
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBioactive compounds from the seeds of Amomum tsaoko Crevost et Lemaire, a Chinese spice as inhibitors of sphingosine kinases, SPHK1/2†
Seulah Lee‡
a,
Joo Chan Lee‡a,
Lalita Subedib,
Kyo Hee Chob,
Sun Yeou Kimb,
Hyun-Ju Parka and
Ki Hyun Kim *a
*a
aSchool of Pharmacy, Sungkyunkwan University, Suwon 16419, Republic of Korea. E-mail: khkim83@skku.edu; Fax: +82-31-290-7730; Tel: +82-31-290-7700
bCollege of Pharmacy, Gachon University, 191 Hambakmoero, Yeonsu-gu, Incheon 21936, Republic of Korea
First published on 22nd October 2019
Abstract
Amomum tsaoko Crevost et Lemaire (Zingiberaceae), a traditional Chinese spice also known as “Caoguo” or “tsao-ko,” has been considered to have many health benefits. As part of our continuous efforts to screen natural resources exhibiting potential bioactivity, we examined the seeds of A. tsaoko and found that its EtOH extract inhibited sphingosine kinases 1 and 2 (SPHK1/2). Bioactivity-based analysis and chemical investigation of the EtOH extract led to the isolation and identification of four aliphatic alcohols (1–4), five fatty acids (5–9), 12 phenolics (10–21), and four terpenoids (22–25), including four new compounds, an acetylated aliphatic alcohol (2), a fatty acid (5), and two phenolics (10–11). In addition, compound 1 was isolated for the first time from natural sources in this study. The structures of all compounds were elucidated based on spectroscopic analysis, including 1D and/or 2D NMR and HR-ESIMS as well as LC/MS analysis. A recently developed method using competing enantioselective acylation (CEA) coupled with LC/MS analysis was applied for the assignment of absolute configuration of compound 5. The absolute configurations of compounds 10 and 11 were determined using ECD calculations. All of the compounds (1–25) isolated from the active fraction were evaluated for their SPHK1/2 inhibitory effects at the concentration of 10 μM. Aliphatic alcohols 2–4, fatty acids 7 and 9, and phenolic compounds 13–15 and 21 showed inhibition against the activity of SPHK1 up to 20% and aliphatic alcohols 2 and 4, fatty acid 8, and phenolic compounds 10, 11, 18, and 22 showed inhibition against the activity of SPHK2 up to 40% compared with the control. Compound 2 showed the highest potency to inhibit SPHK1 enzymatic activity, by 59.75%, and compound 22 showed the highest potency in inhibiting the activity of SPHK2, by 22.75%, in comparison with the control, where both exhibited higher inhibition compared to those of positive controls. Docking modeling studies were conducted to suggest the binding mode of 2 and 22 in the substrate-binding pocket of SPHK1 and SPHK2, respectively.
Introduction
Sphingolipids are key components of the cell membrane that are required for the generation of signals that play important roles in human health as well as in diseases.1 Sphingosine, which is the fundamental base of sphingolipids, is an interconvertible metabolite of ceramides. Through the actions of sphingosine kinases 1 and 2 (SPHK1/2), sphingosine can be interconverted to sphingosine 1 phosphate (S1P), which functions in numerous cellular and physiological processes.2 Hence, SPHK1/2 are considered rate limiting agents for the function of S1P, or its precursors, sphingosines and ceramides. Inhibition of SPHK1/2 can thus inhibit the function of S1P or result in the accumulation of sphingosines or ceramides, which can be beneficial in certain conditions. However, research exploring SPHK1/2 modulators from natural products or as synthetic/semisynthetic compounds has been limited. SPHK1 functions in cell survival and apoptosis, and thus SPHK1 inhibitors have potential to induce apoptosis and anti-proliferative effects in cancer cells.3 The precise roles of SPHK2 have not yet been defined because of its various functions according to cell type. In some cells, SPHK2 decreases cell cycle arrest, thus inhibition of SPHK2 induces cell cycle arrest and apoptosis and thus may exert anticancer effects.4,5 Since sphingosines and ceramides play important roles in cell cycle arrest and apoptosis and S1P functions in cell proliferation, survival, differentiation, and angiogenesis, the inhibition of SPHK1/2 may induce cell cycle arrest and apoptosis by reducing S1P levels and increasing sphingosines and ceramides, which may be beneficial for the treatment of various cancers.6,7 SPHK1/2 inhibitors can thus exhibit anti-cancer activities directly or indirectly through sphingosine metabolism.8 In addition, a recent report showed that SPHK1/2 both play equal roles in mediating the mitogenic actions of insulin.9 Studies in neuroinflammation also revealed reduced SPHK1 and increased SPHK2 in Alzheimer's disease and Huntington's disease.10 Roles for S1P in chronic inflammation, infection, autoimmunity, and in allergic reactions have also been previously reported.11Amomum tsaoko Crevost et Lemaire (Zingiberaceae), which is also known as “Caoguo” or “tsao-ko,” is used as food and spice in Asian cuisine and is considered to have many health benefits for the treatment of stomach disorders, digestive disorders, and throat infections.12,13 Studies have demonstrated that A. tsaoko extracts have various pharmacological properties, including anti-microbial, anti-viral, anti-oxidant, anti-obesity, anti-inflammatory, and anti-proliferative activities.14–21 Phytochemical analysis of A. tsaoko identified a broad range of constituents, including diarylheptanoids, fatty acids, flavonoids, monoterpenes, phenolic metabolites, and steroids. Monoterpenes identified from A. tsaoko were reported to have anti-inflammatory and anti-Trichomonas vaginalis properties,12,19 while diphenylheptanes showed antioxidant, anti-tumor, anti-inflammatory, and neuroprotective effects,22,23 and benzaldehydes and cycloterpenals exhibited antiproliferative and anti-inflammatory effects.13,14 However, the effects of the crude extract or the constituents of A. tsaoko on SPHK1/2 activity have not been investigated.
In this study, as part of our continuous efforts to screen natural resources exhibiting potential bioactivity and to characterize the responsible compounds,24–30 we examined the effects of an ethanol (EtOH) extract of A. tsaoko seeds on SPHK1/2 activity and found that the EtOH extract exhibited inhibitory effects on SPHK1/2. Bioactivity-based analysis and chemical investigation of the EtOH extract led to the isolation and identification of 25 compounds (1–25). Compound 1 was identified from natural sources for the first time and an acetylated aliphatic alcohol (2), a fatty acid (5), and two phenolics (10–11) were identified as new compounds based on structural analysis using spectroscopic techniques including 1D and 2D nuclear magnetic resonance (NMR) and high-resolution electrospray ionization mass spectrometry (HR-ESIMS) as well as LC/MS analysis. The absolute configurations of new compounds were determined by quantum chemical electronic circular dichroism (ECD) calculations and the application of the recently developed method, competing enantioselective acylation (CEA) coupled with LC/MS analysis. Here we report the isolation of compounds 1–25 from the seeds of A. tsaoko, the structural elucidation of the isolated compounds, and the bioactivity of the isolates with regard to SPHK1/2 inhibition.
Results and discussion
Extracts were prepared from dried A. tsaoko seeds using 50% EtOH. The inhibitory effects of the EtOH extract on SPHK1/2 were examined, and the results revealed that 100 μg mL−1 of EtOH extract inhibited SPHK1 and SPHK2 up to 47% and 55%, respectively, compared to the control (Fig. 1). To identify the bioactive constituents of A. tsaoko seeds responsible for the SPHK1/2 inhibitory effect, we first fractionated the EtOH extract into hexane (HX)-soluble and water (H2O)-soluble fractions and examined which fraction was enriched with compounds responsible for SPHK1/2 inhibition (Fig. 1). The HX fraction showed significant inhibition of SPHK1 and SPHK2 by 39% and 67%, respectively, compared to the control (Fig. 1). The water fraction also exhibited inhibitory effect, however as it did not show any significant peaks when analyzed by LC-MS, chemical analysis was not further carried out. These observations indicated that the SPHK1/2 inhibitory effects of A. tsaoko seeds were attributable to bioactive constituents in the HX fraction.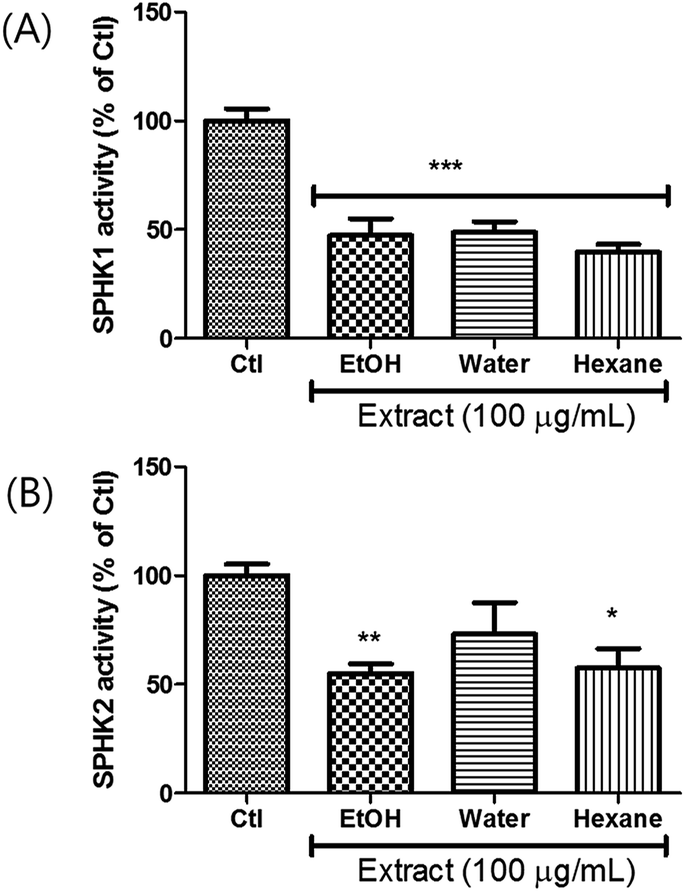 | ||
| Fig. 1 Effects of EtOH extract of A. tsaoko seeds, water-soluble and hexane-soluble fractions on inhibiting the activity of SPHK1 and SPHK2. SPHK1/2 were treated with the extract or the fractions (100 μg mL−1), and SPHK1/2 activity was evaluated based on the overall level of converted ADP. Enzymatic activity of (A) SPHK1 and (B) SPHK2. All data are presented as the mean ± standard error of the mean of independent experiments. *p < 0.05, **p < 0.01, and ***p < 0.001 indicate significant differences compared with the control group. | ||
To identify the active constituents responsible for the SPHK1/2 inhibitory effects of A. tsaoko seeds, we examined the HX fraction using repeated column chromatography and HPLC purification. Four aliphatic alcohols (1–4), five fatty acids (5–9), 12 phenolics (10–21), and four terpenoids (22–25) were isolated (Fig. 2). Of note, among the isolates, an acetylated aliphatic alcohol (2), a fatty acid (5), and two phenolics (10–11) were identified as new compounds, and compound 1 was isolated for the first time from natural sources.
 | ||
| Fig. 2 Chemical structures of compounds 1–25 isolated from the seeds of A. tsaoko. | ||
Compound 1 was isolated as colorless oil. The molecular formula was established as C14H22O4 from the molecular ion peak [M + Na]+ at m/z 277.1414 (calcd for C14H22O4Na, 277.1416) in the positive-ion mode of HR-ESIMS. The IR spectrum showed an absorption band of a carbonyl (1620 cm−1) group. The 1H NMR spectrum of 1 (Table 1) showed signals of an acetyl group at δH 2.06 (3H, s), olefinic methines at δH 5.56 (1H, dt, J = 15.0, 6.5 Hz) and 5.75 (1H, dt, J = 15.0, 7.0 Hz), and an oxygenated methylene at δH 4.50 (2H, d, J = 6.5 Hz). The 13C NMR (Table 1) showed 7 carbon signals, which were comprised of an acetyl group, olefinic carbons, and an oxygenated carbon, determined by the aid of HSQC analysis. Through other 2D NMR techniques, such as HMBC and 1H–1H COSY (Fig. 3), compound 1 was determined to be an acetylated aliphatic alcohol that is structurally symmetrical, in consideration of HR-ESIMS data. The two acetyl groups were shown to be at each end of compound 1 by the HMBC correlations of H-1 and H-10 with each carbonyl carbon (Fig. 3). The HMBC correlations of H-1/C-2 and C-3 as well as H-10/C-8 and C-9 were also observed, which led to the assignment of the location of double bonds. Further analysis with 1H–1H COSY confirmed the connections of C-3/C-4/C-5/C-6/C-7/C-8, and the gross structure of compound 1 was completely elucidated (Fig. 2). This structure was determined as 1,10-diacetate-2,8-decadiene-1,10-diol and named acetoxytsaokol A. A previous study reported the synthesis of compound 1,31 and the current study represents the first identification of this compound from natural sources.
| Position | 1 | 2 | 5 | |||
|---|---|---|---|---|---|---|
| δH | δC | δH | δC | δH | δC | |
| a Signal multiplicity and coupling constants (Hz) are in parentheses. The assignments were based on HSQC, HMBC, and TOCSY/1H–1H COSY experiments; 1H and 13C NMR were measured using NMR spectrometers operating at 700 MHz (1H) and 175 MHz (13C) for compounds 1 and 2, and 600 MHz (1H) and 150 MHz (13C) for compound 5.b Overlapped. | ||||||
| 1 | 4.50 d (6.5) | 65.2 | 4.52 d (6.5) | 65.4 | 170.9 | |
| 2 | 5.56 dt (15.0, 6.5) | 124.0 | 5.58 dt (15.0, 6.5) | 124.1 | 5.83 d (15.5) | 124.1 |
| 3 | 5.75 dt (15.0, 7.0) | 136.2 | 5.78 dt (15.0, 6.5) | 136.5 | 6.83 dt (15.5, 7.0) | 146.5 |
| 4 | 2.06 m | 32.0 | 2.06 mb | 32.1 | 2.22 dt (7.0, 8.0) | 31.0 |
| 5 | 1.40 m | 28.4 | 1.42 dt (7.0, 3.5) | 28.8 | 1.33 mb | 24.8 |
| 6 | 1.40 m | 28.4 | 1.42 dt (7.0, 3.5) | 28.5 | 1.58 mb | 27.9 |
| 7 | 2.06 m | 32.0 | 2.06 mb | 32.2 | 5.43 dt (11.0, 7.5) | 130.6 |
| 8 | 5.75 dt (15.0, 7.0) | 136.2 | 5.71 dt (15.0, 6.5) | 133.2 | 6.04 t (11.0) | 128.5 |
| 9 | 5.56 dt (15.0, 6.5) | 124.0 | 5.65 dt (15.0, 6.0) | 129.3 | 6.51 dd (15.0, 11.0) | 125.0 |
| 10 | 4.50 d (6.5) | 65.2 | 4.10 d (6.0) | 63.9 | 5.65 dd (15.0, 7.0) | 136.2 |
| 11 | 4.10 q (7.0) | 71.9 | ||||
| 12 | 1.48 m, 1.54 mb | 36.9 | ||||
| 13 | 2.26 m | 26.6 | ||||
| 14 | 1.33 mb | 31.5 | ||||
| 15 | 1.33 mb | 22.3 | ||||
| 16 | 0.93 t (7.0) | 12.9 | ||||
1-Me![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) O O |
170.9 | 171.0 | ||||
 |
2.06 s | 21.0 | 2.08 s | 21.2 | ||
| 10-MeO |
170.9 | |||||
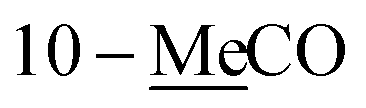 |
2.06 s | 21.0 | ||||
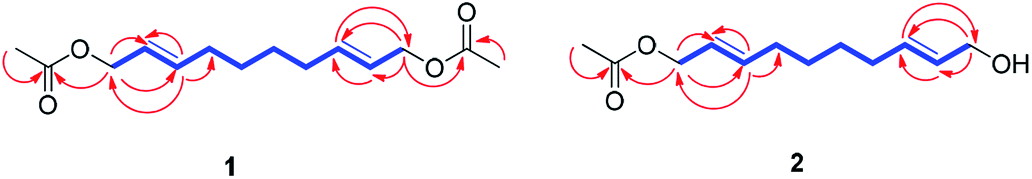 | ||
| Fig. 3 1H–1H COSY/TOCSY (blue bold lines) and key HMBC (red arrows) correlations for compounds 1–2. | ||
Compound 2 was isolated as colorless oil. The molecular formula was established as C12H20O3 from the molecular ion peak [M + Na]+ at m/z 235.1310 (calcd for C12H20O3Na, 235.1310) in the positive-ion mode of HR-ESIMS. The IR spectrum showed absorption bands of hydroxy (3408 cm−1) and carbonyl (1680 cm−1) functional groups. The 1H NMR spectrum of 2 (Table 1) showed signals for an acetyl group (δH 2.08 (3H, s)), four olefinic methines (δH 5.58 (1H, dt, J = 15.0, 6.5 Hz), 5.65 (1H, dt, J = 15.0, 6.0 Hz), 5.71 (1H, dt, J = 15.0, 6.5 Hz), 5.78 (1H, dt, J = 15.0, 6.5 Hz)), and two pairs of oxygenated methylenes (δH 4.52 (2H, d, J = 6.5 Hz), 4.10 (2H, d, J = 6.0 Hz)). The 13C NMR of compound 2 (Table 1) showed 12 carbon signals attributable to an acetyl group, four olefinic carbons, and two oxygenated carbons, which were determined by the aid of HSQC analysis. Further 2D NMR analysis including HMBC and TOCSY experiments revealed that compound 2 was an acetylated alcohol with a similar structure as compound 1, with the only difference in the absence of an acetyl group. The HMBC correlations of H-1 with the carbonyl carbon confirmed the attachment of an acetyl group at C-1 (Fig. 3). In the HMBC spectrum, H-1 correlated with C-2 and C-3 and H-10 correlated with C-8 and C-9, which led to the assignment of the location of double bonds. The linkages between C-3/C-4/C-5/C-6/C-7/C-8 were confirmed by the analysis of TOCSY correlations. The gross structure of compound 2 was completely elucidated as 1-acetate 2,8-decadiene-10-ol and named tsaokol A (Fig. 2).
Compound 5 was isolated as an amorphous gum. The molecular formula was established as C16H26O3 from the molecular ion peak [M − H]− at m/z 265.1795 (calcd for C16H25O3, 265.1804) in the negative-ion mode of HR-ESIMS. The IR spectrum showed absorption bands of hydroxy (3415 cm−1) and conjugated carbonyl (1720 cm−1) functional groups. The 1H NMR spectrum of 5 (Table 1) showed signals for a methyl group at δH 0.93 (3H, t, J = 7.0 Hz), an oxygenated methine at δH 4.10 (1H, q, J = 7.0 Hz), and six olefinic methines (δH 5.43 (1H, dt, J = 11.0, 7.5 Hz), 5.65 (1H, dd, J = 15.0, 7.0 Hz), 5.83 (1H, d, J = 15.5 Hz), 6.04 (1H, t, J = 11.0 Hz), 6.51 (1H, dd, J = 15.0, 11.0 Hz), 6.83 (1H, dt, J = 15.5, 7.0 Hz)). The 13C NMR of compound 5 (Table 1) showed 16 carbon signals comprised of a methyl group, a carbonyl group, an oxygenated carbon, and six olefinic carbons. The disparity in the chemical shifts of the olefinic pair C-2 (δC 124.1)/C-3 (δC 146.5) suggested the existence of a carbonyl group next to the double bond, and HMBC correlations between H-2/C-1 and H-3/C-1 further confirmed the presence of an α,β-unsaturated carbonyl group. Along with this evidence, the signal of a terminal methyl group at δH 0.93 was indicative of a fatty acid. The positions of the double bonds were confirmed as C-2, C-7, and C-9 by MS/MS analysis. The MS2 of 5 (265 → full-scan) yielded m/z 153 [M − H]− and 113 [M − H]−, m/z 195 [M − H]−, and m/z 165 [M − H]−, as shown in Fig. 4. The HMBC correlations of H-11 with C-9 and C-10 confirmed that the oxygenated methine was located next to the olefinic pair C-9/C-10 (Fig. 5), with the coupling constants of H-10 (dd, J = 15.0, 7.0 Hz) further confirming the position of C-11 next to C-10. The HMBC correlations of H-10/C-9 and C-8, H-9/C-10, C-8, and C-7, H-8/C-10, C-9, and C-7, and H-7/C-9, C-8, C-6, and C-5 further confirmed the locations of the remaining olefinic pairs (Fig. 5). The coupling constants of H-9 (dd, J = 15.0, 11.0 Hz), H-8 (t, J = 11.0 Hz), and H-7 (dt, J = 11.0, 7.5 Hz) confirmed the olefinic pairs C-7/C-8 as cis and C-9/C-10 as trans forms. To assign the absolute configuration of compound 5, a recently developed chemical-derivative method, CEA coupled with LC/MS analysis,38 was used. Using HBTM catalysts, the reaction rates of the parallel reactions were compared by LC/MS. For each parallel acylation reaction, two sets of 5 (each 0.2 mg) and S- and R-HBTM catalysts (each 0.1 mg) were used. Samples of each reaction were quantitatively analyzed by LC/MS for the measurement of the reaction rate catalyzed by S- and R-HBTM. As a result of the CEA reaction, the acylated derivative (5A, [M + H]+ peak at m/z 323), esterified by propionic anhydride in the hydroxyl group at C-11, was expected (Fig. 6). This was directly observed in LC/MS analysis, in which the peak for the anticipated derivative was detected in samples of both parallel reactions. These data indicated that the esterification reaction with S-HBTM was faster than that of R-HBTM, by comparing the peak areas of the acylated derivatives (Fig. 6 and S20†), which suggested compound 5 as (11R)-hydroxyhexadeca-(2E,7Z,9E)-trienoic acid (Fig. 6).32 We further referred to properties of chemical substances with similar chemical equivalence as compound 5. The values of specific rotation were compared, [α]25D +17.9° for compound 5 and [α]25D +13° for methyl-(10R)-hydroxyhexadeca-(7Z,11E,13Z)-trienoate,33 and confirmed compound 5 as (11R)-hydroxyhexadeca-(2E,7Z,9E)-trienoic acid. Thus, the structure of compound 5 was completely assigned as shown in Fig. 2.
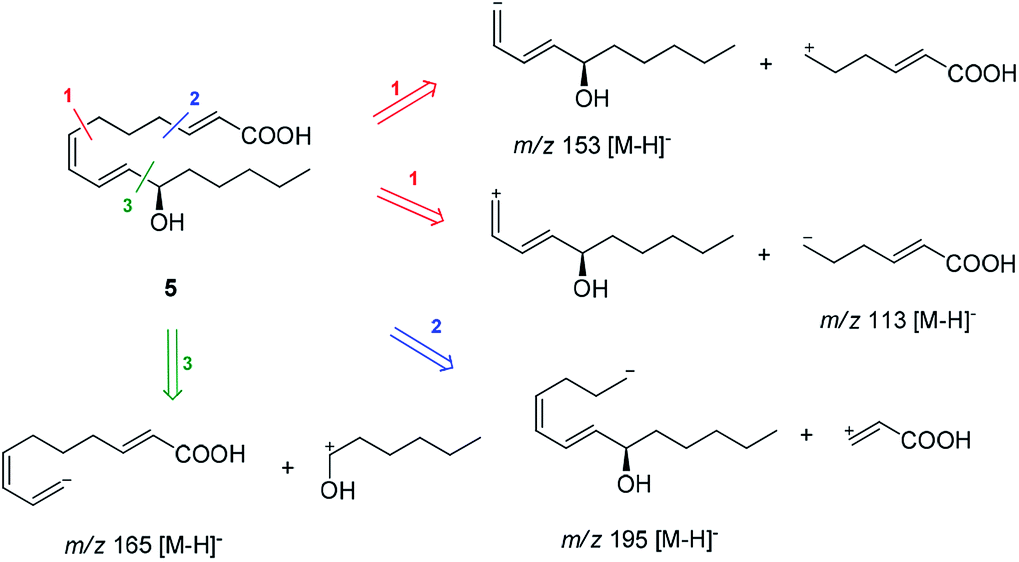 | ||
| Fig. 4 MS/MS analysis of compound 5 (MS2 265 → full-scan). | ||
 | ||
| Fig. 5 1H–1H COSY (blue bold lines) and key HMBC (red arrows) correlations for compounds 5 and 10–11. | ||
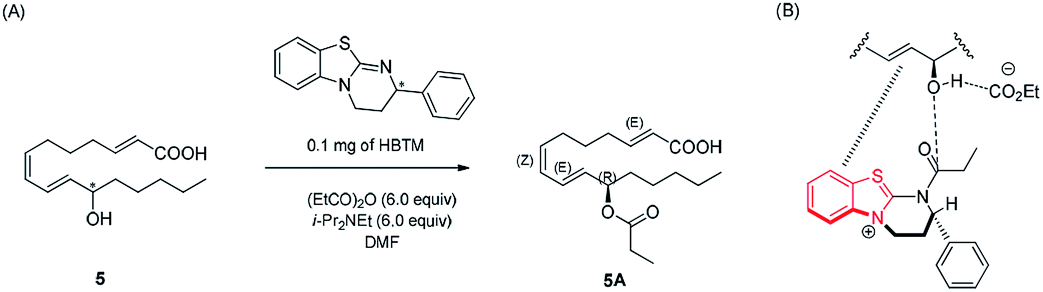 | ||
| Fig. 6 (A) CEA reaction for the determination of absolute configuration of compound 5. (B) Proposed favorable transition state of compound 5 in the reaction. | ||
Compound 10 was isolated as an amorphous gum. The molecular formula was established as C11H16O4 from the molecular ion peak [M − H]− at m/z 211.0962 (calcd for C11H15O4, 211.0970) in the negative-ion mode of HR-ESIMS. The IR spectrum displayed absorption bands of hydroxy (3341 cm−1) and aromatic (1450 cm−1) functional groups. The 1H NMR spectrum of 10 (Table 2) showed signals for a methyl group at δH 1.38 (3H, d, J = 6.5 Hz), a methoxy group at δH 3.19 (3H, s), two relatively deshielded methoxy groups at δH 3.84 (6H, s), an oxygenated methine at δH 4.25 (1H, q, J = 6.5 Hz), and two aromatic methines at δH 6.59 (2H, s). The 13C NMR data showed a total of 11 carbon signals (Table 2), comprised of a methyl, three methoxy, an oxygenated methine, and six aromatic carbons. The methyl group showed HMBC correlations with C-1 and C-1′, and the chemical shifts of H-1 (δH 4.25) and H-2 (δH 1.38) and their coupling constant (J = 6.5 Hz) suggested the presence of an oxygenated ethyl moiety attached to the aromatic ring. The HMBC correlations of OMe-1/C-1 revealed the attachment of a methoxy group at C-1 (Fig. 5). Furthermore, the HMBC correlations of H-1/C-2′ and C-6′ were observed, along with that of H-2′/C-1, C-1′, C-3′, and C-4′ and H-6′/C-1, C-1′, C-5′, and C-4′. Additionally, the HMBC correlations of the two relatively deshielded methoxy groups each with C-3′ and C-5′ confirmed the location of the methoxy groups attached to the aromatic ring (Fig. 5). The structure of compound 10 was determined to be 2,6-dimethoxy-4-[1-methoxyethyl]-phenol. The planar structure of compound 10 as well as its S isomer, 2,6-dimethoxy-4-[(1S)-1-methoxyethyl]-phenol, was previously reported.34 The values of specific rotation were compared, −67.0° for the S isomer and +34.5° for compound 10, and indicated that these are stereoisomers. Thus, quantum chemical ECD calculations, which is one of the most widely used methods for determining chirality,35 were performed to confirm the stereochemistry. As shown in Fig. 7, the calculated ECD curves of 10a (1R) and 10b (1S) show apparent opposite Cotton effects at 227 nm and the experimental ECD curve of 10 matched well with the calculated curve of 10a. Thus, compound 10 was confirmed to be 2,6-dimethoxy-4-[(1R)-1-methoxyethyl]-phenol (Fig. 2).
| Position | 10 | 11 | ||
|---|---|---|---|---|
| δH | δC | δH | δC | |
| a Signal multiplicity and coupling constants (Hz) are in parentheses. The assignments were based on HSQC, HMBC, and 1H–1H COSY experiments; NMR data were measured using NMR spectrometers operating at 850 MHz for 10 and 800 MHz for 11. | ||||
| 1 | 4.25 q (6.5) | 80.8 | 3.96 t (6.5) | 85.7 |
| 2 | 1.38 d (6.5) | 23.8 | 1.61 m, 1.78 m | 30.4 |
| 3 | 0.85 t (7.5) | 9.2 | ||
| 1′ | 134.9 | 132.5 | ||
| 2′ | 6.59 s | 104.2 | 6.55 s | 103.5 |
| 3′ | 149.4 | 147.8 | ||
| 4′ | 135.8 | 134.6 | ||
| 5′ | 149.4 | 147.8 | ||
| 6′ | 6.59 s | 104.2 | 6.55 s | 103.5 |
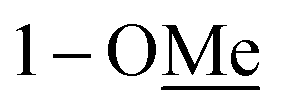 |
3.19 s | 56.1 | 3.18 s | 55.2 |
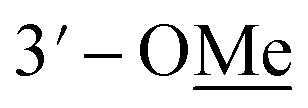 |
3.84 s | 56.3 | 3.83 s | 55.3 |
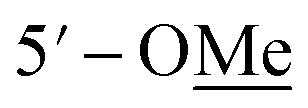 |
3.84 s | 56.3 | 3.83 s | 55.3 |
 | ||
| Fig. 7 Comparison of the experimental and calculated ECD spectra of 10 and 11. | ||
Compound 11 was isolated as an amorphous gum. The molecular formula was established as C12H18O4 from the molecular ion peak [M + Na]+ at m/z 249.1102 (calcd for C12H18O4Na, 249.1103) in the positive-ion mode of HR-ESIMS. The IR spectrum showed absorption bands of hydroxy (3397 cm−1) and aromatic (1448 cm−1) functional groups. The 1H and 13C NMR data of 11 (Table 2) were nearly identical to those of compound 10, except for the signals for the alkyl chain, in which the proton signal of the oxygenated methine of 11 was triplet (δH 3.96 (1H, t, J = 6.5 Hz)) instead of quartet, and the signal for the methyl group was also triplet (δH 0.85 (3H, t, J = 7.5 Hz)). Consistent with the above evidence, an extra carbon signal in the alkyl chain region, a methylene carbon (δC 30.4), was observed in HSQC spectrum. This was confirmed by other 2D NMR including HMBC and 1H–1H COSY (Fig. 5). The HMBC correlations of H-1/C-1′, C-2′, C-6′, C-2 and C-3, H-2/C-1′, C-1 and C-3, and H-3/C-1 and C-2 were observed, and the cross-peaks between H-1/H-2/H-3 in 1H–1H COSY spectrum confirmed the existence of a propyl chain (Fig. 5). The planar structure of compound 11 was previously reported as a synthetic product,36 however the absolute configuration at C-1 was not determined. To confirm the absolute configuration, the ECD spectrum of 11 was compared with the computed ECD spectra of 10a and 10b. The ECD curve of compound 11 exhibited the identical pattern with those of 10 and 10a (Fig. 7), and the specific rotation of 11 ([α]25D +17.5°) was comparable to that of compound 10 ([α]25D +34.5°), which led to the assignment of the absolute configuration as R. Accordingly, compound 11 was elucidated to be 2,6-dimethoxy-4-[(1R)-1-methoxypropyl]-phenol (Fig. 2).
By comparing NMR spectroscopic and physical data with previously reported data, along with LC/MS analysis, the known compounds were identified as (2E)-1-acetate 2-dodecen-1-ol (3),37 (2E,8E)-2,8-decadiene-1,10-diol (4),38 (9S,10E,12Z)-9-hydroxy-10,12-octadecadienoic acid (6),39 (9S,6Z,10E,12Z)-9-hydroxy-6,10,12-octadecatrienoic acid (7),40 (2E)-2-dodecenoic acid (8),41 (2E)-2-tetradecenoic acid (9),42 2,6-dimethoxy-phenol (12),43 2,6-dimethoxy-4-methyl-phenol (13),44 2,6-dimethoxy-4-(methoxymethyl)-phenol (14),45 2,6-dimethoxy-4-(2-propen-1-yl)-phenol (15),46 1-(4-hydroxy-3,5-dimethoxyphenyl)-1-propanone (16),47 3,3′,5,5′-tetramethoxy-[1,1′-biphenyl]-4,4′-diol (17),48 4-hydroxy-3-methoxy-benzaldehyde (18),49 1-(4-hydroxy-3-methoxyphenyl)-ethanone (19),50 1-(4-hydroxy-3-methoxyphenyl)-1-propanone (20),51 3,4-dimethoxy-benzoic acid (21),52 8-hydroxy-2,6-dimethyl-1,6-octadien-3-one (22),53 (2E,6E)-8-(acetyloxy)-2,6-dimethyl-2,6-octadienal (23),54 (3S,6E)-3,7,11-trimethyl-1,6,10-dodecatrien-3-ol (24),55 and (3E)-4-[(1S,4aS,8aS)-decahydro-5,5,8a-trimethyl-2-methylene-1-naphthalenyl]-3-buten-2-one (25).56
All of the isolated compounds (1–25) were tested for cell cytotoxicity in BV2 microglial cells, and no significant cell death was observed at the concentration of 10 μM of each compound (data not shown). Then, in order to identify the bioactive ingredients contributing to the SPHK1/2 inhibitory effects of A. tsaoko seeds, SPHK1/2 inhibition activity of the isolated compounds was evaluated at the concentration of 10 μM (Table 3). The results revealed that compounds 2–4, 7, 9, 13–15, and 21 inhibited the activity of SPHK1 up to 20% compared to the control. Among these compounds, compound 2 showed the most potent inhibition against SPHK1 enzymatic activity by approximately 40% in comparison with the control, with higher inhibition compared to the positive control, resveratrol. Our results indicate that the newly identified compound 2 possesses the highest potency in SPHK1 inhibition, indicating its potential usefulness in the treatment of various disorders. In the case of SPHK2, most of the compounds showed significant inhibition (Table 3). In particular, compounds 4, 8, 10, 11, and 18 inhibited SPHK2 activity by 40–50%, and compounds 2 and 22 showed the most potent inhibition, by approximately 70% compared to the control, with higher inhibition compared to the positive control, SKI-II (Table 3). Although the identification of compound 22 has been previously reported, the current study is the first to demonstrate its inhibitory effects against SPHK2 activity. Compounds 1, 2, and 4 are aliphatic alcohols sharing similar structures, with the only difference in the presence of acetyl groups. Comparison of the relative activity levels of these three compounds suggested that there might be a structure–activity relationship, in this case being the presence of acetyl groups. Both acetyl and hydroxyl groups at the end of the chains of these aliphatic alcohols appear to play a role in SPHK1/2 inhibition. Collectively, our results suggest that compound 2 (tsaokol A), a new compound with the highest potency to inhibit both SPHK1 and SPHK2, may be a promising candidate for further drug discovery as an SPHK1/2 inhibitor.
| Compound | SPHK1 activity (% of Ctl) | SPHK2 activity (% of Ctl) | Compound | SPHK1 activity (% of Ctl) | SPHK2 activity (% of Ctl) |
|---|---|---|---|---|---|
| a SPHK1 and SPHK2 enzyme activity inhibition assay performed with 10 μM of compounds. SPHK inhibition was calculated as percentage (%) of untreated control. The results are expressed as mean ± SD; resveratrol and SKI-II were used as positive controls. | |||||
| 1 | 81.89 ± 7.24 | 83.33 ± 12.35 | 15 | 70.19 ± 14.18 | 79.37 ± 11.22 |
| 2 | 59.75 ± 4.14 | 25.40 ± 4.49 | 16 | 100.97 ± 17.64 | 113.49 ± 10.10 |
| 3 | 74.09 ± 9.27 | 122.22 ± 14.55 | 17 | 96.11 ± 8.93 | 79.37 ± 13.47 |
| 4 | 72.14 ± 14.96 | 58.20 ± 6.61 | 18 | 111.35 ± 14.48 | 35.71 ± 3.37 |
| 5 | 98.89 ± 9.93 | 69.84 ± 11.00 | 19 | 101.53 ± 13.39 | 58.73 ± 17.96 |
| 6 | 81.34 ± 17.80 | 107.94 ± 17.96 | 20 | 85.79 ± 16.14 | 65.08 ± 4.49 |
| 7 | 73.54 ± 8.45 | 105.56 ± 21.33 | 21 | 74.09 ± 15.47 | 79.89 ± 24.41 |
| 8 | 92.91 ± 10.66 | 46.03 ± 2.24 | 22 | 82.94 ± 10.34 | 22.75 ± 2.42 |
| 9 | 71.73 ± 1.88 | 68.78 ± 13.50 | 23 | 92.62 ± 19.44 | 121.43 ± 10.10 |
| 10 | 86.63 ± 13.71 | 61.11 ± 21.33 | 24 | 91.64 ± 15.33 | 76.72 ± 18.60 |
| 11 | 106.96 ± 10.66 | 47.62 ± 2.24 | 25 | 99.86 ± 10.64 | 67.46 ± 19.08 |
| 12 | 86.28 ± 7.98 | 96.83 ± 17.96 | Resveratrol | 85.43 ± 3.82 | |
| 13 | 77.51 ± 6.20 | 72.49 ± 25.86 | SKI-II | 75.35 ± 9.64 | |
| 14 | 73.54 ± 21.97 | 116.67 ± 23.57 | |||
Polyunsaturated fatty acids showed anti-inflammatory potency via inhibition of SPHK1 in a cecal ligation and puncture-induced sepsis model.57 This study was in agreement with our results demonstrating that compounds 5–9 showed good potency to inhibit SPHK1 and/or SPHK2. Compounds 6 and 7 potently inhibited SPHK1, while compounds 5 and 8 inhibited SPHK2. Phenolic compounds, such as epigallocatechin-3-O-gallate (EGCG) and curcumin, are also reported to possess inhibitory activity against SPHK1.58,59 Our results are consistent with these studies, as the isolated phenolic compounds showed significant inhibition of sphingosine kinases. Among the phenolic compounds we identified, compounds 12, 14, 15, and 21 significantly inhibited SPHK1, while compounds 10, 11, 13, and 17–20 showed inhibition against SPHK2 enzymatic activity. Furthermore, most of the phenolic compounds showed higher potency in inhibiting SPHK2. Terpenoids are less known for their ability to inhibit sphingosine kinases and for their involvement in sphingolipid metabolism. Compound 22, a monoterpene, showed potent inhibition of SPHK1/2, particularly SPHK2. Collectively, these results demonstrate that the previously known compounds as well as the newly identified compounds, including compounds 2, 5, and 10–11, exhibit high ability to inhibit SPHK1/2.
To simulate the inhibition mechanism of 2 and 22, which showed the most potent inhibition against SPHK1/2, we constructed docking models of 2 and 22 in the active site of SPHK1/2. For molecular docking analysis of 2, the X-ray crystal structure of SPHK1 (PDB id: 3VZB)60 complexed with the substrate sphingosine was used for the receptor. Trans-resveratrol, which is known as a substrate-competitive SPHK1 inhibitor,61 was also docked into the sphingosine-binding pocket for comparison (Fig. 8A). For the docking of 22 and the reference molecule SKI-II, a homology model of SPHK2 was generated using SPHK1 as a template, which is highly consistent in amino acid sequences with SPHK2.
 | ||
| Fig. 8 Proposed docking pose of 2 and 22 in the active site of SPHK1 (PDB id: 3VZB) and SPHK2 (Homology model based on 3VZD) respectively. (A) Docking poses of 2 and resveratrol were overlapped with the X-ray pose of sphingosine in SPHK1. (B) Docking pose of 22 and SKI-II are shown in the substrate-binding site of SPHK2 homology model. Only hydrogen bonds with key residue (SPHK1: Asp178, SPHK2: Asp 308) were presented by red dotted. Carbon atoms are colored yellow (amino acid), magenta (2), cyan (22), silver (sphingosine), blue-green (resveratrol) and orange (SKI-II); nitrogen is blue; oxygen is red; hydrogen is light grey. | ||
The X-ray structure of SPHK1 complexed inhibitor revealed key interactions between SPHK1 and the inhibitor: (i) the hydrogen bond between hydrophilic group of the inhibitor and Asp178 of helix α7, (ii) hydrophobic interactions in the J-shaped pocket consisting of nonpolar residues (e.g. Phe192, Ile174, Met272, and Phe288).60 These key interactions facilitate the maintenance of the closed conformation of lipid gate consisting of helices α7 and α8, and then inhibit the binding of sphingosine within the substrate binding site of SPHK1. In the binding mode of 2 shown in Fig. 8A, terminal hydroxyl group of 2 forms a hydrogen bond with the side chain carboxylate group of Asp178, where the phenolic moiety of resveratrol and the 3-hydroxyl group of sphingosine also form hydrogen bonds. Compound 2 and resveratrol fit in the hydrophobic J-shaped pocket which is occupied by sphingosine in the X-ray structure. In the substrate-binding pocket of SPHK2, 22 and SKI-II (a reference inhibitor of SPHK2) bind to the hydrophobic pocket, forming hydrogen bonds with Asp304, a key residue corresponding to Asp178 in SPHK1 (Fig. 8B). Our docking results support the biological assay data identifying both 2 and 22 as potent inhibitors against SPHK1 and SPHK2, respectively, and suggest that they may act as substrate-competitive inhibitors.
Conclusion
In conclusion, our study demonstrates for the first time the SPHK1/2 inhibitory effects of A. tsaoko. Bioactivity-based analysis and chemical investigation of the EtOH extract of A. tsaoko seeds led to successful isolation of the active ingredients responsible for this inhibitory activity. Of note, our chemical investigation led to the identification of four new compounds, an acetylated aliphatic alcohol (2), a fatty acid (5), and two phenolics (10–11). Compound 1 was isolated for the first time from natural sources. Among the isolated compounds, aliphatic alcohols 2–4, fatty acids 7 and 9, and phenolic compounds 13–15 and 21 inhibited the activity of SPHK1 up to 20%, each at the concentration of 10 μM, and compound 2 exhibited the most potent inhibition against SPHK1 enzymatic activity (by 59.75% compared to the control) with higher inhibition compared to the positive control, resveratrol. Aliphatic alcohol 4, fatty acid 8, and phenolic compounds 10, 11, and 18 inhibited approximately SPHK2 activity by 40–50% and compounds 2 and 22 showed the highest potency in inhibiting SPHK2 activity by approximately 70% compared with the control group. Both compounds showed inhibition higher than that of the positive control, SKI-II. The docking studies supported the biological data for both 2 and 22 as potent inhibitors against SPHK1 and SPHK2, respectively. Although SPHK1/2 inhibition by A. tsaoko and its bioactive constituents need to be further validated, our findings provide experimental evidence supporting the potential application of A. tsaoko for inhibition of cancer cell proliferation and growth or in other diseases involving sphingosine metabolisms.Experimental section
General experimental procedures
Optical rotations were measured using a JASCO P-2000 polarimeter (JASCO, Easton, MD, USA). Ultraviolet (UV) spectra were acquired on an Agilent 8453 UV-visible spectrophotometer (Agilent Technologies, Santa Clara, CA, USA). Electronic circular dichroism (ECD) spectra were measured on a JASCO J-1500 spectropolarimeter (JASCO). Infrared (IR) spectra were recorded on an IFS-66/s FT-IR spectrometer (Bruker, Karlsruhe, Germany). Nuclear magnetic resonance (NMR) spectra were measured using a Bruker AVANCE III (Bruker). High-resolution (HR)-electrospray ionization (ESI) mass spectra were recorded on Waters Xevo G2 QTOF mass spectrometer and Synapt G2 HDMS quadrupole time-of-flight (TOF) mass spectrometer (Waters Corp., Milford, CT, USA). Tandem mass spectrometry (MS/MS) was performed using Acquity UPLC™ system (Waters) coupled to a Waters Acquity Xevo G2 Q-TOF system (Waters). Preparative high-performance liquid chromatography (HPLC) was conducted using a Waters 1525 binary HPLC pump with Waters 996 photodiode array detector (Waters) and an Agilent Eclipse C18 column (250 × 21.2 mm, 5 μm; flow rate: 5 mL min−1) (Agilent Technologies). Semi-preparative HPLC was conducted with a Shimadzu Prominence HPLC System with SPD-20A/20AV series Prominence HPLC UV-vis detectors (Shimadzu, Tokyo, Japan) and a Phenomenex Luna C18 column (250 × 10 mm, 5 μm; flow rate: 2 mL min−1) (Phenomenex, Torrance, CA, USA). LC/MS analysis was performed on an Agilent 1200 series HPLC system with a diode array detector and 6130 Series ESI mass spectrometer using an analytical Kinetex C18 100 Å column (100 mm × 2.1 mm i.d., 5 μm; flow rate: 0.3 mL min−1) (Phenomenex). Silica gel 60 (230–400 mesh; Merck, Darmstadt, Germany), RP-C18 silica gel (Merck, 230-400 mesh) and silica Sep-Pak Vac 6 cc cartridges (Waters) were used for column chromatography. The packing material for molecular sieve column chromatography was Sephadex LH-20 (Pharmacia, Uppsala, Sweden). Thin-layer chromatography (TLC) was performed with precoated silica gel F254 plates and RP-C18 F254s plates (Merck) and spots were detected under UV light or by heating after spraying with anisaldehyde–sulfuric acid. The 3D molecular modeling was performed using ChemBioDraw Ultra and Avogadro.Chemicals and reagents
All reactions were conducted in capped vials, under dimethylformamide (DMF) as an organic solvent, with air, at room temperature. All solvents and reagents, including DMF, N,N-diisopropylethylamine, and propionic anhydride, were freshly dried and distilled. The S- and R-homobenzotetramisole (HBTM) organic catalysts were purchased from Sigma-Aldrich (St. Louis, MO, USA). Fetal bovine serum (FBS), penicillin–streptomycin (PS), Dulbecco's modified Eagle's medium (DMEM), and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) powder were purchased from Invitrogen (Carlsbad, CA, USA). SK I-II and resveratrol were purchased from Sigma Aldrich.Sample material
Dried A. tsaoko seeds were purchased from Hanherb (Gyeonggi-do, Korea) and were identified by one of the authors (S. Y. Kim). A voucher specimen (SKKU CG-2017-07) has been deposited in the herbarium of the School of Pharmacy, Sungkyunkwan University, Suwon, Korea.Extraction and isolation of compounds
Dried A. tsaoko seeds (1.0 kg) were extracted with 50% EtOH at room temperature (24 h × 3 times). The EtOH extract was filtered and concentrated using a rotary evaporator. The lyophilized powder was kept in the refrigerator at −4 °C until use. The resulting powder (50.0 g) was suspended in distilled water (700 mL) and MeOH (30 mL). The suspension was further subjected to solvent partition using hexane, which yielded HX-soluble and H2O-soluble fractions.The HX-soluble fraction (2.1 g) was fractionated using silica gel column chromatography with a gradient solvent system of hexane/EtOAc (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 → 1:1), yielding 9 subfractions (H1–H9). Subfraction H1 (132.1 mg) was purified by preparative HPLC (MeOH/H2O, 80:20 → 100:0) to afford 6 subfractions (H11–H16). Subfraction H15 (18.4 mg) was purified using silica Sep-Pak with the gradient solvent system of hexane/EtOAc (100:1 → 1:1) to yield compound 3 (2.2 mg). Subfraction H2 (91.6 mg) was subjected to RP-C18 column eluted with the gradient solvent system of MeOH/H2O (80:20 → 100:0), which afforded 6 subfractions (H21–H26). Subfraction H25 (23.9 mg) was purified using silica Sep-Pak with the gradient solvent system of CH2Cl2/MeOH (100:1 → 1:1) and yielded compound 24 (0.8 mg). Subfraction H26 (19.5 mg) was purified using semi-preparative HPLC (MeOH/H2O, 82:18) to furnish compound 25 (tR 38.0 min, 0.4 mg). Subfraction H4 (435.2 mg) was fractionated with preparative HPLC (MeOH/H2O, 60:40 → 80:20), which afforded 8 subfractions (H41–H48). Subfraction H46 (30.5 mg) was purified using semi-preparative HPLC (MeCN/H2O, 57:43) to afford compound 1 (tR 22.0 min, 5.1 mg). Subfraction H5 (145.3 mg) was separated with preparative HPLC (MeOH/H2O, 60:40 → 100:0) to yield 6 subfractions (H51–H56). Subfraction H51 (7.7 mg) was purified using semi-preparative HPLC (MeOH/H2O, 55:45), and compounds 16 (tR 19.5 min, 0.4 mg) and 11 (tR 36.5 min, 0.3 mg) were obtained. Subfraction H52 (5.7 mg) was also purified using semi-preparative HPLC with the solvent system of MeOH/H2O (63:37) to give compound 23 (tR 31.0 min, 0.8 mg), and subfraction H56 (43.8 mg) was purified with MeOH/H2O (83:17, 0.01% formic acid, v/v) to give compound 9 (tR 27.0 min, 3.1 mg). Subfraction H6 (118.8 mg) was fractionated with preparative HPLC (MeOH/H2O, 55:45 → 100:0) to yield 6 subfractions (H61–H66). Subfraction H61 (19.5 mg) was purified using semi-preparative HPLC (MeOH/H2O, 48:52) and compounds 14 (tR 18.5 min, 1.1 mg), 12 (tR 21.0 min, 4.1 mg), 10 (tR 24.0 min, 0.5 mg), 21 (tR 27.5 min, 1.0 mg), 13 (tR 30.5 min, 0.9 mg), and 17 (tR 33.0 min, 1.1 mg) were obtained. Subfraction H62 (5.7 mg) was also purified using semi-preparative HPLC, with the solvent system of MeOH/H2O (56:44) to give compound 15 (tR 36.0 min, 0.8 mg). Subfraction H7 (220.4 mg) was separated with preparative HPLC (MeOH/H2O, 60:40 → 100:0) to give 6 subfractions (H71–H76). Subfraction H71 (24.4 mg) was purified using semi-preparative HPLC (MeCN/H2O, 20:80) to yield compounds 18 (tR 22.0 min, 2.3 mg), 19 (tR 24.5 min, 1.3 mg), and 20 (tR 42.0 min, 1.8 mg). Subfraction H75 (27.6 mg) was also purified using semi-preparative HPLC with the solvent system of MeOH/H2O (80:20, 0.01% formic acid, v/v) to give compound 8 (tR 39.0 min, 6.7 mg). Subfraction H8 (490.9 mg) was fractionated with preparative HPLC (MeOH/H2O, 50:50 → 100:0) and 6 subfractions were obtained (H81–H86). Subfraction H83 (34.5 mg) was purified using semi-preparative HPLC (MeOH/H2O, 44:56, 0.01% formic acid, v/v) to yield compounds 22 (tR 36.0 min, 0.8 mg) and 4 (tR 38.0 min, 1.5 mg). Subfraction H84 (175.2 mg) was also purified using semi-preparative HPLC with the solvent system of MeOH/H2O (60:40), which yielded two subfractions (H841–H842). Subfraction H841 (56.5 mg) was further purified using semi-preparative HPLC (MeOH/H2O, 60:40) to give compound 2 (tR 36.5 min, 8.2 mg). Subfraction H85 (55.1 mg) was also purified using semi-preparative HPLC with the solvent system of MeCN/H2O (47:53), which yielded compound 7 (tR 50.0 min, 1.2 mg). Subfraction H9 (555.3 mg) was subjected to RP-C18 column eluted with MeOH/H2O (50:50 → 100:0), and 8 subfractions were obtained (H91–H98). Subfraction H96 (26.6 mg) was purified using semi-preparative HPLC (MeOH/H2O, 67:33) to give compound 5 (tR 52.0 min, 1.4 mg). Subfraction H97 (53.5 mg) was separated on a Sephadex LH-20 column using a solvent system of CH2Cl2/MeOH (20:80), and two fractions were obtained (H971–H972). Fraction H972 (16.7 mg) was purified using semi-preparative HPLC (MeOH/H2O, 72:28) and compound 6 (tR 60.0 min, 1.6 mg) was obtained.
:1 → 1:1), yielding 9 subfractions (H1–H9). Subfraction H1 (132.1 mg) was purified by preparative HPLC (MeOH/H2O, 80:20 → 100:0) to afford 6 subfractions (H11–H16). Subfraction H15 (18.4 mg) was purified using silica Sep-Pak with the gradient solvent system of hexane/EtOAc (100:1 → 1:1) to yield compound 3 (2.2 mg). Subfraction H2 (91.6 mg) was subjected to RP-C18 column eluted with the gradient solvent system of MeOH/H2O (80:20 → 100:0), which afforded 6 subfractions (H21–H26). Subfraction H25 (23.9 mg) was purified using silica Sep-Pak with the gradient solvent system of CH2Cl2/MeOH (100:1 → 1:1) and yielded compound 24 (0.8 mg). Subfraction H26 (19.5 mg) was purified using semi-preparative HPLC (MeOH/H2O, 82:18) to furnish compound 25 (tR 38.0 min, 0.4 mg). Subfraction H4 (435.2 mg) was fractionated with preparative HPLC (MeOH/H2O, 60:40 → 80:20), which afforded 8 subfractions (H41–H48). Subfraction H46 (30.5 mg) was purified using semi-preparative HPLC (MeCN/H2O, 57:43) to afford compound 1 (tR 22.0 min, 5.1 mg). Subfraction H5 (145.3 mg) was separated with preparative HPLC (MeOH/H2O, 60:40 → 100:0) to yield 6 subfractions (H51–H56). Subfraction H51 (7.7 mg) was purified using semi-preparative HPLC (MeOH/H2O, 55:45), and compounds 16 (tR 19.5 min, 0.4 mg) and 11 (tR 36.5 min, 0.3 mg) were obtained. Subfraction H52 (5.7 mg) was also purified using semi-preparative HPLC with the solvent system of MeOH/H2O (63:37) to give compound 23 (tR 31.0 min, 0.8 mg), and subfraction H56 (43.8 mg) was purified with MeOH/H2O (83:17, 0.01% formic acid, v/v) to give compound 9 (tR 27.0 min, 3.1 mg). Subfraction H6 (118.8 mg) was fractionated with preparative HPLC (MeOH/H2O, 55:45 → 100:0) to yield 6 subfractions (H61–H66). Subfraction H61 (19.5 mg) was purified using semi-preparative HPLC (MeOH/H2O, 48:52) and compounds 14 (tR 18.5 min, 1.1 mg), 12 (tR 21.0 min, 4.1 mg), 10 (tR 24.0 min, 0.5 mg), 21 (tR 27.5 min, 1.0 mg), 13 (tR 30.5 min, 0.9 mg), and 17 (tR 33.0 min, 1.1 mg) were obtained. Subfraction H62 (5.7 mg) was also purified using semi-preparative HPLC, with the solvent system of MeOH/H2O (56:44) to give compound 15 (tR 36.0 min, 0.8 mg). Subfraction H7 (220.4 mg) was separated with preparative HPLC (MeOH/H2O, 60:40 → 100:0) to give 6 subfractions (H71–H76). Subfraction H71 (24.4 mg) was purified using semi-preparative HPLC (MeCN/H2O, 20:80) to yield compounds 18 (tR 22.0 min, 2.3 mg), 19 (tR 24.5 min, 1.3 mg), and 20 (tR 42.0 min, 1.8 mg). Subfraction H75 (27.6 mg) was also purified using semi-preparative HPLC with the solvent system of MeOH/H2O (80:20, 0.01% formic acid, v/v) to give compound 8 (tR 39.0 min, 6.7 mg). Subfraction H8 (490.9 mg) was fractionated with preparative HPLC (MeOH/H2O, 50:50 → 100:0) and 6 subfractions were obtained (H81–H86). Subfraction H83 (34.5 mg) was purified using semi-preparative HPLC (MeOH/H2O, 44:56, 0.01% formic acid, v/v) to yield compounds 22 (tR 36.0 min, 0.8 mg) and 4 (tR 38.0 min, 1.5 mg). Subfraction H84 (175.2 mg) was also purified using semi-preparative HPLC with the solvent system of MeOH/H2O (60:40), which yielded two subfractions (H841–H842). Subfraction H841 (56.5 mg) was further purified using semi-preparative HPLC (MeOH/H2O, 60:40) to give compound 2 (tR 36.5 min, 8.2 mg). Subfraction H85 (55.1 mg) was also purified using semi-preparative HPLC with the solvent system of MeCN/H2O (47:53), which yielded compound 7 (tR 50.0 min, 1.2 mg). Subfraction H9 (555.3 mg) was subjected to RP-C18 column eluted with MeOH/H2O (50:50 → 100:0), and 8 subfractions were obtained (H91–H98). Subfraction H96 (26.6 mg) was purified using semi-preparative HPLC (MeOH/H2O, 67:33) to give compound 5 (tR 52.0 min, 1.4 mg). Subfraction H97 (53.5 mg) was separated on a Sephadex LH-20 column using a solvent system of CH2Cl2/MeOH (20:80), and two fractions were obtained (H971–H972). Fraction H972 (16.7 mg) was purified using semi-preparative HPLC (MeOH/H2O, 72:28) and compound 6 (tR 60.0 min, 1.6 mg) was obtained.
ε) = 200 (3.7) nm; IR (KBr) νmax: 3385, 2944, 2832, 1620, 1029 cm−1; (+)-HR-ESIMS m/z 277.1414 [M + Na]+ (calcd for C14H22O4Na, 277.1416); for 1H and 13C NMR spectroscopic data, see Table 1.ε) = 205 (3.6) nm; IR (KBr) νmax: 3408, 2946, 2832, 1680, 1027 cm−1; (+)-HR-ESIMS m/z 235.1310 [M + Na]+ (calcd for C12H20O3Na, 235.1310); for 1H and 13C NMR spectroscopic data, see Table 1.ε) = 225 (3.9) nm; IR (KBr) νmax: 3415, 2942, 2836, 1720, 1029 cm−1; (−)-HR-ESIMS m/z 265.1795 [M − H]− (calcd for C16H25O3, 265.1804); for 1H and 13C NMR spectroscopic data, see Table 1.ε) = 210 (3.9) nm; ECD (MeOH) λmax (Δε) = 227 (−61.5) nm; IR (KBr) νmax: 3341, 2944, 2824, 1450, 1021 cm−1; (−)-HR-ESIMS m/z 211.0962 [M − H]− (calcd for C11H15O4, 211.0970); for 1H and 13C NMR spectroscopic data, see Table 2.ε) = 210 (3.9) nm; ECD (MeOH) λmax (Δε) = 229 (−319.5) nm; IR (KBr) νmax: 3397, 2942, 2828, 1448, 1029 cm−1; (+)-HR-ESIMS m/z 249.1102 [M + Na]+ (calcd for C12H18O4Na, 249.1103); for 1H and 13C NMR spectroscopic data, see Table 2.MS/MS analysis of compound 5
Stock solution of compound 5 was prepared by dissolving 0.1 mg of sample in 200 μL DMSO. The solution was further diluted with DMSO, filtered through a 0.2 μm hydrophobic PTFE filter, and finally analyzed by LC/MS/MS, Acquity UPLC™ system (Waters) coupled to a Waters Acquity Xevo G2 Q-TOF system (Waters). Analysis was performed by injection of 5 μL of the sample using an Agilent ZORBAX Eclipse Plus C18 column (100 mm × 2.1 mm, 1.8 μm) set at 30 °C. The mobile phase consisting of formic acid in H2O [0.3% (v/v)] (A) and formic acid in acetonitrile [0.3% (v/v)] (B) was delivered at a flow rate of 0.2 mL min−1 by applying the following programmed gradient elution: 0–15.0 min, 10–30% (B); 15.0–15.5 min, 30–100% (B); 15.5–20.0 min, 100% (B); 20.0–20.5 min, 100–10% (B); 20.5–25.5 min, 10% (B). The MS system was equipped with ESI source and operated in both negative and positive ionization mode with data acquiring range from 100 to 600 m/z.Experimental procedure to determine the absolute configuration of 5
ECD calculation
To obtain the conformational differences between 10a (1R) and 10b (1S), computational density-functional-theory (DFT) calculations were carried out. The first structural-energy minimizations of 10a and 10b were performed using Avogadro 1.2.0 with the UFF force field. The ground state geometries of 10a and 10b were established by Tmolex 4.3.1 with the DFT settings (B3-LYP functional, M3 grid size), geometry-optimization options (energy: 10−6 Eh, gradient norm: |dE/dxyz| = 10−3 Eh/b), and the basis set def-SV(P) for all atoms. The calculated ECD spectra of the optimized structures of 10a and 10b were acquired at the B3LYP/DFT functional settings with the basis set def-TZVPP for all atoms. The obtained CD spectra were simulated by overlying each transition (eqn (1)), where σ is the width of the band at 1/e height and ΔEi and Ri are the excitation energies and rotatory strengths for transition i, respectively. In the present study, the value of σ was 0.10 eV.
 | (1) |
Cell cytotoxicity
MTT assay was performed to analyze the cytotoxicity of the isolated compounds. The experiment was performed as described previously with slight modification.62 Briefly, BV2 microglial cells were seeded at the density of 4 × 104 cells per well in a 96 well plate and incubated overnight. The cells were treated with the compounds for 24 h, then the conditioned medium from the treated cells were removed and the cells were incubated with MTT solution (0.5 mg mL−1) for approximately 1 h with complete light protection. MTT solution was removed and the live cells stained blue with MTT solution were exposed with 200 μL of DMSO, which resulted in purple formazan coloured solution. The change in colour was analysed spectrophotometrically by measuring absorbance at 570 nm in the microplate reader (Molecular Devices; San Jose, CA, USA).SPHK1/2 activity inhibition assay
High throughput screening of SPHK inhibition was performed in a 384-well plate. A 5× concentration of each extract (500 μg mL−1) and compounds (50 μM) were prepared in distilled water (DW), resulting in final concentrations as 100 μg mL−1 for extract and 10 μM for compound. DMSO at 5% was used as a control. First, 2 μL of 5× compound, 2 μL of 5X reaction buffer, and 2 μL of 5× sphingosine kinase solution was mixed in each well and pre-incubated for 30 min. We used 2X ATP/ADP as a standard. Next, 2 μL of 5× sphingosine solution and 2 μL of 5× ATP solution was added in each well, except the standard well, and the reaction was started and incubated for 2 h. When the sphingosine substrate comes in contact with ATP in the presence of SPHK1 or SPHK2, S1P is produced together with adenosine diphosphate (ADP). The ADP2 FI assay detection reagents kit was then used for the detection of fluorescence based on ADP. The fluorescence intensity determines the overall level of ADP that is converted with the treatment of compounds, which eventually determines the effect of compounds in inhibiting the activity of SPHK1/2. Detection solution was prepared according to the manufacturer's instructions, and 10 μL was added to each well, followed by incubation at room temperature for 1 h. The fluorescence intensity was measured at 590 nm excitation and 620 nm emission. The amount of SPHK1/2 was calculated as percentage of control.Homology modeling of SPHK2
Since an X-ray crystal structure of SPHK2 has not been published, we built a SPHK2 homology model using homology modeling module in Schrodinger's Maestro 11.6. First, 618 amino acid sequences of human SPHK2 (Accession number: AAH10671.1) were downloaded from the NCBI protein database (https://www.ncbi.nlm.nih.gov/protein). To find a template within the NCBI PDB database, BLAST searching module was used and SPHK1 (PDB id: 3VZD) was detected which showed 58% identity with SPHK2. Amino acid sequences of SPHK2 were aligned with those of human SPHK1 by the Multiple Alignment method and the knowledge-based homology modeling was applied for building a homology model of SPHK2 (Fig. S31†).Molecular docking analyses of 2 and 22 on SPHK1/2
The computational docking was carried out on the Schrodinger's Maestro 11.6 software with Window 10 professional K operating system. First, structures of compounds (2, 22, resveratrol, and SKI-II) were sketched using 2D sketch module of Schrodinger's Maestro 11.6 and several tautomers and ionized states of ligands were generated by the ligprep module. OPLS3e force field was applied to optimize the structure of ligand. Prior to docking performance, the X-ray crystal structure of SPHK1 (PDB id: 3VZB) having 2 Å resolution was downloaded from the Protein Data Bank (http://www.rcsb.org). Sequentially, receptor proteins (SPHK1 and SPHK2 homology model) were prepared by assigning bond orders, removing original hydrogens, generating het states, removing duplicated chains and water molecules, and minimizing in the OPLS3e force field using the protein preparation wizard module. Compounds 2 and 22 were docked into the substrate binding site of SPHK1 (PDB id: 3VZB) and SPHK2 homology model, respectively. The reference molecules including sphingosine, resveratrol, and SKI-II, were also docked under the same condition for comparison. The binding site for SPHK1 was defined by a cube grid box based on a native ligand (sphingosine), and that for SPHK2 was defined by selecting all the amino acid residues in the substrate binding pocket. Glide/XP dockings were performed by default parameters and up to 10 conformers per ligand were generated. Binding affinity of each docking pose of ligand was calculated by Glide-Dock score. To select a plausible docking model from the docking output, not only binding energy but also intermolecular interactions (H-bond, π–π interaction) between key residues and docking poses of control were also considered by visual inspection.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (2018R1A2B2006879) and the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (NRF-2014M3A9B6069338). We would like to thank Dr Young Hye Kim (KBSI) for the ESIMS analysis. This research was supported by the Bio & Medical Technology Development Program of the National Research Foundation of Korea (NRF) funded by the Korean government (MSIT) (NRF-2012M3A9C4048775). This work was also supported by the Nano Convergence Industrial Strategic Technology Development Program (20000105, Development of Cosmeceutical Material Platform using Organo-Nano Complexes based on Natural Active Compounds) funded by the Ministry of Trade, Industry & Energy (MOTIE, Korea).Notes and references
- S. Borodzicz, K. Czarzasta, M. Kuch and A. Cudnoch-Jedrzejewska, Lipids Health Dis., 2015, 14, 55 CrossRef.
- M. Maceyka, K. B. Harikumar, S. Milstien and S. Spiegel, Trends Cell Biol., 2012, 22, 50–60 CrossRef CAS.
- D. Shida, K. Takabe, D. Kapitonov, S. Milstien and S. Spiegel, Curr. Drug Targets, 2008, 9, 662–673 CrossRef CAS.
- E. Lee, J. Jung, D. Jung, C. S. Mok, H. Jeon, C. S. Park, W. Jang and Y. Kwon, Anti Cancer Agents Med. Chem., 2017, 17, 1689–1697 CAS.
- L. Xu, L. Jin, B. Yang, L. Wang, Z. Xia, Q. Zhang and J. Xu, Oncotarget, 2017, 19, 2384–2394 Search PubMed.
- B. Ogretmen and Y. A. Hannun, Nat. Rev. Cancer, 2004, 4, 604–616 CrossRef CAS.
- S. Spiegel and S. Milstien, Nat. Rev. Mol. Cell Biol., 2003, 4, 397–407 CrossRef CAS.
- L. Bryan, T. Kordula, S. Spiegel and S. Milstien, Biochim. Biophys. Acta, 2008, 1781, 459–466 CrossRef CAS PubMed.
- L. Dai, Y. Qi, J. Chen, D. Kaczorowski, W. Di, W. Wang and P. Xia, Mol. Endocrinol., 2013, 28, 197–207 CrossRef.
- A. Di Pardo and V. Maglione, Front. Neurosci., 2018, 12, 249 CrossRef.
- M. Aoki, H. Aoki, R. Ramanathan, N. C. Hait and K. Takabe, Mediators Inflammation, 2016, 2016, 8606878 Search PubMed.
- M. Dai, C. Peng, F. Peng, C. Xie, P. Wang and F. Sun, Pharm. Biol., 2015, 54, 445–450 CrossRef.
- X. Yang, P. Küenzi, I. Plitzko, O. Potterat and M. Hamburger, Planta Med., 2009, 75, 543–546 CrossRef CAS.
- S. S. Hong, J. H. Lee, Y.-H. Choi, W. Jeong, E.-K. Ahn, S. H. Lym and J. S. Oh, Tetrahedron Lett., 2015, 56, 6681–6684 CrossRef CAS.
- S.-S. Moon, J.-Y. Lee and S.-C. Cho, J. Nat. Prod., 2004, 67, 889–891 CrossRef CAS.
- Y. Yang, R.-W. Yan, X.-Q. Cai, Z.-L. Zheng and G.-L. Zou, J. Sci. Food Agric., 2008, 88, 2111–2116 CrossRef CAS.
- Y. Yang, Y. Yue, Y. Runwei and Z. Guolin, Bioresour. Technol., 2010, 101, 4205–4211 CrossRef CAS.
- B. Li, H.-J. Choi, D.-S. Lee, H. Oh, Y.-C. Kim, J.-Y. Moon, W.-H. Park, S.-D. Park and J.-E. Kim, Am. J. Chin. Med., 2014, 42, 1229–1244 CrossRef.
- K. Y. Lee, S. H. Kim, S. H. Sung and Y. C. Kim, Planta Med., 2008, 74, 867–869 CrossRef CAS.
- H. Liu, Q. Yan, D. Zou, X. Bu, B. Zhang, X. Ma, A. Leng, H. Zhang, D. Li and C. Wang, Phytochem. Lett., 2018, 28, 111–115 CrossRef CAS.
- J. G. Kim, H. Jang, T. P. L. Le, H. R. Hong, M. K. Lee, J. T. Hong, D. Lee and B. Y. Hwang, J. Nat. Prod., 2019, 82, 1886–1892 CrossRef CAS.
- T.-T. Zhang, C.-L. Lu and J.-G. Jiang, J. Funct. Foods, 2015, 18, 423–431 CrossRef CAS.
- T.-T. Zhang, C.-L. Lu and J.-G. Jiang, Plant Foods Hum. Nutr., 2016, 71, 450–453 CrossRef CAS.
- S. Lee, D. Lee, T. S. Jang, K. S. Kang, J.-W. Nam, H.-J. Lee and K. H. Kim, Molecules, 2017, 22, 1583 CrossRef.
- J. S. Yu, E. Moon and K. H. Kim, Bioorg. Chem., 2017, 74, 122–125 CrossRef CAS.
- S. Kim, H. M. So, H.-S. Roh, J. Kim, J. S. Yu, S. Lee, S. Seok, C. Pang, K.-H. Baek and K. H. Kim, RSC Adv., 2017, 7, 35297–35304 RSC.
- S. Lee, S. Lee, H.-S. Roh, S.-S. Song, R. Ryoo, C. Pang, K.-H. Baek and K. H. Kim, Cells, 2018, 7, 116 CrossRef CAS.
- H. M. So, H. J. Eom, D. Lee, S. Kim, K. S. Kang, I. K. Lee, K.-H. Baek, J. Y. Park and K. H. Kim, Arch. Pharmacal Res., 2018, 41, 815–822 CrossRef CAS.
- J. S. Yu, H.-S. Roh, K.-H. Baek, S. Lee, S. Kim, H. M. So, E. Moon, C. Pang, T. S. Jang and K. H. Kim, J. Ginseng Res., 2018, 42, 562–570 CrossRef.
- S. Lee, D. Lee, S. O. Lee, J.-Y. Ryu, S.-Z. Choi, K. S. Kang and K. H. Kim, J. Funct. Foods, 2017, 32, 27–36 CrossRef CAS.
- M. Yoshihiko, H. Hideyuki and O. Motohiro, Toso Kenkyu Gijutsu Hokoku, 2009, 53, 19–26 Search PubMed.
- S. R. Lee, H. B. Park and K. H. Kim, Anal. Chem., 2018, 90, 13212–13216 CrossRef CAS.
- L. Previtera, D. Merola and P. Monaco, Phytochemistry, 1985, 24, 1838–1840 CrossRef CAS.
- A. Nagatsu, T. Sugitani, Y. Mori, H. Okuyama, J. Sakakibara and H. Mizukami, Nat. Prod. Res., 2004, 18, 231–239 CrossRef CAS.
- C. Diedrich and S. Grimme, J. Phys. Chem. A, 2003, 107, 2524–2539 CrossRef CAS.
- J.-A. Jiang, C. Chen, J.-G. Huang, H.-W. Liu, S. Cao and Y.-F. Ji, Green Chem., 2014, 16, 1248–1254 RSC.
- S. S. Hong, J. H. Lee, Y.-H. Choi, W. Jeong, E.-K. Ahn, S. H. Lym and J. S. Oh, Tetrahedron Lett., 2015, 56, 6681–6684 CrossRef CAS.
- B.-C. Hong, H.-C. Tseng and S.-H. Chen, Tetrahedron, 2007, 63, 2840–2850 CrossRef CAS.
- S. V. Naidu, P. Gupta and P. Kumar, Tetrahedron, 2007, 63, 7624–7633 CrossRef CAS.
- M. Hamberg, J. Chem. Soc., Perkin Trans. 1, 1993, 24, 3065–3072 RSC.
- A. M. Pomini, P. L. R. Cruz, C. Gai, W. L. Araujo and A. J. Marsaioli, J. Nat. Prod., 2009, 72, 2125–2129 CrossRef CAS.
- Y. Ortin and P. Evans, Synth. Commun., 2013, 43, 1404–1412 CrossRef CAS.
- T. Ritter, K. Stanek, I. Larrosa and E. M. Carreira, Org. Lett., 2004, 6, 1513–1514 CrossRef CAS.
- S. Steinbeiss, C. M. Schmidt, K. Heide and G. Gleixner, J. Anal. Appl. Pyrolysis, 2005, 75, 19–26 CrossRef.
- K. Takeda, T. Terauchi, K. Shin, M. Ino, H. Shibata and M. Yonaga, Bioorg. Med. Chem. Lett., 2012, 22, 4756–4761 CrossRef CAS.
- X. Ren, X. She, K. Peng, Y. Su, X. Xie, X. Pan and H. Zhang, J. Chin. Chem. Soc., 2004, 51, 969–974 CrossRef CAS.
- J. C. del Rio, A. Gutierrez, M. Hernando, P. Landin, J. Romero and A. T. Martinez, J. Anal. Appl. Pyrolysis, 2005, 74, 110–115 CrossRef CAS.
- Y. Y. Wan, Y. M. Du and T. Miyakoshi, Chin. Chem. Lett., 2008, 19, 333–336 CrossRef CAS.
- A. A. Al-Mutairi, F. E. M. El-Baih and H. M. Al-Hazimi, J. Saudi Chem. Soc., 2010, 14, 287–299 CrossRef CAS.
- B. Kale, A. Shinde, S. Sonar, B. Shingate, S. Kumar, S. Ghosh, S. Venugopal and M. Shingare, Tetrahedron Lett., 2010, 51, 3075–3078 CrossRef CAS.
- J. Ito, F.-R. Chang, H.-K. Wang, Y. K. Park, M. Ikegaki, N. Kilgore and K.-H. Lee, J. Nat. Prod., 2001, 64, 1278–1281 CrossRef CAS PubMed.
- A. G. Pinkus, J. A. Kautz and P. Ahobila-Vajjula, J. Chem. Crystallogr., 2002, 32, 149–152 CrossRef CAS.
- R. Iriye, T. Toya, J. Makino, R. Aruga, Y. Doi, S. Handa and H. Tanaka, Agric. Biol. Chem., 1988, 52, 989–996 CAS.
- D. H. R. Barton and D. Crich, Tetrahedron, 1985, 41, 4359–4364 CrossRef CAS.
- J. W. Choi, K. H. Kim, I. K. Lee, S. U. Choi and K. R. Lee, Nat. Prod. Sci., 2009, 15, 44–49 CAS.
- K. H. Kim, J. W. Choi, S. U. Choi, E.-K. Seo and K. R. Lee, Bull. Korean Chem. Soc., 2010, 31, 1035–1037 CrossRef CAS.
- P. Puneet, C. T. Yap, L. Wong, Y. Lam, D. R. Koh, S. Moochhala, J. Pfeilschifter, A. Huwiler and A. J. Melendez, Science, 2010, 328, 1290–1294 CrossRef CAS.
- Z. H. Li, Y. F. Tao, L. X. Xu, H. Zhao, X. L. Li, F. Fang, Y. Wu, J. Lu, Y. H. Li, W. W. Du, J. L. Ren, Y. P. Li, Y. Y. Xu, X. Feng, J. Wang, W. Q. He and J. Pan, Am. J. Transl. Res., 2016, 8, 4548–4563 CAS.
- J. Huang, K. Huang, T. Lan, X. Xie, X. Shen, P. Liu and H. Huang, Mol. Cell. Endocrinol., 2013, 365, 231–240 CrossRef CAS.
- Z. Wang, X. Min, S. H. Xiao, S. Johnstone, W. Romanow, D. Meininger, H. Xu, J. Liu, J. Dai, S. An, S. Thibault and N. Walker, Structure, 2013, 21, 798–809 CrossRef CAS.
- K. G. Lim, A. I. Gray, S. Pyne and N. J. Pyne, Br. J. Pharmacol., 2012, 166, 1605–1616 CrossRef CAS.
- L. Subedi, J. H. Lee, S. Yumnam, E. Ji and S. Y. Kim, Cells, 2019, 8, 194 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: HRESIMS, 1D and 2D NMR spectra of compounds 1–2, 5, and 10–11. See DOI: 10.1039/c9ra07988b |
| ‡ Both authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |