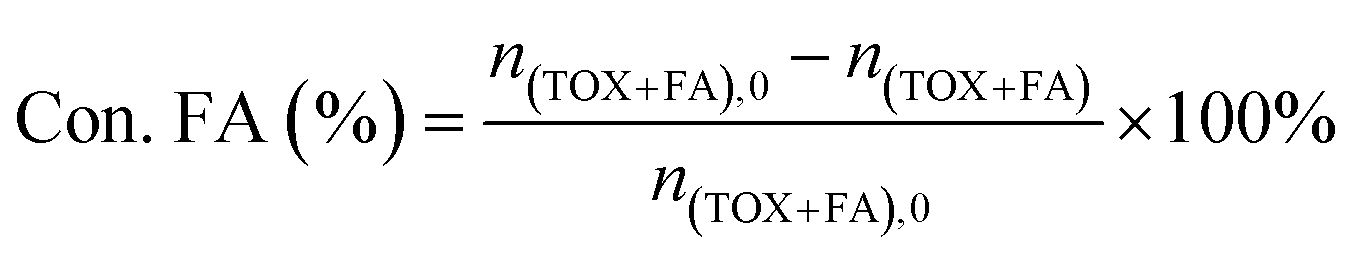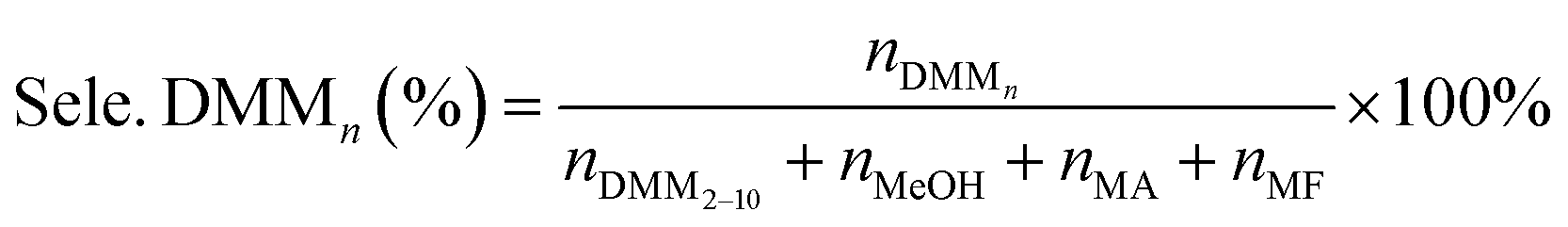DOI:
10.1039/C9RA07765K
(Paper)
RSC Adv., 2019,
9, 40662-40669
Novel polymeric acidic ionic liquids as green catalysts for the preparation of polyoxymethylene dimethyl ethers from the acetalation of methylal with trioxane
Received
25th September 2019
, Accepted 1st December 2019
First published on 9th December 2019
Abstract
A series of micro–mesoporous polymeric acidic ionic liquids (PAILs) have been successfully synthesized and subsequently characterized using Fourier transform-infrared spectroscopy, N2 adsorption–desorption isotherms, scanning electron microscopy and thermogravimetry. Furthermore, the catalytic performance of the synthesized PAILs was investigated for the acetalation of methylal (DMM1) with 1,3,5-trioxane (TOX), micro–mesoporous PAILs copolymerized by divinylbenzene with cations and anions exhibited moderate to excellent catalytic activities for the acetalation. In particular, VIMBs–AMPs–DVB, with higher specific surface area (25.51 m2 g−1) and total pore volume (0.15 cm3 g−1) displayed an elevated conversion of formaldehyde (82.2%) and selectivity for polyoxymethylene dimethyl ethers (CH3O(CH2O)nCH3; PODEn or DMMn) n = 3–8 (52.6%) at 130 °C, 3.0 MPa for 8 h. Moreover, the influence of various reaction parameters was investigated by employing VIMBs–AMPs–DVB as the catalyst and it demonstrated high thermal stability and easy recovery.
1. Introduction
Polyoxymethylene dimethyl ethers (CH3O(CH2O)nCH3, n ≥ 2; PODEn or DMMn) are considered as viable diesel additives due to their qualities of improving the efficiency of combustion and decreasing the pollutant emissions.1–3 Since their physicochemical characteristics are similar to those of diesel, they can be used in contemporary diesel engines without any hardware alterations. DMM3–8 display higher oxygen content and cetane numbers (CN), which reduce the particulate and NOx emissions during the combustion process.2–4 On the other hand, DMM2–5 are non-toxic,5 colorless, and volatile liquids, which are miscible with most common organic compounds. All of these attributes validate the suitability of these chemicals as eco-friendly industrial solvents.
DMMn are generally produced by acetalation of a methyl-group donor (methanol or methylal (DMM1)) with compounds providing the –CH2O– segment (1,3,5-trioxane (TOX), paraformaldehyde (PF) and formaldehyde (FA)) in the presence of acidic catalysts. Various acidic ionic liquids (AILs) were previously employed as catalysts for the synthesis of DMMn. Q. Wu et al.6 reported –SO3H functionalized Brønsted AILs as efficient catalysts for the preparation of DMMn from DMM1 and TOX, among which [PyBs]HSO4 showed high activity with 91.1% conversion of TOX and 70.9% selectivity for DMM3–8, respectively. The same research group studied the correlation between the carbon chain length of alkyl groups connected with AILs and their catalytic activity. It was found that [C6IMBs]HSO4 showed the best catalytic activity and the selectivity of DMM3–8 was 57.9%.7 Most recently, our team has developed a process catalyzed by AILs,8–12 which showed excellent catalytic activity and reusability. Although AILs possess high acid density, negligible volatility, and uniform catalytic sites, a few drawbacks such as high viscosity and easy losses in polar solvents limit their widespread industrial applications. Numerous attempts were made to overcome these drawbacks including adjusting the molecular structure and melting point.
Polymeric AILs (PAILs), which combine the desired catalytic properties of ILs and polymer feature of insolubility in commonly used organic solvents making them easily separated and recycled, have been investigated extensively in several recent studies. The concentration of AILs on the surface of PAILs is higher compared with other supported AILs. The most common type of PAILs are those bearing pendant AILs and acid groups in a polymer chain, prepared from the polymerization of an AILs monomer or a precursor, including imidazolium −SO3H heteropolyanion,13 poly(4-vinylpyridine),14–16 copolymerization of AILs oligomers with divinylbenzene (DVB),17–20 copolymerization of AILs oligomers with resorcinol-FA (RF resin),21 and polymerization of 2-acrylamido-2-methyl-1-propanesulfonic acid with butylamine or 2-(N,N-dimethylamino)ethyl methacrylate.22 For example, Y. Leng13 reported an acidic polymeric hybrid obtained from the polymerization of –SO3H functionalized polymeric ILs-cations with heteropolyanions. Additionally, they used the polymeric hybrid as an excellent catalyst for the esterification of alcohols with carboxylic acids. X. Z. Liang18–20 have fabricated a novel class of solid PAILs via the copolymerization of AILs oligomers with DVB, which showed high efficiencies (Y = 99%) for biodiesel production from waste. X. Z. Liang21 copolymerized AILs oligomers with RF resin and an investigation of their catalytic activities for the acetalization of various carbonyl compounds with diols revealed an average yield of over 99.0%. Most reported PAILs were polymerized by double bond groups on cations, while the anions bonded with cations only by ionic bonds. Since the acid sites on the surface fell off easily, the catalytic activity decreased more or less during reuse and hence the stabilities of these catalysts need to be improved.
Herein, we report a set of novel micro–mesoporous PAILs copolymerized by cations and anions, that is vinyl –SO3H functionalized imidazolium or pyridine zwitterions with 2-acrylamide-2-methylpropanesulfonic acid; vinyl –SO3H functionalized imidazolium or pyridine zwitterions with 2-acrylamide-2-methylpropanesulfonic acid and DVB. Fourier transform infrared (FT-IR) spectroscopy, N2 adsorption–desorption (Brunauer–Emmett–Teller, BET) isotherm, scanning electron microscopy (SEM) and thermogravimetry analysis (TG) were then employed to characterize the catalyst. The catalytic activities of the novel PAILs were investigated through the acetalization of TOX with DMM1. Moreover, the effects of various reaction parameters and the catalyst recyclability were also explored.
2. Experimental
2.1 General
The chemicals of 1,4-butylene sulfone (>98 wt%, Energy Chemical), N-vinyl imidazole (98 wt%, Energy Chemical), 4-vinyl pyridine (>95 wt%, TCI (Shanghai) Development Co., Ltd), and divinylbenzene (DVB, 80 wt%, Energy Chemical) were all analytical grade and were purified by distillation before use. 2-Acrylamide-2-methylpropanesulfonic acid (AMPs, 99 wt%) and azobisisobutyronitrile (AIBM, >98 wt%) were purchased from Energy Chemical. DMM2 (>99.5%), DMM3 (>99.5%), DMM4 (>99.5%) and DMM5 (>99.5%) were isolated and purified via rectification. The remaining chemicals used were of analytical grade and were used without further treatment.
2.2 Catalyst synthesis
2.2.1 Synthesis of vinyl –SO3H functionalized imidazolium or pyridine zwitterions. In a 250 mL round-bottom flask, 1,4-butylene sulfone (0.1 mol, 13.6 g) was added dropwise into a solution of N-vinyl imidazole (0.1 mol, 9.4 g) or 4-vinyl pyridine (0.1 mol, 10.5 g) in anhydrous toluene (50 mL) at room temperature under stirring, and then heated at 60 °C for 24 h. After cooling to room temperature, the white solid was filtered, washed thoroughly with toluene (50 mL ×3) and dried in vacuum. 1-Vinyl-3-sulfonic butyl imidazolium zwitterions (VIMBs) or 4-vinyl-N-sulfonic butyl pyridine zwitterions (VPyBs) was obtained and further characterized.
VIMBs. 1H NMR (400 MHz, D2O): δ 1.65–1.72 (m, 2H), 1.94–2.01 (m, 2H), 2.87 (t, J = 8.0 Hz, 2H), 4.22 (t, J = 6.0 Hz, 2H), 5.33 (d, J = 8.0 Hz, 1H), 5.70 (d, J = 16.0 Hz, 1H), 7.03 (q, J = 8.0 Hz, 1H), 7.52 (s, 1H), 7.70 (s, 1H), 8.99 (s, 1H). 13C NMR (100 MHz, D2O): δ 20.90, 27.92, 49.28, 50.00, 109.31, 119.54, 122.75, 123.20, 134.47. Anal. calcd for C9H14N2O3S: C, 46.94; H, 6.13; N, 12.17; S, 13.92. Found: C, 47.25; H, 5.92; N, 12.18; S, 14.18.
VPyBs. 1H NMR (400 MHz, CD3OD): δ 1.68–1.76 (m, 2H), 2.04–2.11 (m, 2H), 2.88 (t, J = 8.0 Hz, 2H), 4.50 (t, J = 6.0 Hz, 2H), 5.87 (d, J = 12.0 Hz, 1H), 6.34 (d, J = 20.0 Hz, 1H), 6.86 (q, J = 9.3 Hz, 1H), 7.94 (d, J = 8.0 Hz, 2H), 8.63 (d, J = 8.0 Hz, 2H). 13C NMR (100 MHz, D2O): δ 20.85, 29.19, 49.95, 60.31, 124.64, 127.02, 132.02, 143.96, 153.75. Anal. calcd for C11H15NO3S: C, 54.75; H, 6.27; N, 5.81; S, 13.29. Found: C, 53.96; H, 6.13; N, 5.33; S, 13.16.
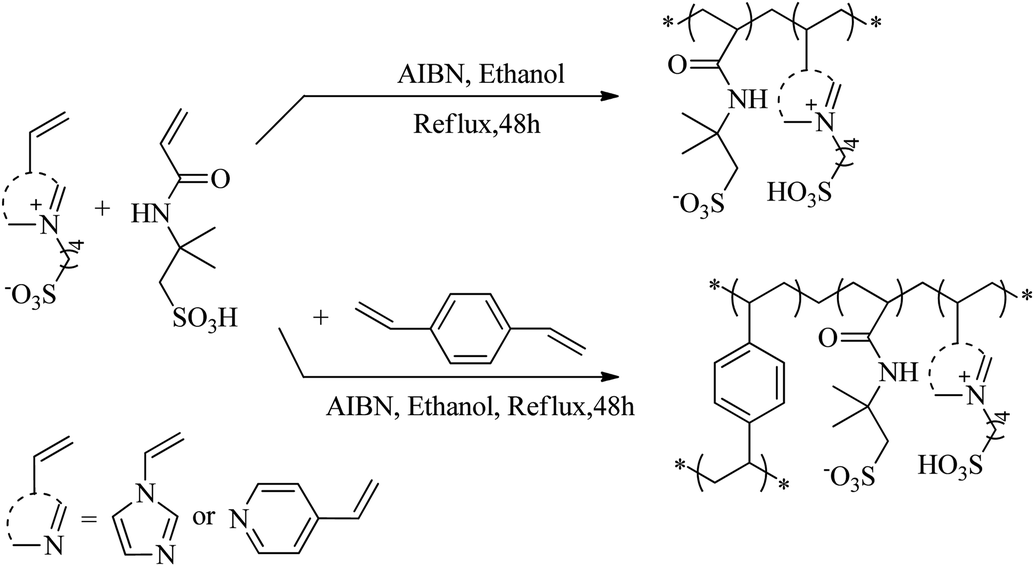
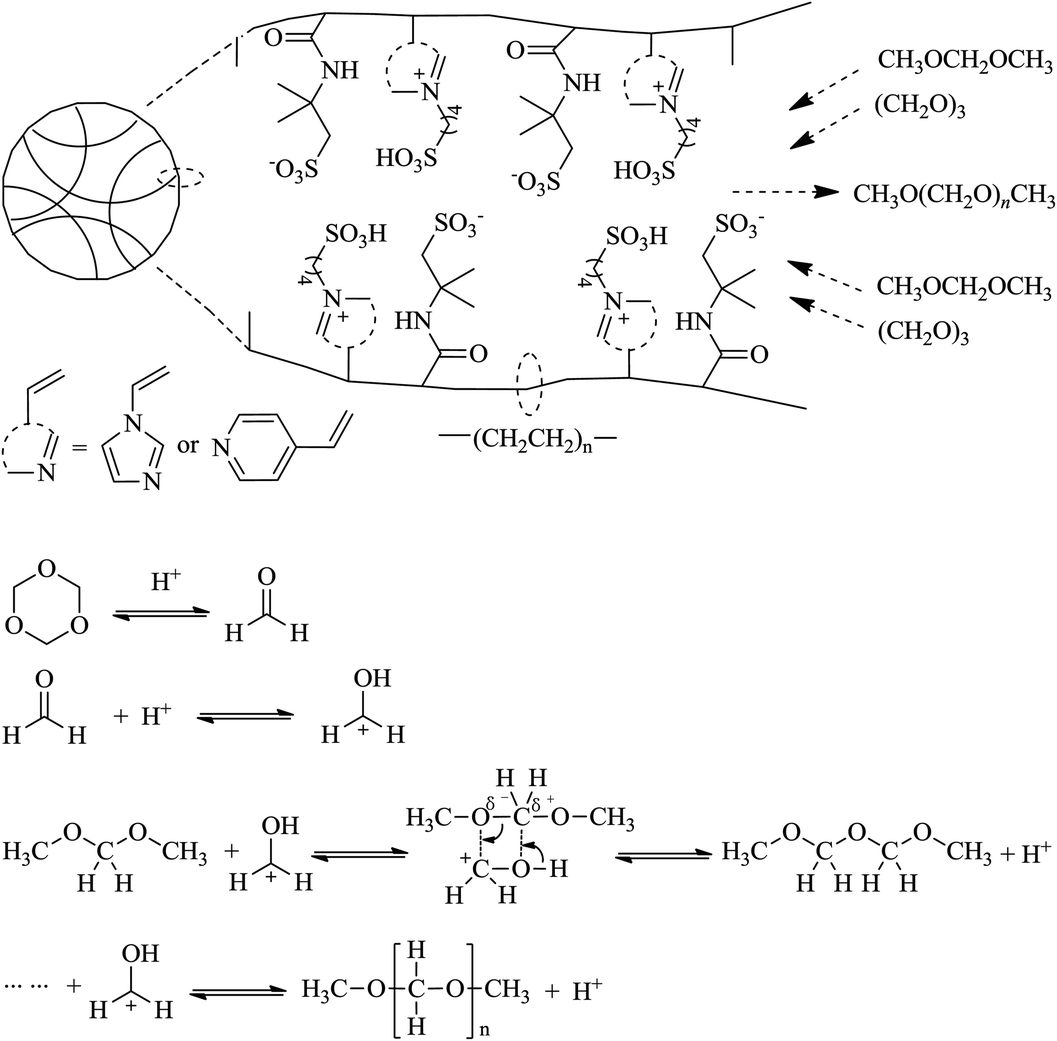
2.2.2 Synthetic method for PAILs. PAILs were prepared according to the literature reports.13–21 As shown in Scheme 1, VIMBs or VPyBs (0.1 mol), AMPs (0.1 mol), AIBM (3.2 wt%), or DVB (0.1 mol), were mixed together in 200 mL ethanol in a round-bottom flask under stirring, and heated gradually to reflux for an additional 48 h. Then, the mixture was kept for another 12 h at room temperature to form an organic gel. The white powder obtained after filtration was washed thoroughly with ethanol (100 mL ×3), ethyl acetate (100 mL ×3) and ether (100 mL ×1) at room temperature. This solid was dried in vacuum (70 °C, 5.3 kPa) for 12 h to eliminate the residual volatiles and moisture.
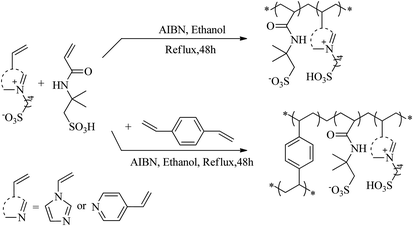 |
| | Scheme 1 Synthetic route of PAILs. | |
2.3 Characterization of catalyst
FT-IR spectra were acquired on a Nicolet NEXUS 870 system (USA) with a scan range of 400–4000 cm−1 using anhydrous KBr as standard. The BET isotherms were recorded on Micromeritics ASAP2010 instrument (USA). The pore size and volumes distributions were calculated by the Barrett–Joyner–Halenda (BJH) technique and the surface areas were estimated using the BET method. SEM images were recorded on a Hitachi SU8020 microscope (Tokyo, Japan) to assess the particle dimensions and morphology. The elemental microanalysis was performed by means of the Vario EL cube system (Elementar Analysensysteme, Germany). TG analysis was conducted on the Netzsch STA 449 F3 simultaneous thermal analysis system (Selb, Germany) between 50 and 800 °C in an N2 atmosphere. The acid capacity of PAILs (50 mg) was measured through acid–base titration with NaCl solution (50 mL; 2 mol L−1) as an ion-exchange agent at room temperature for more than 24 h, and then the filtrate was titrated by NaOH solution (0.01 mol L−1).
2.4 Typical acetalation reaction
The catalytic activities of PAILs were evaluated using the acetalization of DMM1 with TOX. DMM1 (0.24–0.42 mol), TOX (0.1 mol), and PAILs were added into a 100 mL stainless-steel autoclave, which was further flushed three times with nitrogen (2.0 MPa). The solution was heated to the required temperatures and stirred at 600 rpm. After the reaction, the resulting mixture was subjected to Agilent 7890A/5975C gas chromatography/mass spectrometry (GC/MS) and Agilent 6890 GC containing a SE-54 capillary column (30 m × 0.25 mm × 0.33 μm) and a FID detector, the column oven temperature was maintained at 60 °C for 10 min, then increased to 260 °C at a rate of 10 °C min−1, and held for 10 min. The internal standard method using furanidine was adopted for the quantification of TOX, DMMn, methanol, and methyl formate. The contents of FA was estimated through titration with sodium sulfite method provided by ASTM D2194-02 (2012),23 and the concentration of formic acid was determined by acid–base titration with strong base can refer to GB/T 2093-2011. The following equations were used to calculate the FA conversion (Con. FA) and product selectivity (Sele. DMMn).
FA, formaldehyde; MF, methyl formate; MA, formic acid.
3. Results and discussion
3.1 Catalyst characterization
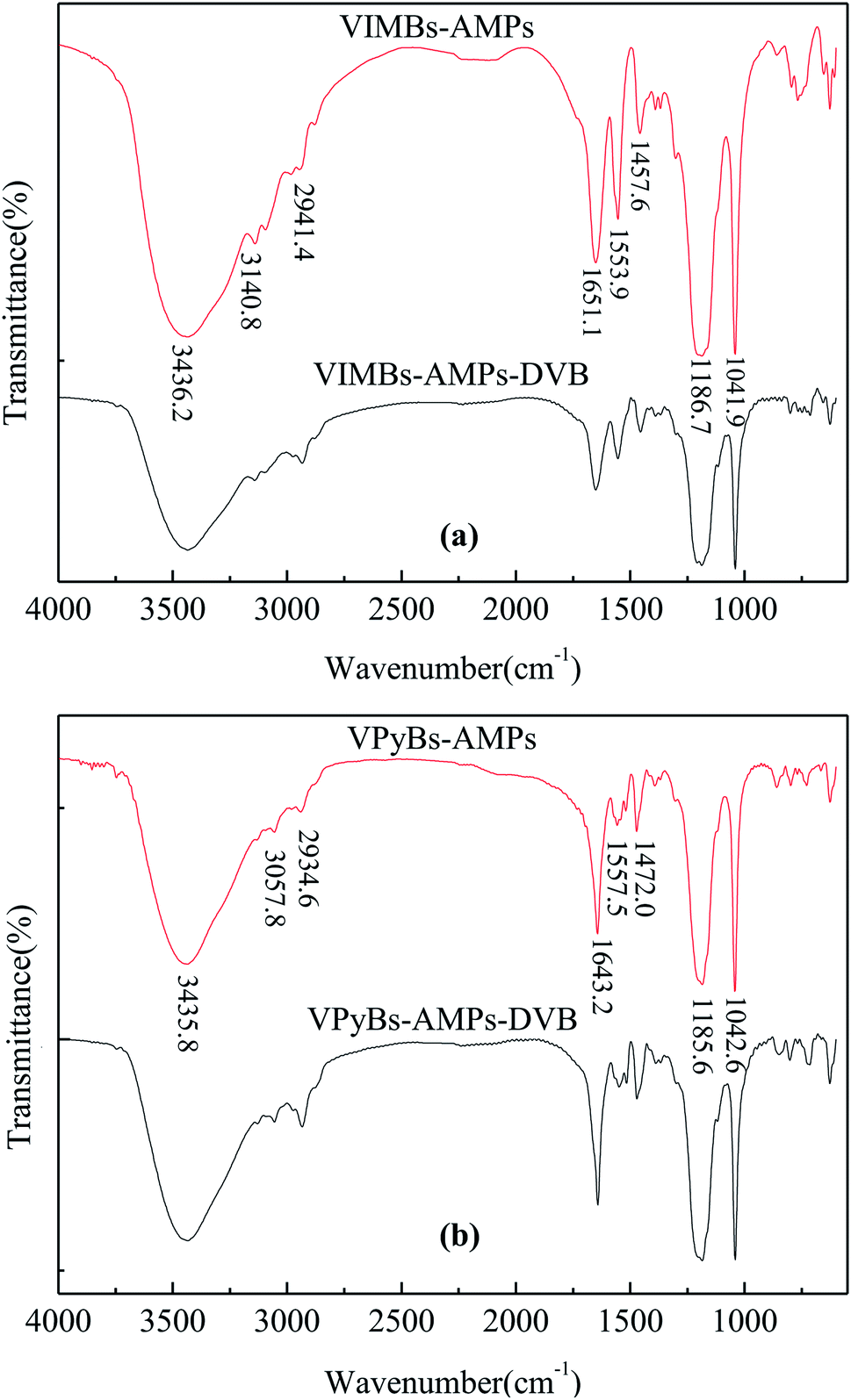
3.1.1 FT-IR analysis. As shown in Fig. 1(a), the FT-IR absorption peaks at about 1041.9 and 1186.7 cm−1 were characteristic of –SO3H.24 The peaks at 3140.8, 1553.9 and 1457.6 cm−1 were stretching vibrations of imidazole C–H, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and CN,25,26 whereas a broad band around 3436.2 cm−1 was ascribed to the stretching vibration of hydrogen bonds of ILs with physisorbed water which overlap with the characteristic peak of N–H.25,27 In addition, the CO and C–H stretching vibration of amide group were also observed at 1651.1 and 2941.4 cm−1.28,29 The results indicated that the vinyl in the precursor of ILs was polymerized and the ILs or acid groups as pendant group have been embedded in polymer chain. Similar results were obtained in Fig. 1(b); the peaks by 3057.8, 1557.5 and 1472.0 cm−1 were attributed to pyridine C–H, CC and CN stretching vibrations, whereas distinctive peaks of –SO3H moiety were observed at 1185.6 and 1042.6 cm−1. The bands around 1643.2, 2934.6 and 3435.8 cm−1 were attributed to CO, C–H, N–H of amide group and hydrogen bonds of ILs with physisorbed water, respectively, suggesting ILs structure had been incorporated in the PAILs.
C and CN,25,26 whereas a broad band around 3436.2 cm−1 was ascribed to the stretching vibration of hydrogen bonds of ILs with physisorbed water which overlap with the characteristic peak of N–H.25,27 In addition, the CO and C–H stretching vibration of amide group were also observed at 1651.1 and 2941.4 cm−1.28,29 The results indicated that the vinyl in the precursor of ILs was polymerized and the ILs or acid groups as pendant group have been embedded in polymer chain. Similar results were obtained in Fig. 1(b); the peaks by 3057.8, 1557.5 and 1472.0 cm−1 were attributed to pyridine C–H, CC and CN stretching vibrations, whereas distinctive peaks of –SO3H moiety were observed at 1185.6 and 1042.6 cm−1. The bands around 1643.2, 2934.6 and 3435.8 cm−1 were attributed to CO, C–H, N–H of amide group and hydrogen bonds of ILs with physisorbed water, respectively, suggesting ILs structure had been incorporated in the PAILs.
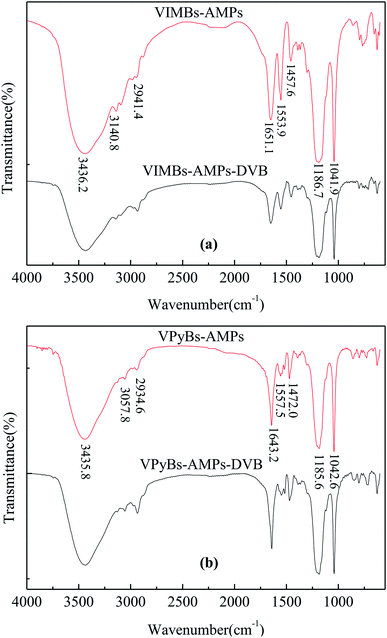 |
| | Fig. 1 FT-IR spectra of PAILs. (a) Imidazolium-based PAILs, (b) pyridinium-based PAILs. | |
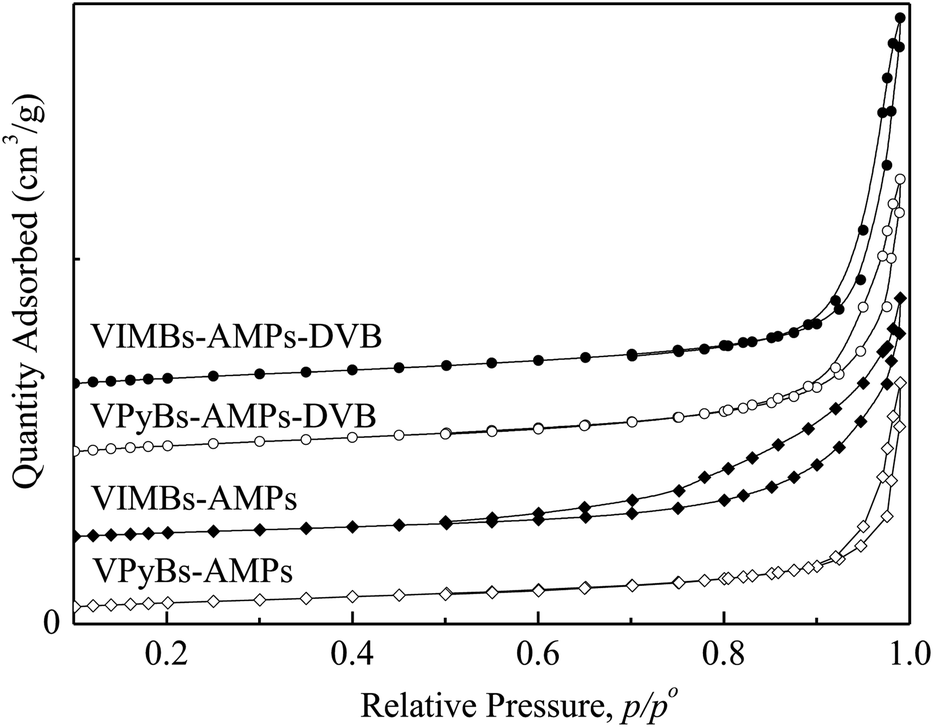
3.1.2 BET isotherms. N2 physisorption was used to evaluate the textural properties of PAILs that synthesized in ethanol under reflux. From the results depicted in Table 1 and Fig. 2, one can see that highly cross-linked network was gradually formed in the process of ILs polymerization, where ethanol acts as not only solvent but also template.30 With the aid of ethanol, the PAILs exhibited abundant disordered nanopores with uniform pore sizes ranging from 1.7 to 200 nm. As shown in Fig. 2, all the BET isotherms of PAILs were of typical type IV with hysteresis loops at the pressures P/P0 = 0.55–1.0, distinctive of mesoporous materials.30,31 The mesoporous (2–50 nm) occupancy rate of PAILs copolymerized by cations and anions was more than 75.29% and the mesoporous rate of PAILs copolymerized by DVB with cations and anions was 66.56–73.72% (Table 2), at the same time, PAILs copolymerized by DVB with cations and anions showed a higher BET surface area and pore volume (Table 1).
Table 1 BET surface properties of PAILs
| Sample |
BET surface area (m2 g−1) |
Total pore volume (cm3 g−1) |
Average pore width (nm) |
| VIMBs–AMPs |
18.87 |
0.09 |
19.55 |
| VPyBs–AMPs |
21.56 |
0.08 |
15.54 |
| VIMBs–AMPs–DVB |
25.51 |
0.15 |
21.96 |
| VPyBs–AMPs–DVB |
32.57 |
0.11 |
13.81 |
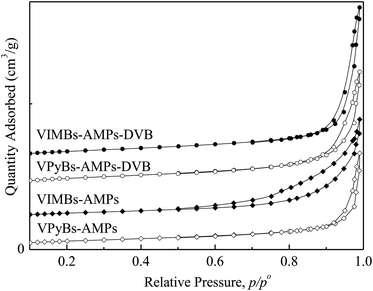 |
| | Fig. 2 N2 adsorption–desorption isotherms of PAILs. | |
Table 2 BJH adsorption pore distribution
| Sample |
Pore distribution under different pore sizes (%) |
| >50 nm |
2–50 nm |
<2 nm |
| VIMBs–AMPs |
2.85 |
91.29 |
5.87 |
| VPyBs–AMPs |
4.11 |
75.29 |
20.60 |
| VIMBs–AMPs–DVB |
7.88 |
73.72 |
18.40 |
| VPyBs–AMPs–DVB |
2.88 |
66.56 |
30.56 |
3.1.3 SEM studies. The SEM images of PAILs were illustrated in Fig. 3. It was found that the synthesized VIMBs–AMPs–DVB and VPyBs–AMPs–DVB were of nearly spherical shape with the sizes about 0.1–0.2 μm (Fig. 3(c) and (d)); moreover, the particles were connected with each other without obvious boundary, which indicates the existence of abundant pores. The primary polymer particles are interconnected to form a cross-linked network structure. Mesoporous and macroporous may be formed by interstitial spaces between primary particles after monomer polymerization. However, the SEM image (Fig. 3(a) and (b)) illustrated amorphous structure for VIMBs–AMPs and VPyBs–AMPs.
 |
| | Fig. 3 The SEM images of PAILs. (a) VIMBs–AMPs, (b) VPyBs–AMPs, (c) VIMBs–AMPs–DVB, (d) VPyBs–AMPs–DVB. | |
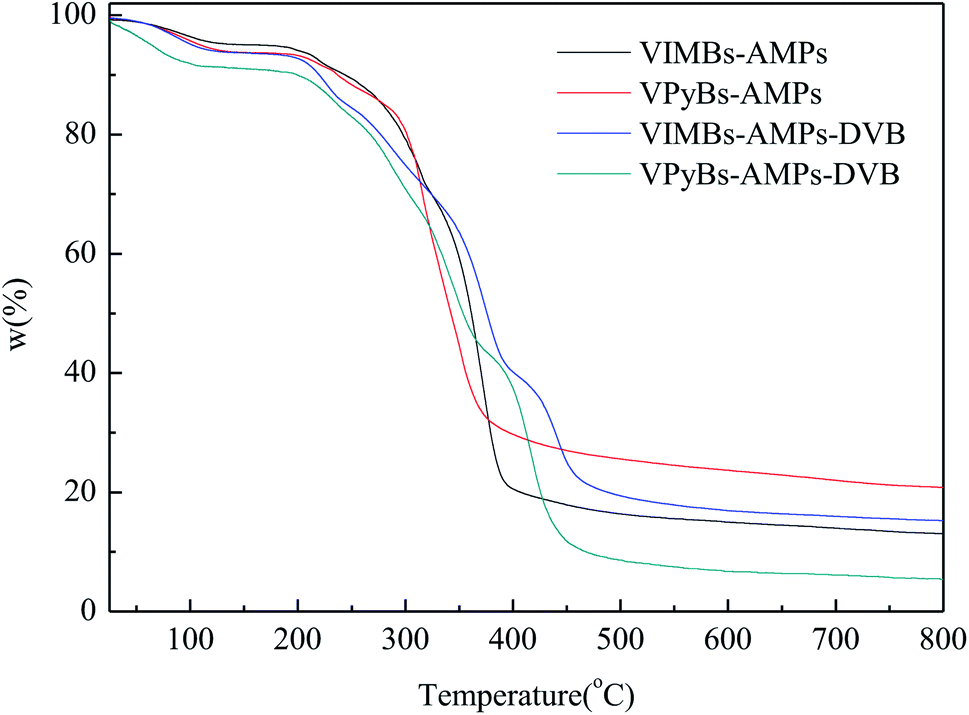
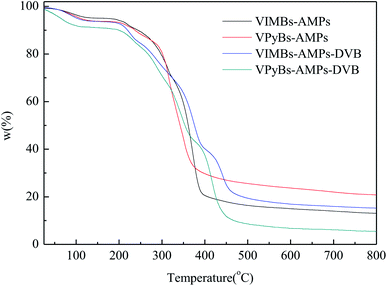
3.1.4 TG analysis. The thermal stability of PAILs was measured via TG, and the weight losses were illustrated in Fig. 4. The small weight loss at the first stage occurred before 150 °C due to the release of adsorption water, solvent, and then monomer of polymer that confined inside some pore channels, whereas the drastic weight loss in the range 210–450 °C was resulted from the decomposition of PAILs prepared by copolymerization of cations and anions. This fact revealed that the tested PAILs demonstrated excellent thermal stability and could, therefore, be applied as catalysts.
 |
| | Fig. 4 TG of PAILs. | |
3.2 Catalytic performance of the synthesized PAILs
The acetalation of DMM1 with TOX was chosen as the model reaction to evaluate and compare the catalytic performance of PAILs. The effects of various catalysts on the acetalation reaction were studied under the following conditions: 6.0 wt% catalyst, DMM1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :HCHO (TOX) = 1.2:1 mol ratio, 140 °C, 3.0 MPa and 6 h; the results were summarized in Table 3. When PAILs VIMBs–AMPs and VPyBs–AMPs were added into the reaction system as the catalyst, the substrate TOX could not be converted smoothly and only 59.8–71.4% conversion of FA and 36.4–41.3% selectivity for DMM3–8 were obtained (entries 1 and 2, Table 3). In contrast, PAILs copolymerized by DVB with cations and anions, for example VIMBs–AMPs–DVB (entry 3, Table 3) and VPyBs–AMPs–DVB (entry 4, Table 3), exhibited moderate to excellent catalytic activities (75.4–80.5% FA conversion and 42.3–48.5% DMM3–8 selectivity). It was noteworthy that VIMBs–AMPs–DVB and VPyBs–AMPs–DVB obtained from copolymerization of DVB with AILs, which with higher specific surface area and total pore volume displayed better catalytic efficiency.
:HCHO (TOX) = 1.2:1 mol ratio, 140 °C, 3.0 MPa and 6 h; the results were summarized in Table 3. When PAILs VIMBs–AMPs and VPyBs–AMPs were added into the reaction system as the catalyst, the substrate TOX could not be converted smoothly and only 59.8–71.4% conversion of FA and 36.4–41.3% selectivity for DMM3–8 were obtained (entries 1 and 2, Table 3). In contrast, PAILs copolymerized by DVB with cations and anions, for example VIMBs–AMPs–DVB (entry 3, Table 3) and VPyBs–AMPs–DVB (entry 4, Table 3), exhibited moderate to excellent catalytic activities (75.4–80.5% FA conversion and 42.3–48.5% DMM3–8 selectivity). It was noteworthy that VIMBs–AMPs–DVB and VPyBs–AMPs–DVB obtained from copolymerization of DVB with AILs, which with higher specific surface area and total pore volume displayed better catalytic efficiency.
Table 3 Influence of catalyst structure on the acetalation reaction of DMM1 with TOXa
| Entry |
Catalyst |
Conversion of FA (%) |
Mass selectivity (%) |
Carbon balance (%) |
| DMM2–8 |
DMM3–8 |
DMM9–10 |
MF |
MeOH |
| Reaction conditions: catalyst: 6.0 wt%, DMM1:HCHO(TOX) = 1.2:1 mol ratio, 140 °C, 3.0 MPa, 6 h. |
| 1 |
VIMBs–AMPs |
59.8 |
94.2 |
36.4 |
0 |
2.9 |
2.0 |
97.5 |
| 2 |
VPyBs–AMPs |
71.4 |
95.1 |
41.3 |
0 |
2.0 |
2.2 |
95.2 |
| 3 |
VIMBs–AMPs–DVB |
80.5 |
96.6 |
48.5 |
0 |
0.7 |
1.9 |
95.1 |
| 4 |
VPyBs–AMPs–DVB |
75.4 |
95.1 |
42.3 |
0 |
2.1 |
2.0 |
95.2 |
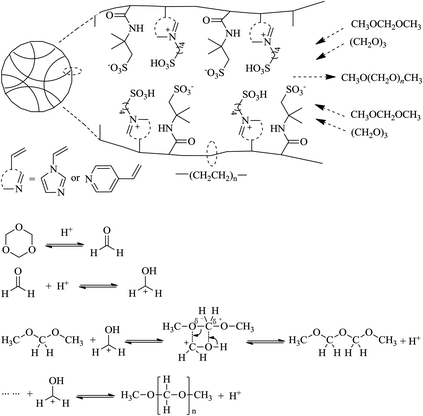
The results indicated that the pore structure played a dominant role in the catalytic reaction. As shown in Scheme 2, the reactants diffuse into the pores of PAILs and interact with the –SO3H functional groups which act as the reactive sites. The conversion of TOX to FA and the sequential chain propagation reaction of FA insertion into lower DMMn species to yield higher DMMn species occurred on the –SO3H sites. After the reaction, DMMn diffused away from the pores of the catalyst.32,33
 |
| | Scheme 2 Plausible mechanistic pathway for acetalation of TOX with DMM1 catalyzed by PAILs. | |
VIMBs–AMPs–DVB was selected as the catalyst to investigate the influence of various reaction factors, and the results were summarized in Table 4. Catalyst loading was confirmed as one of the principal factors; the conversion of FA steadily increased from 70.4% to 80.5% as the catalyst loading was increased from 2 to 6 wt% (entries 1–3, Table 4). However, a further increase in the amount of catalyst resulted in a decline in this conversion. The selectivity for DMM3–8 reached a maximum (48.5%) when the loading of VIMBs–AMPs–DVB was 6 wt% (entry 3, Table 4). These results indicated that excess acid may promote the generation of methanol, FA, methyl formate and other byproducts. The mol ratio of DMM1:HCHO was a key factor for achieving the higher catalytic performance; the selectivity for DMM3–8 gradually reduced from 58.8% to 43.0% when the ratio of DMM1:HCHO was enhanced from 0.8:1 to 1.4:1 (entries 3 and 5–7, Table 4) and the conversion of FA approached the peak value (80.5%) at DMM1:HCHO = 1.2:1 (entry 3, Table 4). Moreover, the catalytic activity was also affected by the temperature; while the reaction temperature was gradually increased to 130 °C from 120 °C, the peak value for the conversion (79.5%) and selectivity (53.2%) was observed (entry 9, Table 4). Further increasing the temperature led to a slight reduction in conversion and selectivity (entries 6, 10, Table 4). The impact of reaction time, ranging from 2 to 8 h, on the acetalation reaction was also investigated (Table 4, entries 9 and 11–13). The FA conversion enhanced gradually by prolonging the reaction time and reached 82.7% at 8 h. Moreover, the most favorable reaction time is determined to be 6 h, at which the selectivity for DMM3–8 was 53.2%.
Table 4 Screening reaction parameters for the acetalation reaction of DMM1 with TOX catalyzed by VIMBs–AMPs–DVB
| Entry |
Catalyst (wt%) |
DMM1:HCHO (mol ratio) |
Temperature (°C) |
Time (h) |
Conversion of FA (%) |
Mass selectivity (%) |
Carbon balance (%) |
| DMM2 |
DMM3–8 |
DMM9–10 |
MF |
MeOH |
| 1 |
2 |
1.2 |
140 |
6 |
70.4 |
55.6 |
41.7 |
0 |
1.3 |
1.0 |
94.7 |
| 2 |
4 |
1.2 |
140 |
6 |
79.5 |
49.1 |
47.2 |
0 |
1.7 |
1.3 |
96.1 |
| 3 |
6 |
1.2 |
140 |
6 |
80.5 |
48.1 |
48.5 |
0 |
0.7 |
1.9 |
95.1 |
| 4 |
8 |
1.2 |
140 |
6 |
78.1 |
46.0 |
47.6 |
0 |
2.7 |
2.2 |
96.0 |
| 5 |
6 |
0.8 |
140 |
6 |
78.9 |
36.2 |
58.8 |
0.1 |
1.8 |
1.7 |
94.1 |
| 6 |
6 |
1 |
140 |
6 |
79.4 |
41.9 |
52.6 |
0 |
2.4 |
1.6 |
98.2 |
| 7 |
6 |
1.4 |
140 |
6 |
79.7 |
51.3 |
43.0 |
0 |
2.7 |
1.9 |
98.4 |
| 8 |
6 |
1 |
120 |
6 |
73.9 |
45.1 |
52.0 |
0 |
1.4 |
0.8 |
98.1 |
| 9 |
6 |
1 |
130 |
6 |
79.5 |
42.6 |
53.2 |
0 |
2.0 |
1.2 |
95.5 |
| 10 |
6 |
1 |
150 |
6 |
79.8 |
42.7 |
51.7 |
0 |
2.1 |
2.0 |
95.9 |
| 11 |
6 |
1 |
130 |
2 |
65.4 |
49.9 |
46.8 |
0 |
1.8 |
0.9 |
98.1 |
| 12 |
6 |
1 |
130 |
4 |
75.1 |
45.7 |
52.2 |
0 |
0.1 |
1.0 |
95.1 |
| 13 |
6 |
1 |
130 |
8 |
82.2 |
43.6 |
52.6 |
0 |
1.8 |
1.1 |
92.5 |
3.3 Recyclability of the catalysts
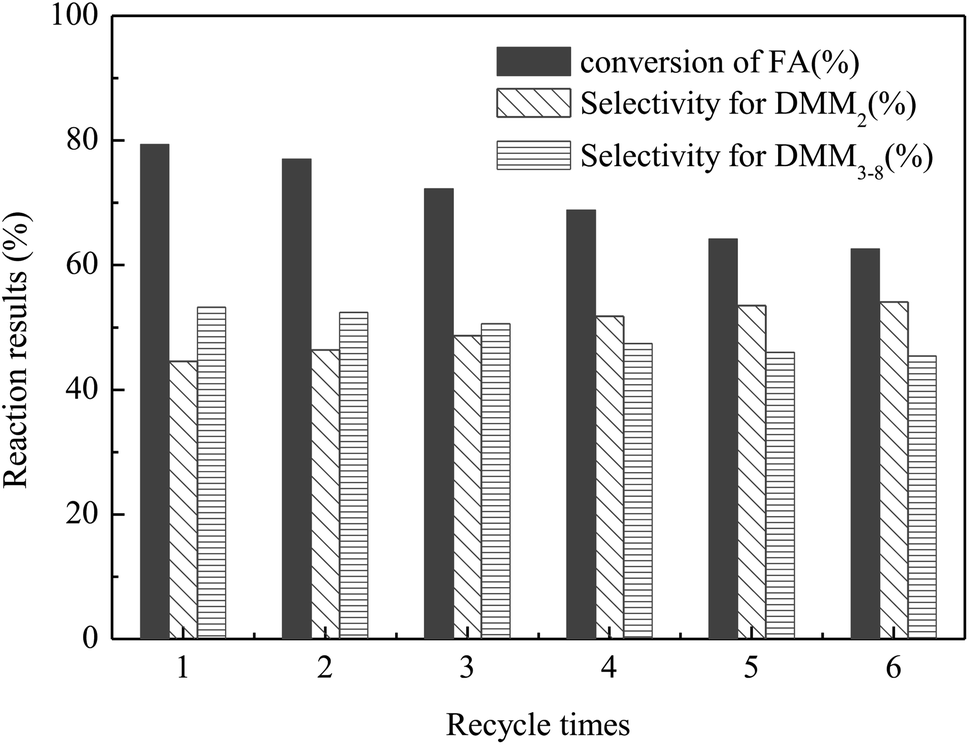
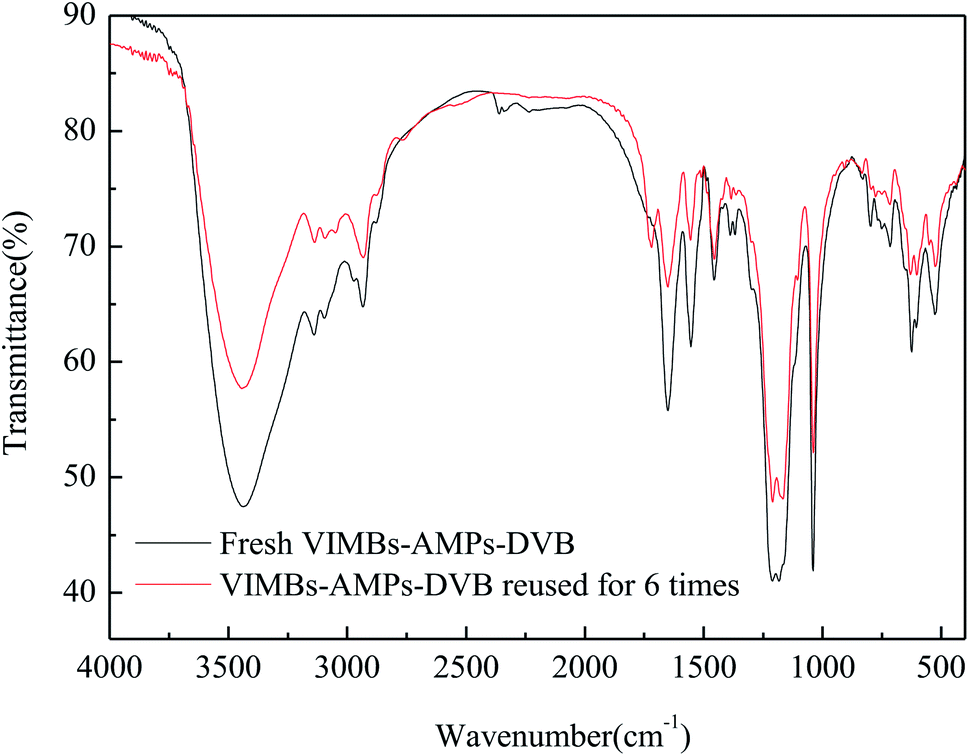
The stability and recyclability of a catalyst are important factors for its industrial application. Thus, the recyclability of VIMBs–AMPs–DVB in the acetalation was tested under the optimized reaction conditions i.e., 130 °C, DMM1:HCHO = 1:1 mol ratio, 6 wt% catalyst and 6 h, and the results were presented in Fig. 5. The catalyst could be recovered from the reaction system by a simple centrifugation and straight away utilized in the next run. The results showed that the catalytic performance of VIMBs–AMPs–DVB gradually decline since the first catalytic reaction, with the conversion of FA from 79.5% down to 62.6% after six times recycling. Additionally, the structure of the catalyst after the sixth run was compared with that of fresh VIMBs–AMPs–DVB using FT-IR analysis (Fig. 6) and SEM (Fig. 7). The structure of catalyst was destroyed after acetalation of DMM1 with TOX, mainly because the amido bond in PAILs can be destroyed under high temperature.29 Acid–base titrations were also performed to estimate the acid capacities of the freshly prepared and reused catalyst (Table 5). Subsequent to the six runs, the acid capacity of VIMBs–AMPs–DVB was rapidly reduced from 1.24 mmol g−1 to 0.68 mmol g−1. The main reason is that –SO3H fell off due to the breaking of amido bond, resulting in a decrease in acid capacity and catalytic activity. According to these comparative experiments, it was concluded that the prepared PAILs with lower reusability and stability in the acetalation reaction systerm. Therefore, the PAILs catalysts need to be further optimized.
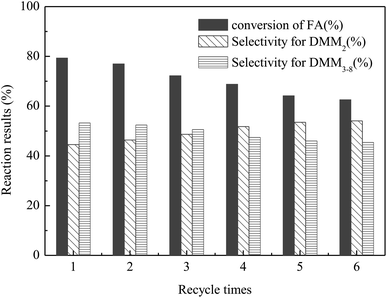 |
| | Fig. 5 The recycle test of VIMBs–AMPs–DVB in the acetalation reaction of DMM1 with TOX. Reaction conditions: VIMBs–AMPs–DVB: 6wt%, DMM1:HCHO(TOX) = 1:1 mol ratio, 130 °C, 3.0 MPa, 6 h. | |
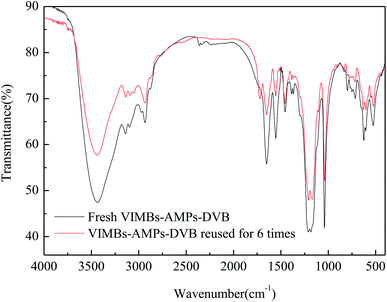 |
| | Fig. 6 FT-IR spectra comparison of the fresh and the six times reused VIMBs–AMPs–DVB. | |
 |
| | Fig. 7 The SEM images of fresh and the six times reused VIMBs–AMPs–DVB. (a) Fresh VIMBs–AMPs–DVB, (b) VIMBs–AMPs–DVB reused for 6 times. | |
Table 5 Surface acidity of VIMBs–AMPs–DVB (mH)
| Catalysts |
Acid capacity (mmol g−1) |
| 1 |
2 |
Average |
| VIMBs–AMPs–DVB |
1.23 |
1.24 |
1.24 |
| VIMBs–AMPs–DVB (6) |
0.67 |
0.68 |
0.68 |
4. Conclusion
In summary, we have fabricated novel micro–mesoporous PAILs for the acetalization of TOX with DMM1 to synthesize polyoxymethylene dimethyl ethers. The PAILs copolymerized by DVB with cations and anions displayed superior catalytic activity and selectivity, together with the advantages of easy separation and recovery. In particular, VIMBs–AMPs–DVB exhibited the greatest catalytic activity, giving a high FA conversion (82.2%) and DMM2–8 selectivity (96.2%). Moreover, the properties of catalysts were characterized in detail using FT-IR spectroscopy, BET isotherms, SEM and TG. Altogether, it was demonstrated that PAILs might be significant and promising catalysts for the acetalation reaction.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Project No. 21673259).
Reference
- J. Burger, M. Siegert, E. Ströfer and H. Hasse, Fuel, 2010, 89, 3315–3319 CrossRef CAS.
- L. Pellegrini, M. Marchionna, R. Patrini, C. Beatrice, N. D. Giacomo and C. Guido, SAE [Tech. Pap.], 2012, 01, 1053–1069 Search PubMed.
- L. Pellegrini, R. Patrini and M. Marchionna, SAE [Tech. Pap.], 2014, 01, 1951–1967 Search PubMed.
- L. Lautenschütz, D. Oestreich, P. Seidenspinner, U. Arnold, E. Dinjus and J. Sauer, Fuel, 2016, 173, 129–137 CrossRef.
- A. Peter, S. M. Fehr, V. Dybbert, D. Himmel, I. Lindner, E. Jacob, M. Ouda, A. Schaadt, R. J. White, H. Scherer and I. Krossing, Angew. Chem., 2018, 130, 9605–9608 CrossRef.
- Q. Wu, M. Wang, Y. Hao, H. S. Li, Y. Zhao and Q. Z. Jiao, Ind. Eng. Chem. Res., 2014, 53, 16254–16260 CrossRef CAS.
- Q. Wu, W. J. Li, M. Wang, Y. Hao, T. H. Chu, J. Q. Shang, H. S. Li, Y. Zhao and Q. Z. Jiao, RSC Adv., 2015, 5, 57968–57974 RSC.
- H. Y. Song, R. Y. Li, F. X. Jin, Z. Li and J. Chen, Mol. Catal., 2018, 455, 179–187 CrossRef CAS.
- H. Y. Song, M. R. Kang, F. X. Jin, G. Q. Wang, Z. Li and J. Chen, Chin. J. Catal., 2017, 38, 853–861 CrossRef CAS.
- J. Chen, H. Y. Song, C. G. Xia and Z. H. Tang, US Pat., 8 344 183 B2, 2013.
- J. Chen, H. Y. Song, C. G. Xia and Z. Li, US Pat., 8 816 131 B2, 2014.
- C. G. Xia, H. Y. Song, J. Chen and Z. Li, US Pat., 8 987 521 B2, 2015.
- Y. Leng, P. P. Jiang and J. Wang, Catal. Commun., 2012, 25, 41–44 CrossRef CAS.
- J. K. Wang, Y. X. Zong, R. G. Fu, Y. Y. Niu, G. R. Yue, Z. J. Quan, X. C. Wang and Y. Pan, Ultrason. Sonochem., 2014, 21, 29–34 CrossRef CAS PubMed.
- W. Y. Li, Y. X. Zong, J. K. Wang and Y. Y. Niu, Chin. Chem. Lett., 2014, 25, 575–578 CrossRef CAS.
- A. R. Kiasat, A. Mouradzadegun and S. J. Saghanezhad, Chin. J. Catal., 2013, 34, 1861–1868 CrossRef CAS.
- J. X. Wu, Y. L. Gao, W. Zhang, A. Tang, Y. Y. Tan, Y. Men and B. Tang, Chem. Eng. Process., 2015, 93, 61–65 CrossRef CAS.
- X. Z. Liang, Appl. Catal., A, 2013, 455, 206–210 CrossRef CAS.
- X. Z. Liang, Energy, 2013, 63, 103–108 CrossRef CAS.
- X. Z. Liang, Ind. Eng. Chem. Res., 2013, 52, 6894–6900 CrossRef CAS.
- X. Z. Liang, Kinet. Catal., 2013, 54, 724–729 CrossRef CAS.
- X. J. Yang, Y. X. Fang, X. M. Li, K. Zhang, Y. D. Cui, B. N. Zhang and G. Q. Yin, e-Polym., 2014, 14, 335–343 CAS.
- Y. Y. Zheng, Q. Tang, T. F. Wang and J. F. Wang, Chem. Eng. Sci., 2015, 134, 758–766 CrossRef CAS.
- J. M. Miao, H. Wan and G. F. Guan, Catal. Commun., 2011, 12, 353–356 CrossRef CAS.
- Z. M. Li, Y. Zhou, D. J. Tao, W. Huang, X. S. Chen and Z. Yang, RSC Adv., 2014, 4, 12160–12167 RSC.
- H. H. Zhao, N. Y. Yu, Y. Ding, R. Tan, C. Liu, D. H. Yin, H. Y. Qiu and D. L. Yin, Microporous Mesoporous Mater., 2010, 136, 10–17 CrossRef CAS.
- Q. Zhang, H. Su, J. Luo and Y. Y. Wei, Green Chem., 2012, 14, 201–208 RSC.
- N. Sahiner, A. O. Yasar and N. Aktas, Renewable Energy, 2017, 101, 1005–1012 CrossRef CAS.
- H. Y. Gao, Y. M. Zhou, X. L. Sheng, S. Zhao, C. Zhang, J. S. Fang and B. B. Wang, Appl. Catal., A, 2018, 552, 138–146 CrossRef CAS.
- Y. L. Zhang, S. Wei, F. J. Liu, Y. C. Du, S. Liu, Y. Y. Ji, T. Yokoi, T. Tatsumi and F. S. Xiao, Nano Today, 2009, 4, 135–142 CrossRef CAS.
- S. I. Cho, S. D. Choi, J. H. Kim and G. J. Kim, Adv. Funct. Mater., 2004, 14, 49–54 CrossRef CAS.
- Y. Y. Zheng, Q. Tang, T. F. Wang, Y. H. Liao and J. F. Wang, Chem. Eng. Technol., 2013, 36, 1951–1956 CrossRef CAS.
- F. Wang, G. L. Zhu, Z. Li, F. Zhao, C. G. Xia and J. Chen, J. Mol. Catal. A: Chem., 2015, 408, 228–236 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Fuxiang Jin,
Meirong Kang and
Jing Chen*
,
Fuxiang Jin,
Meirong Kang and
Jing Chen*