
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRetracted Article: LncRNA ZEB2-AS1 regulates the drug resistance of acute myeloid leukemia via the miR-142-3p/INPP4B axis
Kai Wang *,
Jing Dai,
Tao Liu,
Qiong Wang and
Yingxu Pang
*,
Jing Dai,
Tao Liu,
Qiong Wang and
Yingxu Pang
Department of Hematology, Zhoukou Central Hospital, No. 26, East Renmin Road, Zhoukou, 466000, Henan, China. E-mail: lusulizcgkn@163.com; Tel: +86-394-8521603
First published on 2nd December 2019
Abstract
Dysregulation of long noncoding RNAs (lncRNAs) has been reported to participate in the process of chemoresistance in multiple cancers, including acute myeloid leukemia (AML). LncRNA zinc finger E-box binding homeobox 2 antisense RNA 1 (ZEB2-AS1) has been reported to be up-regulated in AML. However, the biological role of ZEB2-AS1 remains to be determined. Quantitative real time polymerase chain reaction (qRT-PCR) was used to detect the levels of ZEB2-AS1, miR-142-3p and inositol polyphosphate-4-phosphatase type II B (INPP4B). The cell viability and apoptosis were examined by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay and flow cytometry, respectively. Western blotting was applied to analyze levels of BCL2 apoptosis regulator (Bcl-2), BCL2 associated X, apoptosis regulator (Bax), cleaved-caspase-3 and INPP4B. The interaction among ZEB2-AS1, miR-142-3p and INPP4B was verified by dual-luciferase reporter assay and RNA pull-down assay. The levels of ZEB2-AS1 and INPP4B were significantly elevated in AML and chemo-resistance tissues, as well as in THP-1 and THP-1/ADR cells. ZEB2-AS1 elevated the IC50 of ADR, and suppressed cell apoptosis of AML cells, while ZEB2-AS1 increased Bcl-2 expression and decreased the levels of Bax and cleaved-caspase-3. ZEB2-AS1 could enhance the resistance in THP-1 and THP-1/ADR cells. ZEB2-AS1 could sponge miR-142-3p, and ZEB2-AS1 reduced the promotion effect of miR-124-3p on the sensitivity of AML cells. Furthermore, IPNN4B was revealed as a target gene of miR-142-3p. More interestingly, suppression of IPNN4B by shRNA reversed the inhibitory effect of ZEB2-AS1 on the sensitivity of AML cells. LncRNA ZEB2-AS1 promoted ADR resistance of AML via regulating INP4B expression by sponging miR-142-3p, providing a novel therapeutic target for drug resistance of AML.
1. Introduction
Acute myeloid leukemia (AML) is a malignant disease caused by myeloid hematopoietic stem cells. The disease might occur at any stage of human life and the incidence rate of AML increases with age,1 and has attracted increasing attention of medical researchers in worldwide. The occurrence of cancer is usually the result of the mutual regulation of multiple genes,2 rather than the control of a single gene, which undoubtedly increases the difficulty of finding an effective treatment for cancer. For example, relevant studies had reported that AML was associated with gene mutations of DNMT3A, TET2, IDH1 and IDH2.3 In addition, AML is resistant to drugs during treatment, but the mechanism is still unclear.4 Hence, this study focuses on the regulation of drug resistance in AML.LncRNAs (long non-coding RNAs) are regulated during cell differentiation and mis-regulated in cancers. The mis-regulation of lncRNAs was involved in occurrence, development and the drug resistance of various cancers. For instance, EMT-regulating lncRNAs contributed to the drug resistance in cancer cells.5 MALAT-1 and HULC regulated the drug resistance via activating PI3K/AKT pathway by sponging miRNAs.6,7 HOTAIR and UCA1 were discovered to enhance the drug resistance of gastric cancer and colorectal cancer, respectively.8,9 In addition, there were 20 lncRNAs that could promote the leukemia cell proliferation in mouse AML model by shRNA screening, while silencing of different lncRNAs led to different expression patterns in vitro.10 Among them, zinc finger E-box binding homeobox 2 (ZEB2) antisense RNA 1 (ZEB2-AS1) has been proved to relate to the poor prognosis of AML,11 but the effects of ZEB2-AS1 in the drug resistance in AML has not been studied. Therefore, this paper conducts a related experiment on the resistance of ZEB2-AS1 in AML.
MicroRNAs (MiRNAs), 21–23 nt long, are widely presented in eukaryotes. MiRNAs play an important role in the regulation of gene expression and development of organisms through targeting mRNA and regulating the transcription of mRNA.12 MiR-124-3p is a specific miRNA in hematopoietic system, showing low expression in AML patients. Clinical pathological data indicated that the survival rate was low in AML patients with the down-regulation of miR-142-3p.13 Silencing miR-142-3p promoted the proliferation and metastasis of AML cells and inhibited apoptosis,14,15 while up-regulation of miR-142-3p could increase the sensitivity of AML cells to chemotherapy.16 Accumulating researches proved that miR-142-3p may participated in the drug sensitivity of AML.
Inositol polyphosphate 4-phosphatase type II (INPP4B) is a negative regulatory factor in phoinositide 3 kinase (PI3K) signaling pathway, which can inhibit the activation of protein kinase B (AKT) and play an anti-cancer effect.17 INPP4B kncokdown inhibited the proliferation of npm1-mutated AML cells, whereas over-expression of INPP4B achieved AML cell proliferation by activating SGK3.18 A previous study has confirmed that high expression of INPP4B was correlated with autophagy-related proteins in AML.19 INPP4B was highly expressed in AML patients with poor prognosis and overexpression of INPP4B enhanced the drug resistance in AML treatment,20,21 which was caused by DNA repair pathway.22 Thus, INPP4B might be related to the drug resistance of AML.
In this paper, we detected the expression of ZEB2-AS1, miR-142-3p and INPP4B. Moreover, we investigated the function roles of ZEB2-AS1 and explored the underlying mechanism of ZEB2-AS1 in the drug resistance of AML. We attempted to find a novel therapeutic target for AML.
2. Materials and methods
2.1 Sample collection
AML bone marrow tissues (n = 52) and normal tissues (n = 20) were collected from patients in Zhoukou Central Hospital. Among the 52 AML patients, 23 patients were chemo-sensitive and the other 29 patients were chemo-resistant. All experiments were performed in accordance with the guidelines “National Institutes of Health”. Experiments were approved by the ethics committee at Zhoukou Central Hospital, and all patients had submitted written informed consent.2.2 Cell culture and transfection
THP-1 (human AML cell line) and HS-5 (human bone marrow stromal cell line) were purchased from the cell bank of Chinese Academy of Sciences (Shanghai, China). ADR resistant cell line (THP-1/ADR) was purchased from institute of the Haematology, Chinese Academy of Medical Sciences (Tianjin, China). All cells were cultivated in Roswell Park Memorial Institute-1640 (RPMI-1640) medium (Gibco, Carlsbad, CA, USA) added with 10% fetal bovine serum (FBS; Gibco) and 10% penicillin (100 U mL−1)-streptomycin (100 μg mL−1) mixed solution at 37 °C with 5% CO2. THP-1/ADM cells were added ADR (APExBIO Technology, Austin, TX, USA) with a final concentration of 2 μg mL−1.The transfection of AML cells was carried out using Lipofectamine 2000 (Yeasen Bio, Shanghai, China). Empty pcDNA vector (pcDNA) and pcDNA-ZEB2-AS1 (ZEB2-AS1), shRNA-negative control (sh-NC) and shRNA against ZEB1-AS1 (sh-ZEB2-AS1), miR-142-3p mimic or inhibitor (miR-142-3p or anti-miR-142-3p) and their corresponding negative control (miR-NC or anti-miR-NC) were synthesized by Genepharma (Shanghai, China).
2.3 Quantitative real-time PCR (qRT-PCR)
RNA was extracted with TRIzol reagent (Invitrogen, Carlsbad, CA, USA), and M-MLV Reverse Transcriptase (Invitrogen) was used for reverse transcription according to the manufacturer's instructions. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was regarded as the internal reference. The 2−ΔΔCt method was used for the detection of the abundance of ZEB2-AS1, miR-142-3p and INPP4B. Primers used in this paper were as follows: ZEB2-AS1 (forward 5′-CCGTGGGCATGCTGAAT-3′ and reverse 5′-CTGCTGGCAAGCGGAACT-3′); miR-142-3p (forward 5′-CGCCGTGTAGTGTTTCCTAC-3′ and reverse 5′-GTCGTATCCAGTGCAGGGTC-3′); INPP4B (forward 5′-GGAAAGTGTGAGCGGAAAAG-3′ and reverse 5′-CGAATTCGCATCCACTTATTG-3′); GAPDH (forward 5′-GGTCTCCTCTGACTTCAACA-3′ and reverse 5′-GTGAGGGTCTCTCTCTTCCT-3′).2.4 ADR sensitivity assay
To explore the sensitivity of AML cells to ADR, THP-1 and THP-1/ADR cells at the exponential growth stage were incubated in 96-well plate for two days. Then THP-1 cells (transfected with pcDNA, ZEB2-AS1, anti-miR-NC, anti-miR-142-3p, anti-miR-142-3p + sh-NC, anti-miR-142-3p + sh-ZEB2-AS1, ZEB2-AS1 + sh-NC, ZEB2-AS1 + sh-INPP4B) or THP-1/ADR cells (transfected with sh-NC, sh-ZEB2-AS1, miR-NC, miR-142-3p, miR-142-3p + pcDNA, miR-142-3p + ZEB2-AS1, sh-ZEB2-AS1 + pcDNA, sh-ZEB2-AS1 + INPP4B) were treated with ADR at different concentrations (0.0001, 0.001, 0.01, 0.1, 1, 10, 100 and 1000 μM) for 48 h, respectively. 5 mg mL −1 20 μL MTT was added to every well of the 96-well plate, and the AML cells were cultured for an additional 4 hours. Formazan crystal was dissolved with 150 μL DMSO after discarding the supernatant. The optical density was measured by microplate reader at 490 nm, and IC50 was then calculated.2.5 Flow cytometric analysis
Cell apoptosis was measured by Annexin V-FITC/PI apoptosis detection kit (Solarbio, Beijing, China). Cells were harvested after transfection for 48 h, and stained with 5 μL Annexin V combined FITC and PI for 10 min away from light. Then the apoptotic cells (FITC+, PI+/−) were selected by the flow cytometer (BD Biosciences, San Jose, CA, USA). The experiments were repeated three times.2.6 Western blot
The total protein was quantified and was separated via SDS-PAGE gel electrophoresis according to the molecular weight and charge density of the protein, and then transferred onto PVDF membranes (Millipore, Billerica, MA, USA). After being blocked the nonspecific binding sites for 1 h, the PVDF membranes were incubated with primary antibodies against Bcl-2 (ab185002, Abcam, Cambridge, MA, USA), Bax (ab32503, Abcam), cleaved-caspase-3 (ab2302, Abcam), INPP4B (ab81269, Abcam) or GAPDH (ab37168, Abcam) overnight at 4 °C, and then incubated with HRP combined secondary antibody (ab6721, Abcam) for 2 h. Finally, protein signal was detected through ECL system (Beyotime, Shanghai, China). In this experiment, GAPDH was regarded as a control.2.7 Dual-luciferase reporter assay
The binding sites between miR-142-3p and lncRNA ZEB2-AS1 or 3′ UTR of INPP4B were predicted by miRcode and Starbase, respectively. To verify the combination between miR-142-3p and lncRNA ZEB2-AS1, the sequences of ZEB2-AS1, including wild-type (WT) binding sites or mutant binding sites, were amplified and inserted to the luciferase gene downstream of pmirGLO vector (Promega, Madison, WI, USA), named as WT-ZEB2-AS1 and MUT-ZEB2-AS1, respectively. Cells were co-transfected with WT-ZEB2-AS1 or MUT-ZEB2-AS1 and miR-142-3p or miR-NC. Luciferase activity was analyzed after 48 h transfection. The binding between miR-142-3p and 3′ UTR of INPP4B was validated by the same way.2.8 RNA pull-down
In this experiment, Bio-control probe (control) and Bio-ZEB2-AS1 probe (bio-labeled probe of ZEB2-AS1) were synthesized by Ribobio (Guangzhou, China). The THP-1 and THP-1/ADR cells were transfected with Bio-control probe or Bio-control probe and cultured for 48 h. Then the cells were lysed and cultured at 4 °C with Streptavidin-Dyna beads overnight. Finally, qRT-PCR was used to determine the enrichment of miR-142-3p.2.9 Statistical analysis
All statistical analysis and images were done by GraphPad Prism 7. Student's t-test and one-way analysis of variance (ANOVA) were used for the analysis of differences between two groups or among multiple groups. All data were expressed as mean ± standard error (SEM). P < 0.05 was statistically significant.3. Results
3.1 ZEB2-AS1 is up-regulated in ADR-resistant AML tissues and cells
To investigate the effect of ZEB2-AS1 in AML, the expression of ZEB2-AS1 was determined by qRT-PCR. The abundance of ZEB2-AS1 was remarkably increased in the BM tissues from AML patients and chemo-resistant AML patients compared with that from normal patients and chemo-sensitive AML patients (Fig. 1A and B). In addition, the expression of ZEB2-AS1 was higher in THP-1 cells than that in human bone marrow stromal cells HS-5, and the expression was further up-regulated in ADR-resistant THP-1 cells (THP-1/ADR) (Fig. 1C). It was also revealed that the high expression level of ZEB2-AS1 was related to the poor prognosis in AML patients via Kaplan–Meier survival analysis (Fig. 1D). Therefore, these findings suggested that the up-regulation of ZEB2-AS1 was related to the ADR-resistance and poor prognosis of AML patients.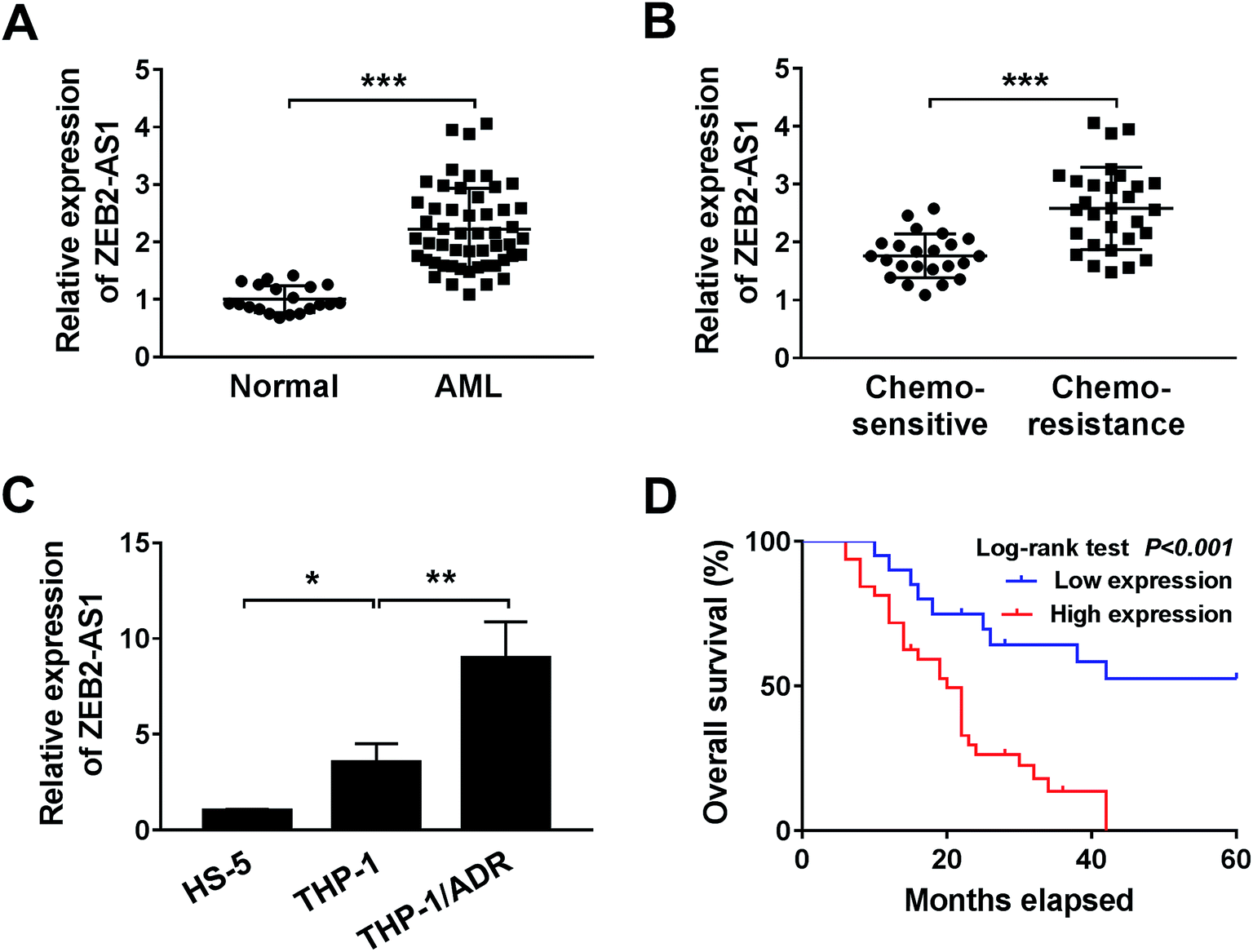 | ||
| Fig. 1 ZEB2-AS1 is up-regulated in ADR-resistant AML tissues and cells. (A and B) ZEB2-AS1 expression in AML tissues was analyzed by qRT-PCR. (A) AML patients (n = 52) and control (n = 20). (B) Chemo-sensitive (n = 23) and chemo-resistance (n = 29). (C) The expression of ZEB2-AS1 was measured by qRT-PCR in HS-5, THP1 and THP1/ADR cells. (D) Kaplan–Meier analysis and log-rank test showed that the expression level of ZEB2-AS1 was related to survival rate. *P < 0.05, **P < 0.01, ***P < 0.001. | ||
3.2 ZEB2-AS1 reduces the cytotoxic effect of ADR in AML cells
In order to study the potential role of ZEB2-AS1 in the chemoresistance of AML cells, the THP-1 cells were transfected with pcDNA or ZEB2-AS1, and HTP-1/ADR cells were transfected with sh-NC or sh-ZEB2-AS1. The data showed that the enrichment of ZEB2-AS1 in THP-1 cells was significantly increased in ZEB2-AS1 group compared with that in the control group, while it was sharply decreased in THP-1/ADR cells transfected with sh-ZEB2-AS1 (Fig. 2A and B). In addition, overexpression of ZEB2-AS1 in THP1 cells elevated the cell viability and the IC50 of ADR (Fig. 2C), while the knockdown of ZEB2-AS1 declined the IC50 of ADR (Fig. 2D). Moreover, the accumulation of ZEB2-AS1 inhibited ADR-mediated apoptosis in THP-1 cells, while the intervention of ZEB2-AS1 accelerated ADR-mediated apoptosis in THP-1/ADR cells (Fig. 2E and F). Western blot assay was conducted to measure the apoptosis-related proteins (Bcl-2, Bax and cleaved-caspase-3) in THP-1 and THP-1/ADR cells transfected with ZEB2-AS1 or sh-ZEB2-AS1, respectively. As showed in Fig. 2G and F, the accumulation of ZEB2-AS1 in THP-1 cells suppressed cell apoptosis, while the interference of ZEB2-AS1 in THP-1/ADR cells promoted cell apoptosis. Collectively, ZEB2-AS1 increased the chemoresistance of THP-1 cells.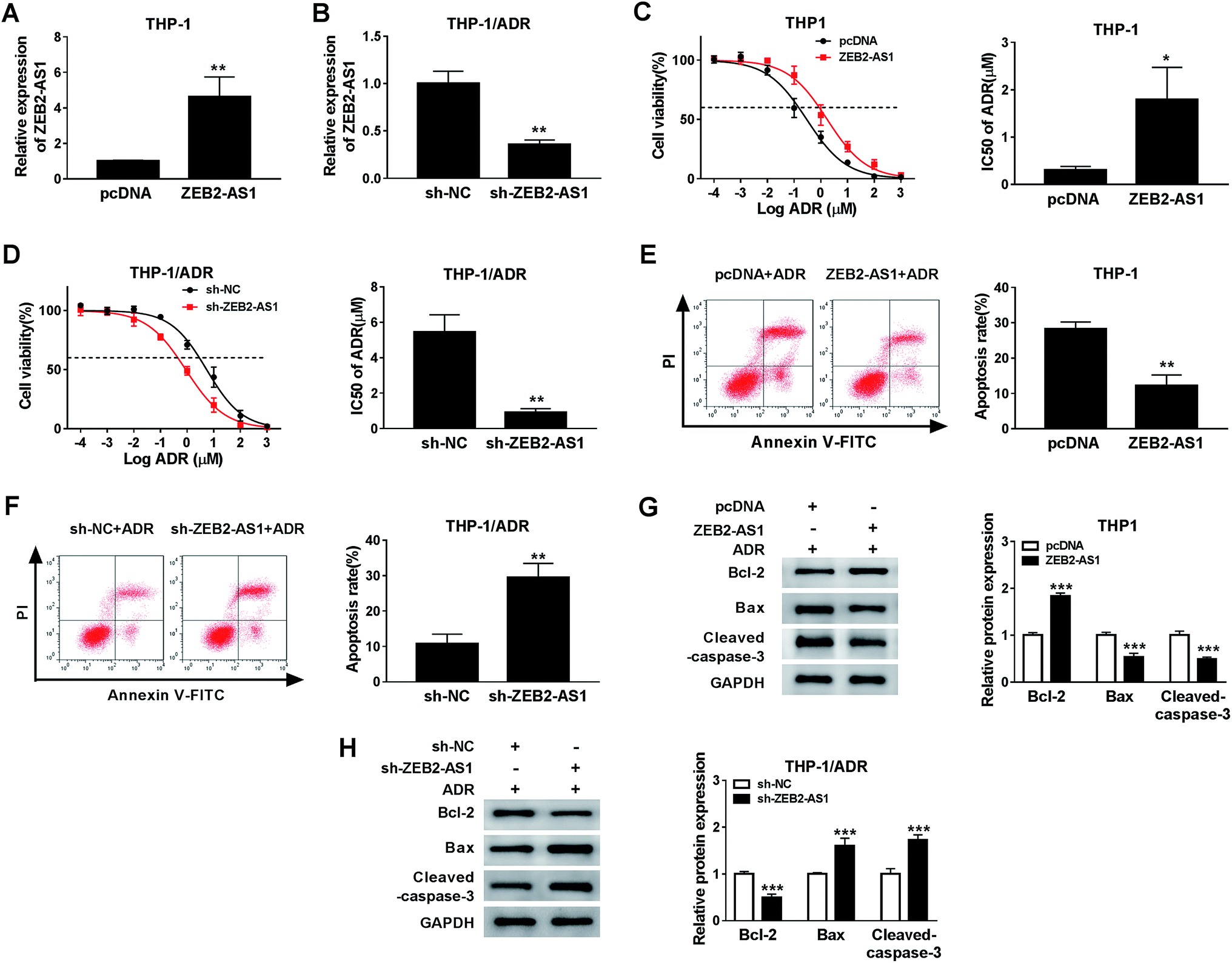 | ||
| Fig. 2 ZEB2-AS1 elevated the ADR resistance in AML cells. (A and B) The level of ZEB2-AS1 was detected in THP1 cells transfected ZEB2-AS1 and THP-1/ADR cells transfected sh-ZEB2-AS1. (C) THP-1 cells (transfected with pcDNA and ZEB2-AS1) and THP-1/ADR cells (sh-NC and sh-ZEB2-AS1) were treated with different concentrations (0.0001, 0.001, 0.01, 0.1, 1, 10, 100 and 1000 μM) of ADR for 48 h, respectively. (C and D) Cell viability, IC50 of ADR were detected by MTT assay. (E and F) Cell apoptosis was measured by floe cytometry. (G and H) Apoptosis-related proteins including Bcl-2, Bax, and cleaved-caspase-3 were detected by western blot. *P < 0.05, **P < 0.01, ***P < 0.001. | ||
3.3 ZEB2-AS1 could sponge miR-142-3p
The specific binding sites between miR-142-3p and ZEB2-AS1 were predicted by miRcode bioinformatic software (Fig. 3A). Luciferase activity was dramatically declined with the accumulation of miR-142-3p in WT-ZEB2-AS1 group compared with that in MUT-ZEB2-AS1 group, suggesting that miR-142-3p was a direct target of ZEB2-AS1 in AML cells. RNA pull-down assay showed that miR-142-3p could bind to ZEB2-AS1 in THP-1 and THP-1/ADR cells (Fig. 3D). In order to investigate the effect of miR-142-3p in AML, the expression of miR-142-3p was detected by qRT-PCR. MiR-142-3p was down-regulated in the BM tissues from AML patients and chemo-resistant AML patients compared with that from normal patients and chemo-sensitive AML patients (Fig. 3E and F). The abundance of miR-142-3p was lower in THP-1 cells than that in HS-5 cells, and the expression was further reduced in THP-1/ADR cells (Fig. 3G). ZEB2-AS1 overexpression significantly reduced the expression of miR-142-3p in THP-1, while the knockdown of ZEB2-AS1 obviously increased the miR-142-3p expression in THP-1/ADR cells (Fig. 3H and I). Taken together, miR-142-3p was a target of ZEB2-AS1, and it was inversely modulated by ZEB2-AS1.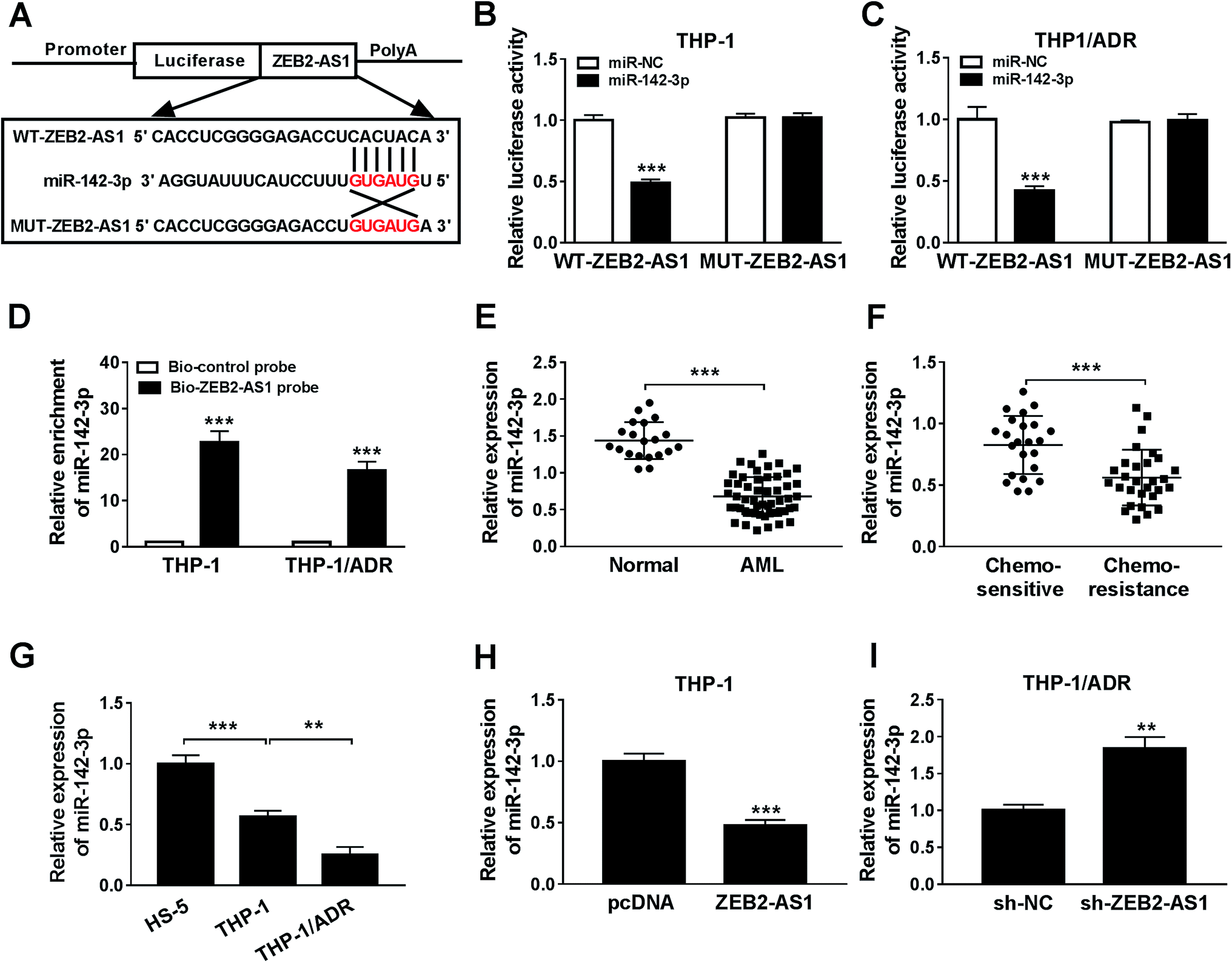 | ||
| Fig. 3 ZEB2-AS1 could sponge miR-142-3p. (A) MiRcode predicted the binding site between ZEB2-AS1 and miR-142-3p. (B–D) Dual luciferase report and RNA pull-down assay were used to verify the target site between ZEB2-AS1 and miR-142-3p, respectively. (E and F) The expression of miR-142-3p were measured in tissues of normal, AML, chemo-sensitive and chemo-resistance patients using qRT-PCR. (G) MiR-142-3p expression of HS-5, THP-1, and THP-1/ADR cells by qRT-PCR. (H and I) QRT-PCR was focused on the correlation between ZEB2-AS1 and miR-142-3p. **P < 0.01, ***P < 0.001. | ||
3.4 MiR-142-3p overexpression improves ADR sensitivity of AML cells
In order to study the relationship between miR-142-3p and ADR sensitivity of AML cells, THP-1 cells were transfected with anti-miR-NC, anti-miR-142-3p, anti-miR-142-3p + sh-NC or anti-miR-142-3p + sh-ZEB2-AS1. We first detected ZEB2-AS1 and found that the knockdown of miR-142-3p in THP-1 cells or miR-142-3p overexpression in THP-1/ADR cells had no significant effect on the expression of ZEB2-AS1 (Fig. 4A and B). As indicated in Fig. 4C, the inhibition of miR-142-3p increased the chemoresistence of ADR in THP-1 cells, and the knockdown of ZEB2-AS1 reversed the effect of miR-142-3p inhibition on the chemoresistence of ADR in THP-1 cells. Meanwhile, the transfection of anti-miR-142-3p suppressed the apoptosis of THP-1 cells, and the intervention of ZEB2-AS1 alleviated the inhibitory effect of miR-142-3p depletion on apoptosis of THP-1 cells (Fig. 4E). Western blot results showed that the abundance of Bcl-2 was increased and the expression of Bax and cleaved-caspase-3 was down-regulated by the transfection of anti-miR-142-3p in THP-1 cells, indicating that miR-142-3p depletion suppressed the apoptosis of THP-1 cells. The co-transfection of anti-miR-142-3p and sh-ZEB2-AS1 abolished the suppressive effect of miR-142-3p depletion on the apoptosis of THP-1 cells (Fig. 4G).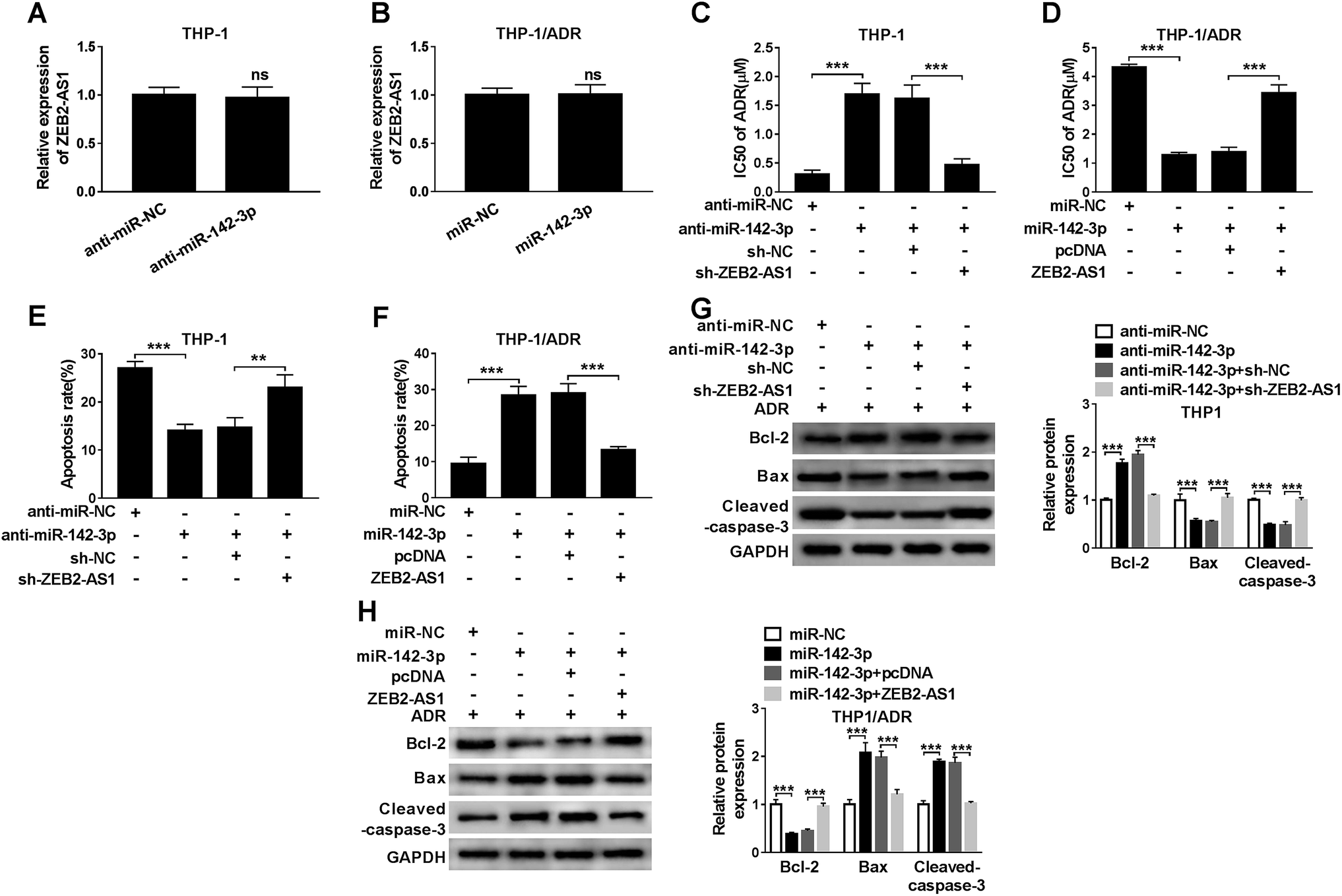 | ||
| Fig. 4 MiR-142-3p overexpression improves ADR sensitivity of AML cells. (A) ZEB2-AS1 expression was measured in THP-1 cells transfected with anti-miR-142-3p or anti-miR-NC by qRT-PCR. (B) ZEB2-AS1 expression was measured in THP-1/ADR cells transfected with miR-142-3p or miR-NC by qRT-PCR. (C and D) THP-1 cells were transfected with anti-miR-142-3p or anti-miR-142-3p + sh-ZEB2-AS1, while THP-1/ADR cells were transfected with miR-142-3p or miR-142-3p + ZEB2-AS1. The IC50 of ADR was evaluated by MTT assay. (E and F) Cell apoptosis was assessed by flow cytometry. (G and H) Western blot assay was performed to measure the expression of apoptosis-related proteins. **P < 0.01, ***P < 0.001. | ||
Besides, the chemoresistance and apoptosis of THP-1/ADR cells transfected with miR-NC, miR-142-3p, miR-142-3p + pcDNA, miR-142-3p + ZEB2-AS1 were measured by MTT assay, flow cytometry and western blot, respectively. As showed in Fig. 4B, the accumulation of miR-142-3p decreased the chemoresistance of ADR in THP-1/ADR, and the overexpression of ZEB2-AS1 reversed the impact of miR-142-3p accumulation on the chemoresistence of ADR in THP-1/ADR cells. Meanwhile, flow cytometry and western blot assay revealed that the overexpression of miR-142-3p promoted the apoptosis of THP-1/ADR and the addition of ZEB2-AS1 attenuated the promoting effect of miR-142-3p overexpression on the apoptosis of THP-1/ADR cells (Fig. 4F and H). These data demonstrated that miR-142-3p overexpression improved ADR sensitivity of AML cells.
3.5 INPP4B directly interacts with miR-142-3p
The relationship between miR-142-3p and INPP4B was predicted via Starbase online software (Fig. 5A). According to the luciferase reporter assay, INPP4B was the target of miR-142-3p (Fig. 5B and C). The mRNA expression of INPP4B was up-regulated in the BM tissues from AML patients and chemo-resistant AML patients compared with that from normal patients and chemo-sensitive AML patients (Fig. 5D and E). In addition, the expression level of INPP4B was the highest in THP-1/ADR cells than that in HS-5 and THP-1 cells (Fig. 5F). To explore the effect of miR-142-3p on INPP4B expression in AML cells, the levels of INPP4B mRNA and protein in THP-1 cells transfected with anti-miR-NC or anti-miR-142-3p were detected. MiR-142-3p inhibitor increased the mRNA and protein levels of INPP4B in THP-1 cells (Fig. 5G and H). Nevertheless, the mRNA and protein levels of INPP4B were decreased in THP-1/ADR cells transfected with miR-142-3p (Fig. 5I and J). In conclusion, the expression of INPP4B was negatively regulated by miR-142-3p.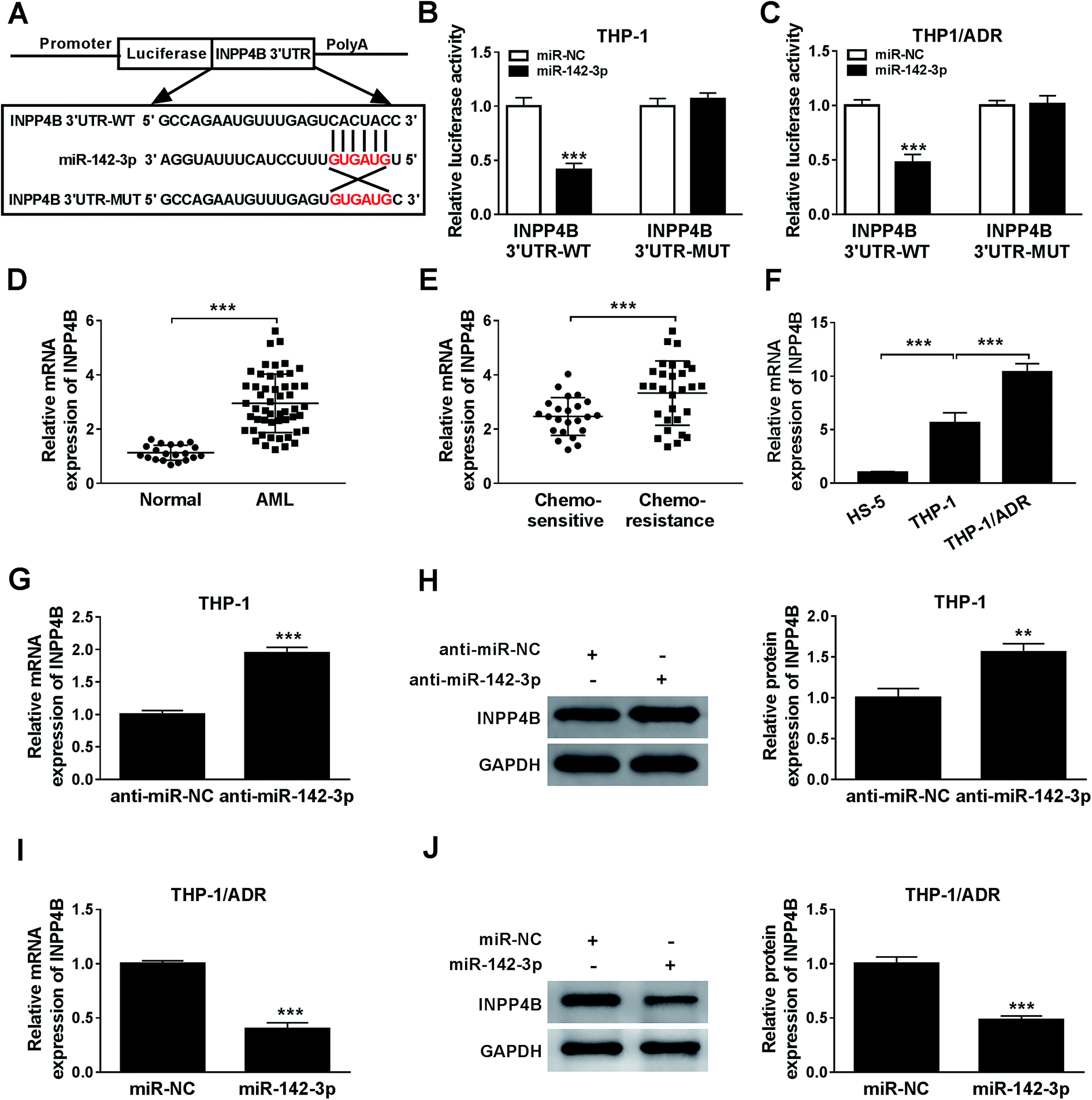 | ||
| Fig. 5 INPP4B is a target of miR-142-3p. (A) StarBase2.0 predicted the potential binding sites between miR-142-3p and INPP4B. (B and C) The luciferase activity was detected by dual luciferase reporter assay. (D and E) The mRNA expression of INPP4B in AML tissues was detected by qRT-PCR. (F) The mRNA expression of INPP4B in AML cells was measured by qRT-PCR. (G–J) QRT-PCR and western blot were carried out to assess the relationship between miR-142-3p and INPP4B in THP-1 and THP-1/ADR cells, respectively. **P < 0.01, ***P < 0.001. | ||
3.6 ZEB2-AS1 promotes the chemoresistance of ADR in AML cells through INPP4B
To illustrate whether INPP4B was involved in ZEB2-AS1 mediated-chemoresistance, THP-1 cells were transfected with pcDNA, ZEB2-AS1, ZEB2-AS1 + sh-NC or ZEB2-AS1 + sh-INPP4B, and THP-1/ADR cells were transfected with sh-NC, sh-ZEB2-AS1, sh-ZEB2-AS1 + pcDNA or sh-ZEB2-AS1 + INPP4B. ZEB2-AS1 overexpression increased the mRNA and protein levels of INPP4B in THP-1 cells, while the addition of sh-INPP4B down-regulated the abundance of INPP4B mRNA and protein (Fig. 6A and B). Similarly, the mRNA and protein levels of INPP4B were reduced with ZEB2-AS1 knockdown, but the phenomenon was reversed with the addition of INPP4B in THP-1/ADR cells (Fig. 6C and D). In addition, IC50 and apoptosis rate were detected by MTT assay and flow cytometry. Overexpression of ZEB2-AS1 increased IC50 of ADR and inhibited apoptosis in THP-1 cells, suggesting that it could reduce sensitivity of THP-1 cells to ADR, this effect was partially restored with IPNN4B knockdown (Fig. 6E and G). ZEB2-AS1 knockdown reduced the IC50 of ADR and increased the apoptosis rate in THP-1/ADR cells, indicating that the interference of ZEB2-AS1 increased sensitivity of THP-1/ADR cells to ADR, which was partially repaired by the addition of IPNN4B (Fig. 6F and H).Apart from this, the changes in apoptotic related proteins (Bcl-2, Bax and cleaved-caspase-3) also confirmed that ZEB2-AS1 up-regulated ADR resistance of AML cells through INPP4B (Fig. 6I and J).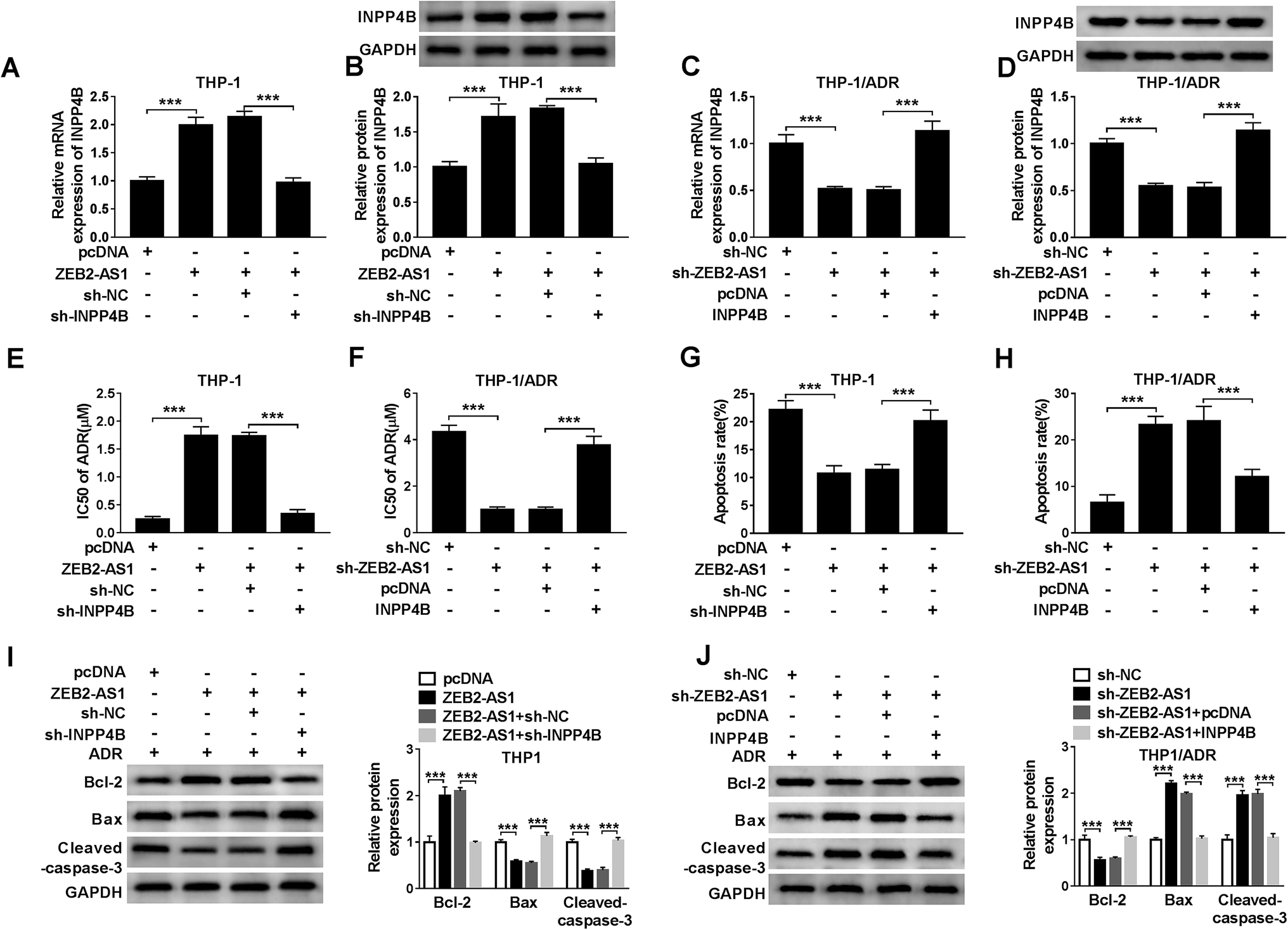 | ||
| Fig. 6 ZEB2-AS1 promotes the chemoresistance of ADR in AML cells through regulating INPP4B. (A–D) QRT-PCR and western blot assays showed that ZEB2-AS1 could regulate the expression of INPP4B. (E and F) IC50 of ADR was measured by MTT assay. (G and H) The apoptosis of AML cells was detected by flow cytometry analysis. (I and J) The apoptosis-related proteins Bcl-2, cleaved-caspase-3 and Bax were detected by western blot. *P < 0.05. | ||
4. Discussion
AML, as a blood cancer, is an acute form of leukemia at the molecular and phenotypic levels.23 Several researchers indicated that the drug resistance is a major problem in the treatment of AML. In recent years, lncRNAs have been proved to exert key roles in RNA transcription and mRNA translation, participating in proliferation, migration and apoptosis of cancer genesis and development. Meanwhile, the effects of lncRNAs in the drug resistance of various cancers have attracted wide attention.24 For example, studies had shown that UCA1 was involved in the regulation of the drug resistance in gastric cancer,25 colorectal cancer26 and AML.27 Small interfering ZEB2 increased the apoptosis of small cell lung cancer cells induced by drugs.28 The expression and molecular mechanism of H19 and ANRIL had been studied in AML, which were regarded as potential prognostic indicators. H19 and ANRIL were up-regulated in AML and H19 and ANRIL deletion inhibited the growth of AML cells.29,30 In this study, high expression of ZEB2-AS1 was accompanied by low survival rate, suggesting that ZEB2-AS1 may be a pathogenic factor of AML. Consistent with the results of H19 on AML cell growth, ZEB2-AS1 overexpression decreased the sensitivity of THP-1 cells, repressed apoptosis and regulated the expression of apoptosis-related proteins, while knockdown of ZEB2-AS1 increased the sensitivity of THP-1/ADR, stimulated apoptosis and expression of apoptosis-related proteins. Taken together, ZEB2-AS1 alleviated the sensitivity of AML and played a crucial role in the resistance of AML.MiRNAs were confirmed to be related with the biomarkers, functions and therapy of cancers. Previous studies indicated that miR-142-3p was down-regulated in cancer cells and the low expression level of miR-142-3p inhibited the differentiation of macrophages in tumor-recruited myeloid and colon cancer cell.31,32 In addition, miR-142-3p was regarded as a new diagnostic marker in AML, and was negatively correlated with HMGB1 expression in drug-resistant AML cells.16 In the present study, we predicted that the putative binding site was predicted between ZEB2-AS1 and miR-142-3p, which was confirmed by luciferase report and RNA pull down analysis. MiR-142-3p was lowly expressed in AML and chemo-resistance tissues, as well as in THP-1 and THP-1/ADR cells. Moreover, ZEB2-AS1 could regulate the expression of miR-142-3p in both THP-1 and THP-1/ADR cells. Overexpression of miR-142-3p inhibited the p-glycoprotein of drug resistant cells and enhanced the sensitivity of AML cells to drugs.16 In agreement with the previous studies, we observed that ZEB2-AS1 enhanced the IC50 of ADR, and suppressed apoptosis and apoptosis-related proteins in AML cells. ZEB2-AS1 functioned as a molecular sponge of miR-142-3p, and improved the sensitivity of AML cells by sponging miR-142-3p.
The inhibition of mRNA degradation or translation might be caused by miRNA regulating gene expression though binding to the 3′ UTR translation region.33 As a member of PI3K, INPP4B was closely related to the AKT pathway.34 SGK3 overexpression was activated by INPP4B in breast cancer.35 Meanwhile, INPP4B overexpression could promote the formation of colonies and elevate the resistance of AML cells.36 These studies suggested that INPP4B was a potential cancer promoter. In this paper, the expression of INPP4B in AML, chemotherapy tissues, and THP-1 and THP-1/ADR cells was higher than that in control groups. According to starBase2.0's prediction, INPP4B was a target of miR-142-3p. INPP4B deletion reversed the promotion effects of ZEB2-AS1 overexpression on the sensitivity of THP-1 cells, while INPP4B overexpression reversed the inhibitory effects of ZEB2-AS1 deletion on the sensitivity of THP-1/ADR cells. INPP4B might be an oncogene, which could induce chemo-resistance in AML patients.
At present, although the drug resistance in cancer is a major problem in medical treatment, several researches have reported the potential regulatory mechanisms of the drug resistance in AML. For example, resveratrol reversed the drug resistance of HL-60/ADR cells in AML by inhibiting the expression of MRP1.37 ZSTK474 had inhibitory effects on HL60/ADR cells, reducing the expression of p-glycoprotein and enhancing the sensitivity of AML.38 The drug resistance in AML was also studied in this paper, showing that ZEB2-AS1 knockdown promoted the apoptosis and THP-1/ADR cells became more sensitive to ADR, which was partially restored by up-regulating IPNN4B. In short, ZEB2-AS1 promoted ADR resistance of AML cells through regulated INPP4B expression by sponging miR-142-3p (Fig. 7).
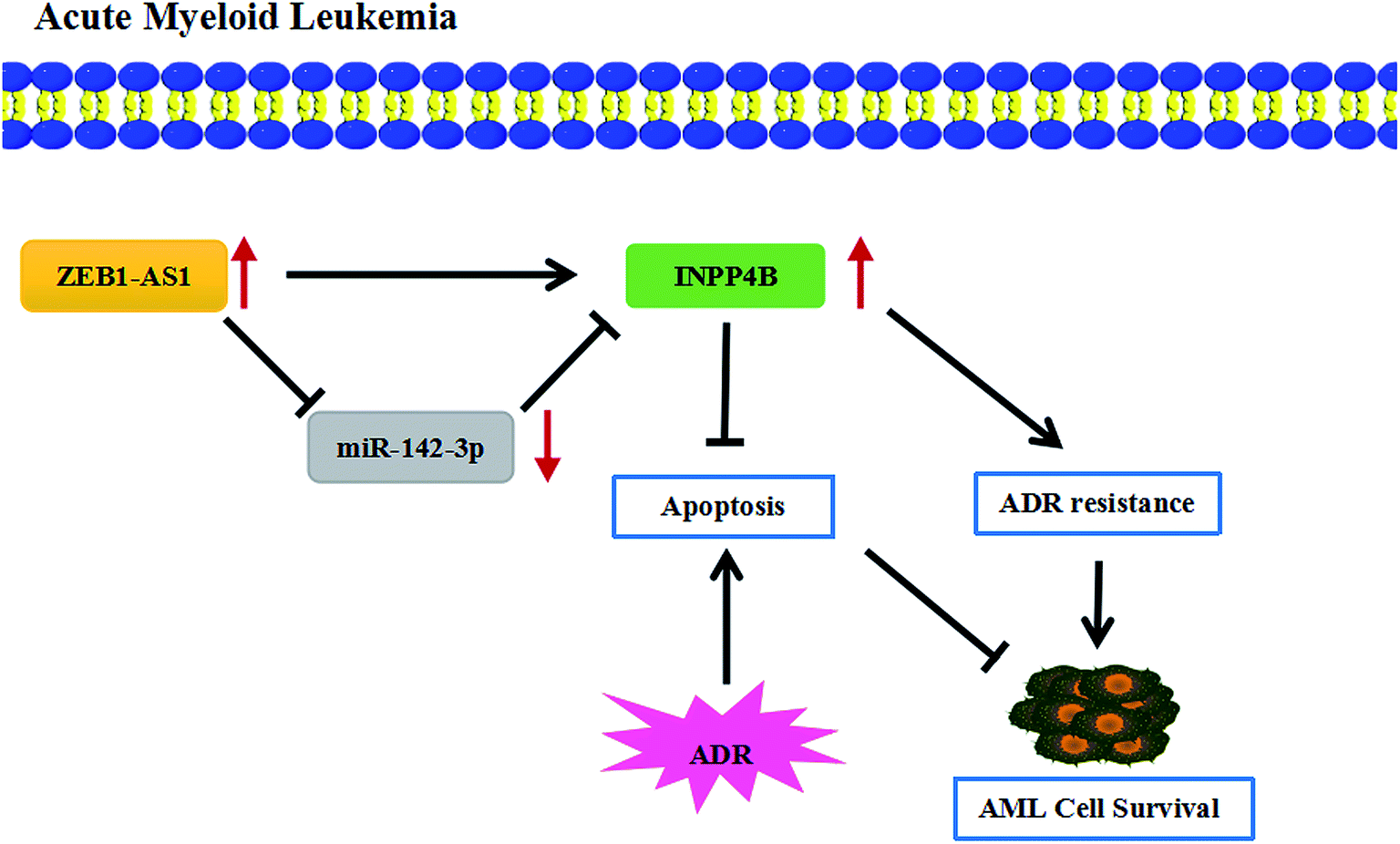 | ||
| Fig. 7 The mechanism diagram of ZEB2-AS1/miR-142-3p/INPP4B axis in AML cells. ↑ indicated that the expression was increased. ZEB2-AS1 regulated cell viability, apoptosis and ADR resistance of AML cells by regulating miR-142-3p/INPP4B axis. | ||
In conclusion, we discovered that ZEB2-AS1 and INPP4B expression were upregulated in chemo-sensitive and AML tissues, as well as in THP-1 and THP-1/ADR cells, while miR-142-3p expression was down-regulated. ZEB2-AS1 knockdown increased the sensitivity of THP-1/ADR cells to ADR. Moreover, ZEB2-AS1 enhanced the sensitivity of AML cells by targeting miR-142-3p. In addition, miR-142-3p directly targeted INPP4B. INPP4B reversed the effects of ZEB2-AS1 on IC50 to ADR, apoptosis and apoptosis-related proteins in AML cells. In a word, ZEB2-AS1 regulated the drug resistance through modulating miR-142-3p/INPP4B axis in AML.
Conflicts of interest
There is no conflict of interest regarding the publication of this paper.References
- C. J. Eaves and R. K. Humphries, N. Engl. J. Med., 2010, 362, 2326–2337 CrossRef CAS PubMed.
- N. B. Hao, Y. F. He, X. Q. Li, K. Wang and R. L. Wang, Oncotarget, 2017, 8, 81572–81582 Search PubMed.
- L. Bullinger, K. Dohner and H. Dohner, J. Clin. Oncol., 2017, 35, 934–946 CrossRef CAS PubMed.
- S. Scholl, B. Spies-Weisshart, A. Klink, L. O. Muegge, H. J. Fricke and A. Hochhaus, Ann. Hematol., 2011, 90, 473–475 CrossRef PubMed.
- H. Richard, F. Stephen, C. Sinead and G. Steven, Cancers, 2017, 9, 38 CrossRef PubMed.
- S. Xu, S. Sui, J. Zhang, N. Bai, Q. Shi, G. Zhang, S. Gao, Z. You, C. Zhan and F. Liu, Int. J. Clin. Exp. Pathol., 2015, 8, 4881–4891 CAS.
- Y. Lu, Y. Li, X. Chai, Q. Kang, P. Zhao, J. Xiong and J. Wang, Gene, 2017, 607, 41–46 CrossRef CAS PubMed.
- J. Yan, Y. Dang, S. Liu, Y. Zhang and G. Zhang, Tumour Immunobiol., 2016, 37, 1–11 CAS.
- Z. Bian, L. Jin, J. Zhang, Y. Yin, C. Quan, Y. Hu, Y. Feng, H. Liu, B. Fei and Y. Mao, Sci. Rep., 2016, 6, 23892 CrossRef CAS PubMed.
- M. J. Delas, L. R. Sabin, E. Dolzhenko, S. R. Knott, E. Munera Maravilla, B. T. Jackson, S. A. Wild, T. Kovacevic, E. M. Stork and M. Zhou, eLife, 2017, 6, e25607 CrossRef PubMed.
- X. Shi, J. Li, L. Ma, L. Wen, Q. Wang, H. Yao, C. Ruan, D. Wu, X. Zhang and S. Chen, Oncol. Lett., 2019, 17, 4935–4947 Search PubMed.
- B. Zacher, K. Abnaof, S. Gade, E. Younesi, A. Tresch and H. Fröhlich, Bioinformatics, 2012, 28, 1714–1720 CrossRef CAS PubMed.
- M. Dahlhaus, C. Roolf, S. Ruck, S. Lange, M. Freund and C. Junghanss, Neoplasma, 2013, 60, 432–438 CrossRef CAS PubMed.
- F. Wang, X. S. Wang, G. H. Yang, P. F. Zhai, Z. Xiao, L. Y. Xia, L. R. Chen, Y. Wang, X. Z. Wang, L. X. Bi, N. Liu, Y. Yu, D. Gao, B. T. Huang, J. Wang, D. B. Zhou, J. N. Gong, H. L. Zhao, X. H. Bi, J. Yu and J. W. Zhang, Mol. Biol. Rep., 2012, 39, 2713–2722 CrossRef CAS PubMed.
- M. C. Trissal, T. N. Wong, J. C. Yao, R. Ramaswamy, I. Kuo, J. Baty, Y. Sun, G. Jih, N. Parikh, M. M. Berrien-Elliott, T. A. Fehniger, T. J. Ley, I. Maillard, P. R. Reddy and D. C. Link, Cancer Res., 2018, 78, 3510–3521 CAS.
- Y. Zhang, Y. Liu and X. Xu, Transl. Oncol., 2017, 10, 410–418 CrossRef PubMed.
- M. Ferron and J. Vacher, Gene, 2006, 376, 152–161 CrossRef CAS PubMed.
- H. Jin, L. Yang, L. Wang, Z. Yang, Q. Zhan, Y. Tao, Q. Zou, Y. Tang, J. Xian, S. Zhang, Y. Jing and L. Zhang, J. Exp. Clin. Cancer Res., 2018, 37, 8 CrossRef PubMed.
- F. Zhang, J. Li, J. Zhu, L. Liu, K. Zhu, S. Cheng, R. Lv and P. Zhang, Biol. Res., 2019, 52, 11–20 CrossRef PubMed.
- S. Rijal, S. Fleming, N. Cummings, N. K. Rynkiewicz, L. M. Ooms, N.-Y. N. Nguyen, T.-C. Teh, S. Avery, J. F. McManus and A. T. Papenfuss, Blood, 2015, 125, 2815–2824 CrossRef CAS PubMed.
- I. Dzneladze, R. He, J. F. Woolley, M. H. Son, M. H. Sharobim, S. A. Greenberg, M. Gabra, C. Langlois, A. Rashid, A. Hakem, N. Ibrahimova, A. Arruda, B. Lowenberg, P. J. Valk, M. D. Minden and L. Salmena, Leukemia, 2015, 29, 1485–1495 CrossRef CAS PubMed.
- P. Wang, D. Ma, J. Wang, Q. Fang, R. Gao, W. Wu, L. Cao, X. Hu, J. Zhao and Y. Li, Tumour Immunobiol., 2016, 37, 12513–12523 CrossRef CAS PubMed.
- A. S. Mer, J. Lindberg, C. Nilsson, D. Klevebring and M. Rantalainen, J. Hematol. Oncol., 2018, 11, 52–64 CrossRef PubMed.
- M. Huarte, Nat. Med., 2015, 21, 1253–1261 CrossRef CAS PubMed.
- Q. Fang, X. Chen and X. Zhi, Med. Sci. Monit., 2016, 22, 3506–3513 CrossRef CAS PubMed.
- Y. N. Yang, R. Zhang, J. W. Du, H. H. Yuan, Y. J. Li, X. L. Wei, X. X. Du, S. L. Jiang and Y. Han, Cancer Cell Int., 2018, 18, 164–174 CrossRef CAS PubMed.
- Q. Li, W. Song and J. Wang, Biomed. Pharmacother., 2019, 109, 1793–1801 CrossRef CAS PubMed.
- S. Fang, X. Zeng, W. Zhu, R. Tang, Y. Chao and L. Guo, Exp. Mol. Pathol., 2014, 96, 438–444 CrossRef CAS PubMed.
- T. F. Zhao, H. Z. Jia, Z. Z. Zhang, X. S. Zhao, Y. F. Zou, W. Zhang, J. Wan and X. F. Chen, Mol. Med. Rep., 2017, 16, 3687–3693 CrossRef CAS PubMed.
- L. Y. Sun, X. J. Li, Y. M. Sun, W. Huang, K. Fang, C. Han, Z. H. Chen, X. Q. Luo, Y. Q. Chen and W. T. Wang, Mol. Cancer, 2018, 17, 127–132 CrossRef PubMed.
- N. Sonda, F. Simonato, E. Peranzoni, B. Calì, S. Bortoluzzi, A. Bisognin, E. Wang, F. M. Marincola, L. Naldini and B. Gentner, Immunity, 2013, 38, 1236–1249 CrossRef CAS PubMed.
- W. W. Shen, Z. Zeng, W. X. Zhu and G. H. Fu, J. Mol. Med., 2013, 91, 989–1000 CrossRef CAS PubMed.
- M. I. Almeida, R. M. Reis and G. A. Calin, Expert Opin. Biol. Ther., 2012, 12, 1153–1159 CrossRef CAS PubMed.
- I. U. Agoulnik, M. C. Hodgson, W. A. Bowden and M. M. Ittmann, Oncotarget, 2011, 2, 321–328 CrossRef PubMed.
- J. A. Gasser, H. Inuzuka, A. W. Lau, W. Wei, R. Beroukhim and A. Toker, Mol. Cell, 2014, 56, 595–607 CrossRef CAS PubMed.
- I. Dzneladze, R. He, J. F. Woolley, M. H. Son, M. H. Sharobim, S. A. Greenberg, M. Gabra, C. Langlois, A. Rashid and A. Hakem, Leukemia, 2015, 29, 1485–1495 CrossRef CAS PubMed.
- Y. K. Guo, M. Shi, Y. L. Qin and Y. J. Li, Zhongguo Shiyan Xueyexue Zazhi, 2017, 25, 736–742 Search PubMed.
- Q. Zhou, Y. Chen, L. Zhang, Y. Zhong, Z. Zhang, R. Wang, M. Jin, M. Gong, Y. Qiu and D. Kong, Oncotarget, 2017, 8, 39064–39076 Search PubMed.
| This journal is © The Royal Society of Chemistry 2019 |