DOI:
10.1039/C9RA07775H
(Paper)
RSC Adv., 2019,
9, 37391-37402
Partially fluorinated copolymers containing pendant piperidinium head groups as anion exchange membranes for alkaline fuel cells†
Received
25th September 2019
, Accepted 11th November 2019
First published on 15th November 2019
Abstract
A new series of partially fluorinated copolymers with varying alkyl side chain length (C3, C6 and C9) and piperidinium head groups have been synthesized and characterized in detail in an effort to improve membrane properties for alkaline fuel cell applications. The copolymers (QPAF4-Cx-pip) provided thin and bendable membranes by solution casting, and achieved high hydroxide ion conductivity up to 97 mS cm−1 in water at 80 °C. Membrane properties such as water absorbability, conductivity, and mechanical properties were tunable with the side chain length. The copolymer main chain and the piperidinium groups were both alkaline stable and the membranes retained high conductivity in 4 M KOH at 80 °C for as long as 1000 h, however, conductivity was lost in 8 M KOH due to Hofmann degradation of the side chain. QPAF4-C3-pip copolymer with the best-balanced properties as anion exchange membrane functioned well in a hydrogen/oxygen alkaline fuel cell to achieve 226 mW cm−2 peak power density at 502 mA cm−2 current density under fully humidified conditions with no back pressure.
1. Introduction
With recent ground breaking developments in hydrogen storage1 and hydrogen generation,2 fuel cells have gained more and more attention as a viable option for sustainable and green energy conversion systems competing with secondary batteries in many fields. The flagship technology, proton exchange membrane fuel cells (PEMFCs), has been commercialized for electric vehicles and residential power generation while PEMFCs still suffer from some shortcomings such as the use of costly precious metal catalysts, limited operating temperature, and insufficient durability. These shortcomings hinder the widespread dissemination of the devices using PEMFCs. As an alternative, anion exchange membrane fuel cells (AEMFCs) can offer several promising advantages.3–5 Under alkaline conditions, non-precious metal catalysts such as Ni and Co are potentially available.6,7 Other benefits include depressed fuel crossover due to reversed ion transport, better catalyst stability, and faster oxygen reduction reaction.8
However, there are some inherent drawbacks to AEMFCs that obstruct their general applicability including relatively low conductivity of hydroxide ion compared to proton and low stability of AEMs under alkaline conditions. The former has been extensively investigated using various polymer structures such as block copolymers,9–11 comb-shaped,12,13 hyperbranched14 or cross-linked polymers.15 All these different approaches have generally focused on maximizing ion exchange capacity (IEC) and promoting proper phase separated morphology, both of which contributed to improved ion conduction. For example, L. Wang and his co-workers developed a grafted LDPE with benzyl trimethylammonium head groups (IEC = 2.54 mequiv. g−1) to achieve up to 208 mS cm−1 of the ion conductivity at 80 °C, 100% RH.4 J. Wang et al. used a two-step process to create a polymer containing rigid terphenyl and N,N-dimethyl piperidinyl segments in the main chain. The resulting polymer membrane had high IEC = 2.37 mequiv. g−1 and low water uptake, and achieved high conductivity (up to 193 mS cm−1 at 95 °C).16 With these and many other efforts, the hydroxide ion conductivity of AEMs has been significantly improved approaching to that of PEMs.
When assessing alkaline stability, we need to distinguish between degradation at the polymer backbone and at the quaternary ammonium functional groups. Former typically takes place at heteroatom linkages in the backbone such as arylene ether, sulphide or sulfone groups, resulting in the cleavage of the polymer main chain and thereby lowering the membranes' mechanical properties.17–19 Therefore, in order to mitigate backbone cleavage, there is a shift towards polymer backbones that are devoid of any those heteroatom linkages20,21 for long-term alkaline stability. We previously developed such a structure; a series of novel perfluoroalkyl and fluorene based copolymers (QPAF4) having no heteroatom linkages in the main chain which provided robust membranes with high elasticity due to the unique combination of perfluoro chain and aromatics. Furthermore, partly thanks to the lack of heteroatom linkages in the polymer backbone, the membranes showed no signs of degradation in 1 M KOH at 80 °C even after 1000 h.22 Regarding the degradation of quaternary ammonium head groups and their vicinity, several pathways have been suggested depending on the substituents and/or the conditions.6 In most cases, the main pathway seems to be either nucleophilic substitution of the hydroxide ion at the α-carbon or Hofmann elimination by hydroxide attack on the β-carbon to the ammonium groups. The simplest method for introducing quaternary ammonium (QA) functionalities onto the polymer is reacting a benzyl halide with tertiary amines (typically, trimethyl amine: TMA).23,24 These membranes show relatively high ion conductivity but suffer from low stability against hydroxide ion attack partly due to the vicinity of electron donating aryl rings and reactive α-carbons.25 To avoid this degradation mechanism, there have been many studies focusing on polymers with pendant alkyl chains to distance the QA groups from the polymer backbone.9,26,27 For example, both conductivity and alkaline stability were improved for polystyrene-based AEMs by replacing C1 spacer with C4 spacer between the polymer backbone and the QA groups.28 The dependence of alkaline stability on alkyl spacer length and the mitigation mechanism of the degradation were investigated in detail by Long et al. using DFT calculations.29 According to their work, activation energy for the hydroxide ion's attack on the alkyl spacer increased with the number of carbon atoms in the spacer, peaking around 4 carbon atoms. They claimed that the alkyl spacers had steric effects and increased Mulliken charge of the β-hydrogen, both lowering the likelihood of Hofmann-elimination. It should be noted here that Parrondo et al., for example, demonstrated contradictory experimental results where benzyl TMA showed superior alkaline stability to hexyl TMA, suggesting that side chain effect on the alkaline stability might depend on the used polymer backbone.30 Therefore, while side chain strategy seems promising in improving the alkaline stability, further investigation is necessary. Alternative main strategy for improving the alkaline stability is replacing TMA with other onium head groups that feature higher half-lives in elevated pH media.25 These include quinuclidinium, piperidinium,31 morpholinium,32 guanidinium,33 phosphonium34 and imidazolium.35 Hetero-cycloaliphatic piperidinium and methylated piperidinium groups exhibited outstanding stability in alkaline solutions. The reason behind the high stability lies in the ring's conformational restrictions inhibiting the formation of transition states for both elimination and substitution reactions.25
In the present study, we combine the alkyl spacer strategy with highly stable hetero-cycloaliphatic QAs in an attempt to improve the performance of our QPAF4 as an AEM.22 A novel series of QPAF4 copolymers with systematically varying interstitial alkyl chain lengths (C3, 6, and 9) on piperidinum groups have been synthesized and characterized in detail. The effects of alkyl chain between the partially fluorinated polymer backbone and the ammonium head groups have been investigated and optimized in terms of the conductivity, alkaline stability and mechanical properties for alkaline fuel cells.
2. Experimental section
Materials and measurement details are included in the ESI†.
2.1. Synthesis of monomers
2,7-Dichlorofluorene. 2,7-Dichlorofluorene was synthesized according to our previous work.36
1,6-Di(3-chlorophenyl)-perfluorohexane (1). 1 was synthesized according to the literature.36
2,7-Dichloro-9,9-bis(3-bromopropyl)-9H-fluorene. 2,7-Dichlorofluorene (3.0 g, 12.8 mmol) and TBAB (600 mg, 1.86 mmol) were dried under vacuum for 30 min. Degassed DMSO (15 mL), 50% (w/w) NaOH aq. (15 mL), 1,3-dibromopropane (13 mL, 90 mmol) were added into the mixture and stirred under N2 for 2 h at r.t. Then, Et2O and deionized water were added into the mixture and stirred for 15 min. The organic layer was separated and dried over Na2SO4 and the solvent was evaporated. The crude product was purified by silica gel column chromatography (CHCl3/hexane = 2/8) to obtain 2,7-dichloro-9,9-bis(3-bromopropyl)-9H-fluorene as a white solid (3.2 g, 53% yield).
2,7-Dichloro-9,9-bis(6-bromohexyl)-9H-fluorene. 2,7-Dichloro-9,9-bis(6-bromohexyl)-9H-fluorene compound was synthesized according to the literature.22
2,7-Dichloro-9,9-bis(9-bromononyl)-9H-fluorene. To a mixture of 2,7-dichlorofluorene (1.0 g, 4.25 mmol), 1,9-dibromononane (8.7 mL, 42.8 mmol) and TBAB (270 mg, 0.84 mmol), 50% (w/w) KOH aq. (1 mL) was added. The mixture was heated to 95 °C and stirred for 2 h. The mixture was then poured into water and extracted with three portions of CH2Cl2 (30 mL). The combined organic layer was dried over Na2SO4, filtered, and evaporated. The crude product was purified via flash silica gel chromatography (hexane, then hexane/CH2Cl2 = 7/1) to obtain 2,7-dichloro-9,9-bis(9-bromononyl)-9H-fluorene as a pale yellow oil (1.4 g, 52% yield).
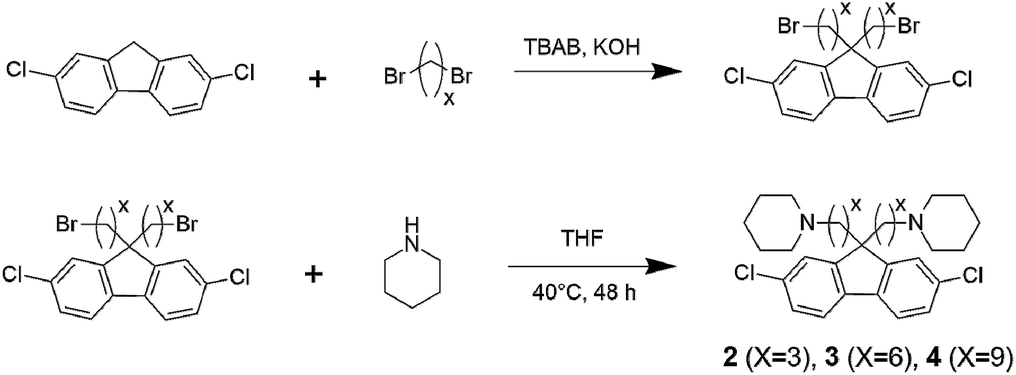
Synthesis of aminated monomers. A typical procedure for the amination of 2,7-dichloro-9,9-bis(9-bromoalkyl)-9H-fluorene is as follows. To a mixture of THF (5.0 mL) and piperidine (5.4 mL, 54.6 mmol), a solution of 2,7-dichloro-9,9-bis(3-bromopropyl)-9H-fluorene (1.3 g, 2.73 mmol) in THF (5.0 mL) was added dropwise, while the temperature was maintained around 0 °C with an ice-bath. After the addition, the mixture was stirred at 40 °C for 48 h. The solvent was evaporated. To the remaining oily product, 0.1 M KOH aq. (50 mL) was added and extracted with three portions of CH2Cl2 (20 mL). The combined organic layer was dried over Na2SO4, filtered and evaporated. Precipitation from THF/water gave 2,7-dichloro-9,9-bis(3-(1-piperidinyl)propane)-9H-fluorene (2) as a white crystalline solid (1.22 g, 92% yield). 2,7-Dichloro-9,9-bis(6-(1-piperidinyl)hexane)-9H-fluorene (3) (89% yield) and 2,7-dichloro-9,9-bis(9-(1-piperidinyl)nonane)-9H-fluorene (4) (95% yield) were obtained from the corresponding bromoalkylated compounds, respectively.
2.2. Polymerization
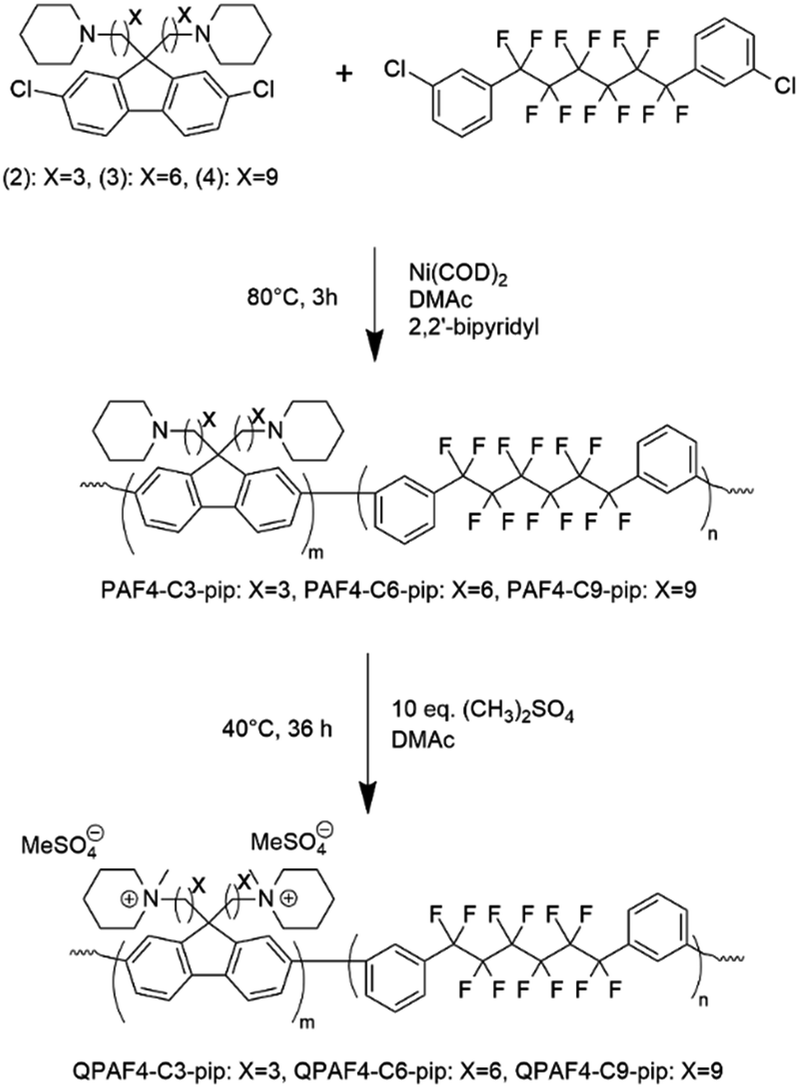
A three neck round bottom flask was equipped with a magnetic stirring bar, a reflux condenser, and nitrogen inlet and outlet. The flask was charged with 1 (725 mg, 1.39 mmol), 2 (436.5 mg, 0.9 mmol), 2,2′-bpy (892 mg, 5.72 mmol), and dry DMAc (6.0 mL). The mixture was heated to 80 °C with stirring. When a homogenous mixture was obtained, Ni(COD)2 (1.573 g, 5.72 mmol) was added to the mixture. The reaction was continued at 80 °C for 3 h. Then, the mixture was allowed to cool to r.t. before pouring dropwise into large excess of a mixture of methanol and conc. HCl (50/50 by volume). The black precipitate turned into a white fibrous solid. The crude product was filtered and washed with methanol/HCl mixture, 0.5 M K2CO3 aq. twice, and ultra-pure water twice. After drying in vacuum at 60 °C overnight, PAF4-C3-pip was obtained as a white fibrous solid (465 mg, 93% yield). PAF4-C6-pip (93% yield) and PAF4-C9-pip (75% yield) were obtained from the monomers 2 and 3, respectively.
2.3. Quaternization and membrane casting
A typical procedure is as follows. PAF4-C3-pip (450 mg, 0.711 mmol of tertiary amino groups) was dissolved in DMAc (5.0 mL) in a glass vial. Once a homogenous solution was obtained by vigorous stirring, dimethyl sulfate (472 mg, 3.55 mmol) was added dropwise to the solution. The reaction was continued at 40 °C for 36 h, and quenched by dropwise addition of the mixture into large excess of ultra-pure water (200 mL) to obtain quaternized QPAF4-C3-pip as a white precipitate. The product was filtered, washed with water, and dried in vacuum at 60 °C overnight to obtain a white solid (467 mg, 97% yield). The obtained white solid was re-dissolved in DMAc (5 mL), filtered, and cast onto a flat glass plate at 45 °C. Drying the solution overnight gave a bendable, slightly yellowish membrane, with a thickness typically ranging between 40–60 μm.
2.4. Ion exchange reaction
Before hydroxide ion conductivity and alkaline stability measurements, the membranes were ion-exchanged to hydroxide ion forms by immersing them in 1 M KOH aq. at 40 °C for 24 h. For TEM observation, membranes were ion-exchanged to tetrachloroplatinate ion forms by immersing in 0.5 M K2PtCl4 aq. at 40 °C for 24 h. For mechanical properties measurements and titration, membranes were ion-exchanged to chloride ion forms by immersing them in a solution containing 10% (w/w) NaCl and 1 M HCl at 40 °C for 24 h. To remove any remaining ions, the membranes were immersed in degassed, ultra-pure water in a tightly sealed container for 24 h at 40 °C before use.
2.5. Preparation of catalystcoated membrane (CCM) and fuel cell operation
A catalyst paste was prepared as follows. A carbon black supported Pt (50 wt%) catalyst (TEC10E50E, Tanaka Kikinzoku Kogyo) was mixed with QPAF4-C6-TMA ionomer (IEC = 2.2 mequiv. g−1) as binder and 6.4 mL of methanol in a ball mill. The dried mass ratio of the QPAF4-C6-TMA binder/catalyst was set to be 0.8. After the ball milling, the obtained paste was applied to both sides of the membranes (QPAF4-C3-pip IEC = 1.57 mequiv. g−1, 44 μm thick and QPAF4-C6-TMA IEC = 1.4 mequiv. g−1, 29 μm thick) via a pulse-swirl-spray technique. The electrode area was 4.4 cm2 and the loading amount of Pt was set to be 0.2 mg cm−2. The obtained CCMs were then ion exchanged in 1.0 M KOH for 48 h and washed with deionized water for 24 h. The CCMs were sandwiched between two gas diffusion layers (GDLs) (anode: carbon cloth, cathode: SGL 29BC for QPAF4-C3-pip and anode: SGL 29BC, cathode SGL 29BC for QPAF4-C6-TMA) with 10 kgf cm−2 for 3 min at r.t. The membrane electrode assemblies (MEAs) were placed into a single cell. Fuel cell measurements were taken at 60 °C with fully humidified hydrogen and oxygen supplied to the anode and the cathode, respectively. The flow rates of the gases were set to be 100 mL min−1 with no back pressure.
3. Results and discussion
3.1. Monomer syntheses
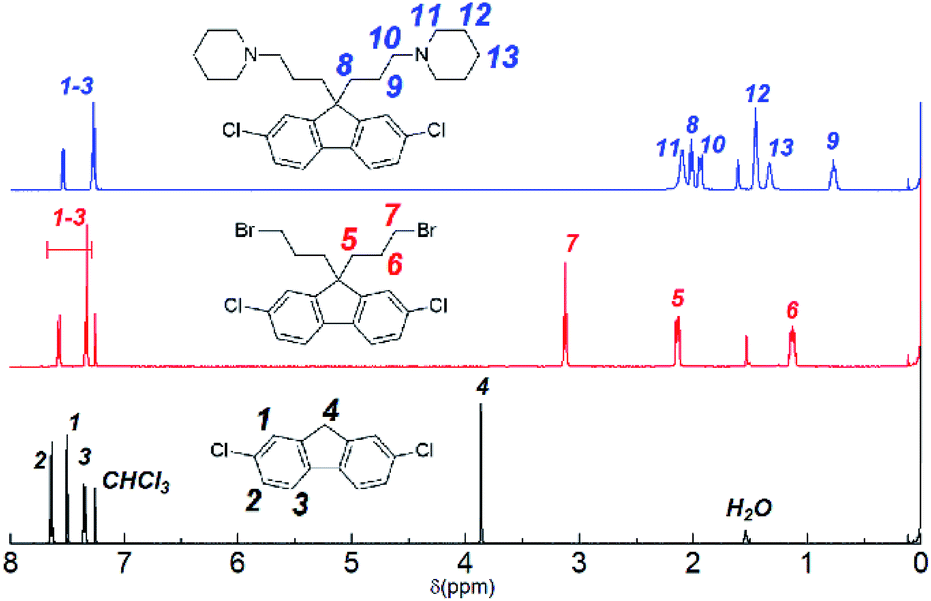
Monomer 1 containing perfluorohexylene groups was synthesized via Ullmann coupling reaction as previously reported.36 Three monomers 2, 3, and 4 containing pendant piperidine head groups with varying alkyl side chain length (x = 3, 6, and 9) were synthesized via two step reactions from 2,7-dichlorofluorene as shown in Scheme 1. In the first step, successful alkylation was confirmed by the disappearance of the peak at 3.87 ppm assignable to the two 9H protons of 2,7-dichlorofluorene in the 1H NMR spectrum (Fig. 1). Appearance of the peaks at 3.13 ppm, 3.30 ppm, and 3.37 ppm (for x = 3, 6, and 9, respectively) that were assignable to the methylene protons adjacent to the bromine groups supported the formation of the bromoalkylated dichlorofluorene. 13C NMR and mass spectra further confirmed the structure of the products (Fig. S1–S4†). The second step was the amination reaction with piperidine, which was also confirmed by 1H and 13C NMR and mass spectra (Fig. 1 and S5–S9†). The methylene proton peaks mentioned above shifted to higher magnetic field, 2.03 ppm for C3, 2.13 ppm for C6, and 2.23 ppm for C6, respectively. The new carbon peaks in the 13C NMR spectra between 25–60 ppm were assignable to piperidine rings. Overall, pure monomers (2, 3, and 4) were obtained in reasonable yields (total ca. 50%).
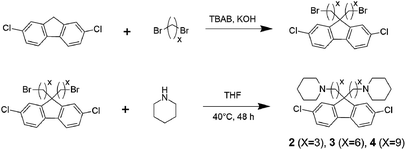 |
| | Scheme 1 Synthesis of piperidine functionalized monomers with different alkyl spacer length (2, 3, and 4). | |
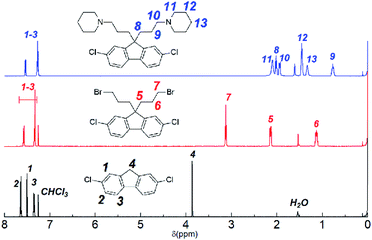 |
| | Fig. 1 1H NMR spectra (in CDCl3) of 2,7-dichlorofluorene (bottom), 2,7-dichloro-9,9-bis(3-bromopropyl)-9H-fluorene (middle), and 2,7-dichloro-9,9-bis(3-(1-piperidinyl)propane)-9H-fluorene (3) (top). | |
3.2. Polymerizations
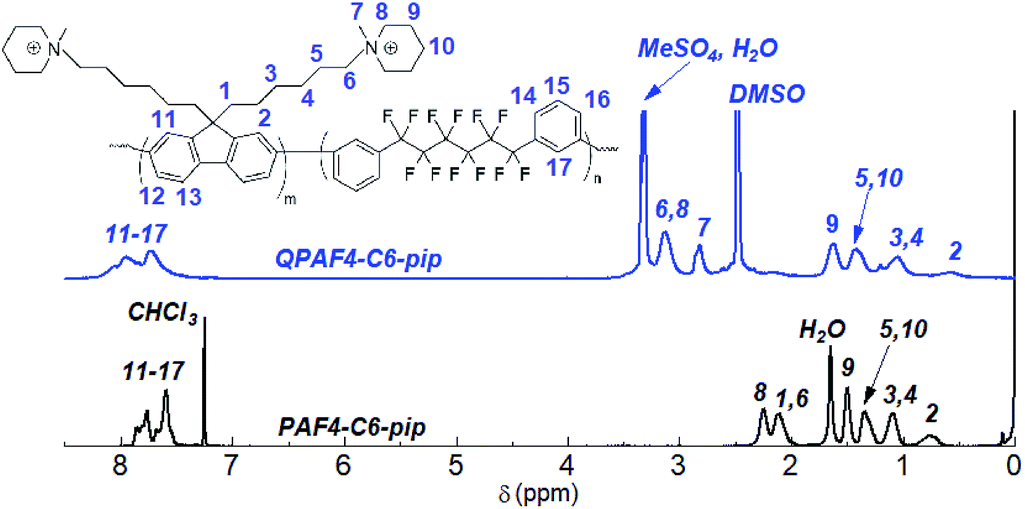
The copolymerization reaction of 1 with 2, 3, or 4 was carried out in the presence of Ni(COD)2 under the conditions similar to our previous work (Scheme 2).22 The feed ratios of the comonomers were determined so that the resulting quaternized copolymers had similar ion exchange capacity (IEC); the target IEC was 1.48–1.80 mequiv. g−1 (Table 1). In all cases, the precursor copolymers were obtained as white fibrous solids in high yields. The molecular weight of the precursor copolymers was estimated by gel permeation chromatography to be Mn = 17.7–20.7 kDa and Mw = 126.4–188.9 kDa with relatively high polydispersity indices (PDI = 7.1–9.1). The copolymers were readily soluble in CHCl3 and DMSO. A typical 1H NMR spectrum is shown in Fig. 2 for PAF4-C6-pip, where all proton peaks were well-assigned to the supposed copolymer structure. From the peak integrals, the composition of the piperidine-containing components was somewhat lower than that of the feed comonomer ratios for 2 and 3 (PAF4-C3-pip and PAF4-C6-pip, respectively), and comparable with for 4 (PAF4-C9-pip). Methylation reaction was carried out with excess dimethyl sulfate in DMAc at 40 °C for 36 h to yield the final quaternized copolymers in methyl sulfate ion forms. Methyl iodide could also be a methylation reagent, however, was not used since following ion exchange reactions were not often efficient. The quaternized copolymers were not soluble in less polar solvents (e.g., CHCl3) but in polar solvents (e.g., DMSO, DMAc, and NMP). In the 1H NMR spectrum (Fig. 2), proton peaks at 2.11 and 2.25 ppm adjacent to the tertiary amine groups in the precursor copolymers shifted to lower magnetic fields (at 3.12 ppm) after the quaternization reaction. A new prominent peak appeared at 2.82 ppm assignable to the methyl groups introduced onto the ammonium groups. The results suggest successful and quantitative quaternization reaction. Casting the quaternized copolymers from DMAc solution provided transparent and ductile membranes. Titration of the obtained membranes revealed that IEC values were 1.57 (C3), 1.48 (C6), and 1.50 (C9) mequiv. g−1, respectively. The titrated IEC values were close to those obtained by 1H NMR spectra and calculated from the feed comonomer ratios.
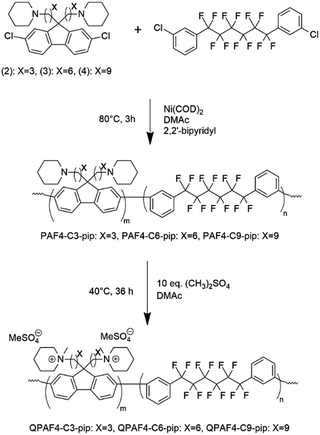 |
| | Scheme 2 Synthesis of QPAF4-C(3, 6 and 9)-pip polymers. | |
Table 1 Polymer composition, target and obtained IECs, molecular weight and yield of QPAF4-C(3, 6 and 9)-pip polymers
| No |
Monomer feed ratio |
Target IEC(mequiv. g−1) |
Obtained compositiona |
Obtained IEC (mequiv. g−1) |
Molecule weight (kDa) |
Yieldc (%) |
| m |
n |
m |
n |
NMR |
Titrationb |
Mn |
Mw |
| Determined by 1H NMR spectra. Determined by Mohr-titration. Overall yield including polymerization and quaternization reactions. |
| QPAF4-C3-pip |
0.8 |
1.54 |
1.80 |
1.0 |
1.80 |
1.63 |
1.57 |
17.7 |
126.4 |
90 |
| QPAF4-C6-pip |
0.8 |
1.68 |
1.50 |
1.0 |
1.92 |
1.47 |
1.48 |
17.6 |
128.0 |
90 |
| QPAF4-C9-pip |
0.8 |
1.60 |
1.48 |
1.0 |
1.64 |
1.42 |
1.50 |
20.6 |
188.9 |
72 |
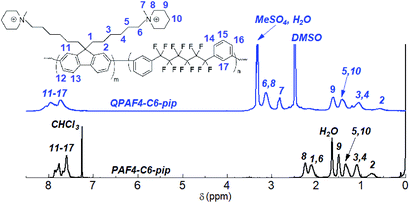 |
| | Fig. 2 1H NMR spectra of PAF4-C6-pip copolymer in CDCl3 and QPAF4-C6-pip quaternized polymer in DMSO-d6. | |
3.3. Morphology
Fig. 3 shows cross-sectional TEM images of QPAF4-C(3, 6 and 9)-pip membranes. The dark areas are the hydrophilic domains stained with tetrachloroplatinate ions, and the bright areas correspond to the hydrophobic domains. The three membranes exhibited similar phase-separated morphology with spherical hydrophilic and hydrophobic domains. The hydrophilic domain sizes increased slightly but noticeably with increasing pendant alkyl spacer length; ca. 1.1 nm, 1.2 nm, and 1.4 nm in diameter for QPAF4-C3-pip, QPAF4-C6-pip, and QPAF4-C9-pip, respectively. These hydrophilic domain sizes were smaller than those of QPAF4-C6-TMA (ca. 1.5 nm) with trimethylammonium head groups22 likely due to the more hydrophobic nature of piperidinium head groups.
 |
| | Fig. 3 TEM images of QPAF4-C(3, 6 and 9)-pip membranes stained with tetrachloroplatinate ions. | |
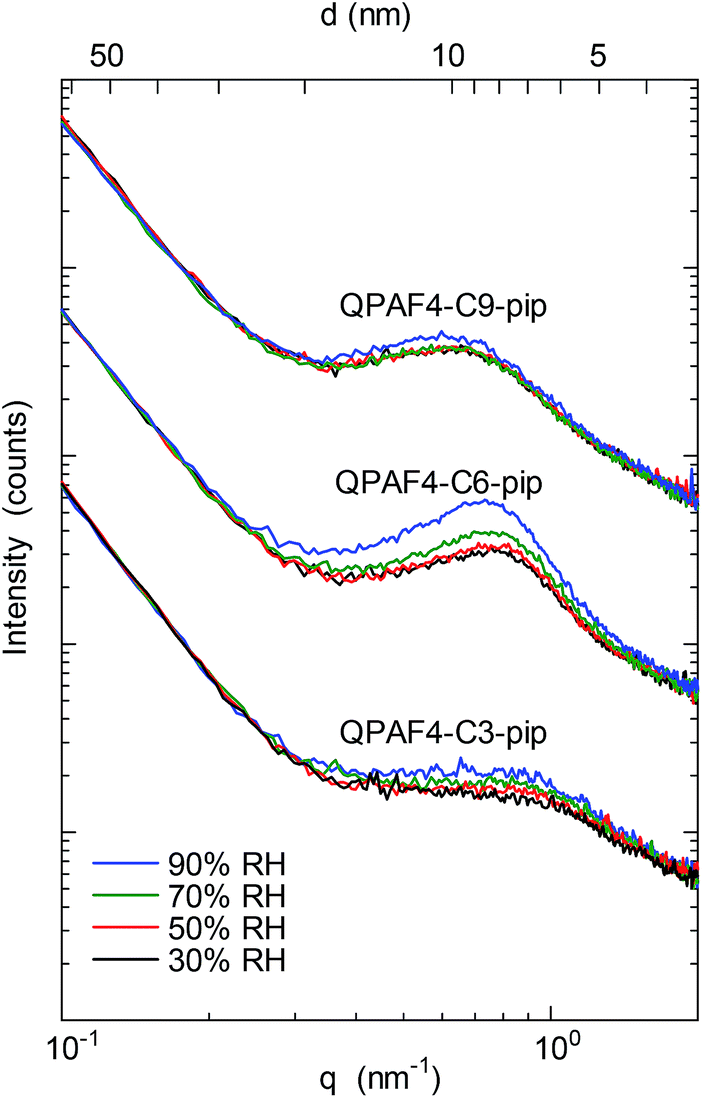
SAXS patterns of the QPAF4-C(3, 6 and 9)-pip membranes (in Cl− ion forms) at different relative humidity are shown in Fig. 4. A clear peak was observed at q (scattering vector) = 0.83 nm−1 or d (d-spacing) = 7.5 nm for C3, q = 0.7 nm−1 or d = 9.0 nm for C6, and q = 0.57 nm−1 or d = 11.0 nm for C9, respectively. For comparison, QPAF4-C6-TMA showed ca. q = 0.7 nm−1 or d = 8 nm d-spacing.22 Peak positions for QPAF4-Cx-pip membranes did not shift significantly between 30–70% RH, however, at 90% RH a slight shift towards higher d-spacing was observed (Fig. S11†) possibly due to increased water absorption. Since in all cases the peaks developed more as increasing the humidity (Fig. S12†), the d-spacing would correspond to the periodic distance between the hydrophilic domains. It is not well-understood why the regularity of the hydrophilic domains increased as increasing the humidity. Overall, the SAXS patterns revealed an important tendency of increasing inter-domain spacing between hydrophilic clusters by increasing the alkyl spacer length in the polymers. This tendency is further discussed with TEM images to create a comprehensive morphological model (Fig. 5).
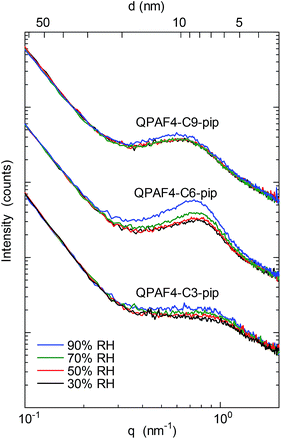 |
| | Fig. 4 SAXS patterns of QPAF4-C(3, 6 and 9)-pip membranes in Cl− ion forms at 30%, 50%, 70% and 90% relative humidity. | |
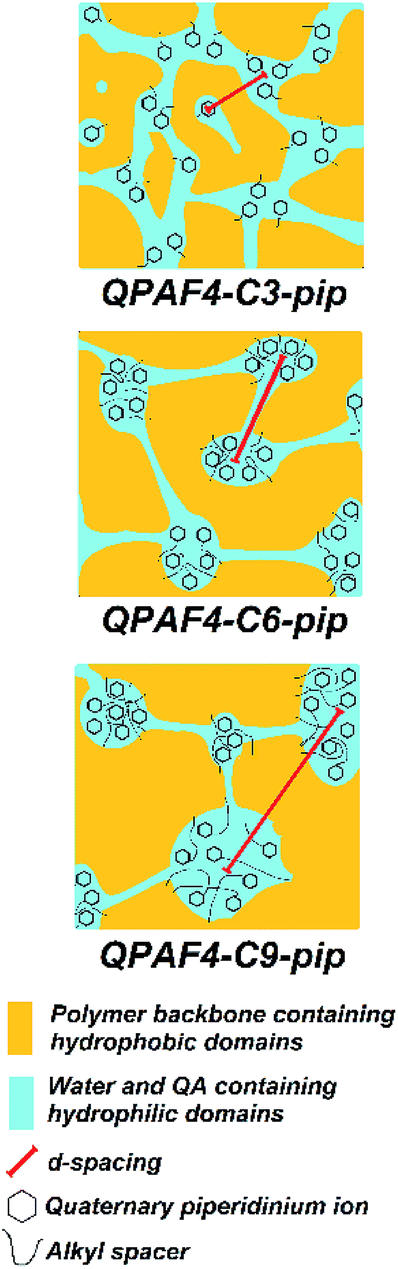
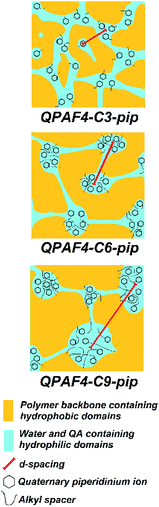 |
| | Fig. 5 Possible morphological models of QPAF4-C(3, 6 and 9)-pip polymer membranes under wet conditions based on TEM images and SAXS patterns. | |
The larger inter-domain spacing with longer pendant chain length is not contradictory to the above TEM images.37 From the titrated IECs (Table 1) and the number of hydrophilic clusters (dark regions) counted on the TEM images (Fig. 3), average number of quaternary ammonium groups per hydrophilic cluster was roughly estimated for these membranes. The average number was 1531 for QPAF4-C3-pip, 1842 for QPAF4-C6-pip and 2337 for QPAF4-C9-pip, respectively. The increase was most likely due to the alkyl spacers' contribution to the mobility of the ammonium head groups allowing the formation of larger, more distant clusters. The short alkyl spacers would restrict the hydrophilic domain size, however, seemed to facilitate a more uniform morphology. Similar results were reported by Duan et al.38 who claimed that conformational freedom of the quaternary ammonium head groups tethered with longer side chains promoted the formation of larger hydrophilic clusters. Based on the TEM images and SAXS patterns of QPAF4-pip membranes, possible morphology is illustrated in Fig. 5. With increasing the alkyl spacer length, both the average size of the hydrophilic clusters and the distance between the clusters increased. This boosted agglomeration for longer side chains led to a less efficient hydrophilic interconnectivity throughout the membrane that had severely affected, in particular, on hydroxide ion conductivity as discussed below.
3.4. Water uptake and ion conductivity
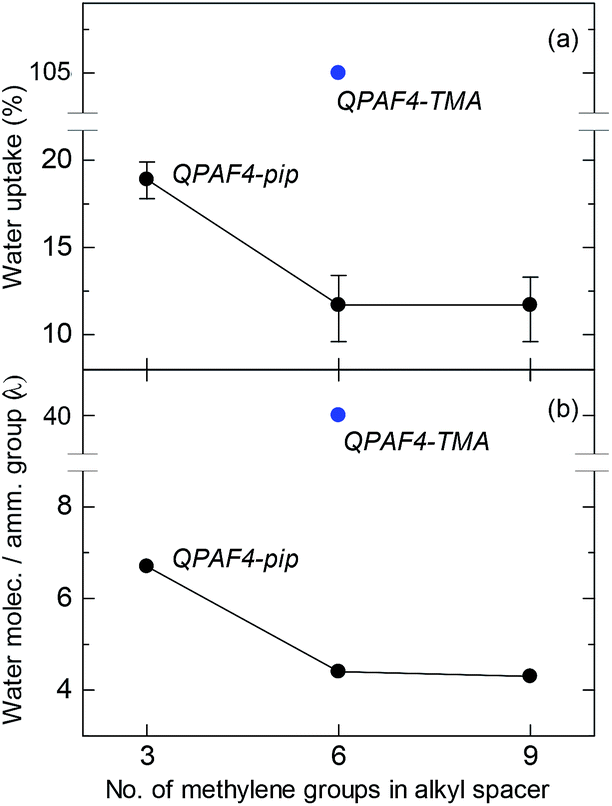
Water uptake and λ (number of absorbed water molecules per ammonium group) are plotted as a function of carbon number in the alkyl spacer in Fig. 6. A significant drop was observed for both values from C3 to C6 alkyl spacer membranes, while C6 and C9 membranes showed similar water absorption. The effect of pendant chain length on water uptake seemed dependent also on the polymer main chain structure. For example, it was reported for poly(phenylene oxide)s (PPOs) with pendant ammonium groups that the water uptake increased from C1 to C5 and then decreased as further increasing the pendant chain length from C5 to C7.39 For poly(ether sulfone) tethered with C2–C12 pendant alkyl chains end-capped with trimethylammonium groups, the maximum water uptake was observed at C6.40 It is recognized that there are two counteracting effects on water absorption; as increasing the pendant chain length, free volume in the polymer membrane increases facilitating water absorption and hydrophobicity of the side chain increases to decrease water absorbing capability (larger free volume with longer side chain is partly supported by SAXS data where larger distance between hydrophilic domains was suggested). In the present case, the water uptake did not show the maximum but simply decreased as increasing the pendant chain length presumably because of more hydrophobic nature of the piperidinium head groups compared with TMA. As illustrated in Fig. 5, the concentration of the ammonium groups in the water filled hydrophilic domains lowered as increasing the side chain length because of the increased amount of aliphatic chains also present in these domains. It is believed that the result of this intricate balance of increased free volume and dilution via alkyl chains resulted in the water uptake behavior.
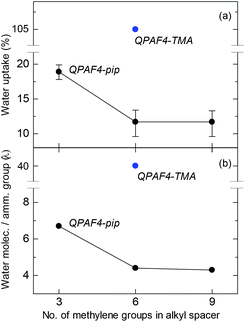 |
| | Fig. 6 (a) Water uptake and (b) λ values at 30 °C for QPAF4-C(3, 6 and 9)-pip membranes as a function of alkyl spacer length. | |
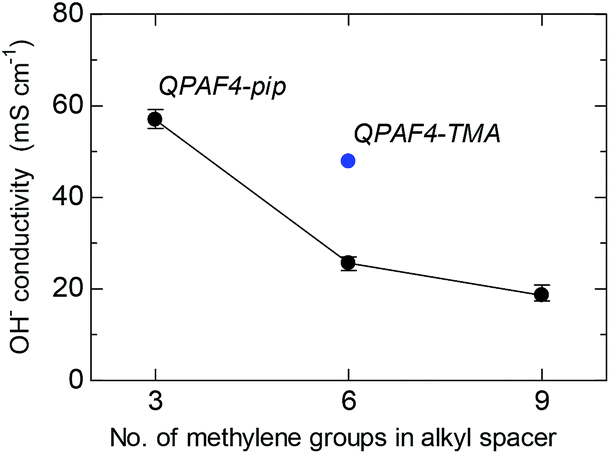
Hydroxide ion conductivity of QPAF4-C(3, 6 and 9)-pip membranes was measured in water at 30 °C and is shown in Fig. 7 as a function of carbon number in the alkyl spacer. The conductivity was 57.0 mS cm−1 for QPAF-C3-pip membrane and decreased as increasing the pendant chain length to 25.6 mS cm−1 and 18.6 mS cm−1 for QPAF4-C6-pip and QPAF4-C9-pip, respectively. The effect of the pendant chain length on the conductivity was very similar to that on water uptake. With longer alkyl spacers, the hydrophilic domains contained a more pronounced agglomeration (Fig. 5) but less inter-connected ionic channels resulting in lower water absorption and ion conductivity. The QPAF4-C6-pip membrane with bulkier and more hydrophobic head groups exhibited lower hydroxide ion conduction than that of QPAF4-C6-TMA membrane (48.7 mS cm−1).
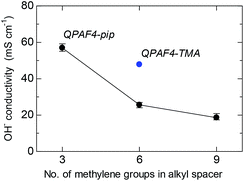 |
| | Fig. 7 Hydroxide ion conductivity of QPAF4-C(3, 6 and 9)-pip membranes in water at 30 °C as a function of alkyl spacer length. | |
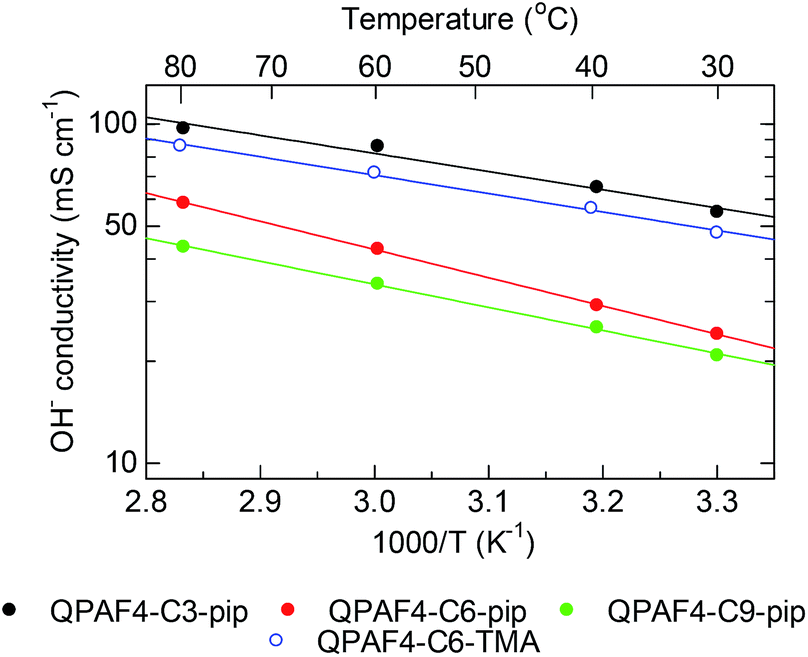
Temperature dependence of the hydroxide ion conductivity of QPAF4-C(3, 6 and 9)-pip membranes is shown in Fig. 8 with that of QPAF4-C6-TMA for comparison. QPAF4-C3-pip showed the highest conductivity (97 mS cm−1) at 80 °C, followed by QPAF4-C6-pip (59 mS cm−1) and QPAF4-C9-pip (43 mS cm−1). These membranes showed an Arrhenius-type temperature dependence of the conductivity within the tested temperature range. Apparent activation energies (Ea) obtained from the slopes were between 10.3-14.8 kJ mol−1, which were in the same range as that of QPAF4-C6-TMA membrane (10.4 kJ mol−1). The results suggest similar ion conduction mechanism (most possibly, Grotthuss mechanism) independent on alkyl spacer length, morphology or the ammonium head groups.
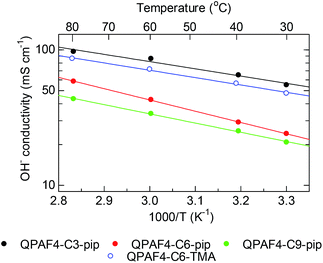 |
| | Fig. 8 Temperature dependence of hydroxide ion conductivity of QPAF4-C(3, 6 and 9)-pip membranes in water. | |
3.5. Mechanical properties
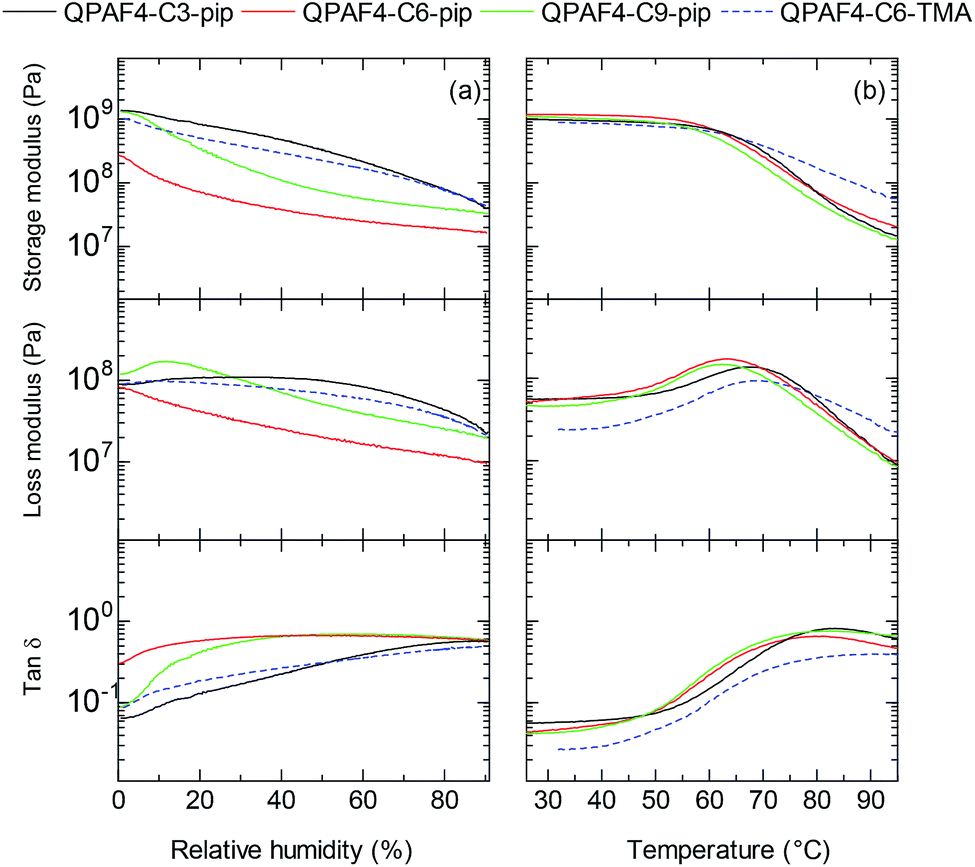
The mechanical properties of the membranes were tested via dynamic mechanical analysis (DMA), where all membranes were analysed in Cl− ion forms to avoid the effect of CO2 from the atmosphere. Storage modulus (E′), loss modulus (E′′) and tan δ (= E′′/E′) were measured as a function of relative humidity at 80 °C (Fig. 9a) and as a function of temperature at 60% RH (Fig. 9b). In the humidity dependence, although the membranes did not exhibit peaks associated with the glass transition (Tα), a clear decrease was observed in both storage and loss moduli. The humidity dependence of E′ and E′′ values was not corresponding to the alkyl spacer length, ammonium head groups, and water absorbing capability of the membranes. In the temperature dependence, the storage moduli of the membranes decreased at ca. 60 °C regardless of their pendant carbon length and ammonium head groups. The loss moduli exhibited a peak for QPAF4-Cx-pip membranes at 62.4 °C, 63.4 °C and 67.5 °C for C9, C6 and C3, respectively, and for QPAF4-C6-TMA at 68.4 °C. It seemed that longer side chain as flexible spacer caused lower transition temperature for QPAF4-Cx-pip membranes. Higher transition temperature of QPAF4-C6-TMA membrane despite its much higher water uptake must be related with the small and compact ammonium head groups. The hydrophilic component (alkyl side chains and ammonium head groups) had somewhat smaller effect on the viscoelastic properties than that on the other membrane properties probably because the hydrophobic main chains were also contributable.
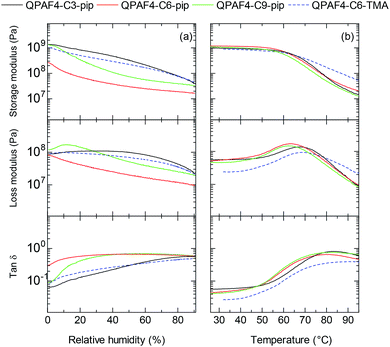 |
| | Fig. 9 DMA curves of QPAF4-C(3, 6, 9)-pip and QPAF4-TMA membranes: (a) temperature dependence at 60% RH, (b) humidity dependence at 80 °C. | |
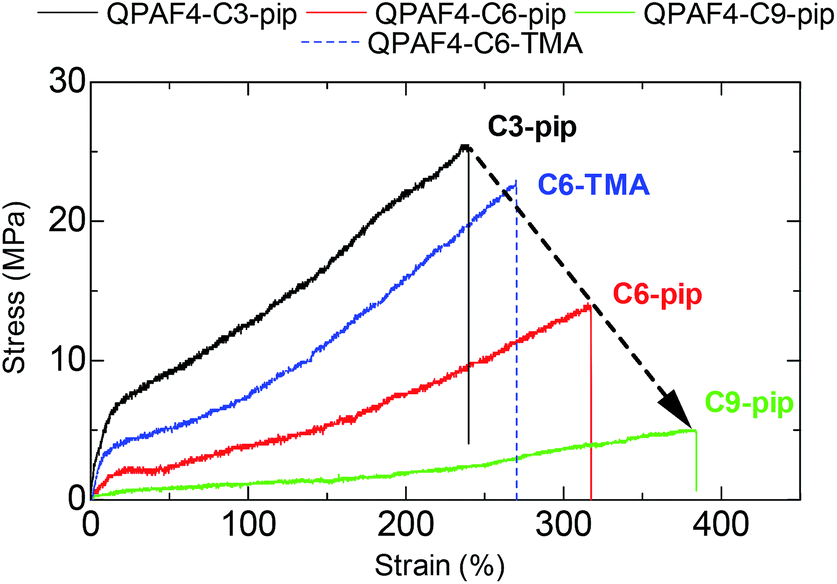
Tensile strengths of the membranes were determined via stress versus strain (SS) measurements at 80 °C and 60% RH in Cl− forms (Fig. 10). The side chain length greatly affected the tensile strength of the membranes; maximum stress decreased and elongation at break increased as increasing the side chain length. The highest elongation at break was achieved with QPAF4-C9-pip (384%) and the highest yield stress was achieved with QPAF4-C3-pip (25.5 MPa). Similar results were reported for poly(arylene ether ketone)s with pendant multi cation system41 and with imidazole functionalized long-side chain structure.42 It is considered that longer flexible side chains functioned as internal plasticizers by increasing free volume. As discussed above, the longer side chains formed larger, less inter-connected hydrophilic domains (Fig. 5) within the membrane, which in turn could have caused similar morphological effect on the hydrophobic domains to account for the smaller maximum stress with longer side chain length. Therefore, changing the morphology via varying the side chains not only affected the water uptake and ion conductivity but the mechanical properties of the membranes as well. Comparison with QPAF4-C6-TMA revealed that the bulky piperidinium head groups also increased the free volume causing smaller yield stress and higher elongation of the membranes. Overall, the stretching properties of the QPAF4 membranes were controllable with the hydrophilic components without altering the viscoelastic properties and their humidity/temperature dependence.
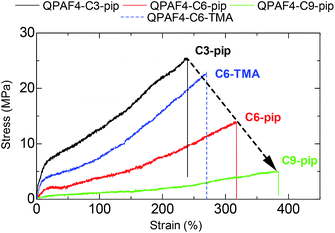 |
| | Fig. 10 Stress vs. strain curves of QPAF4-C(3, 6, 9)-pip and QPAF4-C6-TMA membranes at 80 °C and 60% RH. | |
3.6. Alkaline stability
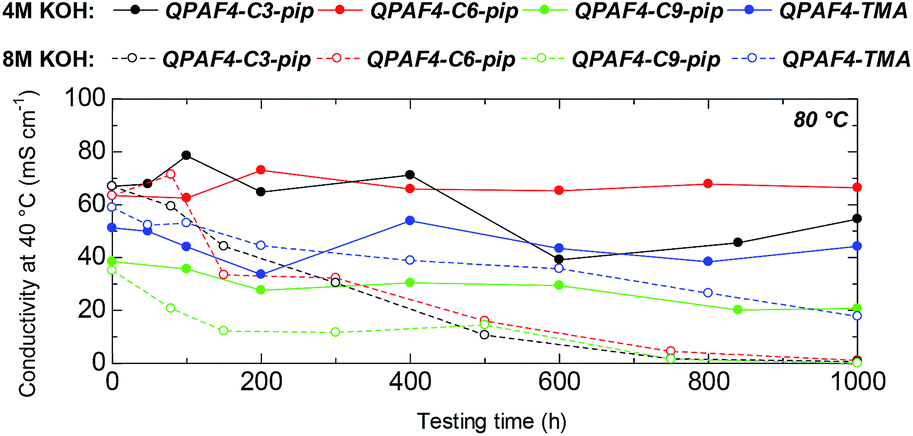
Alkaline stability of the membranes was tested in 4 M and 8 M KOH at 80 °C for 1000 h (Fig. 11). In 4 M KOH, QPAF4-C6-pip showed the highest stability with no practical loss of the hydroxide ion conductivity (100% remaining of the conductivity). The stability was followed by QPAF4-C3-pip with 81% remaining of the conductivity and QPAF4-C9-pip with 54% remaining of the conductivity, respectively. For comparison, QPAF4-C6-TMA exhibited 86% remaining of the conductivity. Under the same conditions, QPAF4-C6-pip seemed more alkaline stable than QPAF4-C6-TMA.
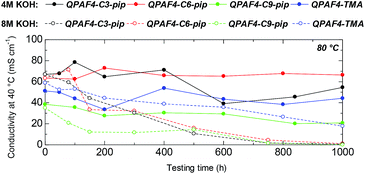 |
| | Fig. 11 Time course of OH− conductivity of QPAF4-C(3, 6 and 9)-pip and QPAF4-C6-TMA membranes in 4 M and 8 M KOH at 80 °C. | |
In 8 M KOH, the hydroxide ion conductivity significantly decreased with testing time for all three QPAF4-Cx-pip membranes. The conductivity after 1000 h was 0.6 mS cm−1 (1% remaining), 1.1 mS cm−1 (2% remaining), and 0.3 mS cm−1 (1% remaining) for QPAF4-C3-pip, –C6-pip, and –C9-pip, respectively. The large loss of the conductivity was a result of the decrease in IEC (Table 2). QPAF4-C6-TMA achieved 30% remaining after 1000 h revealing better alkaline stability than QPAF4-Cx-pip membranes. Compared to the previous work on alkaline stability of small molecules in which quaternary piperidinium compound showed longer half-life than tetramethylammonium ion in 10 M KOH at 160 °C,25 alkaline stability of QPAF4 was not improved with the piperidinium head groups. Jannasch et al. also compared chemical stability in 2 M NaOH at 90 °C between poly(arylene fluorene) polymers having trimethylammonium and piperidinium groups attached to the fluorene groups via C6 spacers and found similar results of TMA having superior stability.43 It was argued that the alkylene spacer inflicts conformational strain on the piperidinyl ring which initiated ring opening elimination most likely at the β-protons.
Table 2 Young's modulus and IEC of the pristine and post alkaline stability (in 8 M KOH after 1000 h) QPAF4-C(3, 6 and 9)-pip membranes
| No |
IECa (mequiv. g−1) |
Young's modulus (MPa) |
Yield stress (MPa) |
Elongation (%) |
| Initial |
Post-test |
Initial |
Post-test |
Initial |
Post-test |
Initial |
Post-test |
| Determined by Mohr-titration. |
| QPAF4-C3-pip |
1.57 |
0.37 |
38.3 |
302.9 |
25.5 |
21.1 |
240 |
196 |
| QPAF4-C6-pip |
1.48 |
0.38 |
4.4 |
8.3 |
14.1 |
15.4 |
317 |
325 |
| QPAF4-C9-pip |
1.50 |
0.51 |
1.2 |
33.2 |
5.0 |
12.6 |
384 |
112 |
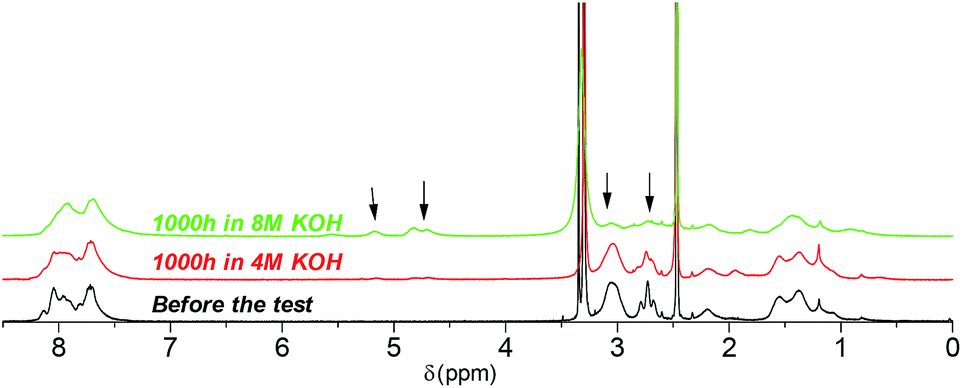
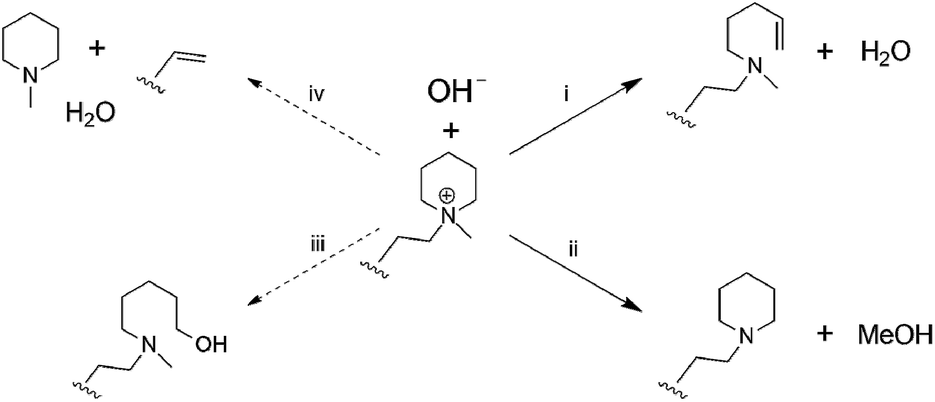
Post-test membranes were subjected to 1H and 19F NMR analyses to investigate the degradation mechanism. Unfortunately, since some membranes became insoluble after the test, only QPAF4-C3-pip after 4 M and 8 M KOH stability tests and QPAF4-C6-pip after 4 M KOH stability test were available. Fig. 12 compares the 1H NMR spectra of the pristine QPAF4-C3-pip and the post-test membranes. The sample after the stability test in 4 M KOH showed small changes corresponding to 20% loss of IEC, which explained high conductivity retention (Fig. 11). In the spectrum of the sample after 8 M KOH stability test, peaks appeared at 4.5–4.8 ppm and 5.3 ppm that are typical for protons connected to unsaturated carbon centres. This is indicative of degradation via Hofmann-elimination. Since the peak at 2.2 ppm assignable to the alkyl spacer (methylene protons adjacent to the nitrogen atoms) did not decrease, ring opening Hofmann elimination is probable ((i) in Fig. 13). Decreased intensity of the peaks at 2.6–2.8 ppm and 2.9–3.2 ppm also supports ring-opening Hofmann elimination via loss of methylene protons located next to the quaternary ammonium groups.
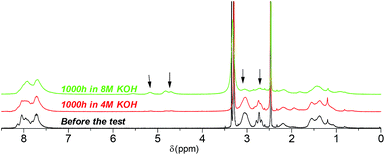 |
| | Fig. 12 1H NMR spectra of QPAF4-C3-pip (in Cl− forms, DMSO-d6) before and after the alkaline stability tests in different concentration of KOH aqueous solution at 80 °C. | |
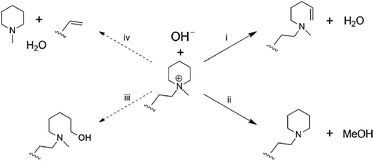 |
| | Fig. 13 Possible hydroxide ion induced degradation mechanisms of the piperidinium end-capped alkyl spacers. (i) Ring opening Hofmann-elimination, (ii) demethylation, (iii) SN2 of hydroxide ion, and (iv) chain scission of alkyl spacer via Hofmann-elimination. | |
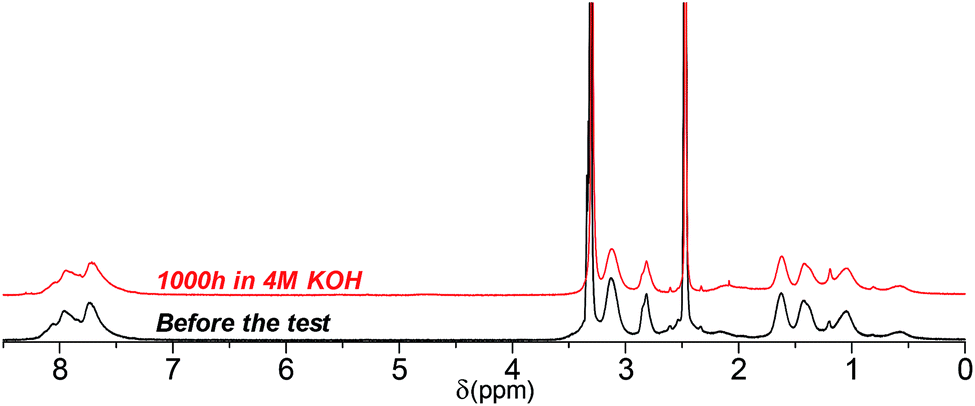
Similar to its C3 counterpart, 1H NMR spectrum of QPAF4-C6-pip showed no or minor signs of degradation after 1000 h in 4 M KOH (Fig. 14). Under these conditions, the loss of IEC after the test was merely 5%. The result was in accordance with the high conductivity remaining as discussed above (Fig. 11). Unfortunately, since QPAF4-C6-pip membrane after 1000 h in 8 M KOH was insufficiently soluble in both DMSO-d6 and CDCl3, NMR analysis was unavailable. The change in solvent solubility is indicative of degradation involving structural changes in the side chain (ring-opening Hofmann elimination) since the membrane did not show changes in flexibility or overall physical appearance which are more related with the polymer main chains. This idea was supported by the fact that the post-test samples, regardless of the testing conditions, showed no changes in the 19F NMR spectra; the perfluoroalkylene moiety in the main chains were highly robust under strongly alkaline conditions (Fig. S13†). The change in the solubility was possibly caused by cross-linking reactions through the side chains, where unsaturated carbon bonds were likely to undergo coupling reactions under heated conditions.44 QPAF4-C9-pip membrane could have experienced this type of in situ cross-linking reaction more than the C3 and C6 analogues during prolonged exposure to heat under alkaline stability test conditions. It is probable that the longer side chains having larger conformational freedom had higher chances of the cross-linking reaction.
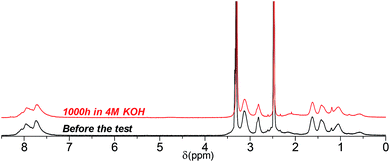 |
| | Fig. 14 1H NMR spectra of QPAF4-C6-pip (in Cl− forms, DMSO-d6) before and after the alkaline stability test in 4 M KOH at 80 °C. | |
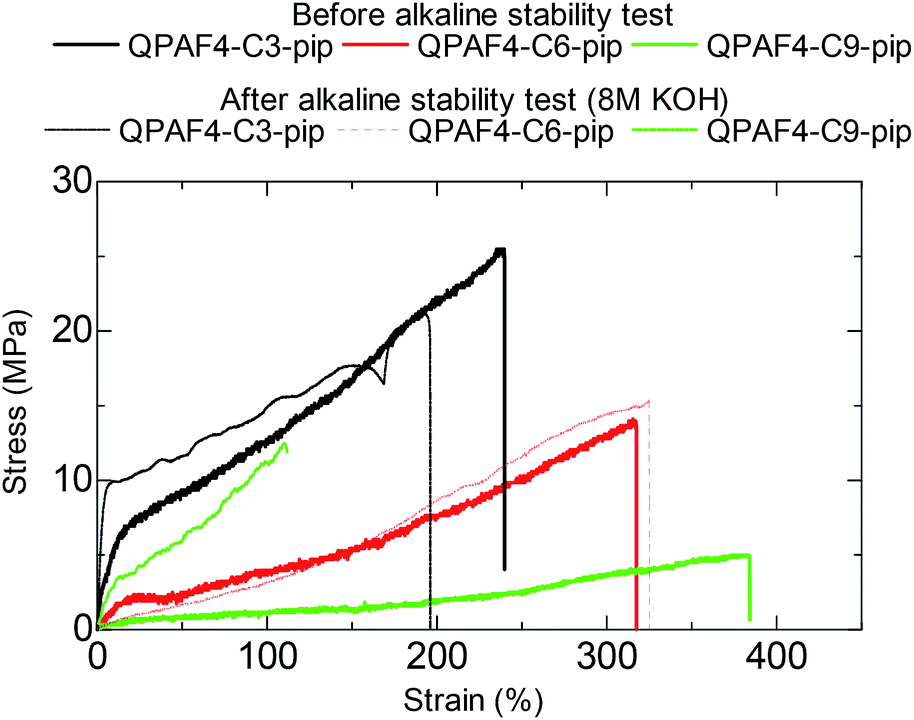
Fig. 15 shows the stress vs. strain curves of the pristine and post-test (in 8 M KOH after 1000 h) membranes. As an example, the post-test QPAF4-C9-pip showed considerably lower elongation (112%) and higher yield stress (12.6 MPa) and higher Young's modulus (33.2 MPa) compared to those (384%, 5.0 MPa, and 1.2 MPa, respectively) of the pristine membrane. These changes in tensile properties also suggest possible cross-linking to some extent.45 In addition, loss of quaternary ammonium groups inevitably caused lower water absorbability which should result in increased Young's modulus by making the membrane stiffer and less elastic.46 QPAF4-C3 and 6-pip membranes exhibited much smaller changes in SS curves after the alkaline stability test, indicating smaller degree of the cross-linking reaction. It was likely that for QPAF4 membranes chemical structure of the polymer was a more crucial factor than the membrane morphology in alkaline stability.
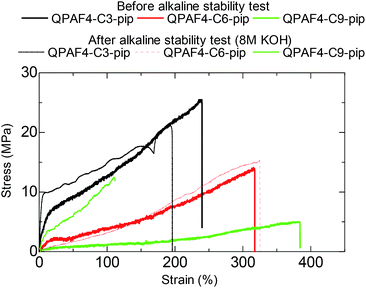 |
| | Fig. 15 Stress vs. strain curves of QPAF4-C(3, 6 and 9)-pip membranes before and after the alkaline stability test in 8 M KOH for 1000 h. | |
3.7. Fuel cell performance
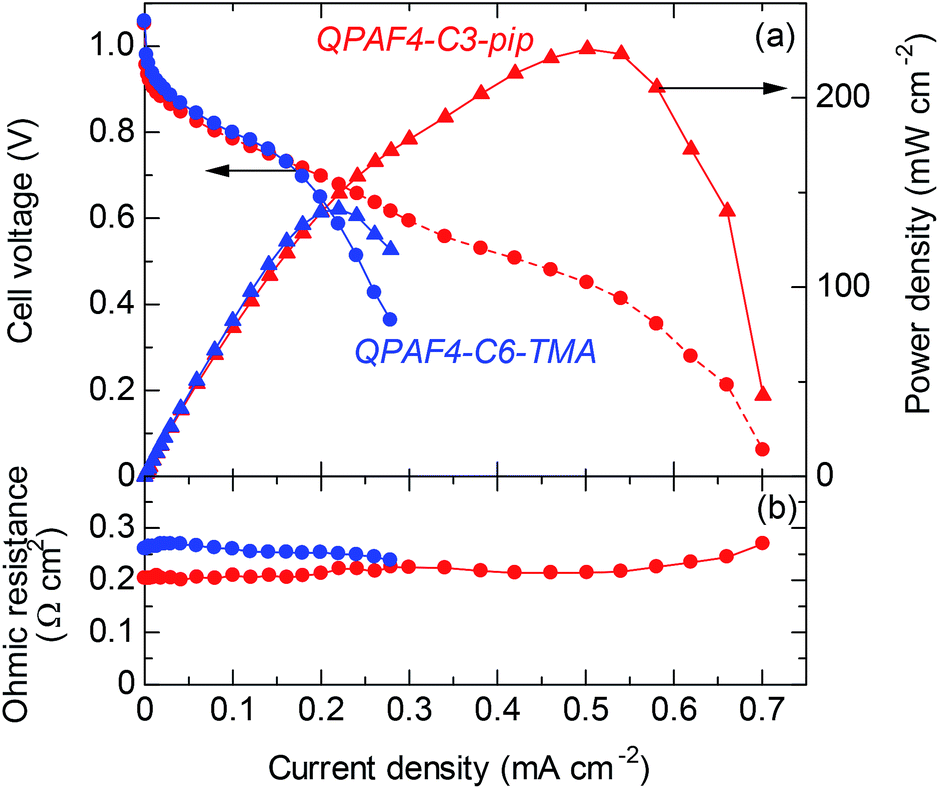
For fuel cell performance evaluation, QPAF4-C3-pip was chosen as it exhibited the highest hydroxide ion conductivity. The cell voltage and power density of the cell operated at 60 °C with fully humidified hydrogen and oxygen are plotted as a function of current density in Fig. 16a. Data for QPAF4-C6-TMA membrane cell are also included for comparison. The open circuit voltage (OCV) was as high as ca. 1.05 V for both cells, suggesting similarly high gas impermeability of the QPAF4-C3-pip and QPAF4-C6-TMA membranes. (Please note that hydrogen and oxygen permeabilities of QPAF4-C6-TMA membrane (in Cl− form) were 2.81 × 10−9 cm3 cm s−1 cm−2 cmHg−1 and 1.73 × 10−9 cm3 cm s−1 cm−2 cmHg−1, respectively, at 80 °C and 90% RH as reported in our previous paper.22) The ohmic resistance was ca. 0.21 Ω cm2 for QPAF4-C3-pip cell (increased slightly at higher current density) and ca. 0.26 Ω cm2 for QPAF4-C6-TMA cell. The membrane resistances calculated from the hydroxide ion conductivity at 60 °C in water (Fig. 8) and the thickness of the membrane were 0.066 Ω cm2 for QPAF4-C3-pip, and 0.073 Ω cm2 for QPAF4-C6-TMA, respectively. The ohmic resistances of the cells were more than 3 times higher than those calculated from the conductivity, most probably because of the lower ion conductivity of the membranes in humidified conditions than in water. The IV curves were similar for both cells at low current density (<0.2 A cm−2). At higher current density, QPAF4-C3-pip cell exhibited higher performance since it utilized carbon cloth as anode gas diffusion layer (GDL) compared to carbon paper GDL for QPAF4-C6-TMA cell. The results indicate that carbon cloth GDL had better mass (‘water’ in the present case) transport capability resulting in lower diffusion polarization at the anode. The maximum power density was 226 mW cm−2 at 502 mA cm−2 for QPAF4-C3-pip cell and 141 mW cm−2 at 220 mA cm−2 for QPAF4-C6-TMA cell respectively. The fuel cell performance of the QPAF4-C3-pip cell was reasonably high compared with state-of-the-art AEMFCs reported in the literature47–49 considering that no backpressure for hydrogen and oxygen supplied to the cell and reasonably low Pt loading (0.2 mg cm−2) used for the electrodes.
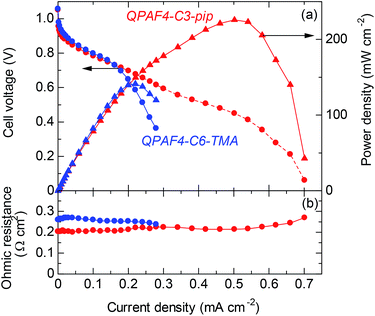 |
| | Fig. 16 (a) Cell voltage and power density and (b) ohmic resistance as a function of the current density of QPAF4-C3-pip (44 μm) and QPAF4-C6-TMA (29 μm) alkaline fuel cells operated at 60 °C and 100% RH. | |
4. Conclusions
In the present study, effect of interstitial alkyl spacer length and hetero-cycloaliphatic quaternary ammonium (piperidinium) groups were evaluated for partially fluorinated copolymers (QPAF4) as anion exchange membranes for alkaline fuel cell applications. Water uptake decreased with increasing alkyl spacer length, especially from C3 (19%) to C6 (12%) due to increased hydrophobicity. Similarly, QPAF4-C6-pip membrane exhibited much lower water affinity than QPAF4-C6-TMA because of stronger hydrophobicity of piperidinium head groups. With increasing side chain length, hydrophilic/hydrophobic phase separation developed more because of more freedom in molecular motion for the piperidinium head groups. The hydroxide ion conductivity also increased tendentiously (from 19 mS cm−1 to 59 mS cm−1 at 30 °C, for C9-pip to C3-pip, respectively) with decreasing alkyl spacer length. Mechanical properties such as stress/stretching curves were tunable via changing the side chain length. Elongation jumped from 240% to 384%, yield stress dropped from 25.5 MPa to 5.0 MPa when shifting alkyl spacer from C3 to C9. For C3 and C6 alkyl spacer polymers, hydroxide ion induced degradations were minimal even under harsh conditions (4 M KOH, 80 °C, 1000 h). However, the use of piperidinium as QA instead of TMA led to severer degradation, especially in 8 M KOH, due to susceptibility of β-hydrogens to Hofmann-elimination. An alkaline fuel cell using QPAF4-C3-pip as membrane and electrode binder exhibited low ohmic resistance and high fuel cell performance reflecting high hydroxide ion conductivity of the membrane. The obtained maximum power density was not among the best reported in the literature, however, reasonably high taking into consideration low platinum catalyst loading and no backpressure operation.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was partly supported by Japan Society for the Promotion of Science (JSPS) through a Grant-in-Aid for Scientific Research (KAKENHI JP18H02030, JP18H05515, JP18K19111) and Japan/Swiss joint research project (JRP), by Japan Science and Technology Agency (JST) through Japan/China joint research project (SICORP, JPMJSC18H8), and by New Energy and Industrial Technology Development Organization (NEDO) of Japan through the fund for “Advanced Research Program for Energy and Environmental Technologies”. The authors thank Tosoh Finechem Co. for supplying 1,6-diiodo-dodecafluorohexane.
Notes and references
- L. Morris, J. J. Hales, M. L. Trudeau, P. Georgiev, J. P. Embs, J. Eckert, N. Kaltsoyannis and D. M. Antonelli, Energy Environ. Sci., 2019, 12, 1580–1591 RSC.
- S. Tembhurne, F. Nandjou and S. Haussener, Nat. Energy, 2019, 4, 399–407 CrossRef.
- Y. Oshiba, J. Hiura, Y. Suzuki and T. Yamaguchi, J. Power Sources, 2017, 345, 221–226 CrossRef CAS.
- L. Wang, M. Bellini, H. A. Miller and J. R. Varcoe, J. Mater. Chem. A, 2018, 6, 15404–15412 RSC.
- T. J. Omasta, A. M. Park, J. M. LaManna, Y. Zhang, X. Peng, L. Wang, D. L. Jacobson, J. R. Varcoe, D. S. Hussey, B. S. Pivovar and W. E. Mustain, Energy Environ. Sci., 2018, 11, 551–558 RSC.
- J. R. Varcoe, P. Atanassov, D. R. Dekel, A. M. Herring, M. A. Hickner, P. A. Kohl, A. R. Kucernak, W. E. Mustain, K. Nijmeijer, K. Scott, T. Xu and L. Zhuang, Energy Environ. Sci., 2014, 7, 3135–3191 RSC.
- G. Borisov, H. Penchev, K. M. Dimitrova, F. Ublekov, E. Lefterova, V. Sinigersky and E. Slavcheva, Mater. Lett., 2019, 240, 144–146 CrossRef CAS.
- M. Niu, C. Zhang, G. He, F. Zhang and X. Wu, Int. J. Hydrogen Energy, 2019, 44, 15482–15493 CrossRef CAS.
- C. X. Lin, X. Q. Wang, L. Li, F. H. Liu, Q. G. Zhang, A. M. Zhu and Q. L. Liu, J. Power Sources, 2017, 365, 282–292 CrossRef CAS.
- E. Kim, S. Lee, S. Woo, S. H. Park, S.-D. Yim, D. Shim and B. Bae, J. Power Sources, 2017, 359, 568–576 CrossRef CAS.
- B. Bae, K. Miyatake and M. Watanabe, Macromolecules, 2010, 43, 2684–2691 CrossRef CAS.
- N. Li, T. Yan, Z. Li, T. Thurn-Albrecht and W. H. Binder, Energy Environ. Sci., 2012, 5, 7888–7892 RSC.
- N. Li, Y. Leng, M. A. Hickner and C.-Y. Wang, J. Am. Chem. Soc., 2013, 135, 10124–10133 CrossRef CAS PubMed.
- Q. Ge, X. Liang, L. Ding, J. Hou, J. Miao, B. Wu, Z. Yang and T. Xu, J. Membr. Sci., 2019, 573, 595–601 CrossRef CAS.
- J. Zhou, M. Ünlü, I. Anestis-Richard and P. A. Kohl, J. Membr. Sci., 2010, 350, 286–292 CrossRef CAS.
- J. Wang, Y. Zhao, B. P. Setzler, S. Rojas-Carbonell, C. B. Yehuda, A. Amel, M. Page, L. Wang, K. Hu, L. Shi, S. Gottesfeld, B. Xu and Y. Yan, Nat. Energy, 2019, 4, 392–398 CrossRef CAS.
- D. Chen and M. A. Hickner, ACS Appl. Mater. Interfaces, 2012, 4, 5775–5781 CrossRef CAS PubMed.
- S. Kim, J. Yan, B. Schwenzer, J. Zhang, L. Li, J. Liu, Z. G. Yang and M. A. Hickner, Electrochem. Commun., 2010, 12, 1650–1653 CrossRef CAS.
- C. Fujimoto, D.-S. Kim, M. Hibbs, D. Wrobleski and Y. S. Kim, J. Membr. Sci., 2012, 423, 438–449 CrossRef.
- J. Fan, S. Willdorf-Cohen, E. M. Schibli, Z. Paula, W. Li, T. J. G. Skalski, A. T. Sergeenko, A. Hohenadel, B. J. Frisken, E. Magliocca, W. E. Mustain, C. E. Diesendruck, D. R. Dekel and S. Holdcroft, Nat. Commun., 2019, 10, 2306 CrossRef PubMed.
- J. Fan, A. G. Wright, B. Britton, T. Weissbach, T. J. G. Skalski, J. Ward, T. J. Peckham and S. Holdcroft, ACS Macro Lett., 2017, 6, 1089–1093 CrossRef CAS.
- H. Ono, T. Kimura, A. Takano, K. Asazawa, J. Miyake, J. Inukai and K. Miyatake, J. Mater. Chem. A, 2017, 5, 24804–24812 RSC.
- M. M. Hossain, L. Wu, X. Liang, Z. Yang, J. Hou and T. Xu, J. Power Sources, 2018, 390, 234–241 CrossRef CAS.
- A. M. A. Mahmoud, A. M. M. Elsaghier, K. Otsuji and K. Miyatake, Macromolecules, 2017, 50, 4256–4266 CrossRef CAS.
- M. G. Marino and K. D. Kreuer, ChemSusChem, 2014, 8, 513–523 CrossRef PubMed.
- J. Han, Q. Liu, X. Li, J. Pan, L. Wei, Y. Wu, H. Peng, Y. Wang, G. Li, C. Chen, L. Xiao, J. Lu and L. Zhuang, ACS Appl. Mater. Interfaces, 2015, 7, 2809–2816 CrossRef CAS PubMed.
- A. M. A. Mahmoud and K. Miyatake, J. Mater. Chem. A, 2018, 6, 14400–14409 RSC.
- L. Wang and M. A. Hickner, Soft Matter, 2016, 12, 5359–5371 RSC.
- H. Long, K. Kim and B. S. Pivovar, J. Phys. Chem. C, 2012, 116, 9419–9426 CrossRef CAS.
- J. Parrondo, M.-S. J. Jung, Z. Wang, C. G. Arges and V. Ramani, J. Electrochem. Soc., 2015, 162, 1236–1242 CrossRef.
- H.-S. Dang and P. Jannasch, J. Mater. Chem. A, 2017, 5, 21965–21978 RSC.
- X. Yan, R. Deng, Y. Pan, X. Xu, I. E. Hamouti, X. Ruan, X. Wu, C. Hao and G. He, J. Membr. Sci., 2017, 533, 121–129 CrossRef CAS.
- T. A. Sherazi, S. Zahoor, R. Raza, A. J. Shaikh, S. A. R. Naqvi, G. Abbas, Y. Khan and S. Li, Int. J. Hydrogen Energy, 2015, 40, 786–796 CrossRef CAS.
- Y. Liu, B. Zhang, C. L. Kinsinger, Y. Yang, S. Seifert, Y. Yan, C. M. Maupin, M. W. Liberatore and A. M. Herring, J. Membr. Sci., 2016, 506, 50–59 CrossRef CAS.
- M. R. Hibbs, J. Polym. Sci., Part B: Polym. Phys., 2013, 51, 1736–1742 CrossRef CAS.
- H. Ono, J. Miyake, S. Shimada, M. Uchida and K. Miyatake, J. Mater. Chem. A, 2015, 3, 21779–21788 RSC.
- Q. Yang, L. Li, X. L. Gao, H. Y. Wu, F. H. Liu, Q. G. Zhang, A. M. Zhu, C. H. Zhao and Q. L. Liu, J. Membr. Sci., 2019, 578, 230–238 CrossRef CAS.
- X. Duan, C. Wang, T. Wang, X. Xie, X. Zhou and Y. Ye, J. Membr. Sci., 2019, 573, 64–72 CrossRef CAS.
- H.-S. Dang and P. Jannasch, Macromolecules, 2015, 48, 5742–5751 CrossRef CAS.
- C. X. Lin, X. L. Huang, D. Guo, Q. G. Zhang, A. M. Zhu, M. L. Ye and Q. L. Liu, J. Mater. Chem. A, 2016, 4, 13938–13948 RSC.
- L. Li, C. X. Lin, X. Q. Wang, Q. Yang, Q. G. Zhang, A. M. Zhu and Q. L. Liu, J. Membr. Sci., 2018, 553, 209–217 CrossRef CAS.
- J. Liao, J. Zhu, S. Yang, N. Pan, X. Yu, C. Wang, J. Li and J. Shen, J. Membr. Sci., 2019, 574, 181–195 CrossRef CAS.
- T. H. P. Ham, J. Olsson, A. Allushi and P. Jannasch, Workshop on Ion Exchange Membranes for Energy Applications 2019 DLR Institute of Networked Energy Systems, http://lup.lub.lu.se/record/7c827707-e442-4038-ac59-0d3b24530df2, 2019.
- L. Wu, Q. Pan, J. R. Varcoe, D. Zhou, J. Ran, Z. Yang and T. Xu, J. Membr. Sci., 2015, 490, 1–8 CrossRef CAS.
- D. Koronka, A. M. A. Mahmoud and K. Miyatake, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 1059–1069 CrossRef CAS.
- J. Pan, S. Lu, Y. Li, A. Huang, L. Zhuang and J. Lu, Adv. Funct. Mater., 2010, 20, 312–319 CrossRef CAS.
- N. Chen, C. Long, Y. Li, C. Lu and H. Zhu, ACS Appl. Mater. Interfaces, 2018, 10, 15720–15732 CrossRef CAS PubMed.
- M. Niu, C. Zhang, G. He, F. Zhang and X. Wu, Int. J. Hydrogen Energy, 2019, 44, 15482–15493 CrossRef CAS.
- J. Liu, X. Yan, L. Gao, L. Hu, X. Wu, Y. Dai, X. Ruan and G. He, J. Membr. Sci., 2019, 581, 82–92 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra07775h |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *bc
*bc