
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn situ interfacial surface modification of hydrophilic silica nanoparticles by two organosilanes leading to stable Pickering emulsions†
Yafit Itzhaik Alkotzer‡
ab,
Franziska Grzegorzewski‡ab,
Eduard Belausovc,
Einat Zelingerd and
Guy Mechrez *ab
*ab
aDepartment of Food Sciences, Institute of Postharvest and Food Sciences, Agricultural Research Organization (ARO), Volcani Center, 68 HaMaccabim Road, Rishon Letzion 7505101, Israel. E-mail: guyme@volcani.agri.gov.il; Tel: +972-3-9683990
bAgro-Nanotechnology and Advanced Materials Center, Agricultural Research Organization (ARO), Volcani Center, 68 HaMaccabim Road, Rishon Letzion 7505101, Israel
cDepartment of Ornamental Plants and Agricultural Biotechnology, Institute of Plant Sciences, Agricultural Research Organization (ARO), Volcani Center, 68 HaMaccabim Road, Rishon Letzion 7505101, Israel
dThe Interdepartmental Equipment Unit, The Robert H. Smith Faculty of Agriculture, Food and Environment, The Hebrew University of Jerusalem, POB 12, Rehovot 7610001, Israel
First published on 2nd December 2019
Abstract
Oil-in-water Pickering emulsions are stabilized by in situ functionalization of hydrophilic silica nanoparticles with two organosilane precursors of opposite polarity, dodecyltriethoxysilane (DTES) and 3-(aminopropyl)triethoxysilane (APTES), in a two-step emulsification procedure. The modification of the silica nanoparticles is verified by Fourier transform infrared (FTIR) spectroscopy analysis. The stabilization of the oil droplets by silica is confirmed by tracing the localization of the colloidal silica nanoparticles at the oil–water interface, as observed by confocal fluorescence microscopy. In comparison to modification of the silica nanoparticles prior to the emulsification, in situ functionalization of silica with both organosilanes achieves enhanced emulsion stability and homogeneity, by forming a polysiloxane network between the silica nanoparticles, through polymerization of the organosilanes in the presence of water. The polysiloxane network fixes the silica in place as solid shells around the emulsion droplets, in structures called colloidosomes. These colloidosome shell structures are visualized using confocal microscopy and cryogenic scanning electron microscopy, the latter method successfully enables the direct observation of the silica nanoparticles embedded in the polysiloxane matrix around the oil droplets. Stabilizing the Pickering emulsion droplets and forming silica-based colloidosome shells is dependent on the extent of the hydrolysis and polycondensation reaction of the two organosilanes.
Introduction
Mixing of two immiscible liquids (for example, water and oil) by emulsification is enabled either by surfactants, amphiphilic molecules that self-assemble at the liquid–liquid interface and lower the surface tension of the emulsion droplets, or by solid colloidal particles that adsorb at the surface of the emulsion droplets by a mechanism of partial wetting of the solid by the two liquids.1,2 The latter case, known as a Pickering emulsion,3,4 has gained considerable attention in recent years. In comparison to surfactant-based emulsions (classical emulsions), Pickering emulsions show improved stability, low toxicity, adjustable permeability, and diverse functionality according to a variety of particles available, making the emulsions suitable for many applications in biomedical and food sciences.5–9 One such application route is emulsion templating, leading to the fabrication of robust microcapsules with colloidal particle shells, known as colloidosomes.10–15 Initially self-assembled at the interface of the Pickering emulsion droplets, the colloidal particles are then converted to a close-packed solid shell either by thermal annealing,11,16 gelation,17 covalent cross-linking,14,18 or polymerization,13,19 resulting in robust superstructures that can withstand the removal of the liquid template. The controlled and tunable characteristics of Pickering emulsions make the colloidosomes especially promising in the field of microencapsulation, by achieving permeable, size-flexible, and compatible solid capsules for active ingredients such as living cells,20–22 enzymes,23–25 and drug molecules.26Pickering emulsions can be either oil-in-water (o/w) or water-in-oil (w/o), according to the wettability of the solid particles at the oil–water interface, expressed by the contact angle θ which the particle makes with the interface.27 For hydrophilic particles, the aqueous phase wets the particles better than the oil phase, thus making water the continuous phase, and oil the dispersed phase. In this case the contact angle is < 90°, and o/w emulsions will occur. For hydrophobic particles, the oil phase is the better wetting liquid, θ is > 90°, and w/o emulsions will form. When θ is close to 90°, the particle is the most effectively adsorbed at the interface, making it an ideal Pickering stabilizer.2,28
One of the most commonly used stabilizing agent in Pickering emulsions is silica, which is a biocompatible, low-cost material, suitable for large-scale synthesis and biomedical applications.29,30 Hydrophilic by nature of their OH surface groups (silanol groups), pure silica particles can stabilize o/w emulsions with polar oils that have a limited solubility in water.31,32 However, silica particles can be easily made more hydrophobic by versatile surface chemistry modification routes to achieve optimum partial wetting conditions by water and oil.28,33,34 One such route is by chemical grafting of organosilanes to the silica surface; the choice of the organosilane reactant and the extent of the reaction determine the degree of surface hydrophobicity and functionalization.1,28,35 Heterogeneous modification of colloidal silica particles with both hydrophilic and hydrophobic organosilanes prior to emulsification was found to significantly improve the emulsion stability towards coalescence, compared to homogeneously modified particles.33 Colloidosomes were produced from the initial state of Pickering emulsions by interfacial reaction of the surface silanol groups of the silica stabilizer with organosilanes that undergo hydrolysis and condensation reactions in water, resulting in a stable solid shell around the emulsion droplets, by forming either a polysiloxane network between the particles,22 or a thin continuous silica layer, which acts as a binder for the silica particles at the interface.19
In this study we present a method to prepare stable, homogenous, toluene-in-water Pickering emulsions, stabilized by silica nanoparticles, which are grafted with two trialkoxysilane agents during the emulsification process (in situ functionalization). Both organosilane precursors, dodecyltriethoxysilane (DTES) and 3-(aminopropyl)triethoxysilane (APTES), chosen due to their opposite polarity, are necessary to achieve stability of the emulsion droplets, by forming a polysiloxane network between the silica nanoparticles through hydrolysis and polymerization of their ethoxy groups in the presence of water (Fig. 1). We found that in situ functionalization with these two organosilanes is preferable in comparison to other approaches, such as prior modification of the silica nanoparticles, or the employment of only one organosilane agent during emulsification, thus resulting in enhanced emulsion stability and improved homogeneity of the emulsion droplets. These emulsion characteristics, attributed to the polysiloxane network between the silica nanoparticles, lead to the formation of silica-based colloidosomes, which are visualized by confocal laser scanning microscopy (CLSM) and cryogenic-scanning electron microscopy (cryo-SEM). The silica-stabilized Pickering emulsions are studied here in terms of stability and droplet size distribution by confocal microscopy, and the functionalized silica nanoparticles are characterized by Fourier transform infrared spectroscopy (FTIR). Schematic illustration of the research concept is depicted in Fig. 1.
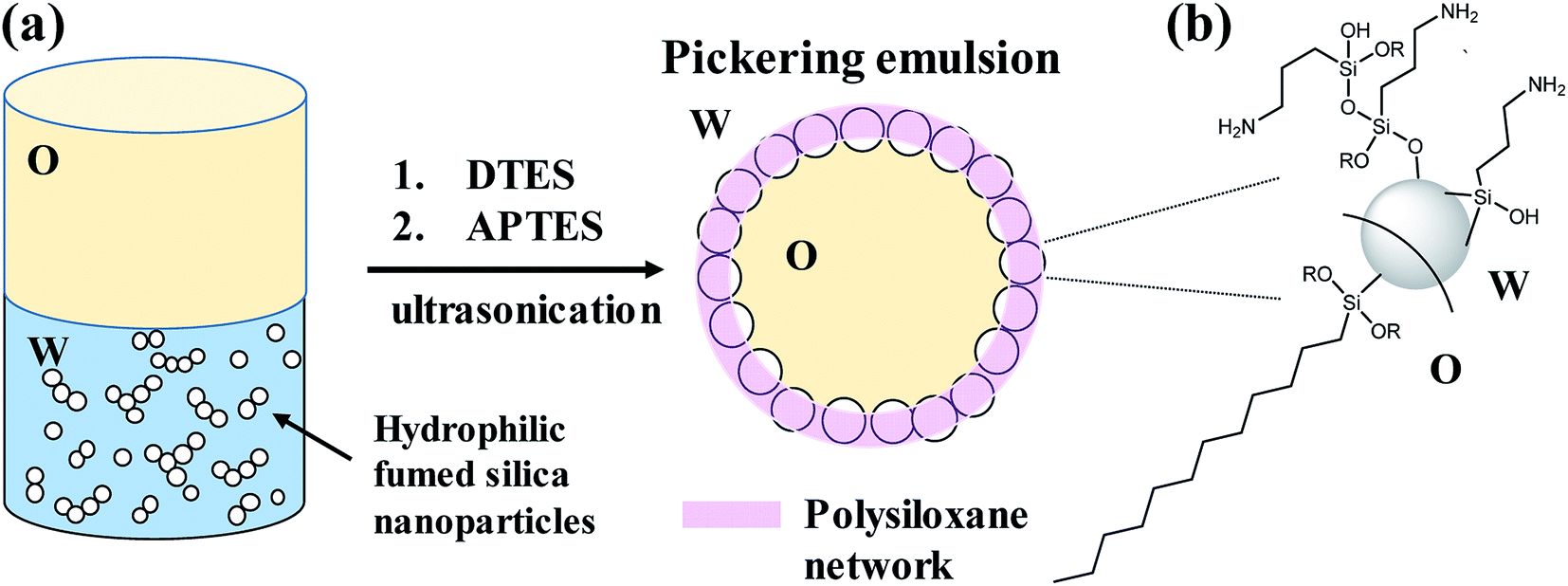 | ||
| Fig. 1 Schematic illustration of the research concept: (a) stabilization of toluene-in-water Pickering emulsion by hydrophilic fumed silica nanoparticles occurs by in situ functionalization of the silica surface with two organosilanes of opposite polarity, APTES and DTES. A polysiloxane network forms between the silica nanoparticles as a result of the hydrolysis and polymerization of the organosilanes in the presence of water, leading to silica-based colloidosomes formation. (b) Siloxane bonds are formed as the hydrolyzed organosilane molecules are reacting by condensation with the silica surface silanol groups, or with each other (one possible route of APTES polymerization is illustrated). | ||
Results and discussion
Formation of Pickering emulsions by in situ functionalization of silica
Hydrophilic Aerosil® 300 fumed silica consists of primary nanoparticles of 7 nm average diameter, merged together in the form of aggregates, which are held together in the powder as agglomerates as a result of physical interactions, such as van der Waals forces or hydrogen bonds. Dispersing the silica powder by ultrasonication prior to emulsification breaks the agglomerates and separates them into aggregated silica units of 146 ± 2 nm average size, according to dynamic light scattering (DLS) measurements of an aqueous 2% w/v silica dispersion.1% w/v dispersed silica aggregates were reacted under ultrasonic field with two organosilanes of opposite polarity, DTES and APTES, in a biphasic system of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v water and toluene, generating a homogeneous, densely packed, toluene-in-water emulsion (Fig. 2a and b). Droplet size distribution analysis, done by measuring the droplet diameters from confocal microscopy images, showed that the o/w emulsion is composed of droplets ranging from 2 to 35 μm in diameter, with the majority at the 2–20 μm size range, and an average droplet size diameter of 11.4 ± 6.4 μm (Fig. 2c). The functionalization of the silica nanoparticles with DTES and APTES was verified by FTIR analysis (Fig. 2d). The functionalized nanoparticles were separated from the emulsion, and then purified prior to the FTIR measurements in order to remove unattached and physically adsorbed reagents from product, and ensure analysis of the organosilanes that are explicitly attached to the surface by covalent bonding. The FTIR spectra clearly show the vibrational modes of the CH2, CH3 bonds at 2855–2960 cm−1 forming after DTES reaction (step 1 in emulsification process, green curve in Fig. 2d), in comparison to pristine SiO2 (blue curve in Fig. 2d). Following APTES reaction (step 2 in emulsification process), the vibrational modes of the NH2 bond at 1547 cm−1, together with those of the Si–CH2 bond at 1412 cm−1, and the CH2, CH3 bonds at 2855–2960 cm−1 appear (red curve in Fig. 2d), thus confirming a successful covalent attachment of the organosilanes to the silica surface.36,37
:1 v/v water and toluene, generating a homogeneous, densely packed, toluene-in-water emulsion (Fig. 2a and b). Droplet size distribution analysis, done by measuring the droplet diameters from confocal microscopy images, showed that the o/w emulsion is composed of droplets ranging from 2 to 35 μm in diameter, with the majority at the 2–20 μm size range, and an average droplet size diameter of 11.4 ± 6.4 μm (Fig. 2c). The functionalization of the silica nanoparticles with DTES and APTES was verified by FTIR analysis (Fig. 2d). The functionalized nanoparticles were separated from the emulsion, and then purified prior to the FTIR measurements in order to remove unattached and physically adsorbed reagents from product, and ensure analysis of the organosilanes that are explicitly attached to the surface by covalent bonding. The FTIR spectra clearly show the vibrational modes of the CH2, CH3 bonds at 2855–2960 cm−1 forming after DTES reaction (step 1 in emulsification process, green curve in Fig. 2d), in comparison to pristine SiO2 (blue curve in Fig. 2d). Following APTES reaction (step 2 in emulsification process), the vibrational modes of the NH2 bond at 1547 cm−1, together with those of the Si–CH2 bond at 1412 cm−1, and the CH2, CH3 bonds at 2855–2960 cm−1 appear (red curve in Fig. 2d), thus confirming a successful covalent attachment of the organosilanes to the silica surface.36,37
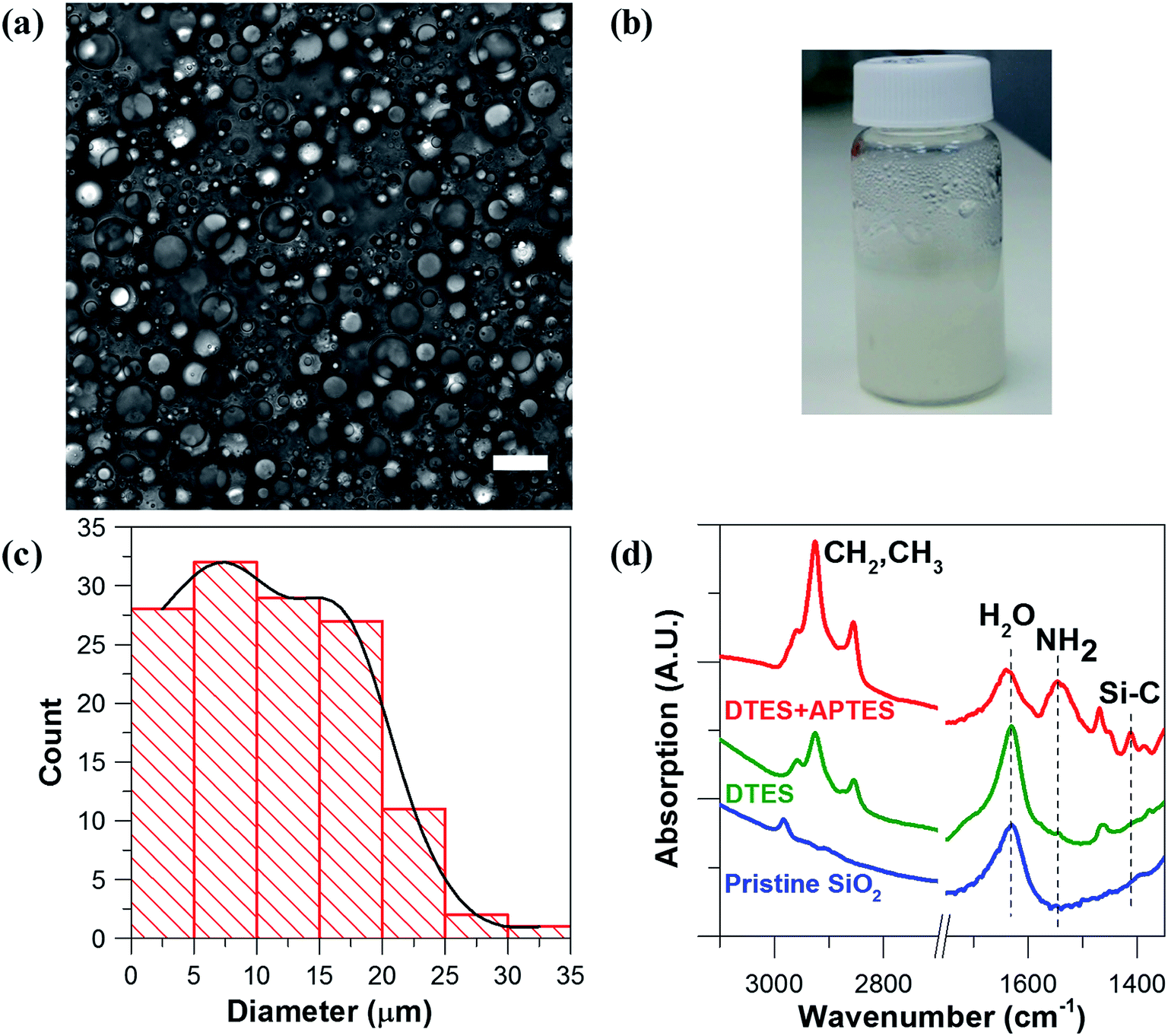 | ||
| Fig. 2 (a) Confocal microscopy image of a 1:1 v/v toluene-in-water (o/w) emulsion, stabilized by 1% w/v Aerosil® 300 silica nanoparticles that were functionalized in situ with DTES and APTES (scale bar 20 μm). (b) The silanization reaction leads to the formation of a stable emulsion with a homogeneous, densely packed, appearance. (c) Droplet size distribution graph of the emulsion droplets, showing the majority of the droplet size in the range of 2–20 μm diameter, with an average droplet size diameter of 11.4 ± 6.4 μm (d) FTIR spectra confirming the functionalization of the silica nanoparticles (pristine, before reaction, blue curve), showing the vibrational modes of the CH2, CH3 bonds after DTES reaction (step 1 in emulsification process, green curve), and of the NH2, Si–C, and CH2, CH3 bonds forming after APTES reaction (step 2 in emulsification process, red curve). | ||
The emulsion was found to be stable to creaming for at least 4 weeks after preparation. However, weekly droplet size analysis showed a broader distribution indicative of droplet coalescence occurring with time, although the majority of the droplets analyzed still remained at the 2–20 μm size range 4 weeks after preparation. The average droplet diameter measured after 4 weeks was 13.2 ± 12.0 μm (Fig. S1 in the ESI†).
The stabilization of the toluene droplets in water by silica nanoparticles was visualized in confocal fluorescence microscopy, by tracing the localization of 6-aminofluorescein-labelled silica nanoparticles (SiO2-dye) at the toluene–water interface and in the major water phase, and of Nile Red in the minor toluene phase, confirming oil-in-water Pickering emulsion formation (Fig. 3). The emulsion was analyzed in confocal microscopy using a z-series stack, outlining the self-assembly of the silica nanoparticles at the oil–water interface (Fig. S2 in the ESI†).
 | ||
| Fig. 3 Confocal microscopy images of 1:1 v/v Pickering toluene-in-water emulsion stabilized by 1% w/v 6-aminofluorescein labelled-silica nanoparticles (SiO2-dye) functionalized by DTES and APTES, and stained with Nile Red (scale bar 20 μm): (a) transmission image of emulsion droplets; (b) green channel of (a), with SiO2-dye particles concentrated around the oil droplets, proving Pickering emulsion formation; (c) red channel of (a) showing the Nile Red signal coming from the toluene droplets, thus proving oil-in-water emulsion formation. | ||
Prior modification of silica with 6-aminofluorescein decreased the surface area of the silica nanoparticles available for DTES and APTES reaction, resulting in an incomplete emulsification of the oil phase, with creaming occurring immediately after preparation. Accordingly, larger emulsion droplets were obtained, due to the decrease in particle concentration.1,2 The labelled silica aggregates were also observed in the major phase, evidently due to inefficient emulsification, as the excess silica that does not participate in emulsion stabilization is assembled in water. Nevertheless, confocal fluorescence microscopy analysis was done successfully on the emulsified volume, as shown in Fig. 3.
The role of DTES and APTES in emulsification
The emulsification of toluene in water using in situ functionalized silica nanoparticles was examined for each step and component in the reaction (for visualization of the experiments described here, see Fig. 4). | ||
| Fig. 4 Images of toluene/water (1:1 v/v) biphasic systems: (a) 1% w/v hydrophilic fumed Aerosil® 300 silica before sonication, and (b) after 30 min sonication. (c) Silica after 10 min reaction with DTES (step 1 in emulsification process), and (d) after additional 20 min (e) DTES-functionalized silica (from step 1) after 20 min reaction with APTES (step 2 in emulsification process). (f) Silica after 10 min reaction with APTES alone, and (g) after additional 20 min (h) DTES without any silica added after 10 min sonication, and (i) the same system after adding APTES and sonicating for additional 20 min. | ||
1% w/v hydrophilic fumed silica were dispersed in a biphasic system of 1:1 v/v toluene and water. The biphasic system was sonicated for 30 min without adding the organosilane reagents; however, no emulsion was formed under these conditions. Agglomeration of the silica nanoparticles in the aqueous phase was observed after sonication, evidenced by the turbidity of the aqueous phase (Fig. 4a and b).
The in situ reactions of the organosilanes with silica were monitored for each reagent separately, and compared to their successive reaction. Surface modification of hydrophilic silica with DTES did not result in emulsification, even after a prolonged reaction of additional 20 min sonication (Fig. 4c and d). On the other hand, employing APTES alone in surface modification did result in o/w emulsion formation after 30 min sonication (Fig. 4f and g), although a significant volume of the toluene was not emulsified, compared to the emulsion formed when DTES was employed prior to APTES reaction (compare Fig. 4e and g). A broad droplet size distribution was observed in APTES-functionalized emulsion (Fig. 4g), with 28% of the measured droplet diameters larger than 35 μm, and an average droplet diameter of 29.0 ± 18.9 μm (Fig. S3 in the ESI†). The emulsion was not stable towards coalescence, as the average droplet diameter increased to 46.4 ± 15.4 μm 4 weeks after preparation.
To rule out a surfactant effect of the organosilanes that may result in a classical emulsion formation, DTES and APTES were reacted successively in a toluene and water biphasic system without the silica nanoparticles. The organosilanes alone did not form an emulsion under these conditions, although turbidity in the aqueous phase was observed after 30 min sonication, and was attributed to the hydrolysis and polycondensation of the trialkoxysilanes in water (Fig. 4h and i).
In general, silanization reaction of trialkoxysilanes proceeds through the hydrolysis of the alkoxy groups into trihydroxyl groups, which then condense with the surface silanol groups, forming a siloxane bond between the organosilane and the silica surface. The extent of the hydrolysis and condensation is highly dependent on the reaction conditions, such as temperature, the organosilane concentration, and the amount of water present.36,38–40 Here, the choice of the organosilane reagents and their combined reaction affect the final emulsion properties, with APTES being the crucial factor in Pickering emulsion formation. The reaction of APTES with the silica surface may follow many possible routes in the presence of water, either by covalent attachment (siloxane bond), or by weak interactions with surface silanols via hydrogen bonds or electrostatic attractions. Furthermore, excess of water leads to polymerization of APTES molecules, resulting in self-assembly of neighboring silanes on the silica surface (horizontal polymerization), or formation of oligomers and polymers of silane in solution (vertical polymerization), which can also attach to the silica surface.36,38,41 On the other hand, DTES, which has three available ethoxy groups as well, is less reactive toward hydrolysis and condensation than APTES, due to the steric hindrance imposed by its long n-alkyl chain.42 Furthermore, DTES is insoluble in water, and its hydrolysis reaction is dependent on the molecules being located at the oil–water interface, i.e., in contact with water, as opposed to APTES, which is soluble both in water and in toluene, and can go through hydrolysis and polymerization at the water phase before attaching to the silica surface. Nevertheless, DTES is clearly an essential component in the emulsification process: according to the results mentioned above, when APTES was preceded by DTES in reaction, the emulsion homogeneity and stability were enhanced, compared to a reaction of APTES or DTES alone.
Prior modification of silica with APTES or DTES
Surface modification of hydrophilic silica by organosilanes is a common method to achieve stable Pickering emulsions, essentially by controlling the wettability of silica toward water and oil with the extent of the grafting. Conventionally, the silica surface modification is done prior to the emulsification process.1,28,33 Here, as a comparison to the in situ silica functionalization, leading to emulsion formation, modification of silica nanoparticles with either DTES or APTES was done prior to emulsification; 1% w/v of the modified nanoparticles, SiO2–APTES or SiO2–DTES, were then dispersed in a 10 mL 1:1 v/v toluene and water biphasic system by 30 min sonication.
Silica modification by DTES was expected to change the surface properties to more hydrophobic, thus enabling stabilization of either o/w or w/o emulsions, depending on the extent of surface functionalization. However, similar to the in situ reaction with DTES alone, emulsification did not occur with SiO2–DTES, and the nanoparticles remained dispersed only in the water phase after reaction. This result indicates that the silica modification by DTES was insufficient, leaving the surface too hydrophilic to stabilize Pickering emulsions. On the other hand, SiO2–APTES nanoparticles were able to stabilize emulsions of toluene-in-water, although a large fraction of the toluene phase was not emulsified (Fig. 5a). The emulsified fraction of toluene in water was analyzed by confocal microscopy, and the emulsion appeared homogeneous, with densely packed droplets (Fig. 5b). Droplet size distribution analysis, done by measuring the droplet diameters from confocal microscopy images, showed that 80% of the droplets are in the range of 5 to 25 μm in diameter (Fig. 5c). However, creaming was observed immediately after preparation, and within a week the average droplet diameter increased by a factor of 2, evidently due to coalescence of the oil droplets (17.8 ± 10.0 μm of the freshly prepared emulsion, compared to 36.7 ± 17.9 μm after one week).
 | ||
| Fig. 5 (a) Prior modification of Aerosil® 300 silica nanoparticles with APTES (SiO2–APTES) leads to an inefficient emulsification of 1:1 v/v toluene in water, with creaming occurring after preparation. (b) Confocal microscopy image of the emulsified volume of toluene in water (middle part of the vial presented in (a), arrow), stabilized by 1% w/v SiO2–APTES nanoparticles (scale bar 20 μm). (c) Droplet size distribution of the o/w emulsion shown in (b). | ||
These findings are in agreement with APTES and DTES reactivity towards silica functionalization, as discussed previously. However, in situ functionalization of silica is superior to prior modification of silica, resulting in enhanced emulsion homogeneity and stability. Furthermore, the in situ functionalization method has the advantage of performing a rapid, two-step procedure in one vessel, leading to successful emulsification of toluene in water without any material lost during procedure steps, whereas prior modification of silica requires the reaction to be performed in an organic medium, then the separation of residual reactants from product, and the redispersion of the product in water before emulsification, all of which makes the latter method more limited and tedious to perform.1
Silica-based colloidosomes from Pickering emulsions
Colloidal silica nanoparticles assembled at the interface of the Pickering emulsion droplets can be linked together by utilizing the silica surface groups in varied chemical reactions to form solid shells around the droplets, in structures called colloidosomes.14,15,19,22 Here in this study, an interfacial reaction linking the silica nanoparticles was enabled by polymerization and condensation of DTES and APTES molecules in the presence of water, resulting in a polysiloxane network formation between the silica nanoparticles, thus fixing them in place as solid shells around the emulsion droplets. These colloidosome structures were visualized using confocal microscopy, as the solvents begin to evaporate from the sample under observation (Fig. 6a and b). In contrast, in emulsions stabilized by SiO2–APTES particles (obtained by prior modification of silica with APTES), the shell structures around the emulsion droplets were not observed under the same drying conditions: the emulsion droplets start to distort and shrink as the silica nanoparticles aggregate together and eventually collapse on one side of the former emulsion droplets (Fig. 6c). These observations reinforce our previous findings of enhanced emulsion properties achieved by in situ silica functionalization, which are evidently due to the polysiloxane network forming at the interface during reaction of DTES and APTES. The polysiloxane network either does not form, or occurs at low level of polymerization, in prior modification of silica. | ||
| Fig. 6 Confocal microscopy images of Pickering emulsion droplets recorded during solvent evaporation under ambient conditions (scale bar 20 μm in all images): (a) and (b) SiO2 colloidal shell structures obtained from in situ DTES-and-APTES-functionalized SiO2-stabilized o/w emulsion. (c) In case of prior modification of silica, in SiO2–APTES-stabilized o/w emulsion, shrinkage of emulsion droplets occurs as the silica nanoparticles start to aggregate and eventually collapse on one side of the droplets. | ||
The nanostructure of the colloidosome shells, templated from o/w emulsions stabilized by in situ functionalized SiO2, was characterized via direct imaging using high resolution cryo-SEM (Fig. 7). This method enabled the direct observation of the emulsion structure by ultrafast cooling of vitrified cryo-SEM specimens; however, due to uneven fracturing occurring during sample preparation, water sublimation had to be done in order to see the nanosized silica particles comprising the colloidosome shells. After sublimation, these silica shells were visibly seen around the emulsion droplet (Fig. 7a and b). Energy-dispersive X-ray spectroscopy (EDX) analysis performed on the colloidosome shells confirmed that the shells contain silicon (22 wt% of the sampled area, vs. O and C signals), whereas the surrounding medium is aqueous with only trace amounts of silicon detected (0.7 wt%, vs. O and C signals). Observing the shell structure more closely revealed that the silica nanoparticles are embedded in the polysiloxane matrix (Fig. 7c). In both cryo-SEM micrographs where the colloidosome shells around the emulsion droplets are fully depicted (Fig. 7a and b), the shells are incomplete, indicating that the polysiloxane matrix forming between the silica nanoparticles had broken due to the forces applied on the sample during fracturing and water sublimation. As opposed to the particulate shell structures shown in Fig. 7a–c, a thick, continuous silica shell is presented in Fig. 7d: here, a hollow shell remained after sample handling (Fig. 7d, inset), providing evidence for a robust colloidosome structure of silica in the polysiloxane matrix, with a thickness of ca. 700 nm. A rigid shell is an important factor in obtaining intact, stable microcapsules after the emulsion template is removed, although this step is not necessary to encapsulate active ingredients, as the encapsulation can occur in solution, providing that the colloidosomes are permeable to the encapsulated species, which in turn are immiscible with the major phase.11,22 Here, the colloidosome shells eventually collapse with the evaporation of the solvents under ambient conditions. However, it appears that the interfacial polysiloxane network between the silica nanoparticles in Pickering emulsions is permeable to small molecules such as the fluorophore Nile Red that diffuses into the emulsion droplets after a few minutes of mixing (see Fig. 3c). Controlling the emulsion formation parameters, for example, the reagent concentrations, the reaction temperature, and the pH of the aqueous phase, may lead to an efficient templating of stable colloidosomes for encapsulation purposes, and is currently the subject of further studies.
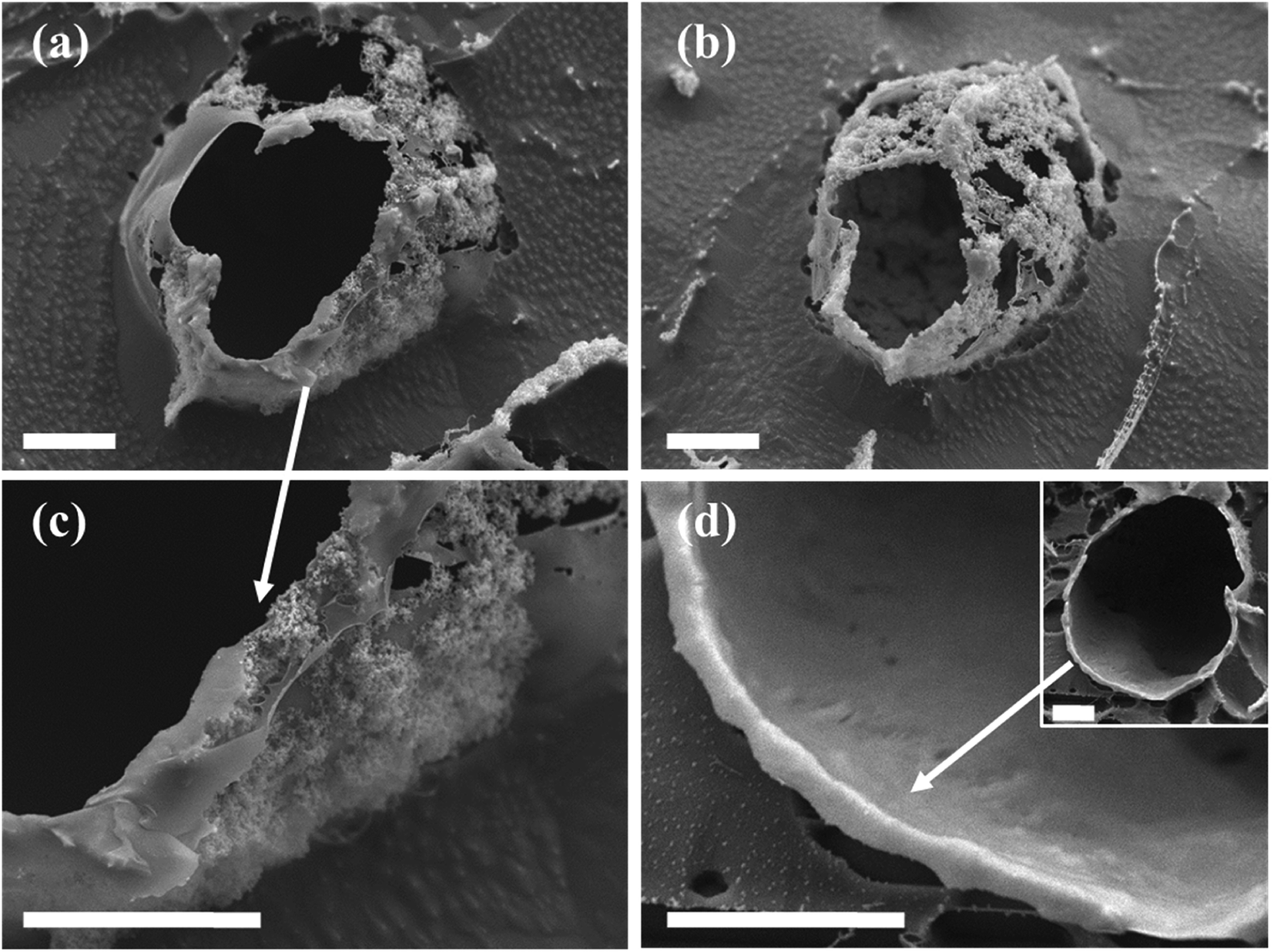 | ||
| Fig. 7 Cryo-SEM images of characteristic colloidosome structures templated from in situ DTES-and-APTES-functionalized SiO2-stabilized o/w emulsion droplets (scale bar 5 μm in all images): (a) and (b) The silica nanoparticles are fixed in place by the polysiloxane matrix, resulting in a shell structure called colloidosomes. (c) A closer look at the colloidal shell reveals that the silica nanoparticles are embedded in the polysiloxane matrix, which is formed during silanization reaction of APTES and DTES with silica. (d) A hollow shell templated from an emulsion droplet (inset), comprised of a thick, continuous, silica-polysiloxane matrix of ca. 700 nm thickness. | ||
Interfacial polysiloxane network formation between the silica nanoparticles
The interfacial reaction of APTES and DTES with silica affects the Pickering emulsion formation by forming a polysiloxane network between the nanoparticles, and stabilizing the oil droplets in the aqueous phase. Furthermore, the silica nanoparticles are fixed in place by the polymeric matrix, forming colloidosome shell structures around the emulsion droplet, as observed in confocal microscopy and cryo-SEM images. Stabilizing the Pickering emulsion droplets and forming silica-based colloidosome shells is dependent on the extent of the hydrolysis and polycondensation reaction of the two organosilanes. It is clear from the experiments described above that in situ reaction results in enhanced polymerization of the two reagents in comparison to prior modification of silica. The difference between the two methods stems from the greater energy invested in the ultrasonication process during in situ reaction, resulting in elevated temperatures around the sonicating probe, thus enhancing the reaction rate of the hydrolysis and condensation of the organosilane reagents.43,44 It should be noted that according to the FTIR spectra shown in Fig. 2d, the vibrational mode at 1389 cm−1, originating from the CH3 bend in the ethoxy moieties of APTES and DTES, is indicative of an incomplete hydrolysis and siloxane condensation.36,37 Nevertheless, there is sufficient hydrolysis and polymerization of APTES and DTES to form a polysiloxane network between the silica nanoparticles by in situ functionalization, as was directly observed by confocal microscopy and cryo-SEM imaging.We found one report where interfacial hydrolysis and self-condensation reactions of two alkylchlorosilanes with silica particles stabilizing w/o Pickering emulsion produced stable microcapsules, as a result of a polyalkylsiloxane shell forming around the emulsion droplets. The two alkylchlorosilanes were chosen due to their different reactivity, as a consequence of their different alkyl chain length, thus restricting the reaction to the interface of the Pickering emulsion droplets, and simulating a complete polysiloxane network formation.22 In correlation to this report, here in this study two trialkoxysilanes, APTES and DTES, with opposite polarity and different reactivity, were employed to functionalize the silica nanoparticles. It is possible that the APTES reaction with silica is affected by the steric hindrance of the long alkyl chain of the DTES, resulting in self-polymerization in water before condensation into the silica surface, or that the presence of the two reagents in the two phasic system limits the reaction to the oil–water interface, thus improving the emulsification efficiency, and resulting in enhanced emulsion properties. Elucidating the mechanism of surface functionalization by DTES and APTES, and their combined effect on the emulsion formation will be further explored in future research. Quantitative analysis needs to be developed in order to estimate the extent of the silica functionalization by the two organosilanes, by determining the silanol and carbon content of the silica nanoparticles,28 and employing other characterization techniques, such as nuclear magnetic resonance (NMR) spectroscopy of 1H NMR in combination with 29Si NMR,45 and thermal gravimetric analysis (TGA) coupled with mass spectrometer (MS).
Conclusions
In this study, we have presented a method to prepare stable, homogenous, toluene-in-water Pickering emulsions, by in situ functionalization of hydrophilic silica nanoparticles with two organosilanes of opposite polarity, DTES and APTES. Employing both organosilane reagents successively in a two-step procedure was found to improve the emulsion stability and homogeneity, in comparison to standard approaches, such as employing only one of the organosilane reagents, or modifying the silica nanoparticles prior to the emulsification. Both APTES and DTES undergo hydrolysis and polycondensation reactions in the presence of water, resulting in a formation of a polysiloxane network between the silica nanoparticles. The mechanism of surface functionalization by DTES and APTES, and their combined effect on the emulsion formation will be further explored in future research.In situ functionalization of silica as a method to prepare o/w Pickering emulsion was found to have several advantages over the more commonly used approach of modifying the silica nanoparticles prior to the emulsification. In the in situ method, the two organosilanes are employed in a rapid, two-step procedure taking place in one vessel, with minimum time invested in the reaction, and without any material lost during procedure steps, by excluding the need for separation and purification of the silica nanoparticles prior to emulsification. Still, the most significant advantage of the in situ functionalization is the enhanced emulsion stability and homogeneity, which are the result of the polysiloxane network forming between the silica nanoparticles during reaction of DTES and APTES. The polysiloxane network either does not form, or occurs at low level of polymerization, in prior modification of silica.
Characterization of the o/w emulsion structure formed by in situ functionalization of silica was successfully done by cryo-SEM imaging. By this method we achieved direct observation of the silica nanoparticles embedded in the polysiloxane network around the oil droplets, in shell structures called colloidosomes. Controlling the extent of the polysiloxane network formation between the silica nanoparticles by optimizing the parameters of the emulsification process, may lead to the production of robust colloidosome structures for encapsulation applications, which is currently the subject of further studies.
Experimental
Materials
Hydrophilic fumed silica powder (Aerosil® 300, 300 m2 g−1 BET area, primary particle diameter 7 nm, according to manufacturer)46 was obtained from Evonik Industries, Germany (see Fig. S4 in the ESI† for SEM characterization of the silica morphology. Full SEM and TEM characterization of Aerosil® fumed silica can be found in ref. 46). 3-(Aminopropyl)triethoxysilane (APTES), 99%, dodecyltriethoxysilane (DTES), technical, 6-aminofluorescein, 95%, succinic anhydride, ≥99%, 2-(N-morpholino)ethanesulfonic acid (MES) hydrate, ≥99.5%, N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC), ≥ 98%, N-N-diisopropylethylamine (EDIPA), Nile Red, technical, were purchased from Sigma-Aldrich, Israel. Acetonitrile (ACN), ethanol, and toluene of analytical grade with purity ≥ 99%, as well as ultra-pure deionized water (ULS/MS grade) were used as received without further purification.Pickering emulsions by in situ functionalization of silica
Prior to emulsification, 2% w/v fumed silica nanoparticles were dispersed in water by an ultrasonic processor (Vibra-Cell™ VCX 750, Sonics, USA) employed with a 13 mm diameter probe tip operating at 25% amplitude and up to 20 W for 10 min. In all sonication processes described henceforth the same operating conditions were employed; during the process the reaction vessel was kept cool using an ice bath.After sonication, 5 mL out of the aqueous silica dispersion were mixed with 5 mL of toluene to give a total volume of 10 mL and an o:w (v/v) ratio of 1:1. The content of the silica nanoparticles in the biphasic system was thus fixed to 1% w/v for all the studied emulsions for the purpose of comparison with other approaches, as described below. For the same reason, the o:w (v/v) ratio of 1:1 was fixed for all the studied emulsions, after obtaining stable, densely packed emulsions by in situ functionalization of silica under these conditions, see results and discussion section. Characterization of the emulsion properties and its dependence on the reaction parameters, for example the silica content in the biphasic system, the reagent concentrations, and the o:w v/v ratio, will be further explored in future research.
Emulsification of 1% w/v SiO2 in o:w 1:1 mixture proceeded in two steps: first, 0.5 mL of 0.2 M DTES in toluene were added to the mixture, followed by sonication for 10 min. Then, 0.5 mL of 0.2 M APTES in water were added, followed by sonication for 20 min. In both steps, hydrolysis of the ethoxy groups of the reactants occurs, resulting in the formation of silanol groups, which in turn covalently attach to the silica surface by condensation with the surface silanol groups, and forming siloxane (Si–O–Si) bonds (silanization reaction). Emulsification with only one of the reactants, either DTES or APTES, was done by adding 0.5 mL of the organosilane precursor (of the same concentration, 0.2 M, in the relevant solvent) to the 10 mL o:w (v/v) 1:1 mixture with 1% w/v SiO2, followed by sonication for 30 min.
All emulsions were then stored under ambient conditions prior to further analysis.
Pickering emulsions by prior modification of silica
Silica nanoparticles were modified prior to emulsification with either APTES or DTES precursors as follows: 1% w/v fumed silica nanoparticles were dispersed in 10 mL toluene (for DTES modification), or 10 mL ethanol (for APTES modification). 0.5 mL of the organosilane precursor were then added to the respective silica dispersion, out of a stock solution of 0.2 M reagent in the relevant solvent. The reaction vessel was then shaken by ELMI Intelli Mixer RM-2M at 70 rpm and 99° rotation for 1 h under ambient conditions. The modified silica nanoparticles were collected by centrifugation (9000 rpm for 10 min at 25 °C, using a Sigma 3-18KS centrifuge from Sigma Laborzentrifugen GmbH, Germany). The separated particles were then rinsed twice with the relevant solvent in order to remove unreacted and physically adsorbed species from product; after each rinsing the particles were separated from solution by centrifugation under the same conditions as specified before. The product, either SiO2–DTES or SiO2–APTES particles, was dried at 35 °C under vacuum for 3 h.1% w/v SiO2–DTES or SiO2–APTES particles were dispersed in a 10 mL water and toluene 1:1 v/v mixture, which was emulsified by sonication for 30 min. The emulsions were then stored under ambient conditions prior to further analysis.
Fluorescent labelling of silica nanoparticles
The covalent attachment of 6-aminofluorescein to silica nanoparticles was carried out in several synthesis steps under ambient conditions. First, 0.5 mL of 0.2 M APTES in water were added to 5 mL of 2% w/v silica aqueous dispersion and the mixture was stirred by a vortex mixer at high speed for 10 min. After reaction the silica nanoparticles were separated from solution by centrifuging at 10286 rpm for 15 min at 25 °C (the same conditions were further employed for the rinsing cycles and the next steps of the synthesis). The silica nanoparticles were rinsed twice with water and once with ACN to remove all traces of reactants from product; after each rinsing the particles were separated from solution by centrifugation. After the first synthesis step, the silica nanoparticles obtained from the silanization reaction were functionalized with NH2 groups on the surface (SiO2–NH2), although the shortened reaction time left the silica surface with sufficient silanol groups for further modifications.
The next step in labelling was carried out through an amidation reaction between the SiO2–NH2 nanoparticles and succinic anhydride to introduce carboxyl groups on the SiO2 surface by forming an N-[3-(triethoxysilyl)propyl]succinamidic acid derivative (SiO2–COOH).47 A stock solution of 70 mg succinic anhydride in 10 mL ACN together with 20 μL EDIPA was prepared for the reaction. 1.6% w/v of SiO2–NH2 nanoparticles were dispersed in the succinic anhydride stock solution, and the dispersion was stirred in vortex mixer at high speed for 3 h. The SiO2–COOH nanoparticles were then separated from reactant solution by centrifugation; rinsing and centrifugation cycles were done twice with ACN and once with water to remove weakly attached reactants from product.
The attachment of the 6-aminofluorescein dye to the SiO2–COOH nanoparticles proceeded by forming an amide bond between the N-[3-(triethoxysilyl)propyl]succinamidic acid derivative and the NH2 group of the dye (SiO2-dye), using EDC as a zero-length cross-linker.48 Stock solutions of 1 mg of 6-aminofluorescein and 100 mg of EDC were prepared separately, both in 10 mL of 0.5 M MES (pH 3.0). 0.1 g of SiO2–COOH nanoparticles were dispersed in 3 mL 0.5 M MES, then 1.5 mL of EDC and 0.5 mL of the dye stock solutions were added to the dispersion. The reaction vessel was stirred by vortex mixer for 1 h. After reaction the SiO2-dye nanoparticles were separated from reactants by centrifugation. In order to verify that excess reactants and weakly attached dye molecules were removed from product, 5–7 rinsing and centrifugation cycles with 0.05 M MES (pH 3.7) were performed; the supernatant solution after each cycle was checked for fluorescence by plate reader analysis (Synergy™ Neo2 Multi-mode Microplate Reader, BioTek Instruments, Inc., Winooski, VT, USA. Excitation and emission wavelengths of the fluorescein dye were set to 488 nm and 520 nm, respectively). The rinsing cycles were continued until there was no fluorescent signal from the supernatant solution.
The SiO2-dye nanoparticles were then dispersed in water, to be further employed in the previously described in situ emulsification process (1% w/v SiO2-dye nanoparticles in 10 mL water and toluene 1:1 v/v mixture). The localization of the fluorescently labelled silica nanoparticles in the Pickering emulsion was done by confocal laser scanning microscopy analysis.
Fluorescent labelling of oil droplets
For verification of o/w emulsion formation, the fluorophore Nile Red, which stains the oil phase, was employed. For this purpose, 100 μL of 0.06 mM Nile Red stock solution in toluene were mixed with 900 μL of the silica-based Pickering emulsion using a vortex mixer at medium speed for 10 min. The distribution of the fluorophore in the oil phase of the emulsion was then observed in confocal laser scanning microscope.Particle size measurements
Size distribution of the 2% w/v silica dispersion was determined using a MasterSizer 2000 (Malvern Panalytical, Malvern/UK). The silica dispersion was sonicated for 10 min before the measurements. 1.5 mL of the dispersion were then transferred to a square cuvette for dynamic light scattering (DLS) measurements. The size distribution was calculated from Mie theory using the silica refractive index of 1.46 for the dispersed phase and 1.33 for water in Zetasizer Software (Version 7.11, Malvern Instruments Ltd., Malvern/UK) for data collection and analysis.Fourier transform infrared (FTIR) spectroscopy analysis
The surface modification of the SiO2 nanoparticles was analyzed by elucidating the molecular structure of the covalently attached organosilanes, using Thermo Scientific™ Nicolet™ iS50 FTIR spectrometer, equipped with attenuated total reflection (ATR) module (smart iTX–Diamond). The output signal was collected with a deuterated triglycine sulfate (DTGS) detector. Each FTIR spectrum represents the average of 256 scans at 4 cm−1 resolution. For these measurements, in situ functionalized SiO2 nanoparticles were separated from emulsion by centrifugation at 10286 rpm for 15 min at 25 °C. The separated particles were then dispersed by vortex twice in water and once in toluene to remove unattached or physically adsorbed organosilanes; after each rinsing the particles were separated from solution by centrifugation (under the same conditions specified above). The silica nanoparticles were dried at 80 °C for 3 h, and then kept in a desiccator until measurement. Hydrophilic fumed silica powder Aerosil® 300 was dried before measurements under the same conditions, and then kept in a desiccator until measurement.
Confocal laser scanning microscopy (CLSM) analysis
Image acquisition was done using a Leica SP8 laser scanning microscope (Leica, Wetzlar, Germany), equipped with a solid state laser with 488 nm light, HC PL APO CS 63x/0.75 objective (Leica, Wetzlar, Germany) and Leica Application Suite X software (LAS X, Leica, Wetzlar, Germany). Imaging of 6-aminofluorescein signal was done using a solid state laser with 488 nm light, and the emission was detected in a range of 500–540 nm. Imaging of Nile Red signal was done using a solid state laser with 552 nm light, and the emission was detected in the range of 580–670 nm. For the analysis, 5 μL were taken from emulsion and drop-cast on a microscopic slide, equipped with a coverslip. Droplet size distribution was analyzed using Fiji software49 by measuring the droplet diameters from confocal microscopy images for each emulsion type. Image acquisition without a coverslip was done to directly observe colloidosome structures, forming by evaporation of emulsion droplets, leaving silica shells on the microscope slide. The process was monitored over time, with a frame rate of 1.7 s.Cryogenic-scanning electron microscopy (cryo-SEM)
Cryogenic-scanning electron microscopy analysis was performed on a JSM-7800F Schottky field-emission scanning electron microscope (Jeol Ltd., Tokyo/Japan), equipped with a cryogenic system (Quorum PP3010, Quorum Technologies Ltd., Laughton/United Kingdom). Liquid nitrogen was used in all heat exchange units of the cryogenic system. A small droplet of emulsion was placed on the sample holder, between two rivets, quickly frozen in liquid nitrogen for a few seconds and transferred to the preparation chamber where it was fractured (at −140 °C). The revealed fractured surface was sublimed at −90 °C for 10 min to eliminate any presence of condensed ice, and then coated with platinum. The temperature of the sample was kept at −140 °C. Images were acquired with a low electron detector (LED) at an accelerating voltage of 5.0 kV and a working distance of 3.9 mm. Energy-dispersive X-ray spectroscopy (EDX) analysis was performed with a LED detector at an accelerating voltage of 5.0 kV and a working distance of 9.9 mm.High-resolution scanning electron microscopy (HRSEM)
The surface morphology of the silica Aerosil® 300 nanoparticles was obtained by a MIRA3 field-emission scanning electron microscope (Tescan, Brno/Czech Republic) with an accelerating voltage of 5.0 kV and a secondary electron (SE) detector. For this analysis, 5 μL of 0.2% w/v fumed silica nanoparticles dispersed in ethanol were drop-cast onto a conductive carbon adhesive tape and dried at 60 °C for 1 h. Prior to imaging, a thin gold-palladium layer was evaporated onto the sample for electric conductivity, in order to avoid surface charging by the electron beam.Abbreviations
| DTES | Dodecyltriethoxysilane |
| APTES | 3-(Aminopropyl)triethoxysilane |
| FTIR | Fourier transform infrared spectroscopy |
| cryo-SEM | Cryogenic scanning electron microscopy |
| o/w | Oil-in-water |
| w/o | Water-in-oil |
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The authors would like to acknowledge the financial support of the Magnet program [SHPS], administered by the Israel Innovation Authority. The authors thank Magen Lahat for HRSEM characterization, and Dr Noga Yaakov for assistance in data analysis.Notes and references
- Y. Chevalier and M.-A. Bolzinger, Colloids Surf., A, 2013, 439, 23–34 CrossRef CAS.
- R. Aveyard, B. P. Binks and J. H. Clint, Adv. Colloid Interface Sci., 2003, 100–102, 503–546 CrossRef CAS.
- W. Ramsden, Proc. R. Soc. London, 1904, 72, 156–164 CrossRef.
- S. U. Pickering, J. Chem. Soc., Trans., 1907, 91, 2001–2021 RSC.
- J. Wu and G.-H. Ma, Small, 2016, 12, 4633–4648 CrossRef CAS.
- J. Frelichowska, M.-A. Bolzinger, J.-P. Valour, H. Mouaziz, J. Pelletier and Y. Chevalier, Int. J. Pharm., 2009, 368, 7–15 CrossRef CAS PubMed.
- C. C. Berton-Carabin and K. Schroën, Annu. Rev. Food Sci. Technol., 2015, 6, 263–297 CrossRef CAS PubMed.
- C. Linke and S. Drusch, Crit. Rev. Food Sci. Nutr., 2018, 58, 1971–1985 CrossRef CAS PubMed.
- N. Yaakov, K. Ananth Mani, R. Felfbaum, M. Lahat, N. Da Costa, E. Belausov, D. Ment and G. Mechrez, ACS Omega, 2018, 3, 14294–14301 CrossRef CAS PubMed.
- O. D. Velev, K. Furusawa and K. Nagayama, Langmuir, 1996, 12, 2374–2384 CrossRef CAS.
- A. D. Dinsmore, M. F. Hsu, M. G. Nikolaides, M. Marquez, A. R. Bausch and D. A. Weitz, Science, 2002, 298, 1006–1009 CrossRef CAS PubMed.
- Y. Liu, X. Chen and J. H. Xin, J. Mater. Sci., 2006, 41, 5399–5401 CrossRef CAS.
- T. Chen, P. J. Colver and S. A. F. Bon, Adv. Mater., 2007, 19, 2286–2289 CrossRef CAS.
- L. A. Fielding and S. P. Armes, J. Mater. Chem., 2012, 22, 11235–11244 RSC.
- K. L. Thompson, M. Williams and S. P. Armes, J. Colloid Interface Sci., 2015, 447, 217–228 CrossRef CAS PubMed.
- H. N. Yow and A. F. Routh, Langmuir, 2009, 25, 159–166 CrossRef CAS PubMed.
- P. F. Noble, O. J. Cayre, R. G. Alargova, O. D. Velev and V. N. Paunov, J. Am. Chem. Soc., 2004, 126, 8092–8093 CrossRef CAS PubMed.
- R. K. Shah, J.-W. Kim and D. A. Weitz, Langmuir, 2010, 26, 1561–1565 CrossRef CAS PubMed.
- H. Wang, X. Zhu, L. Tsarkova, A. Pich and M. Möller, ACS Nano, 2011, 5, 3937–3942 CrossRef CAS PubMed.
- P. H. R. Keen, N. K. H. Slater and A. F. Routh, Langmuir, 2012, 28, 1169–1174 CrossRef CAS PubMed.
- Y. Gong, A. M. Zhu, Q. G. Zhang, M. L. Ye, H. T. Wang and Q. L. Liu, ACS Appl. Mater. Interfaces, 2013, 5, 10682–10689 CrossRef CAS PubMed.
- J. van Wijk, T. Heunis, E. Harmzen, L. M. T. Dicks, J. Meuldijk and B. Klumperman, Chem. Commun., 2014, 50, 15427–15430 RSC.
- M. Li, D. C. Green, J. L. R. Anderson, B. P. Binks and S. Mann, Chem. Sci., 2011, 2, 1739–1745 RSC.
- C. Zhang, C. Hu, Y. Zhao, M. Möller, K. Yan and X. Zhu, Langmuir, 2013, 29, 15457–15462 CrossRef CAS PubMed.
- P. H. R. Keen, N. K. H. Slater and A. F. Routh, Langmuir, 2014, 30, 1939–1948 CrossRef CAS PubMed.
- F. Nan, J. Wu, F. Qi, Q. Fan, G. Ma and T. Ngai, J. Mater. Chem. B, 2014, 2, 7403–7409 RSC.
- P. Finkle, H. D. Draper and J. H. Hildebrand, J. Am. Chem. Soc., 1923, 45, 2780–2788 CrossRef CAS.
- B. P. Binks and S. O. Lumsdon, Langmuir, 2000, 16, 8622–8631 CrossRef CAS.
- A. Liberman, N. Mendez, W. C. Trogler and A. C. Kummel, Surf. Sci. Rep., 2014, 69, 132–158 CrossRef CAS PubMed.
- L. Tang and J. Cheng, Nano Today, 2013, 8, 290–312 CrossRef CAS PubMed.
- J. Frelichowska, M.-A. Bolzinger and Y. Chevalier, Colloids Surf., A, 2009, 343, 70–74 CrossRef CAS.
- B. P. Binks and D. Yin, Soft Matter, 2016, 12, 6858–6867 RSC.
- S. Björkegren, L. Nordstierna, A. Törncrona and A. Palmqvist, J. Colloid Interface Sci., 2017, 487, 250–257 CrossRef PubMed.
- Y. Zhu, J. Jiang, K. Liu, Z. Cui and B. P. Binks, Langmuir, 2015, 31, 3301–3307 CrossRef CAS PubMed.
- B. P. Binks and A. T. Tyowua, Soft Matter, 2016, 12, 876–887 RSC.
- J. Kim, P. Seidler, L. S. Wan and C. Fill, J. Colloid Interface Sci., 2009, 329, 114–119 CrossRef CAS PubMed.
- C.-H. Chiang, H. Ishida and J. L. Koenig, J. Colloid Interface Sci., 1980, 74, 396–404 CrossRef CAS.
- M. Zhu, M. Z. Lerum and W. Chen, Langmuir, 2012, 28, 416–423 CrossRef CAS PubMed.
- Y. Liu, Y. Li, X.-M. Li and T. He, Langmuir, 2013, 29, 15275–15282 CrossRef CAS PubMed.
- R. M. Pasternack, S. Rivillon Amy and Y. J. Chabal, Langmuir, 2008, 24, 12963–12971 CrossRef CAS PubMed.
- A. Y. Fadeev and T. J. McCarthy, Langmuir, 2000, 16, 7268–7274 CrossRef CAS.
- M. Jin, S. Li, J. Wang, M. Liao and Y. Zhao, Appl. Surf. Sci., 2012, 258, 7552–7555 CrossRef CAS.
- K. S. Suslick, M. M. Fang, T. Hyeon and M. M. Mdleleni, in Sonochemistry and Sonoluminescence, ed. L. A. Crum, T. J. Mason, J. L. Reisse and K. S. Suslick, Springer Netherlands, Dordrecht, 1999, pp. 291–320 Search PubMed.
- A. L. R. Costa, A. Gomes and R. L. Cunha, Food Res. Int., 2018, 107, 717–725 CrossRef CAS PubMed.
- G. S. Caravajal, D. E. Leyden, G. R. Quinting and G. E. Maciel, Anal. Chem., 1988, 60, 1776–1786 CrossRef CAS.
- Brochures - AEROSIL® fumed silica, https://www.aerosil.com/product/aerosil/en/services/downloads/Pages/brochures.aspx Search PubMed.
- S. C. Feifel and F. Lisdat, J. Nanobiotechnol., 2011, 9, 59 CrossRef CAS PubMed.
- Z. Grabarek and J. Gergely, Anal. Biochem., 1990, 185, 131–135 CrossRef CAS.
- J. Schindelin, I. Arganda-Carreras, E. Frise, V. Kaynig, M. Longair, T. Pietzsch, S. Preibisch, C. Rueden, S. Saalfeld, B. Schmid, J.-Y. Tinevez, D. J. White, V. Hartenstein, K. Eliceiri, P. Tomancak and A. Cardona, Nat. Methods, 2012, 9, 676–682 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra07597f |
| ‡ These authors contributed equally to the research. |
| This journal is © The Royal Society of Chemistry 2019 |