
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA minimal structural variation can overcome tumour resistance of oxaliplatin: the case of 4,5-dehydrogenation of the cyclohexane ring†
Paride
Papadia‡
 *ab,
Valentina
Gandin‡
c,
Alessandra
Barbanente
bd,
Alessandro G.
Ruello
bd,
Cristina
Marzano
c,
Katia
Micoli
bd,
James D.
Hoeschele
e,
Giovanni
Natile
bd and
Nicola
Margiotta
*bd
*ab,
Valentina
Gandin‡
c,
Alessandra
Barbanente
bd,
Alessandro G.
Ruello
bd,
Cristina
Marzano
c,
Katia
Micoli
bd,
James D.
Hoeschele
e,
Giovanni
Natile
bd and
Nicola
Margiotta
*bd
aDepartment of Biological and Environmental Sciences and Technologies (DiSTeBA), University of Salento, Prov.le Lecce-Monteroni, Centro Ecotekne, 73100 Lecce, Italy. E-mail: paride.papadia@unisalento.it; Tel: +39 832 299263
bCIRCMSB, Consorzio Interuniversitario di Ricerca in Chimica dei Metalli nei Sistemi Biologici, Via Celso Ulpiani 27, 70121 Bari, Italy
cDipartimento di Scienze del Farmaco, Università di Padova, Via Marzolo 5, 35131, Padova, Italy
dDipartimento di Chimica, Università degli Studi di Bari Aldo Moro, Via E. Orabona 4, 70125 Bari, Italy. E-mail: nicola.margiotta@uniba.it; Tel: +39 80 5442759
eDepartment of Chemistry, Eastern Michigan University, 48197 Ypsilanti, MI, USA
First published on 11th October 2019
Abstract
A new family of anticancer compounds has been derived from oxaliplatin by inserting a double-bond between carbons 4 and 5 of the 1,2-diaminocyclohexane ring. Testing against a panel of human tumour cell lines including cervical (A431), ovarian (2008), and colon carcinomas (HCT-15 and LoVo), and two oxaliplatin-resistant clones (LoVo OXP and LoVo MDR) has shown that the new compounds have, in general, equal if not better cytotoxic activity and are able to overcome the oxaliplatin-resistance. Moreover, the oxalato derivative induced lipid droplets increase in LoVo OXP cells thus suggesting the involvement of metabolism stress in its mechanism of action.
1R,2R-Diaminocyclohexane(oxalato)platinum(II), oxaliplatin, initially launched in France in 1996 for the treatment of patients with metastatic carcinoma of the colon and further adopted in Europe in 1999 and in USA in 2002,1 is still one of the most successful cisplatin analogues. Other platinum compounds containing structural variations of the diaminocyclohexane (DACH) carrier ligand have also proven to be endowed with marked cytotoxic activity.2 Among these, kiteplatin, [PtCl2(cis-1,4-DACH)], which incorporates a cis-1,4-DACH instead of a trans-1,2-DACH ligand, is effective in overcoming cisplatin and oxaliplatin resistance.3–6 Similarly to cisplatin and oxaliplatin, kiteplatin also interacts with double strand DNA forming mainly 1,2-d(GpG) cross-links, however kiteplatin adducts are removed with lower efficiency by the cancer cell repair systems.5 Also an oxaliplatin analogue containing just an additional methyl group in position 4 of the cyclohexane ring was shown to harbor improved therapeutic characteristics including significantly reduced adverse effects and reduced vulnerability to resistance mechanisms both in vitro and in vivo.7 Thus, limited structural variations in the carrier ligand can modulate significantly the pharmaceutical behaviour of this family of platinum drugs. We therefore deemed to investigate the consequences of introducing another structurally small, but potentially important, variation in the cyclohexane ring. The ligand selected for the new compounds was the racemic mixture of trans-1,2-diamine-4-cyclohexene (DACHEX) in which an unsaturation is inserted between carbon atoms 4 and 5 of the cyclohexane ring. This structural modification will force the C3–C6 atoms of the ring to be coplanar and constrain the stereochemistry in a more rigid half chair conformation.8 Moreover, the π-electrons will add an unprecedented nucleophilic character to the lipophilic six-carbon ring which in the platinum–DNA adduct is exposed in the major groove.9,10 The presence of a chemically reactive carbon–carbon double bond also opens the door to additional chemical modifications harbouring targeting and/or dual acting moieties.
To assess the biological effects of this localized structural modification, four Pt(II)–DACHEX complexes (Fig. 1), differing in the nature of the leaving groups (two halides or a dicarboxylate) were prepared and assayed for cytotoxicity in vitro: cis-[PtCl2(DACHEX)] (1), cis-[PtI2(DACHEX)] (2), [Pt(CBDCA)(DACHEX)] (3), and [Pt(OXA)(DACHEX)] (4) (CBDCA = 1,1-cyclobutane dicarboxylate, OXA = oxalate).
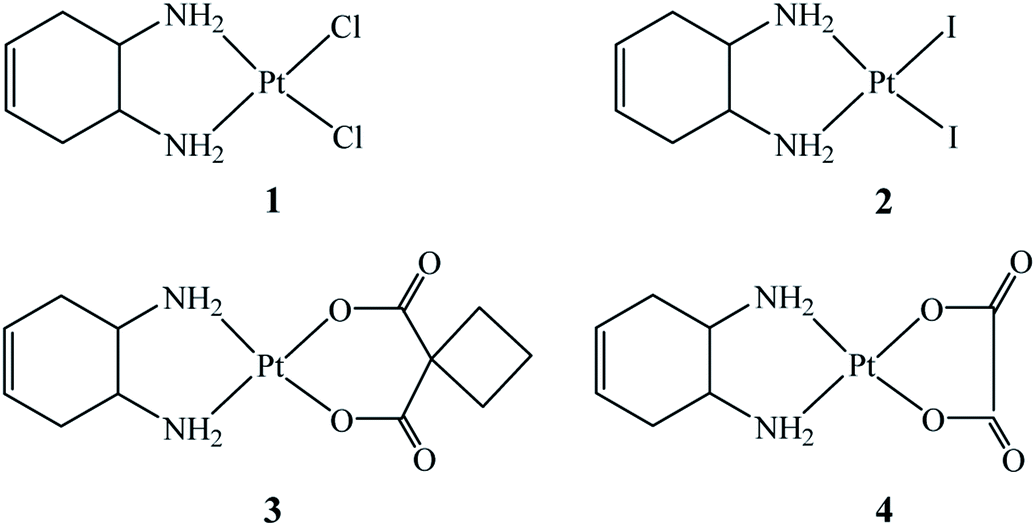 | ||
| Fig. 1 Sketches of the four new complexes with the DACHEX ligand. | ||
The four complexes where fully characterized by elemental analysis, ESI-MS, and NMR (see ESI†).
The dichlorido complex 1 was first reported about forty years ago, but tested only against the leukaemia L1210 cell line and found to be 18 times less active than oxaliplatin.11 We prepared complex 1 by direct reaction of racemic trans-1,2-diamine-4-cyclohexene (see Scheme 1 in ESI†) with K2[PtCl4]. The diiodido complex cis-[PtI2(DACHEX)] (2) was obtained by the classical Dhara synthesis, i.e. activation of K2[PtCl4] with KI (8-fold excess) and subsequent reaction with DACHEX. Pt–diiodido complexes, such as cis-[PtI2(NH3)2]12,13 and cis-[PtI2(1R,2R-DACH)],14 initially neglected vs. the chlorido derivatives, have been recently reinvestigated since some of them possess superior antitumor properties.15 The dicarboxylate derivatives cis-[Pt(CBDCA)(DACHEX)] (3) and cis-[Pt(OXA)(DACHEX)] (4, the direct analogue of oxaliplatin), were synthesized by refluxing cis-[Pt(CBDCA)(DMSO)2] or cis-[Pt(OXA)(DMSO)2] with DACHEX for 2 h. Heating was essential for favouring the substitution of the second DMSO ligand by the second nitrogen of the chelating diamine, as reported in the ESI (Schemes 2 and 3).† The choice of the latter synthetic route was dictated by the failure of all attempts to prepare 3 and 4 by dehalogenation of compound 1 or 2 with a silver salt and subsequent reaction with the corresponding dicarboxylate (coordination of Ag+ to the double bond of cyclohexene was observed).
The cytotoxicity of the newly synthesized compounds (1–4) was evaluated in vitro towards a panel of human cell lines including cervical (A431), ovarian (2008), and colon (HCT-15 and LoVo) carcinomas. The antiproliferative activity was also investigated in two additional colon cancer cell lines which were selected for the resistance to oxaliplatin (LoVo OXP)16 or for the multidrug resistant phenotype (LoVo MDR). The cytotoxicity was evaluated by means of the MTT test after 72 h treatment with increasing concentrations of the tested compounds. IC50 values, calculated from dose–survival curves, are shown in Table 1.
| Compound | IC50 (μM) ± S.D. | |||||||
|---|---|---|---|---|---|---|---|---|
| A431 | 2008 | HCT-15 | LoVo | LoVo OXP | R.F. | LoVo MDR | R.F. | |
| a Cells (3–5 × 103 cells per mL) were treated for 72 h with increasing concentrations of the tested compounds. Cytotoxicity was assessed by the MTT test. IC50 values were calculated by the four parameter logistic model (p < 0.05). S.D. = standard deviation. OXP = oxaliplatin, CDDP = cisplatin. Resistance Factor (R.F.) is defined as IC50 resistant/IC50 parent line. | ||||||||
| 1 | 3.0 ± 0.7 | 3.7 ± 1.1 | 4.0 ± 0.8 | 1.2 ± 0.8 | 2.0 ± 0.6 | 1.7 | 1.0 ± 0.3 | 0.8 |
| 2 | 1.1 ± 0.4 | 1.5 ± 0.5 | 0.9 ± 0.2 | 1.4 ± 0.5 | 2.6 ± 0.6 | 1.9 | 2.2 ± 0.8 | 1.6 |
| 3 | 5.4 ± 0.9 | 10.1 ± 2.3 | 3.4 ± 0.7 | 3.1 ± 1.1 | 3.0 ± 1.0 | 1.0 | 3.0 ± 0.9 | 1.0 |
| 4 | 3.7 ± 0.7 | 2.6 ± 0.5 | 5.2 ± 0.8 | 1.8 ± 0.8 | 3.6 ± 1.0 | 2.0 | 0.8 ± 0.4 | 0.4 |
| CDDP | 2.0 ± 0.8 | 2.3 ± 1.1 | 15.3 ± 2.6 | 7.6 ± 1.5 | 13.1 ± 2.5 | 1.7 | 7.5 ± 1.0 | 1.0 |
| OXP | 3.7 ± 1.0 | 1.7 ± 1.0 | 1.2 ± 0.4 | 1.1 ± 0.6 | 14.3 ± 2.1 | 13.0 | 1.4 ± 0.8 | 1.3 |
All compounds, with the exception of 3, were similar to cisplatin and oxaliplatin against A431 and 2008 cell lines. The lower activity of compound 3 is in line with the lower toxicity of carboplatin (having the same dicarboxylate leaving group). All compounds (comprising compound 3) were superior to cisplatin, but comparable to oxaliplatin, in LoVo and HCT-15 cell lines. This result indicates that the new carrier ligand DACHEX preserves the characteristic of the 1R,2R-DACH ligand present in oxaliplatin in rendering the platinum drug active towards colon carcinoma cell lines. Finally, all new compounds were superior to cisplatin and oxaliplatin in LoVo OXP cell line and superior to cisplatin in LoVo MDR cell line. As expected, among the four complexes containing DACHEX, 1 and 2 resulted the most active because of the presence of the two halido ligands which, as evidenced in LoVo OXP cells, showed the highest uptake (see following discussion) and, once in the cell, are supposed to be hydrolysed faster than the bis-chelated dicarboxylato ligands (CBDCA and OXA) present in compounds 3 and 4, respectively. Moreover, between compounds 1 and 2, the di-iodido derivative 2 was the most active with an average IC50 value of 1.62 μM to be compared with the mean IC50 value of 2.5 μM for compound 1. This result was rather expected since, quite recently, diiodido derivatives of cisplatin and oxaliplatin have proven to be quite active in several solid tumor cell lines (such as colo-rectal cells).12–15 Finally, the CBDCA derivative 3 was the least effective while compound 4, the closest structural analogue of oxaliplatin (containing the same oxalato leaving ligand), possesses a cytotoxic activity slightly higher than that of oxaliplatin (average IC50 = 4.7, 2.9, and 3.9 μM for 3, 4, and OXP, respectively). To investigate the ability of the new complexes to overcome the acquired resistance of human tumor cell lines, two colon cell lines characterized by their resistance to oxaliplatin (LoVo OXP) and to multidrugs (LoVo MDR) were selected. The molecular mechanism involved in oxaliplatin resistance appears to depend upon: (i) decreased cellular accumulation, which is thought to be related to a greater activity of the ATP7B exporter rather than to the activity of P-glycoprotein (P-gp) and multidrug resistance protein 1 (MRP1), and (ii) more efficient repair of oxaliplatin-induced DNA-damage by NER (Nucleotide Excision Repair).17 LoVo OXP cells (derived from LoVo cells grown in the presence of increased concentration of oxaliplatin) were about 13-fold more resistant to oxaliplatin than parental cells.16 The data reported in Table 1 clearly indicate that all compounds possess resistant factors much lower than that of oxaliplatin (R.F. of 1.7, 1.9, 1.0, and 2.0 for 1, 2, 3, and 4, respectively vs. R.F. of 13.0 for OXP), thus confirming their ability to overcome the oxaliplatin-resistance. In Table 1 are also reported the results obtained with the multidrug resistant colon carcinoma subline LoVo MDR in which the resistance to doxorubicin,18 a drug belonging to the MDR spectrum, is associated with an overexpression of drug transporters, such as the 170 kDa P-gp.19 All platinum derivatives tested against this cell line showed a similar response as for the parental subline (R.F. = 0.8, 1.6, 1.0, 0.4, 1.0, and 1.3 for 1, 2, 3, 4, cisplatin, and OXP, respectively) thus suggesting that these platinum complexes are not P-gp substrates. A first conclusion that can be drawn from the above results is that the different shape of the diamine used in this work (DACHEX) does not significantly alter the antitumor effect in comparison to oxaliplatin, however all complexes are able to overcome the oxaliplatin-resistance. This latter result evidences the importance of the DACHEX ligand in determining the activity of 4 in comparison to oxaliplatin against oxaliplatin-resistant cells.
With the aim to correlate the cytotoxic potential with some property of the complexes, such as their ability to enter cancer cells and to target DNA, we evaluated cellular uptake and DNA platination levels of LoVo OXP cells treated for 24 h with equimolar concentrations (2.5 μM) of tested compounds. The platinum levels were quantified by means of GF-AAS analysis. As depicted in Fig. 2, all DACHEX Pt(II) complexes were more effective than OXP in entering cancer cells and binding to DNA. In particular, a direct and linear correlation was found (R2 = 0.98) between the cytotoxic activity and the cellular uptake of the various compounds (Fig. 2a and a′). Interestingly, the cellular uptake of 4 resulted to be much higher than that of oxaliplatin, which differs only for the absence of the double bond in the cyclohexane ring and hence has a similar lipophilicity. At the present stage of the investigation we can only hypothesize an active/facilitated transport or a more efficient accumulation (due to failure of the exporter proteins) for the DACHEX-containing complexes. The results also showed a rough relationship between cellular uptake and DNA platination (Fig. 2c).
 | ||
| Fig. 2 (a) Cellular uptake and correlation between cytotoxicity and cellular platinum levels (a′) in drug-treated LoVo OXP cells. LoVo OXP cells were incubated for 24 h with 2.5 μM of tested complexes. The amount of cellular Pt was estimated by GF-AAS. (b) Nuclear DNA platination levels in drug-treated LoVo OXP cells. LoVo OXP were incubated for 24 h with 2.5 μM of tested complexes. The amount of DNA-bound Pt was estimated by GF-AAS. (c) Correlation between cellular uptake and DNA-platination levels. | ||
Cisplatin and oxaliplatin are known to form similar adducts with DNA and, specifically, 60–65% intrastrand GG, 25–30% intrastrand AG, 5–10% intrastrand GNG, and 1–3% interstrand GG crosslinks.20 However, the adducts formed by the two clinically used Pt-drugs could be differently recognized by mismatch repair proteins and, in general, damage-recognition proteins. For instance, some of these proteins have been found to bind with greater affinity to cisplatin-GG adducts than to oxaliplatin-GG adducts, probably because of the different steric and hydrophilic/lipophilic character of the non-leaving ligands (two ammines in cisplatin and 1R,2R-DACH in oxaliplatin).21 The differential recognition of the GG adducts could be responsible for the differences in cytotoxicity and tumour selectivity of the two drugs. Although until recently the assumption has been made that oxaliplatin kills cells by eliciting a DNA-damage response, as does cisplatin, there has not been satisfactory explanation for the unique clinical and side-effect profiles of oxaliplatin. For instance, within the panel of cell lines used in this work, cisplatin was much less effective toward colon cell lines HCT-15 and LoVo (IC50, on average, one order of magnitude greater) than toward cervical (A431) and ovarian (2008) cell lines; in contrast oxaliplatin was equally active (comparable IC50) towards all four tumor cell lines. Therefore it has been proposed that oxaliplatin could act also through a mechanism of action fundamentally distinct from that of cisplatin.22 In a seminal work it was demonstrated that, at clinically relevant concentrations, oxaliplatin can cause ribosome biogenesis stress, whereas cisplatin does not.23 A DNA-damage independent mechanism of cell death is also consistent with previous observations that cisplatin- or oxaliplatin-resistant cell lines often do not exhibit cross-resistance.24 To better characterize the structural and ultrastructural changes induced by the newly synthesized Pt(II) DACHEX derivatives, LoVo OXP cells were exposed to IC50 of 1 and 4 for 36 h and then subjected to TEM analysis. 1-treated LoVo OXP cells revealed the induction of a massive DNA condensation and the induction of mitochondrial swelling (increase in size and decrease in turbidity) and loss of typical internal ultrastructure, i.e. cristae (Fig. 3b and c), compared to control cells (Fig. 3a). These features are consistent with DNA damage and induction of apoptosis. In contrast, cells treated with 4 did not show the classical signs of DNA condensation or mitochondrial damage; however, a substantial increase in lipid droplets production was observed (Fig. 3d and e), thus suggesting the involvement of a lipid metabolism stress in the cell death. Similar results were obtained with OXP at IC75 concentrations, where an increase of lipid droplets formation was evidenced. Thus, while the data relative to 4 (oxalate “leaving” group) are in line with previously reported data on oxaliplatin showing ribosome stress biogenesis, those relative to 1 (chloride leaving ligands) are in accord with DNA damage, as for cisplatin. The above experiment indicates that the carrier ligand is not the only determinant of the mechanism of action of platinum drugs since compounds 1 and 4, although having the same carrier ligand, can exert different types of toxicity which, most likely, depends upon the different types of leaving ligands. Of course, this does not mean that the carrier ligand does not play a role. Indeed, the same experiment performed using oxaliplatin, showed the same morphological changes as those observed for 4, however oxaliplatin required a dose (IC75) much greater than that of 4 (IC50) in this cell line model, indicating that in oxaliplatin-resistant cells the leaving group can orient towards the mechanism of toxicity while the carrier ligand can determine the dose required to produce a given cytotoxic effect.
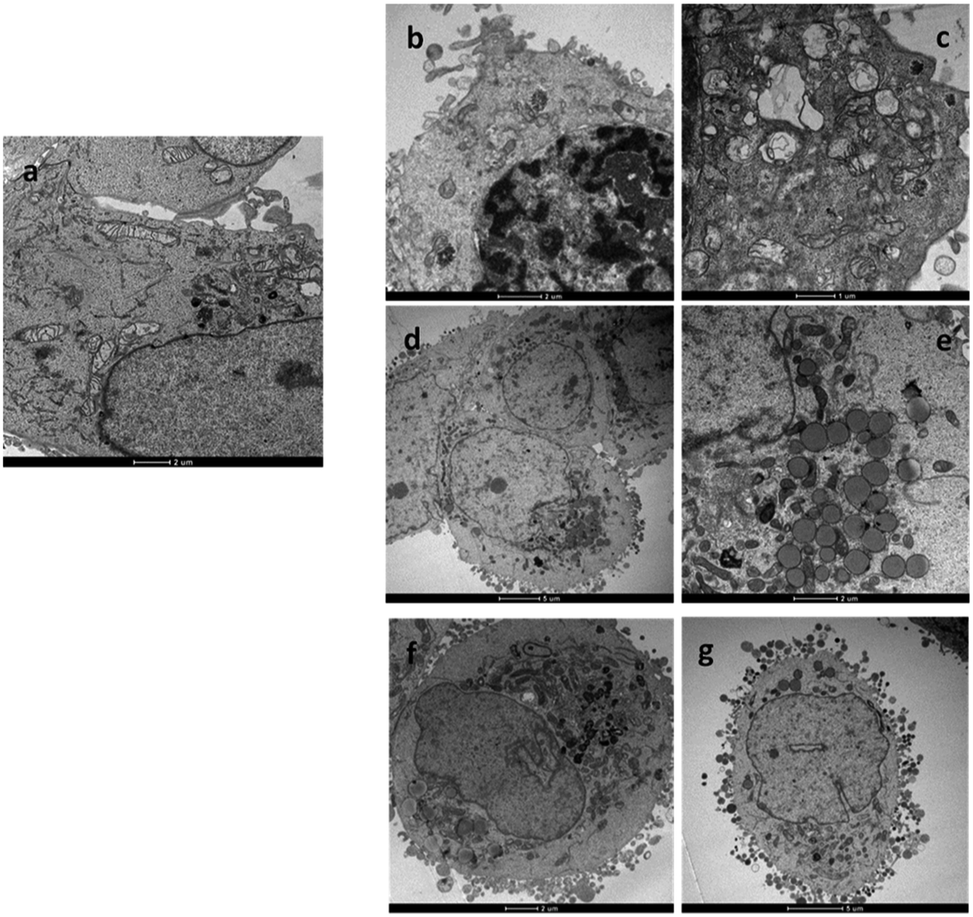 | ||
| Fig. 3 TEM analysis. Micrographs of LoVo OXP cells after 36 h incubation with (a) no drug (control); (b) and (c) IC50 of 1; (d) and (e) IC50 of 4; (f) and (g) IC75 of OXP. | ||
In conclusion, inspired by the strong anti-cancer activity of oxaliplatin, four new oxaliplatin analogues having the carrier ligand trans-1,2-diamino-4-cyclohexene and different leaving ligands (Cl in 1, I in 2, CBDCA in 3, and OXA in 4) have been synthesized, characterized, and tested in vitro. The dicarboxylate derivatives 3 and 4 could not be prepared from compounds 1 and 2 by dehalogenation with a silver salt and subsequent reaction with the corresponding dicarboxylate (coordination of Ag+ to the double bond of cyclohexene takes place) but required a different procedure contemplating displacement of the DMSO ligands in the precursor complex cis-[Pt(O,O′-dicarboxylate)(DMSO)2] by trans-1,2-diamine-4-cyclohexene. The new compounds had cytotoxic activity similar to those of cisplatin and oxaliplatin in cervical (A431) and ovarian (2008) human cancer cell lines (slightly lower only for 3); were more active than cisplatin, and equally active as oxaliplatin, in colon HCT-15 and LoVo carcinomas; and, most importantly, were able to overcome the oxaliplatin-resistance in LoVo OXP cells. Finally, all platinum compounds were effective against multi-drug resistant cancer cells suggesting that platinum complexes are not P-gp substrates. The cellular uptake showed a linear correlation with the cancer cell killing effects. However, since there was also a quasi-linear correlation between drug uptake and DNA platination, this does not mean that for all complexes the major toxic event takes place in the nucleus. Indeed, the electron microscopy studies clearly showed a different mechanism of action of DACHEX complex 1 (bearing Cl− leaving ligands as cisplatin) with respect to complex 4 (bearing oxalate leaving ligand as OXP). In particular, 4 induces lipid droplets increase thus suggesting the involvement of metabolism stress rather than DNA damage. These data are consistent with recent reports suggesting that oxaliplatin, unlike cisplatin and carboplatin, can kill cells not necessarily through the DNA-damage response mechanism but rather through alternative mechanisms such as induction of ribosome biogenesis stress. However, this does not mean that the carrier ligand does not play a role. Indeed, same morphological changes were observed for 4 and for oxaliplatin but the dose required for oxaliplatin was greater (IC75) than that required for 4 (IC50) indicating that while the leaving group can orient towards the mechanism of toxicity, the carrier ligand can determine the dose required do produce a given cytotoxic effect. Most importantly, we think that the influence on the lipid metabolism by the oxaliplatin analog 4 represents an important and peculiar feature of the DACHEX ligand. So far compounds 1–4 have been prepared using the racemic mixture of trans-1,2-diamino-4-cyclohexene: we plan to prepare compounds with individual enantiomers to see if there are differences between the R,R and the S,S enantiomers as observed in the case of oxaliplatin. The investigation should also be extended to in vivo and toxicity experiments, paying attention to neurotoxicity, which is the most serious side effect of oxaliplatin.
Conflicts of interest
The authors have no conflicts of interest to declare.Acknowledgements
The Universities of Salento, Bari, and Padova, and the Italian Ministero dell'Università e della Ricerca (MIUR) are gratefully acknowledged. Paride Papadia would like to acknowledge financial support from the MIUR for ex-60% funds. The Authors would like to thank the Department of Biological and Environmental Sciences and Technologies (DiSTeBA), University of Salento, for covering the full Open Access publication fees.References
- J. Graham, M. Muhsin and P. Kirkpatrick, Nat. Rev. Drug Discovery, 2004, 3, 11 CrossRef CAS.
- M. J. Cleare and J. D. Hoeschele, Bioinorg. Chem., 1973, 2, 187–210 CrossRef CAS.
- R. Ranaldo, N. Margiotta, F. P. Intini, C. Pacifico and G. Natile, Inorg. Chem., 2008, 47, 2820–2830 CrossRef CAS.
- J. Kasparkova, T. Suchankova, A. Halamikova, L. Zerzankova, O. Vrana, N. Margiotta, G. Natile and V. Brabec, Biochem. Pharmacol., 2010, 79, 552–564 CrossRef CAS.
- V. Brabec, J. Malina, N. Margiotta, G. Natile and J. Kasparkova, Chem.–Eur. J., 2012, 18, 15439–15448 CrossRef CAS.
- N. Margiotta, C. Marzano, V. Gandin, D. Osella, M. Ravera, E. Gabano, J. A. Platts, E. Petruzzella, J. D. Hoeschele and G. Natile, J. Med. Chem., 2012, 55, 7182–7192 CrossRef CAS.
- U. Jungwirth, D. N. Xanthos, J. Gojo, A. K. Bytzek, W. Korner, P. Heffeter, S. A. Abramkin, M. A. Jakupec, C. G. Hartinger, U. Windberger, M. Galanski, B. K. Keppler and W. Berger, Mol. Pharmacol., 2012, 81, 719–728 CrossRef CAS.
- F. A. L. Anet and M. Z. Haq, J. Am. Chem. Soc., 1965, 87, 3147–3150 CrossRef CAS.
- S. T. Mutter, N. Margiotta, P. Papadia and J. A. Platts, J. Biol. Inorg Chem., 2015, 20, 35–48 CrossRef CAS.
- N. Margiotta, E. Petruzzella, J. a. Platts, S. T. Mutter, R. J. Deeth, R. Ranaldo, P. Papadia, P. a. Marzilli, L. G. Marzilli, J. D. Hoeschele and G. Natile, Dalton Trans., 2015, 44, 3544–3556 RSC.
- J. M. Hill, L. M. Hall, R. J. Speer, H. J. Ridgway, D. P. Stewart and A. D. Newman, J. Clin. Hematol. Oncol., 1977, 7, 231–242 Search PubMed.
- T. Marzo, S. Pillozzi, O. Hrabina, J. Kasparkova, V. Brabec, A. Arcangeli, G. Bartoli, M. Severi, A. Lunghi, F. Totti, C. Gabbiani, A. G. Quiroga and L. Messori, Dalton Trans., 2015, 44, 14896–14905 RSC.
- D. Musumeci, C. Platella, C. Riccardi, A. Merlino, T. Marzo, L. Massai, L. Messori and D. Montesarchio, Dalton Trans., 2016, 45, 8587–8600 RSC.
- D. Cirri, S. Pillozzi, C. Gabbiani, J. Tricomi, G. Bartoli, M. Stefanini, E. Michelucci, A. Arcangeli, L. Messori and T. Marzo, Dalton Trans., 2017, 46, 3311–3317 RSC.
- T. Parro, M. A. Medrano, L. Cubo, S. Muñoz-Galván, A. Carnero, C. Navarro-Ranninger and A. G. Quiroga, J. Inorg. Biochem., 2013, 127, 182–187 CrossRef CAS.
- V. Gandin, M. Pellei, F. Tisato, M. Porchia, C. Santini and C. Marzano, J. Cell. Mol. Med., 2012, 16, 142–151 CrossRef CAS.
- P. Noordhuis, A. C. Laan, K. van de Born, N. Losekoot, I. Kathmann and G. J. Peters, Biochem. Pharmacol., 2008, 76, 53–61 CrossRef CAS.
- M. L. Slovak, G. A. Hoeltge, W. S. Dalton and J. M. Trent, Cancer Res., 1988, 48, 2793–2797 CAS.
- C. Wersinger, G. Rebel and I. H. Lelong-Rebel, Amino Acids, 2000, 19, 667–685 CrossRef CAS PubMed.
- D. Wang and S. J. Lippard, Nat. Rev. Drug Discovery, 2005, 4, 307–320 CrossRef CAS.
- Y. Jung and S. J. Lippard, Chem. Rev., 2007, 107, 1387–1407 CrossRef CAS.
- L. Gatti and P. Perego, in Platinum and Other Heavy Metal Compounds in Cancer Chemotherapy: Molecular Mechanisms and Clinical Applications, ed. A. Bonetti, R. Leone, F. M. Muggia and S. B. Howell, Humana Press, Totowa, NJ, 2009, pp. 115–124 Search PubMed.
- P. M. Bruno, Y. Liu, G. Y. Park, J. Murai, C. E. Koch, T. J. Eisen, J. R. Pritchard, Y. Pommier, S. J. Lippard and M. T. Hemann, Nat. Med., 2017, 23, 461 CrossRef CAS.
- O. Rixe, W. Ortuzar, M. Alvarez, R. Parker, E. Reed, K. Paull and T. Fojo, Biochem. Pharmacol., 1996, 52, 1855–1865 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Material and methods; synthesis of the platinum compounds; NMR characterization of the compounds; biological investigation. See DOI: 10.1039/c9ra07760j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |