slowly added dropwise into the HA solution. After 30 min, the mixture was stirred in the dark at room temperature for 24 h. At the end of the reaction, the solution was transferred into a dialysis bag (MWCO: 3.5 kDa) and allowed to dialyze for 48 h with frequent exchanges of deionized water to remove water-soluble byproducts. The composition of HA–CUR was confirmed using a 1H-NMR spectrometer and FT-IR. The grafting rate of CUR to HA was determined.Preparation of HA–CUR-NPs
The freeze-dried powder of HA–CUR was weighed into an aqueous solution, and the solution was ultrasonicated for 3 min in an ice bath at 60 W (active every 2 s with an interval of 3 s) to obtain HA–CUR self-assembled NPs. The CUR content in NPs was determined via HPLC (Agilent 1260, CA, USA).Preparation of HA–CUR/DOX-NPs and HA–CUR/PTX-NPs
HA–CUR/DOX-NPs were prepared using the probe ultrasonication method. Briefly, 5 mg of DOX·HCl were reacted with a two-molar excess of triethylamine in methanol to obtain DOX free base. Then, 1 mg of DOX was dissolved in 0.5 ml of methanol and slowly injected into 1 ml of HA–CUR-NPs (weight ratio of DOX and CUR was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) with stirring for 24 h at room temperature. The organic solvent in the solution was removed using a rotary evaporator. Then, the solution was further sonicated for 5 min in an ice bath at 100 W (active every 2 s with an interval of 3 s). HA–CUR/PTX-NPs were also prepared as described previously.
:1) with stirring for 24 h at room temperature. The organic solvent in the solution was removed using a rotary evaporator. Then, the solution was further sonicated for 5 min in an ice bath at 100 W (active every 2 s with an interval of 3 s). HA–CUR/PTX-NPs were also prepared as described previously.
Particle size and zeta potential
The size distribution, zeta potential, and polydispersity index (PDI) of HA–CUR/DOX-NPs were measured at 25 °C using a Zetasizer Nano ZS90 instrument (Malvern Instruments, Worcestershire, UK). Each parameter was measured four times, and the mean values and SDs were calculated.Serum stability
The stability of HA–CUR/DOX-NPs in serum was examined in 10% FBS. NPs were incubated with 10% FBS (v/v) solution at 37 °C for 12, 24, 36, 48, or 60 h. At each time point, their size distribution and zeta potential were measured.CD44 expression analysis in cell lines
A549, HCT-8, and HepG2 cells were harvested and washed with washing buffer (phosphate-buffered saline [PBS] supplemented with 2% FBS, pH 7.4). Cells were incubated with CD44 antibodies on ice for 1 h and then washed three times with washing buffer. After washing, Alexa Fluor 488-conjugated anti-rabbit IgG was applied to the cells as the secondary antibody for 1 h at room temperature. Cells were analyzed using a flow cytometer (BD Biosciences, CA, USA).In vitro cellular uptake
In vitro cytotoxicity
The in vitro cytotoxicity of DOX was evaluated using the MTT assay. Briefly, A549 or A549/ADR cells were seeded into 96-well plates at a density of 1 × 104 cells for 24 h. Then, the cells were incubated with the different formulations (CUR, HA–CUR, DOX, HA–CUR/DOX-NPs) for 24, 48, or 72 h. Afterwards, MTT solution (1 mg ml−1) was added to each well, and the cells were incubated for another 4 h at 37 °C. Formazan crystals were dissolved using DMSO, and the spectrophotometric absorbance at 570 nm was determined using a microplate reader (Spectrophotometer, Thermo Scientific, USA). The results were expressed as the percentage of viable cells relative to untreated control cells.To further prove that the HA–CUR system can reverse MDR, PTX, which has a different anti-tumor mechanism and different structural characteristics, was selected to investigate drug resistance. The cytotoxicity of PTX and HA–CUR/PTX-NPs was also evaluated using the MTT assay as described previously.
Determination of P-gp activity
The activity of P-gp was determined using a fluorometric MDR assay kit. Per the manufacturer's instructions, cells were seeded into 96-well clear flat-bottom black-wall microplates and treated as indicated. Then, the cells were treated with DOX, HA–CUR, DOX + HA–CUR, HA–CUR/DOX-NPs, or verapamil (positive control). After incubation for 24 h, 100 μl of MDR dye loading solution were added to each well, and the plates were incubated at room temperature for 1 h in the dark. The fluorescence of cells was detected using a fluorescence microplate reader (Ex/Em = 490 nm/525 nm). All experiments were performed in triplicate, and the results were compared with the control findings.Western blot assay
After appropriate treatment, cells were lysed in RIPA lysis buffer containing a protease and phosphatase inhibitor cocktail on ice for 30 min and then centrifuged at 13000 × g for 10 min. The protein concentration was determined using a BCA protein assay kit. Proteins (20 μg per well) were separated via SDS-PAGE and then transferred to a polyvinylidene difluoride membrane.
The membranes were blocked with 5% bovine serum albumin (BSA) for 1 h and then incubated with anti-P-gp (1:2500), anti-Bcl-2 (1:1000), anti-Bax (1:1000), or anti-β-actin (1:2000) diluted antibody in 5% BSA containing TBST at 4 °C overnight. After three washes with TBST, membranes were incubated with a secondary antibody (1:20000) for 1 h at room temperature. Finally, the proteins were visualized using a Super Signal West Pico Kit and then imaged using a ChemiDoc™ XRS (Bio-Rad, USA).
Quantitative real-time polymerase chain reaction (qRT-PCR)
P-gp, Bcl-2, and Bax expression in A549/ADR cells was measured via qRT-PCR. In brief, cells were treated with DOX, HA–CUR, DOX + HA–CUR, and HA–CUR/DOX-NPs for 24 h, and then total RNA was extracted from the cells using TRIzol reagent (Thermo Fisher) following the manufacturer's protocol. Reverse transcription of RNA was conducted using a first-strand cDNA synthesis kit (Invitrogen). qRT-PCR was performed using SYBR Green and an Mx3005P qPCR instrument (Agilent) in a three-step program (3 min at 95 °C followed by 40 cycles of 15 s at 95 °C and 1 min at 60 °C). mRNA expression relative to GAPDH expression was calculated using the ΔΔCt method. The primer sequences used for qRT-PCR analysis are listed in Table 1.| Gene | Forward (5′–3′) | Forward (5′–3′) |
|---|---|---|
| P-gp | TTGCTGCTTACATTCAGGTTTCA | AGCCTATCTCCTGTCGCATTA |
| bax | CCCGAGAGGTCTTTTTCCGAG | CCAGCCCATGATGGTTCTGAT |
| Bcl-2 | GGTGGGGTCATGTGTGTGG | CGGTTCAGGTACTCAGTCATCC |
| GAPDH | GGAGCGAGATCCCTCCAAAAT | GGCTGTTGTCATACTTCTCATGG |
Statistical analysis
Statistical analysis was performed using GraphPad Prism 7.0 statistical software (San Diego, CA, USA). The results were expressed as the mean ± SD. Statistical significance was tested using a two-tailed Student's t-test or one-way ANOVA. Significant differences between or among groups are indicated by *p < 0.05, **p < 0.01, or ***p < 0.001.Results
Synthesis and characterization of HA–CUR
HA–CUR was successfully synthesized via esterification between the carboxyl group of HA and hydroxy group of CUR. The synthetic route is shown in Fig. 1A. The structure of HA–CUR was confirmed using 1H-NMR and FTIR. Fig. 1B presents the 1H-NMR spectra of HA, CUR, and HA–CUR. The peaks for HA–CUR at 1.9–2.0 and 6.6–6.9 ppm belonged to HA (acetyl peak, –NHCOCH3) and CUR (aromatic protons), respectively, proving that the HA–CUR conjugate was successfully synthesized. The structure of the HA–CUR conjugate was also characterized by its FTIR spectra (Fig. 1C). Compared with the spectrum of HA, the peaks for HA–CUR at 3411 and 2892 cm−1 became sharper and stronger, and these peaks were attributed to the –OH group and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O functional group, respectively, in the structure of CUR. In addition, new peaks appeared at 1634, 1518, and 1210 cm−1, and the peak at 1518 cm−1 was attributed to the vibration of CC in the benzene ring in curcumin. The formation of an ester bond can be judged by the peaks at 1210 and 1634 cm−1, further confirming that CUR was successfully grafted to HA.10 The grafting yield of curcumin in the polymer of HA–CUR is 7% by comparing the integral of H (6.8 ppm) on the CUR benzene ring with the integral of H (4.8 ppm) on HA.
O functional group, respectively, in the structure of CUR. In addition, new peaks appeared at 1634, 1518, and 1210 cm−1, and the peak at 1518 cm−1 was attributed to the vibration of CC in the benzene ring in curcumin. The formation of an ester bond can be judged by the peaks at 1210 and 1634 cm−1, further confirming that CUR was successfully grafted to HA.10 The grafting yield of curcumin in the polymer of HA–CUR is 7% by comparing the integral of H (6.8 ppm) on the CUR benzene ring with the integral of H (4.8 ppm) on HA.
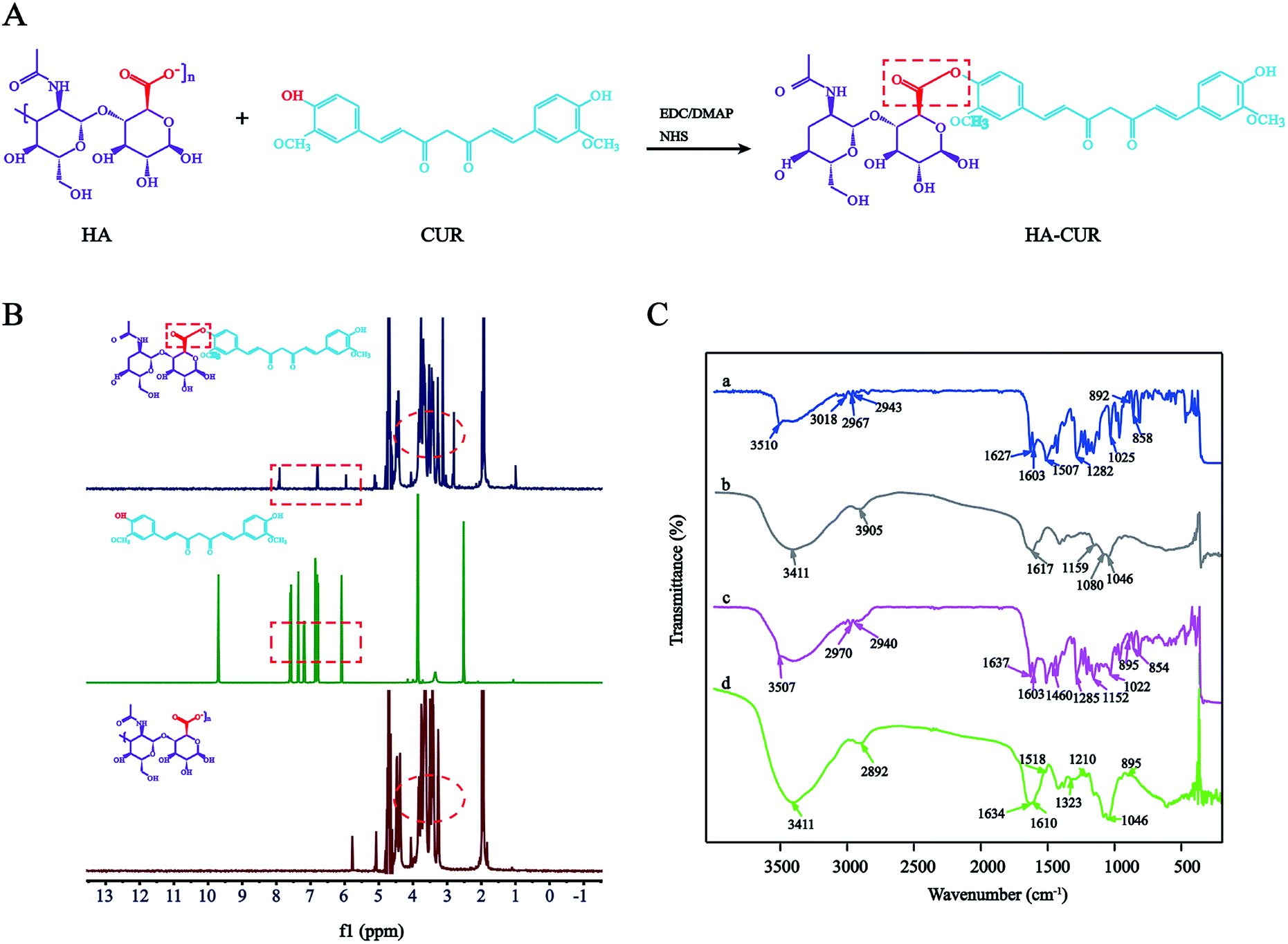 | ||
| Fig. 1 Synthesis and characterization of HA–CUR. (A) Synthetic route of HA–CUR. (B) 1H-NMR spectrum of CUR, HA and HA–CUR. (C) FTIR spectrum of CUR (a), HA (b), HA + CUR (c) and HA–CUR (d). | ||
Pyrene fluorescence probe method were used to determine the CMC of HA–CUR micelles. The results were shown in the Fig. S1.† The abscissa of the mutation point corresponding to the fluorescence ratio is −0.95, we calculated the concentration of HA–CUR micelle is 0.11220 mg ml−1 under this mutation point, means the lowest concentration of HA–CUR forming nanoparticle solution is 0.11220 mg ml−1.
Preparation and characterization of HA–CUR/DOX-NPs
HA–CUR and DOX could self-assemble in an aqueous medium to form NPs. As shown in Fig. 2A–F, HA–CUR and HA–CUR/DOX-NPs formed a uniform dispersion solution. The average particle size of HA–CUR-NPs was 164.9 nm (narrow PDI, 0.116; zeta potential, was −31.2 mV) and the average particle size of HA–CUR/DOX-NPs was 179.8 nm (narrow PDI, 0.117; zeta potential, −21.1 mV). The EE% of DOX in HA–CUR/DOX-NPs was 82.7%, indicating that DOX was well packaged in the NPs.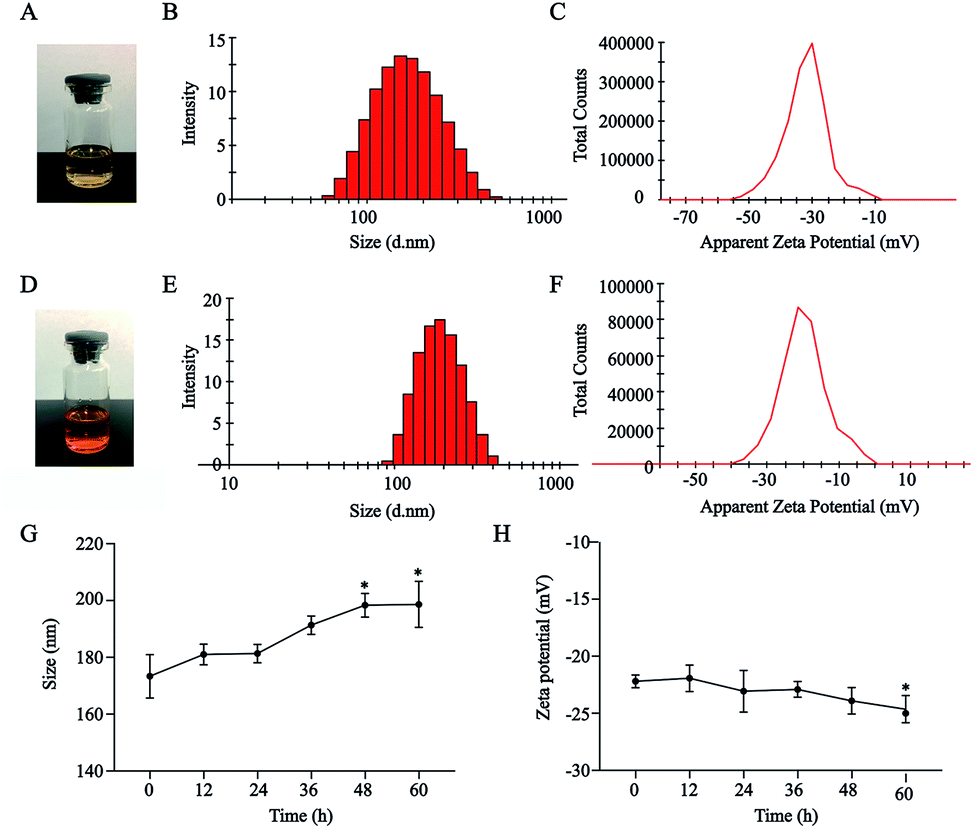 | ||
| Fig. 2 Characterization of HA–CUR and HA–CUR/DOX nanoparticles. (A) The picture of HA–CUR nanoparticle solution. (B) Particle size distribution of HA–CUR nanoparticles. (C) Zeta potential of HA–CUR nanoparticles. (D) The picture of HA–CUR/DOX-NPs solution. (E) Particle size distribution of HA–CUR/DOX-NPs. (F) Zeta potential of HA–CUR/DOX-NPs. (G) Particle size (H) zeta potential stability of HA–CUR/DOX-NPs in 10% serum within 60 h. Data represent mean ± SD (n = 4). *p < 0.05 compared to control group. | ||
To explore the serum stability of HA–CUR/DOX-NPs, we incubated the NPs with 10% BSA for 0, 12, 24, 36, 48, or 60 h. Size and zeta potential were evaluated (Fig. 2F and G). The results indicated that the particle size of HA–CUR/DOX-NPs was slightly increased with prolonged incubation, but there was no obvious precipitation observed. Meanwhile, the zeta potential was slightly decreased because of the slight negative charge of BSA. Therefore, NPs were stable in aqueous BSA solution.
CD44 expression analysis in cell lines
Prior studies illustrated that CD44 is highly expressed on the surface of tumor cells and that it has strong affinity for HA.21 Therefore, we detected CD44 expression on the surface of A549, HCT-8, and HepG2 tumor cells. The results of flow cytometry indicated that CD44 was highly expressed on the surface of A549 and HepG2 cells, with positive rates of 82.8 (Fig. 3A) and 63.6% (Fig. 3C), respectively. However, the rate of CD44 expression on the HCT-8 cell surface was only 5.4% (Fig. 3B). The results were consistent with another report.22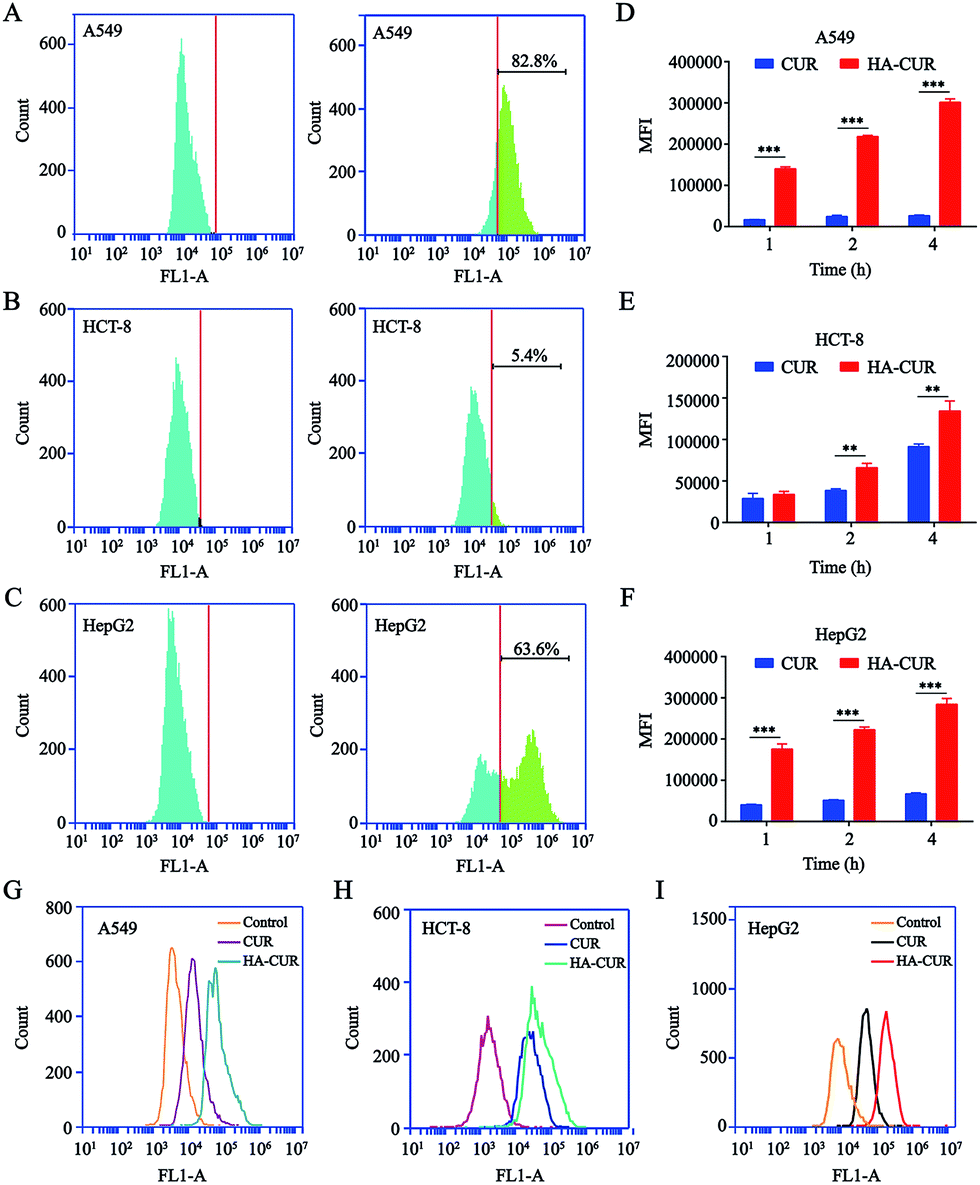 | ||
| Fig. 3 CD44 expression analysis and in vitro cellular uptake of HA–CUR. CD44 expression on (A) A549, (B) HCT-8 and (C) HepG2 cells were determined by flow cytometry. In vitro cellular uptake of CUR and HA–CUR in (D) A549, (E) HCT-8 and (F) HepG2 cells for 1, 2 and 4 h. The uptake of CUR and HA–CUR in (G) A549, (H) HCT-8 and (I) HepG2 cells was detected by flow cytometry for 4 h. The concentration of CUR was 20 μg ml−1. Data represent mean ± SD (n > 5). **p < 0.01 and ***p < 0.001. | ||
To examine whether HA-coated NPs selectively increase the cellular uptake of CUR in A549, HCT-8, and HepG2 cells, cells were treated with CUR and HA–CUR for different times (1, 2, or 4 h). As shown in Fig. 3D–F, based on the quantitative analysis via flow cytometry, the cellular uptake of CUR and HA–CUR increased constantly from 0 to 4 h. HA–CUR uptake by A549 and HepG2 cells was remarkably enhanced by the binding of HA and high expression of CD44 on the cell surface. The lower uptake of HA–CUR in HCT-8 cells was related to the low expression of CD44 on the cell surface. The results in Fig. 3G–I also illustrated that HA–CUR delivered CUR into cells more efficiently than free CUR, and the cellular uptake was positively correlated with the cell surface CD44 expression.
Cellular uptake
Fluorescence microscopy was employed to investigate the cellular uptake and intracellular distribution of HA–CUR/DOX-NPs. The nuclei were stained with DAPI, which emits blue fluorescence. As shown in Fig. 4A and B, after 4 h of incubation, the red fluorescence of DOX following treatment with free DOX and HA–CUR/DOX-NPs was observed in the nuclei of A549 and A549/ADR cells. We also found that HA–CUR/DOX-NPs exhibited stronger fluorescence than free DOX. These results indicated that DOX could be released from HA–CUR/DOX-NPs, followed by its entry into nuclei as free DOX, and CUR can be used to reverse drug resistance in tumor cells, enabling more DOX uptake by cells.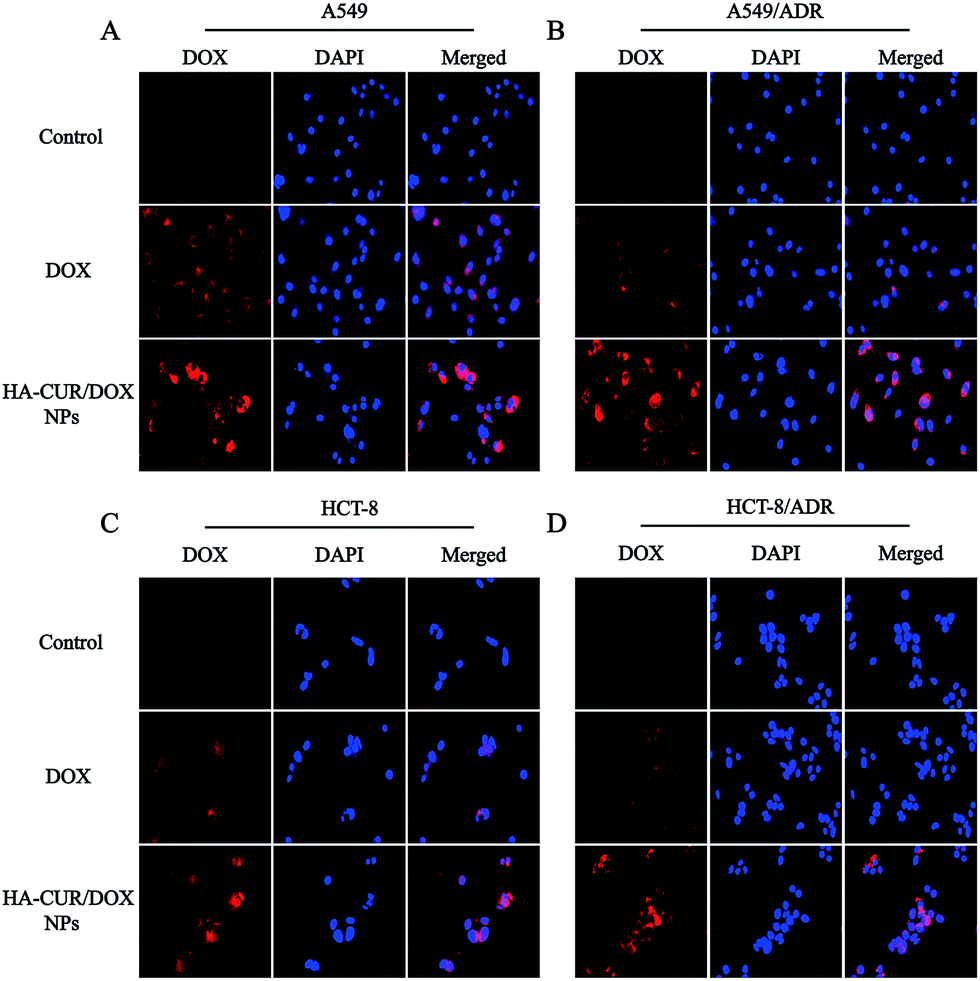 | ||
| Fig. 4 Fluorescence microscopy images of DOX and HA–CUR/DOX-NPs after incubation with 4 h in (A) A549, (B) A549/ADR, (C) HCT-8 and (D) HCT-8/ADR cells. Nuclei stained by DAPI (blue) while DOX showed red fluorescence. For each panel, (1) DOX (red); (2) nuclei stained by DAPI (blue); (3) merged: overlay of (1) and (2). | ||
To verify that CUR can reverse DOX resistance in tumor cells, we also used the HCT-8 cell line and its resistant variant to study the cellular uptake of DOX and HA–CUR/DOX-NPs. As shown in Fig. 4C and D, compared with the findings in the HA–CUR/DOX-NPs group, the fluorescence of free DOX group was distinct weaker, especially in HCT-8/ADR cells with strong MDR activity, in which there was almost no fluorescence in the nuclei of free DOX-treated cells. These results suggested that HA–CUR/DOX-NPs could facilitate efficient DOX release mainly because HA can enhance the binding of NPs to tumor cells and CUR can inhibit the efflux of drugs from tumors, thus increasing the uptake of DOX by drug-resistant tumor cells and increasing the therapeutic effect.23
In vitro cytotoxicity study
The in vitro cytotoxicity of CUR, HA–CUR, DOX, HA–CUR/DOX-NPs, PTX, and HA–CUR/PTX-NPs was evaluated using the MTT assay. Prior research found that CUR, as an MDR-reversing agent, can increase the sensitivity of tumor cells to drugs,24,25 First, we tested the toxicity of CUR and HA–CUR in A549 and A549/ADR cells. As shown in Fig. 5A and B, HA–CUR exhibited stronger cytotoxicity than free CUR, and its cytotoxic effects were concentration- and time-dependent. We considered that the reason for this finding may be that the targeted modification of HA promotes the entry of CUR into tumor cells and increases its biological stability.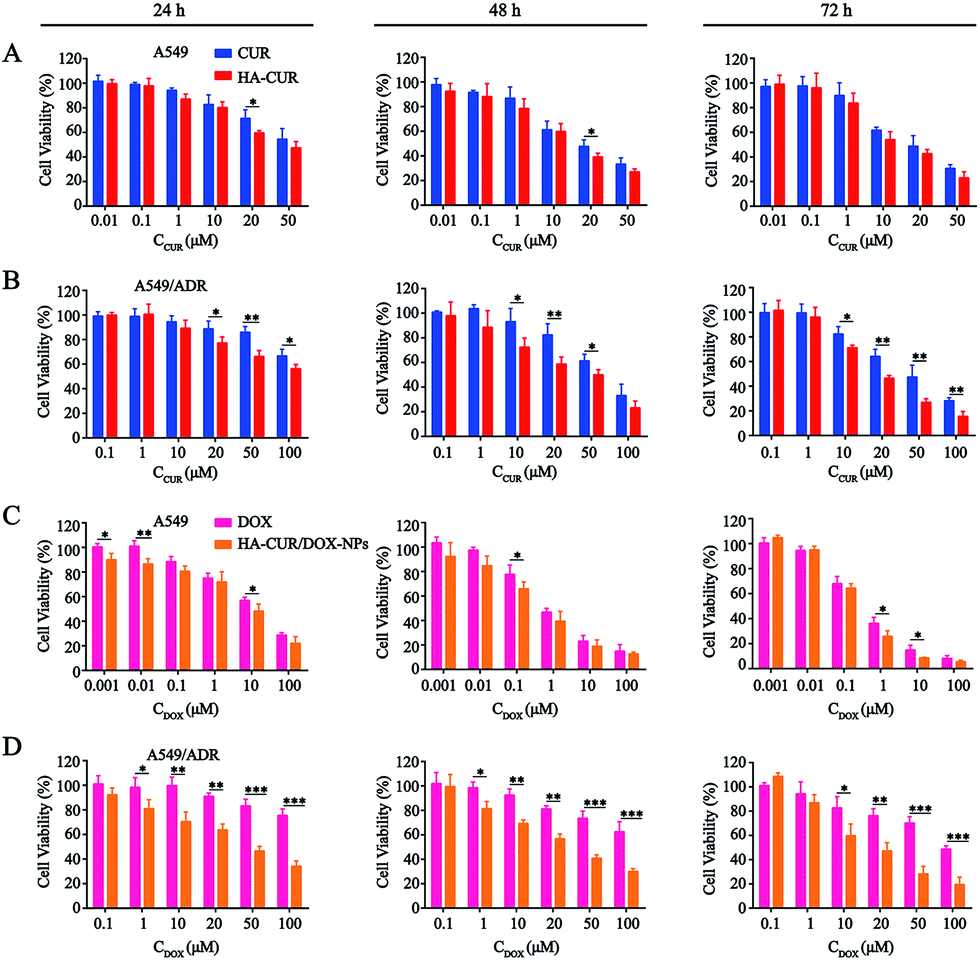 | ||
| Fig. 5 In vitro cytotoxicity of different formulations. The cytotoxicity of CUR and HA–CUR on (A) A549 cells and (B) A549/ADR cells for 24 h, 48 h and 72 h. The cytotoxicity of DOX and HA–CUR/DOX-NPs on (C) A549 cells and (D) A549/ADR cells for 24 h, 48 h and 72 h. Data represent mean ± SD (n > 5). ***p < 0.001, **p < 0.01 and *p < 0.05. | ||
Then, we examined the toxicity of DOX and HA–CUR/DOX-NPs in A549 and A549/ADR cells. Treatment with DOX significantly decreased the viability of A549 cells, whereas it had no obvious effect on the viability of A549/ADR cells. The drug resistance index of A549/ADR cells to DOX was 20.41 (Table 2). The viability of A549 cells was decreased by more than 50% by treatment with 7 μM DOX for 48 h (Fig. 5C), whereas this treatment decreased the viability of A549/ADR cells by approximately 10% (Fig. 5D), indicating that the A549/ADR cells were resistant to DOX. However, HA–CUR/DOX-NPs displayed obviously enhanced cytotoxicity in both A549 and A549/ADR cells.
| Drug | A549 | A549/ADR | RI |
|---|---|---|---|
| a Denoted as IC50 of resistant cell/IC50 of the sensitive cell. | |||
| CUR | 18.09 | 29.37 | 1.62 |
| HA–CUR | 11.37 | 11.9 | 1.05 |
| DOX | 6.14 | 125.3 | 20.41 |
| HA–CUR/DOX-NPs | 2.82 | 9.36 | 3.32 |
| PTX | 8.06 | 103.2 | 12.8 |
| HA–CUR/PTX-NPs | 1.72 | 4.03 | 2.34 |
Prior studies found that tumor cells are prone to MDR after developing resistance to a single chemotherapeutic drug,26 and thus, we also investigated the tolerance of A549 and A549/ADR cells to PTX, as well as the ability of CUR to reverse chemotherapy drug resistance. We then evaluated PTX and HA–CUR/PTX-NPs in A549 and A549/ADR cells. As shown in Fig. 6A and B, PTX significantly decreased the viability of A549 cells in a concentration- and time-dependent manner. Following treatment with 10 μM PTX for 48 h, the survival rate of A549 cells was approximately 55%, whereas PTX decreased the viability of A549/ADR cells to 70%, indicating that A549/ADR cells were resistant to PTX. However, HA–CUR/PTX-NPs significantly enhanced the cytotoxicity of PTX in A549 and A549/ADR cells, possibly because the HA–CUR/PTX-NPs could rapidly release PTX in tumor cells and HA–CUR could prevent PTX efflux as well as enhance its uptake by tumor cells. These results suggest that CUR can reverse the resistance of tumor cells to chemotherapy and that the system can be used to deliver other drugs.
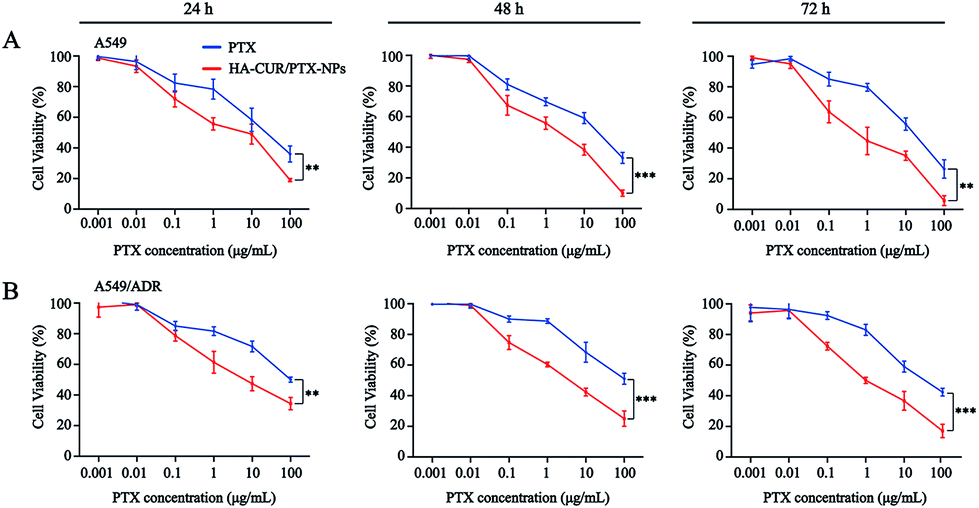 | ||
| Fig. 6 In vitro cytotoxicity of different formulations. The cytotoxicity of PTX and HA–CUR/PTX-NPs on (A) A549 cells and (B) A549/ADR cells for 24 h, 48 h and 72 h. Data represent mean ± SD (n > 5). ***p < 0.001 and **p < 0.01. | ||
HA–CUR/DOX-NPs inhibit P-gp expression and activity
To better understand the potential mechanism by which CUR enhances sensitivity of A549/ADR cells to DOX, P-gp activity and expression were further investigated. P-gp activity was determined using a fluorometric MDR assay kit, in which the P-gp inhibitor verapamil (30 μM) served as positive control.27 As shown in Fig. 7A, the intracellular intensity of the fluorescent P-gp substrate in A549/ADR cells was enhanced by HA–CUR/DOX-NPs treatment, meaning that HA–CUR/DOX-NPs effectively inhibited P-gp activity. In particular, treatment with DOX, HA–CUR, DOX + HA–CUR and HA–CUR/DOX-NPs increased the intracellular fluorescence by 1.77-, 2.77-, 3.1- and 4.3-fold, respectively, versus that in the untreated control, we also used HCT-8/ADR cells to verify this result. Fig. 7B reveals that HA–CUR/DOX-NPs also significantly enhanced the fluorescence intensity in these cells, in line with the results in A549/ADR cells.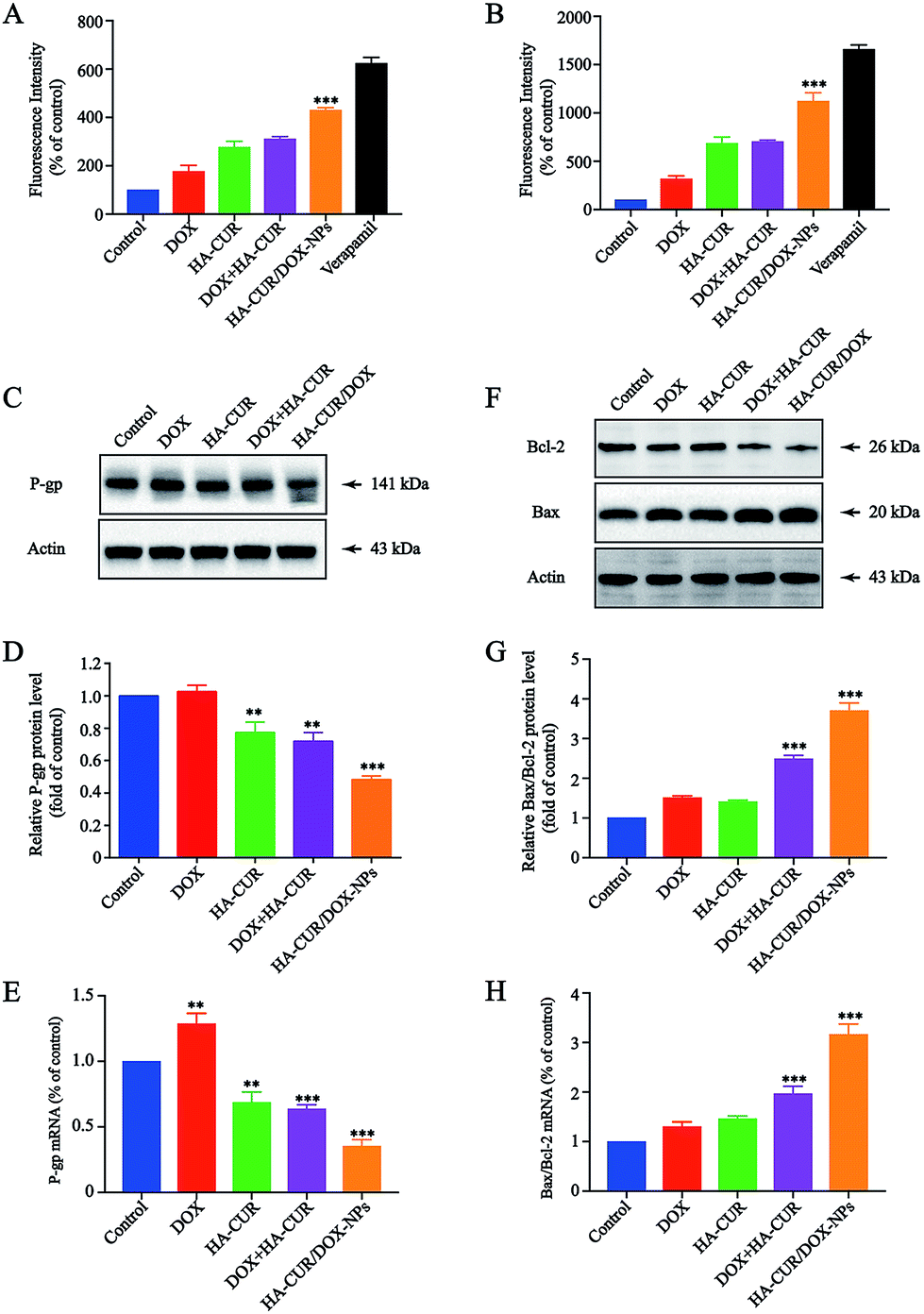 | ||
| Fig. 7 Effect of HA–CUR/DOX-NPs on apoptosis and drug efflux. (A) A549/ADR cells and (B) HCT/ADR cells were treated with different formulations, including DOX, HA–CUR, DOX + HA–CUR, HA–CUR/DOX-NPs and verapamil for 24 h, then P-gp activity was determined using a fluorometric MDR assay kit. The P-gp inhibitor verapamil (30 μM) was served as positive control. (C) P-gp, (F) Bcl-2 and Bax protein expression in A549/ADR cells were detected by western blot. Quantitative analysis of western blotting bands including (D) P-gp, (G) Bax/Bcl-2. qRT-PCR quantitative analysis of the expression of (E) P-gp, (H) Bax/Bcl-2. Data represent mean ± SD (n > 5). ###p < 0.001, ##p < 0.01, #p < 0.05 compared to control group. | ||
P-gp expression was measured via western blotting, as shown in Fig. 7C, HA–CUR/DOX-NPs significantly inhibited the expression of P-gp. The quantitative analysis of P-gp expression is shown in Fig. 7D. The mRNA expression of P-gp was significantly downregulated after treatment with HA–CUR/DOX-NPs, in line with the western blotting results (Fig. 7E). Taken together, we deduced that CUR plays a major role in inhibiting P-gp expression and activity, leading to increased intracellular accumulation of DOX in A549/ADR cells.
HA–CUR/DOX-NPs induced mitochondria-dependent apoptosis
Bcl-2/Bax family proteins play major roles in modulating mitochondria-dependent apoptosis.28 Studies have found that CUR can inhibit the proliferation of tumor cells through the mitochondrial apoptosis program, specifically by upregulating Bax gene expression and downregulating Bcl-2 gene expression.29 Therefore, we further investigated whether HA–CUR/DOX-NPs could affect the mitochondrial apoptosis pathway. Expression of pro-apoptotic Bax and anti-apoptotic Bcl-2 was detected in A549/ADR cells via western blotting and qRT-PCR analysis. Fig. 7F shows that HA–CUR/DOX-NPs increased the expression of pro-apoptotic Bax and decreased the expression of anti-apoptotic Bcl-2 in A549/ADR cells. According to the quantitative analysis results presented in Fig. 7G, Bax/Bcl-2 expression was significantly increased following HA–CUR/DOX-NP treatment. The experimental results of qRT-PCR were consistent with the western blotting data (Fig. 7H). These results implied that HA–CUR/DOX-NPs induce apoptosis through Bax/Bcl-2-mediated mitochondrial dysfunction.Discussion
At present, cancer is a major cause of human death, and the development of MDR to chemotherapy is a major obstacle to the effective treatment of tumors.30 MDR has been found in many cancers such as lung cancer, leukemia, breast cancer, and glioma.31 The causes of MDR in tumor cells are complicated, mainly including overexpression of drug efflux proteins and changes of apoptosis signals related to Bax/Bcl-2.32 Therefore, finding effective treatments to alleviate adverse drug reactions and overcome MDR is necessary for the treatment of cancer.DOX, an anthracycline antibiotic, is one of the most efficacious drugs for the treatment of breast and lung cancers in the clinic.33 However, the clinical application of DOX has been severely hindered because of its narrow therapeutic window and the development of MDR.34 Several strategies have been applied to improve the efficacy of chemotherapeutic agents, such as co-delivery of chemotherapy drugs and chemosensitizers,35 chemical modification,36 and the application of NP-based targeted drug delivery.37 Many studies used chemosensitizers to increase the sensitivity of tumor cells to chemotherapeutic drugs. CUR, as a good sensitizer, can regulate MDR protein expression and inhibit cancer cell proliferation.38,39
Combination therapy is increasingly used to treat cancer.40 Recently, many studies have used nano-delivery systems to co-deliver chemotherapeutics and chemosensitizers for tumor therapy. Wang et al. revealed that the co-delivery of DOX and CUR polymeric micelles can improve anti-tumor efficacy.41 Zhao et al. revealed that the simultaneous delivery of DOX and CUR by DOX/CUR-NPs can enhance the treatment of diethylnitrosamine-induced hepatocellular carcinoma.23 CD44 is highly expressed on the surface of tumor cells, and HA is a specific ligand. Studies have revealed that drug therapy targeting CD44 has certain anti-tumor effects.42
In this study, NPs incorporated with DOX and CUR were prepared to simultaneously deliver both agents to overcome MDR to DOX. First, we successfully synthesized HA–CUR and characterized it using 1H-NMR and FT-IR. Then, we developed HA–CUR and HA–CUR/DOX self-assembling NPs, evaluated their effects against MDR via in vitro cell experiments, and finally explored their anti-tumor mechanism (Scheme 1 and Fig. 1).
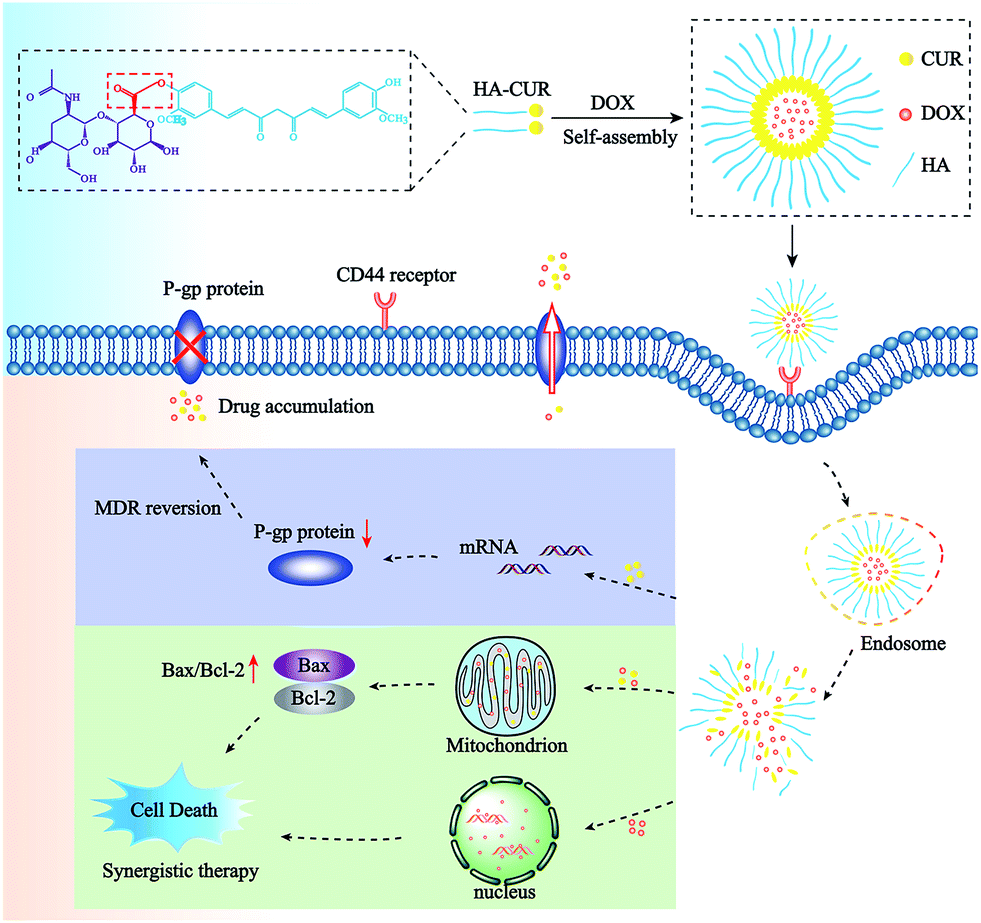 | ||
| Scheme 1 Structure and treatment strategy of HA–CUR/DOX nanoparticles. The HA–CUR/DOX-NPs are formed by HA–CUR and DOX self-assembly. The nanoparticles endocytosed into cells through CD44 receptor on the surface of tumor cells and releasing DOX and CUR. The therapy was based on a synergistic effect involving the DOX and CUR and the mechanism of HA–CUR/DOX-NPs is to inhibit the expression of P-gp and promote tumor cell apoptosis through the mitochondrial apoptosis pathway. | ||
Our NP characterizations found that the average particle size and zeta potential of HA–CUR/DOX-NPs were approximately 180 nm and −21 mV, respectively. The NPs had good encapsulation activity for DOX and superior serum stability, suggesting their potential to increase the clinical applicability of DOX therapy (Fig. 2).
HA is the natural ligand of CD44 and active targeting of HA can enhance the therapeutic effect of drugs in tumors. Xiong et al. revealed that HA modification can facilitate tumor-targeted drug delivery.10 Our results indicated that CD44 was highly expressed on the surface of A549 and HepG2 cells, and HA–CUR uptake was significantly higher than that of free CUR. These results confirm the specific binding of HA with CD44, which can increase the targeting ability of NPs (Fig. 3).
We used fluorescence microscopy to observe the uptake of HA–CUR/DOX-NPs by A549, A549/ADR, HCT-8, and HCT-8/ADR cells. We found that the fluorescence was weak in A549/ADR and HCT-8/ADR cells following treatment with DOX, indicating severe resistance to the drug. After treatment with HA–CUR/DOX-NPs, we observed significant increases of fluorescence, indicating a reversal of MDR by CUR. These results were consistent with another report.19 We found increases in cytotoxicity and decreases in the IC50 and resistance index in cells treated with HA–CUR/DOX-NPs, suggesting the synergistic effects of HA–CUR/DOX-NPs compared with free DOX, which might be related to their passive tumor targeting and reversal of drug resistance. These results were consistent with a previous report (Fig. 4–6).
Previous studies indicated that drug efflux by membrane pumps and defensive cell death mediated by anti-apoptotic proteins are the two most important mechanisms of MDR.43 To further explore the therapeutic mechanism of HA–CUR/DOX-NPs, P-gp activity and expression were initially investigated. We found that HA–CUR/DOX-NPs inhibit both the expression and activity of P-gp, thereby increasing the intracellular level of DOX. The mitochondrial apoptotic pathway associated with Bax/Bcl-2 is closely related to MDR in tumor cells. CUR has been reported to enhance DOX-induced cell death through mitochondrial-mediated apoptosis in tumor cells.44 Meanwhile, we found that HA–CUR/DOX-NPs increased the mRNA and protein levels of Bax/Bcl-2, confirming the role of Bax and Bcl-2 in HA–CUR/DOX-NP-induced apoptosis (Fig. 7).
In conclusion, the synergy effect between DOX and CUR and the mechanism of anti-tumor MDR are the novelty of this research. Our data illustrated that CUR sensitizes resistant cancer cells to DOX-induced apoptosis. Co-assembly of DOX and HA–CUR targeted NPs may represent a novel therapeutic strategy for cancer cells that are resistant to DOX-based therapies, and the delivery system can be applied to other chemotherapy drugs.
Conclusions
We successfully synthesized HA–CUR and developed HA–CUR/DOX self-assembling NPs to reverse MDR in tumor cells. The well-formed HA–CUR/DOX-NPs exhibited good serum stability, favorable biocompatibility, and effective targeting of tumor cells in vitro. Our results indicated that the HA–CUR/DOX-NPs could efficiently facilitate the treatment of drug-resistant tumor cells through combination therapy. HA modification further improved the uptake of NPs by tumor cells through CD44-mediated specific targeting. Additionally, the HA–CUR/DOX-NPs effectively inhibited drug efflux by suppressing the expression and activity of P-gp and promoted tumor cell apoptosis through the mitochondrial apoptosis pathway related to Bax/Bcl-2, thus increasing the therapeutic effect. However, we acknowledge the potential limitations of this research. Further studies are needed to study the effect of the combination treatment using in vivo models of multidrug-resistant cancer. In conclusion, we anticipate that HA–CUR/DOX-NPs can improve the efficacy of treatment for multidrug-resistant tumor cells, and co-delivery of an anticancer drug and CUR using HA-targeted nanocarriers might represent a promising strategy for tumor therapy.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by National Natural Science Foundation of china (No. 81773673), Zhejiang Province Welfare Technology Applied Research Project (No. LGF19H300007) and Key Laboratory of Ningbo, China (No. 2016A22002).References
- Y. Xiao, J. Liu, M. Guo, H. Zhou, J. Jin, J. Liu, Y. Liu, Z. Zhang and C. Chen, Nanoscale, 2018, 10, 12639–12649 RSC
.
- W. Huang, H. Zhao, J. Wan, Y. Zhou, Q. Xu, Y. Zhao, X. Yang and L. Gan, Theranostics, 2019, 9, 3825–3839 CrossRef CAS PubMed
- C. Lim, J. Moon, T. Sim, W. R. Won, E. S. Lee, Y. S. Youn and K. T. Oh, J. Controlled Release, 2019, 295, 164–173 CrossRef CAS PubMed
- M. Wang, J. Wu, Y. Li, F. Li, X. Hu, G. Wang, M. Han, D. Ling and J. Gao, J. Controlled Release, 2018, 288, 34–44 CrossRef CAS PubMed
- D. H. Wang, H. L. Wei, H. S. Zhao, C. Y. Hao, Z. H. Min and J. M. Liu, Pharmacol. Res., 2005, 52, 376–385 CrossRef CAS PubMed
- X. Xie, X. Shao, W. Ma, D. Zhao, S. Shi, Q. Li and Y. Lin, Nanoscale, 2018, 10, 5457–5465 RSC
- M. Chen, F. Song, Y. Liu, J. Tian, C. Liu, R. Li and Q. Zhang, Nanoscale, 2019, 11, 3814–3826 RSC
- J. Liu, Z. Ye, M. Xiang, B. Chang, J. Cui, T. Ji, L. Zhao, Q. Li, Y. Deng, L. Xu, G. Wang, L. Wang and Z. Wang, Biomaterials, 2019, 223, 119475 CrossRef CAS PubMed
- L. Wang, Y. Chang, Y. Feng, X. Li, Y. Cheng, H. Jian, X. Ma, R. Zheng, X. Wu, K. Xu and H. Zhang, Nano Lett., 2019, 19(10), 6800–6811 CrossRef CAS PubMed
- H. Xiong, J. Ni, Z. Jiang, F. Tian, J. Zhou and J. Yao, Biomater. Sci., 2018, 6, 2527–2540 RSC
- Y. Yang, Z. Huang, X. Pu, G. Yin, L. Wang and F. Gao, Cell Proliferation, 2018, 51, e12486 CrossRef PubMed
- J. L. Revalde, Y. Li, B. C. Hawkins, R. J. Rosengren and J. W. Paxton, Biochem. Pharmacol., 2015, 93, 305–317 CrossRef CAS PubMed
- S. Man, L. Zhang, J. Cui, L. Yang, L. Ma and W. Gao, Cell Proliferation, 2018, 51, e12458 CrossRef PubMed
- S. Guerrero, M. Inostroza-Riquelme, P. Contreras-Orellana, V. Diaz-Garcia, P. Lara, A. Vivanco-Palma, A. Cardenas, V. Miranda, P. Robert, L. Leyton, M. J. Kogan, A. F. G. Quest and F. Oyarzun-Ampuero, Nanoscale, 2018, 10, 22612–22622 RSC
- A. Cao, P. Ma, T. Yang, Y. Lan, S. Yu, L. Liu, Y. Sun and Y. Liu, Mol. Pharm., 2019, 16, 2502–2510 CrossRef CAS PubMed
- Y. Byeon, J. W. Lee, W. S. Choi, J. E. Won, G. H. Kim, M. G. Kim, T. I. Wi, J. M. Lee, T. H. Kang, I. D. Jung, Y. J. Cho, H. J. Ahn, B. C. Shin, Y. J. Lee, A. K. Sood, H. D. Han and Y. M. Park, Cancer Res., 2018, 78, 6247–6256 CAS
- Y. Hong, S. Che, B. Hui, Y. Yang, X. Wang, X. Zhang, Y. Qiang and H. Ma, Biomed. Pharmacother., 2019, 112, 108614 CrossRef CAS PubMed
- L. Wang, H. Guan, Z. Wang, Y. Xing, J. Zhang and K. Cai, Mol. Pharm., 2018, 15, 2503–2512 CrossRef CAS PubMed
- M. Yang, L. Yu, R. Guo, A. Dong, C. Lin and J. Zhang, Nanomaterials, 2018, 8(3), 167–184 CrossRef PubMed
- J. B. Hu, S. J. Li, X. Q. Kang, J. Qi, J. H. Wu, X. J. Wang, X. L. Xu, X. Y. Ying, S. P. Jiang, J. You and Y. Z. Du, Carbohydr. Polym., 2018, 193, 268–280 CrossRef CAS PubMed
- M. Y. Kwon, C. Wang, J. H. Galarraga, E. Pure, L. Han and J. A. Burdick, Biomaterials, 2019, 222, 119451 CrossRef CAS PubMed
- T. Wang, J. Hou, C. Su, L. Zhao and Y. Shi, J. Nanobiotechnol., 2017, 15, 7 CrossRef PubMed
- X. Zhao, Q. Chen, Y. Li, H. Tang, W. Liu and X. Yang, Eur. J. Pharm. Biopharm., 2015, 93, 27–36 CrossRef CAS PubMed
- A. H. Abouzeid, N. R. Patel, C. Sarisozen and V. P. Torchilin, Pharm. Res., 2014, 31, 1938–1945 CrossRef CAS PubMed
- C. Sarisozen, A. H. Abouzeid and V. P. Torchilin, Eur. J. Pharm. Biopharm., 2014, 88, 539–550 CrossRef CAS PubMed
- Q. Chen, X. Liu, Z. Luo, S. Wang, J. Lin, Z. Xie, M. Li, C. Li, H. Cao, Q. Huang, J. Mao and B. Xu, J. Cell. Physiol., 2019, 234, 6611–6623 CrossRef CAS PubMed
- S. Wang, A. Wang, M. Shao, L. Lin, P. Li and Y. Wang, Sci. Rep., 2017, 7, 8419 CrossRef PubMed
- L. Suarez, B. Vidriales, J. Garcia-Larana, A. Lopez, R. Martinez, V. Martin-Reina, M. Tormo, J. D. Gonzalez-San Miguel, E. Lavilla, R. Garcia-Boyero, A. Orfao and J. F. San Miguel, Haematologica, 2001, 86, 1287–1295 CAS
- H. Moghtaderi, H. Sepehri, L. Delphi and F. Attari, Bioimpacts, 2018, 8, 185–194 CrossRef CAS PubMed
- C. Mao, F. Li, Y. Zhao, W. Debinski and X. Ming, Theranostics, 2018, 8, 6274–6290 CrossRef CAS PubMed
- B. Shabnam, G. Padmavathi, K. Banik, S. Girisa, J. Monisha, G. Sethi, L. Fan, L. Wang, X. Mao and A. B. Kunnumakkara, Translational Oncology, 2018, 11, 1379–1389 CrossRef PubMed
- Y. Sun, C. Wang, Q. Meng, Z. Liu, X. Huo, P. Sun, H. Sun, X. Ma, J. Peng and K. Liu, J. Cell. Physiol., 2018, 233, 3066–3079 CrossRef CAS PubMed
- J. Liu, D. Chi, S. Pan, L. Zhao, X. Wang, D. Wang and Y. Wang, Int. J. Pharm., 2019, 557, 264–272 CrossRef CAS PubMed
- Y. Xing, J. Zhang, F. Chen, J. Liu and K. Cai, Nanoscale, 2017, 9, 8781–8790 RSC
- J. P. Desale, R. Swami, V. Kushwah, S. S. Katiyar and S. Jain, Nanomedicine, 2018, 13, 2759–2776 CrossRef CAS PubMed
- M. Zhou, L. Li, L. Li, X. Lin, F. Wang, Q. Li and Y. Huang, Acta Pharm. Sin. B, 2019, 9, 615–625 CrossRef PubMed
- G. S. R. Raju, B. Dariya, S. K. Mungamuri, G. Chalikonda, S. M. Kang, I. N. Khan, P. S. Sushma, G. P. Nagaraju, E. Pavitra and Y. K. Han, Semin. Cancer Biol., 2019, 8, 13 Search PubMed
- A. Alemi, J. Zavar Reza, F. Haghiralsadat, H. Zarei Jaliani, M. Haghi Karamallah, S. A. Hosseini and S. Haghi Karamallah, J. Nanobiotechnol., 2018, 16, 28 CrossRef PubMed
- P. Zhang, Z. L. Lai, H. F. Chen, M. Zhang, A. Wang, T. Jia, W. Q. Sun, X. M. Zhu, X. F. Chen, Z. Zhao and J. Zhang, J. Exp. Clin. Cancer Res., 2017, 36, 190 CrossRef PubMed
- Q. S. Wang, L. N. Gao, X. N. Zhu, Y. Zhang, C. N. Zhang, D. Xu and Y. L. Cui, Theranostics, 2019, 9, 6239–6255 CrossRef PubMed
- J. Wang, W. Ma and P. Tu, Macromol. Biosci., 2015, 15, 1252–1261 CrossRef CAS PubMed
- R. Liu, W. Xiao, C. Hu, R. Xie and H. Gao, J. Controlled Release, 2018, 278, 127–139 CrossRef CAS PubMed
- Y. L. Lo, Int. J. Mol. Sci., 2012, 14, 158–176 CrossRef PubMed
- M. Mohajeri and A. Sahebkar, Crit. Rev. Oncol. Hematol., 2018, 122, 30–51 CrossRef PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra08202f |
| This journal is © The Royal Society of Chemistry 2019 |