
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNitrogen aeration alters the spatial distribution and metal adsorption of extracellular polymeric substances in waste-activated sludge
Ying Liu abc,
Wenzhou Lvc,
Siqing Xiaab and
Zhiqiang Zhang*ab
abc,
Wenzhou Lvc,
Siqing Xiaab and
Zhiqiang Zhang*ab
aState Key Laboratory of Pollution Control and Resource Reuse, Key Laboratory of Yangtze River Water Environment, Ministry of Education, College of Environmental Science and Engineering, Tongji University, Shanghai 200092, China. E-mail: zhiqiang@tongji.edu.cn
bShanghai Institute of Pollution Control and Ecological Security, Shanghai 200092, China
cFaculty of Civil and Environmental Engineering, Ningbo University, Ningbo 315211, China
First published on 23rd October 2019
Abstract
Extracellular polymeric substances (EPS) extracted from waste-activated sludge (WAS) have the potential to remove heavy metal ions from wastewater; both the spatial distribution and metal adsorption of EPS from WAS after nitrogen aeration were systematically investigated in this study. Compared with air aeration, nitrogen aeration significantly improved the contents of proteins (PN) and polysaccharides (PS) in the Slime-EPS (S-EPS) and loosely-bound EPS (LB-EPS), significantly increased the PS content, and slightly increased the PN content in the tightly-bound EPS (TB-EPS). The variations in the fluorescence intensities (FI) of the peaks I and II for the S-EPS, LB-EPS and TB-EPS were basically consistent with the abovementioned variations in the concentrations of these EPS. Notably, nitrogen aeration dramatically improved the content of protein-like substances in the LB-EPS. For the same aeration time, the Pb2+ reclamation rates obtained by the LB-EPS extracted from the nitrogen-aerated WAS were much higher than those achieved by the LB-EPS extracted from the air-aerated WAS. The FTIR analyses further indicated that nitrogen aeration improved the contents of the functional groups, especially –OH, –COOH and –NH2, responsible for binding heavy metals, in both the LB-EPS and TB-EPS. The SEM analyses verified that the nitrogen scours contributed to the EPS release, and Pb2+ reclamation was achieved via the attachment of Pb2+ onto the edge of the EPS. The influences of the nitrogen aeration on the spatial distribution and metal adsorption of the EPS in WAS were revealed for the first time in this study. Thus, this study lays the foundation for the application of nitrogen aeration in the resource utilization of WAS.
1. Introduction
With the rapid development of municipal wastewater treatment plants (WWTPs), a significant amount of waste-activated sludge (WAS) has been produced in recent decades.1 Therefore, WAS disposal has become a big issue in the management of WWTPs. Extracellular polymeric substances (EPS) are the main components in the activated sludge, which are a complex mixture of high-molecular-weight biomolecules, including proteins (PN), polysaccharides (PS), humic substances, nucleic acids, and lipids, surrounding the microbial cells.2 The rational utilization of EPS would be a promising approach to reclaim the WAS. The EPS have been reported to be used as a biosorbent for the reclamation of heavy metals from wastewater3,4 because they possess high quantities of binding groups, such as carboxyl, hydroxyl, amino, phosphate, and sulphate, in their compositions. Most of these functional groups are negatively charged at neutral pH, which can bind with specific metal ions to form organometallic complexes. Generally, the biosorbent is always harvested from particular microbes through pure culture cultivation, which expends large amounts of media or nutrition, and the cost is often significantly high. Therefore, cost-effective biosorbents are attracting significant attention all over the world nowadays.2,5WAS produced by WWTPs, containing a significant amount of natural organic macromolecule substances, is regarded as a good source of biosorbents.6 Some types of microorganisms in the WAS can degrade the organic pollutants in wastewater and excrete EPS under aerobic conditions.7 Multiple factors, such as the substrate type, nutrition concentration, cell growth phase, and particular substances, affect the yields of the EPS.8–11 For example, under steady-state conditions, the PN content in the EPS extracted from the sludge, which was used in the treatment of winery or municipal wastewater, was higher than that in the cases of pulp-paper, textile, and petrochemical wastewater.8 This indicated that the origin of the wastewater affected the composition of the EPS extracted from the WAS. Furthermore, the ratio of carbon to nitrogen (C/N) in wastewater can impact the proportion of PN to PS in the EPS. The EPS contains higher PN but lower PS at the C/N ratio of 5. However, the PN content reduced dramatically, whereas the PS content increased when the C/N ratio was increased to 40.9 The EPS yield also strictly depended on the growth phase of the bacterial cells. The effects of the growth phase on the EPS yields varied with the cultivation time. In the exponential growth phase, the EPS production increased with the cultivation time. Conversely, it decreased with the extension of the cultivation time in the stationary phase.10 Moreover, toxic substances in the wastewater affected the PN proportion in the EPS. At a specific low concentration, the increase in the PN content far surpassed that of the other components in the EPS. However, when the toxic substances reached a threshold concentration, the promotion of the EPS yield became less significant.11
The short-time aerobic digestion (STAD) of the activated sludge has been confirmed to be a practical approach for promoting the EPS yields, which is beneficial to both the EPS source and sludge stabilization.7 The extracted EPS has also been found to be a superior biosorbent for the reclamation of metal ions from wastewater.6,12,13 The roles of STAD in the WAS treatment include aerobic digestion and scour elution. Both could influence the sludge characteristics and the EPS yields. Our previous studies showed that with the extension of the aeration time, a certain amount of the EPS was biodegraded by some aerobic microbes, resulting in a decrease in the extracted amount of the EPS from the WAS.14–17 Compared to air aeration, nitrogen aeration introduces not only a scour elution but also an anaerobic environment in the system, which would influence the bacterial communities, microbial metabolites and sludge properties.18 However, how nitrogen aeration affects the production of the EPS from WAS is rarely studied to date. Moreover, the influences of nitrogen aeration on the spatial distribution and metal adsorption of the EPS in WAS need to be elucidated.
In this study, nitrogen was used to sparge the WAS to produce a scour elution in an anaerobic environment. To evaluate its influence on the yields of the EPS from WAS, the nitrogen aeration of WAS was comparatively conducted along with both the subsequent air aeration and the direct air aeration of WAS. Moreover, the influences of nitrogen aeration on the spatial distribution and metal adsorption of the EPS in WAS were systematically investigated.
2. Materials and methods
2.1 Experimental setup
All the experiments were conducted in a fermenter (NBS 115, Eppendorf, Germany) with an effective volume of 7.5 liters. The returned activated sludge was sampled from a wastewater treatment plant situated in Ningbo city, China, which employed an A2/O process to treat the municipal wastewater. The sampled activated sludge was settled for 30 min, and the supernatant was decanted. The concentrated sludge was screened by a sieve with a pore size of 1.2 mm in diameter, and the sludge characteristics were as follows: sludge volume (SV30) of 89–91, mixed liquid suspended solids (MLSS) of 10.5–13.5 g L−1, mixed liquid volatile suspended solids (MLVSS) of 6.0–6.2 g L−1 and pH 6.5–7.0. Moreover, four liters of activated sludge was poured into the jar of the fermenter and aerated by pure nitrogen at the flow rate of 4.0 L min−1. The DO value was determined by a portable DO meter (HQ40d, Hach, USA). During nitrogen aeration, the DO value was almost kept at 0 mg L−1. After nitrogen aeration, to evaluate the effects of oxygen on the changes in the EPS concentration, subsequent air aeration was performed for 4 h. Filtered air instead of nitrogen gas was sparged into the system with the same flow rate, and the DO value was kept at 2–3 mg L−1. Air aeration was also carried out to compare the results with those obtained under nitrogen aeration. Each aeration process continued for 4 h and was executed in triplicate at room temperature. All samples were obtained every hour to analyze the fractions and the compositions of the different layers of EPS.2.2 Extraction of the layers of EPS from WAS
S-EPS: typically, 25 mL homogeneous WAS was added to a 50 mL centrifugal tube by a syringe equipped on the fermenter. The tube was centrifuged at 550 rpm for 15 min, and the supernatant was S-EPS. Then, the excess supernatant was decanted, and a 0.01 mol L−1 NaCl solution was added to achieve a total volume of 25 mL; the tubes were stirred thoroughly, sonicated at 120 W for 2 min, and then centrifuged at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 10 min. The supernatant was LB-EPS. Similarly, the NaCl solution was added to the tube to re-suspend the sludge residue. The mixture was sonicated at the higher power of 210 W for 4 min, followed by the same centrifugation process. The supernatant finally obtained was the TB-EPS.
000 rpm for 10 min. The supernatant was LB-EPS. Similarly, the NaCl solution was added to the tube to re-suspend the sludge residue. The mixture was sonicated at the higher power of 210 W for 4 min, followed by the same centrifugation process. The supernatant finally obtained was the TB-EPS.
2.3 Pb2+ adsorption of the EPS
Herein, 5 mL of extracted LB-EPS or TB-EPS and 1 mL Pb2+ stock solution at a concentration of 1000 mg L−1 were added to a 50 mL centrifugal tube. Then, ultrapure water was added to achieve a total volume of 20 mL. After this, the pH was adjusted to 4.5 with a solution of 0.1 mol L−1 HNO3 and 0.1 mol L−1 NaOH. After vigorously stirring the solution at 25 °C and 200 rpm for 2 hours, centrifugation was conducted at 12000 rpm for 10 min. The supernatant was diluted for further determination. The supernatant obtained after the adsorption of Pb2+ was diluted 10-fold to achieve a final concentration of Pb2+ below 5 mg L−1. The content of Pb2+ was measured by an inductively coupled plasma emission spectrometer (ICP, PQ 9000 Elite, Germany). The Pb2+ reclamation rate (%) was calculated according to eqn (1):| Reclamation rate = [(C0 − Ce)/C0] × 100% | (1) |
2.4 Analytical methods
:200 (w/w). An FTIR spectrometer (Nicolet 5700, USA) was applied to scan the functional groups in the range of 4000–400 cm−1.21000 rpm for 15 min. After discarding the supernatant, the sample was dehydrated by a series of ethanol solutions and freeze-dried by a vacuum freeze dryer.22 Then, 5 mL of the extracted EPS or the Pb2+-adsorbed EPS was pipetted into a tube and freeze-dried by a vacuum freeze dryer. The sludge and EPS surface structure and morphology were observed by SEM (Quanta 250 FEG, USA).3. Results and discussion
3.1 Influences of nitrogen aeration on the spatial distribution of the EPS in WAS
The spatial distribution variations of the EPS extracted from the WAS at different nitrogen aeration times are shown in Fig. 1. During nitrogen aeration for 4 h, the contents of PN and PS in the S-EPS gradually increased, and their increasing proportions were 87% and 72% at 4 h, respectively. Moreover, the contents of PN and PS in the LB-EPS gradually increased, and their increasing proportions reached 128.8% and 71.5% at 4 h, respectively. The contents of PN and PS in the TB-EPS gradually increased, and their increasing proportions reached 12.2% and 50.4% at 4 h, respectively. However, during air aeration, the contents of PN and PS in all the layers of EPS, especially in the S-EPS and LB-EPS, first increased and then decreased over time. The contents of PN and PS in the S-EPS and LB-EPS after nitrogen aeration were markedly higher than those after air aeration; in addition, the PN content in the TB-EPS was slightly increased when compared with the PS content. In our previous study, we speculated that in the first stage of air aeration, the air scours contributed to the increase in the concentration of EPS; however, in the later stage, the biodegradation of the EPS by aerobic microorganisms possibly resulted in a decrease in the concentration of EPS.14 To validate the results, subsequent air aeration was conducted for 4 h after nitrogen aeration for 4 h. The results showed that both the PN and PS contents in all the layers of EPS decreased continuously, and their reduced contents were almost equal to those obtained after direct air aeration. For nitrogen aeration, the mutual action of the anaerobic environment and scour elution contributed to the increase in the contents of PN and PS in all the layers of EPS.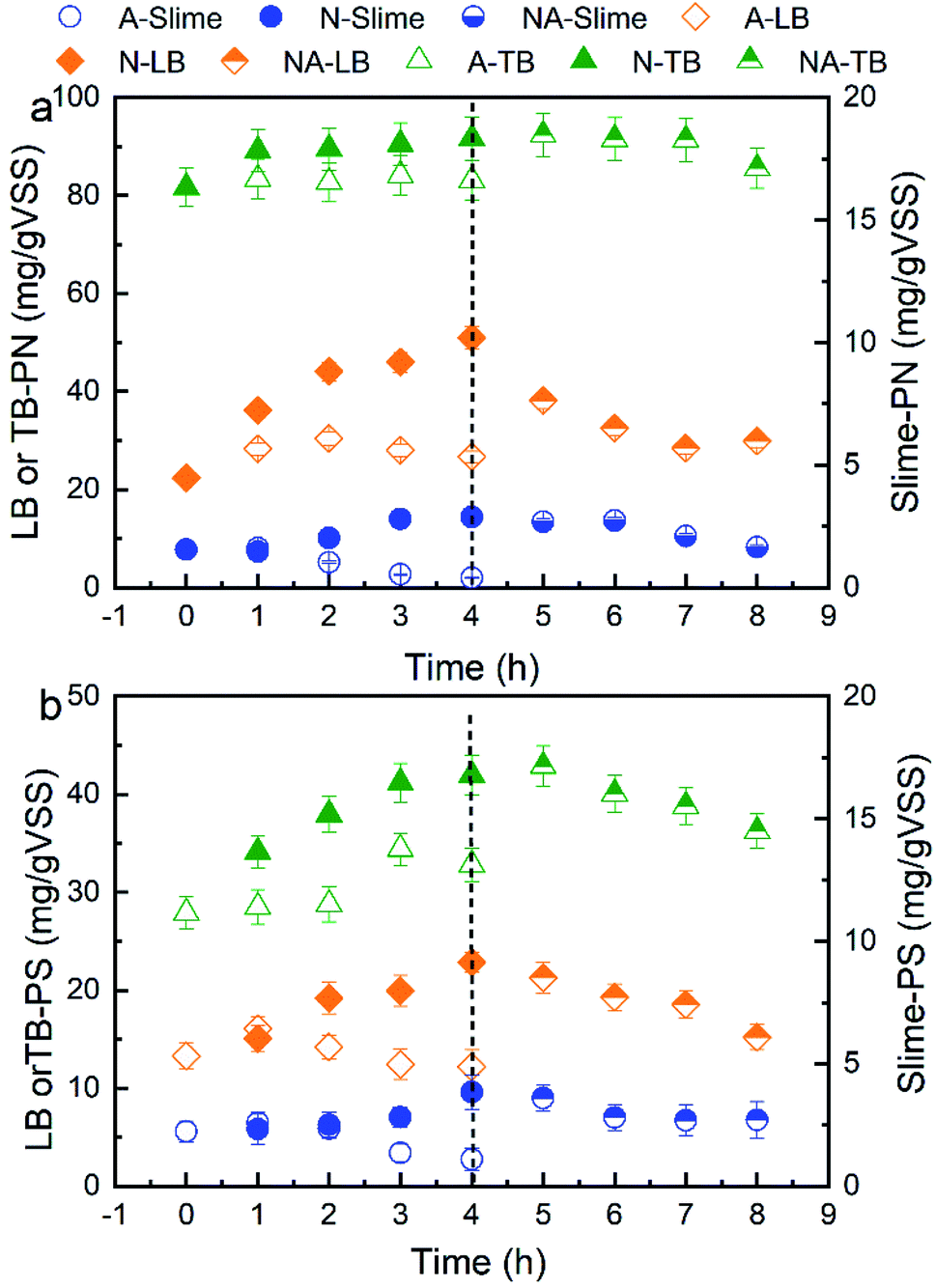 | ||
| Fig. 1 Effects of aeration on the (a) PN and (b) PS contents of the S-EPS, LB-EPS and TB-EPS extracted from WAS. A and N represent the aeration by air or nitrogen, respectively. NA represents the additional aeration after aeration by nitrogen for 4 h. | ||
3.2 Influences of nitrogen aeration on the fluorescent composition of EPS in the WAS
The 3D-EEM fluorescence spectra variations for the S-EPS, LB-EPS, and TB-EPS layers in the WAS obtained by nitrogen aeration or air aeration (4 h, N1–N4) are shown in Fig. 2, 3 and 4, respectively. The results were also compared with those obtained by subsequent air aeration (4 h, NA1–NA4) and direct air aeration (4 h, A1–A4). Herein, two primary peaks can be identified in the fluorescence spectra. Peak I with the Ex/Em of 220/330–350 nm is associated with aromatic protein-like substances, and peak II with the Ex/Em of 280–285/335–355 nm is associated with tryptophan protein-like substances.23–25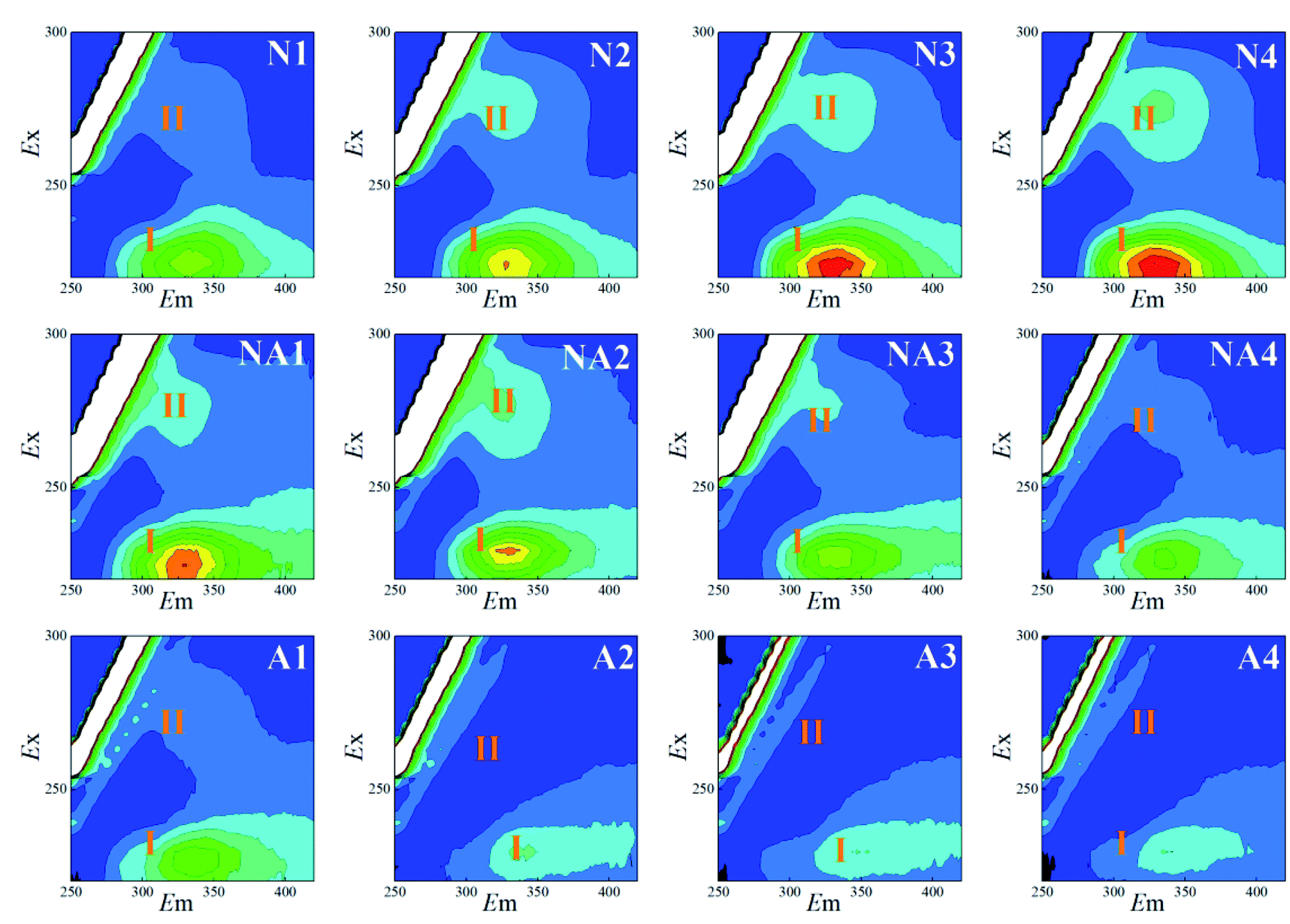 | ||
| Fig. 2 Variations in the 3D-EEM spectra of the S-EPS during aeration. N1–N4: nitrogen aeration for 1–4 h at the intervals of one hour. The same is shown below. NA1–NA4: air aeration after nitrogen aeration for 1–4 h. A1–A4: air aeration for 1–4 h. | ||
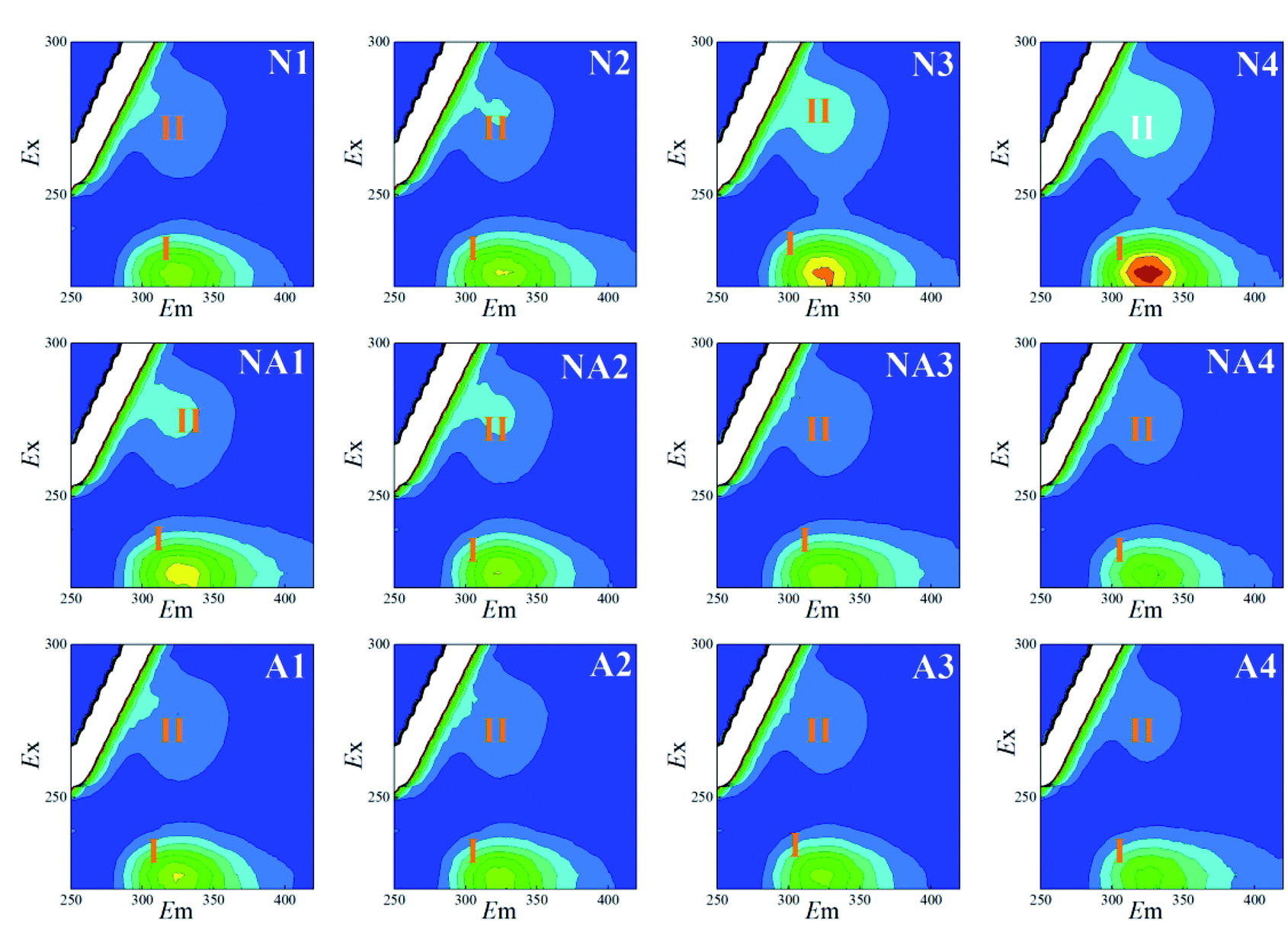 | ||
| Fig. 3 Variations in the 3D-EEM spectra of the LB-EPS during aeration. N1–N4: nitrogen aeration for 1–4 h at the intervals of one hour. The same is shown below. NA1–NA4: air aeration after nitrogen aeration for 1–4 h. A1–A4: air aeration for 1–4 h. | ||
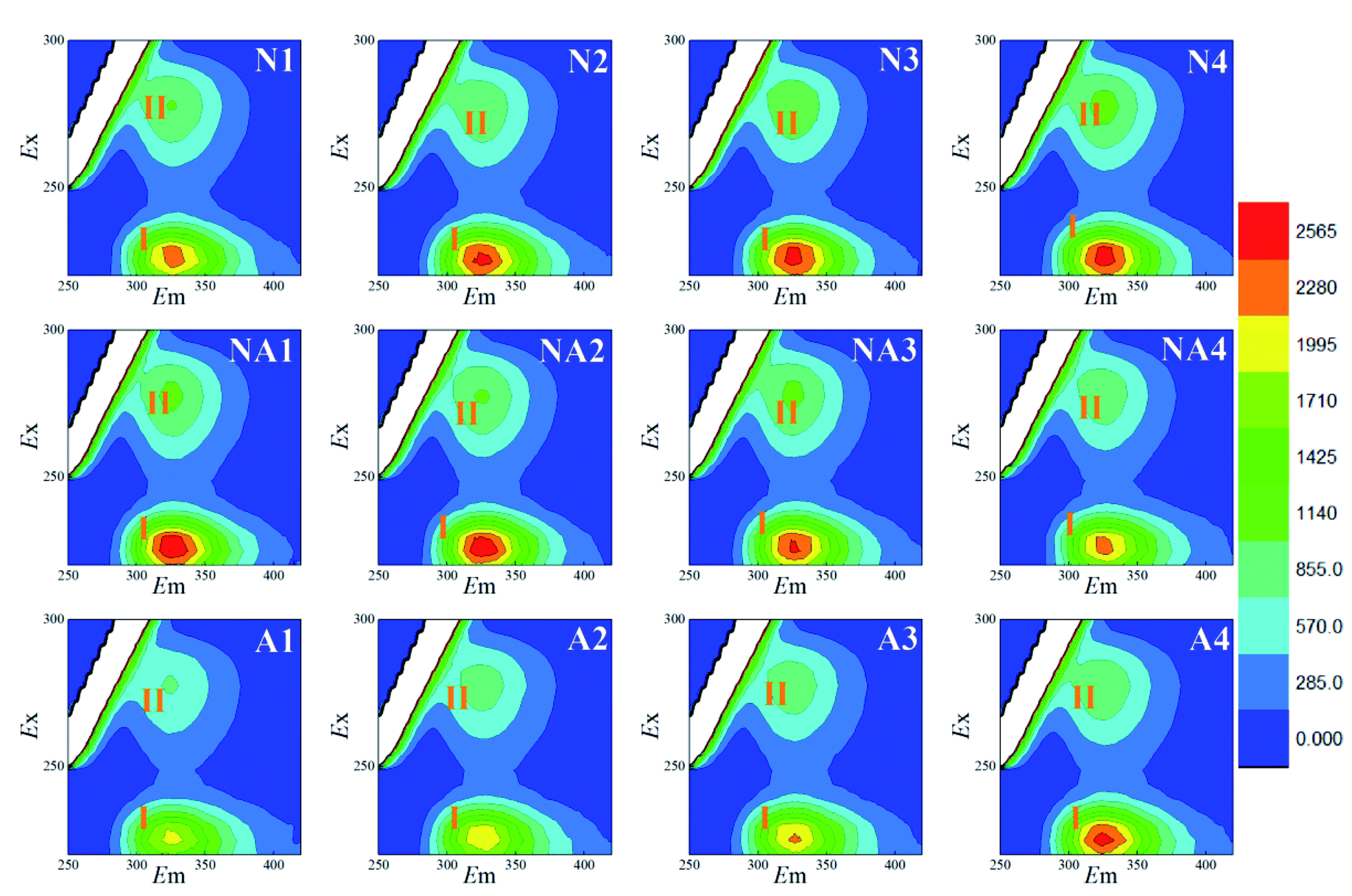 | ||
| Fig. 4 Variations in the 3D-EEM spectra of the TB-EPS during aeration. N1–N4: nitrogen aeration for 1–4 h at the intervals of one hour. The same is shown below. NA1–NA4: air aeration after nitrogen aeration for 1–4 h. A1–A4: air aeration for 1–4 h. | ||
As observed from Fig. 2, the fluorescence signals of the peaks I and II for the S-EPS increased with an increase in the nitrogen aeration time. The fluorescence intensity (FI) values of the peaks I and II for the S-EPS increased by about 86.0% and 156.0% (Table 1) after 4 h of nitrogen aeration, respectively. When subsequent air aeration was conducted, the FI values for the peaks I and II decreased by about 69.2% and 85.8%, respectively, which were consistent with the changes observed in the fluorescence spectra.
| EPS fractions | Fluorescence intensity | ||||
|---|---|---|---|---|---|
| WAS | WAS obtained by 4 h nitrogen aeration | WAS obtained by 4 h nitrogen aeration and subsequent 4 h air aeration | WAS obtained by 4 h air aeration | ||
| Peak I | Slime | 570 | 1060 | 326 | 266 |
| LB | 1087 | 1807 | 921 | 991 | |
| TB | 1515 | 2550 | 1852 | 2117 | |
| Peak II | Slime | 220 | 563 | 80 | 110 |
| LB | 256 | 532 | 300 | 300 | |
| TB | 473 | 960 | 823 | 889 | |
For the LB-EPS (Fig. 3) and TB-EPS (Fig. 4), similar variations in the fluorescence spectra and FI were found. Under nitrogen aeration conditions, the FI values of the peaks I and II for the LB-EPS gradually increased by about 66.2% and 107.8% after 4 h, respectively. When air aeration was used after nitrogen aeration from 5 h to 8 h, the FI values of the peaks I and II gradually decreased and were reduced by about 49.0% and 43.6% at the end, respectively (Table 1). For the TB-EPS, the FI values of the peaks I and II gradually increased by about 68.3% and 103.0% for nitrogen aeration after 4 h, respectively. However, the FI values of the peaks I and II decreased by about 27.4% and 14.3% at the end (Table 1), respectively, when air aeration was carried out after nitrogen aeration from 5 h to 8 h.
By comparing N1–N4 and A1–A4, as shown in Fig. 2–4, it was found that the FI values of the peaks I and II in the case of nitrogen aeration were higher than those in the case of air aeration (Table 1); this indicated that more EPS production could be obtained by the nitrogen aeration of the WAS. Under the anaerobic environment of nitrogen aeration, some microbial cells disintegrated and released the EPS. The aerobic biodegradation of the EPS decreased, and certain microorganisms still secreted some type of EPS.26 Therefore, the anaerobic environment and gas scour might be responsible for the increase in the concentration of EPS.
3.3 Influences of nitrogen aeration on Pb2+ adsorption by the EPS
| Samples | Pb2+ reclamation rate (%) | |
|---|---|---|
| LB-EPS | TB-EPS | |
| a WAS represents the original activated sludge. N-2 h and N-4 h represent the WAS sparged by nitrogen for 2 h and 4 h, respectively. NA-2 h and NA-4 h represent the WAS sparged by nitrogen for 4 h followed by air for 2 h and 4 h, respectively. A-2 h and A-4 h represent the WAS sparged by air for 2 h and 4 h, respectively. | ||
| WAS | 28.2 | 42.4 |
| N-2 h | 54.2 | 62.9 |
| N-4 h | 61.1 | 63.6 |
| NA-2 h | 49.7 | 63.3 |
| NA-4 h | 42.7 | 62.1 |
| A-2 h | 37.6 | 53.7 |
| A-4 h | 39.5 | 61.6 |
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and the deformation vibration of –OH, are commonly found for carboxylates and phenolic hydroxyls, respectively. The adsorption peak between 1190 and 1000 cm−1 indicates the stretching vibration of CC and asymmetric stretching vibration of C–O–C. The adsorption band at 1247 cm−1 is characteristic for the stretching vibration of C–O.
O and the deformation vibration of –OH, are commonly found for carboxylates and phenolic hydroxyls, respectively. The adsorption peak between 1190 and 1000 cm−1 indicates the stretching vibration of CC and asymmetric stretching vibration of C–O–C. The adsorption band at 1247 cm−1 is characteristic for the stretching vibration of C–O.
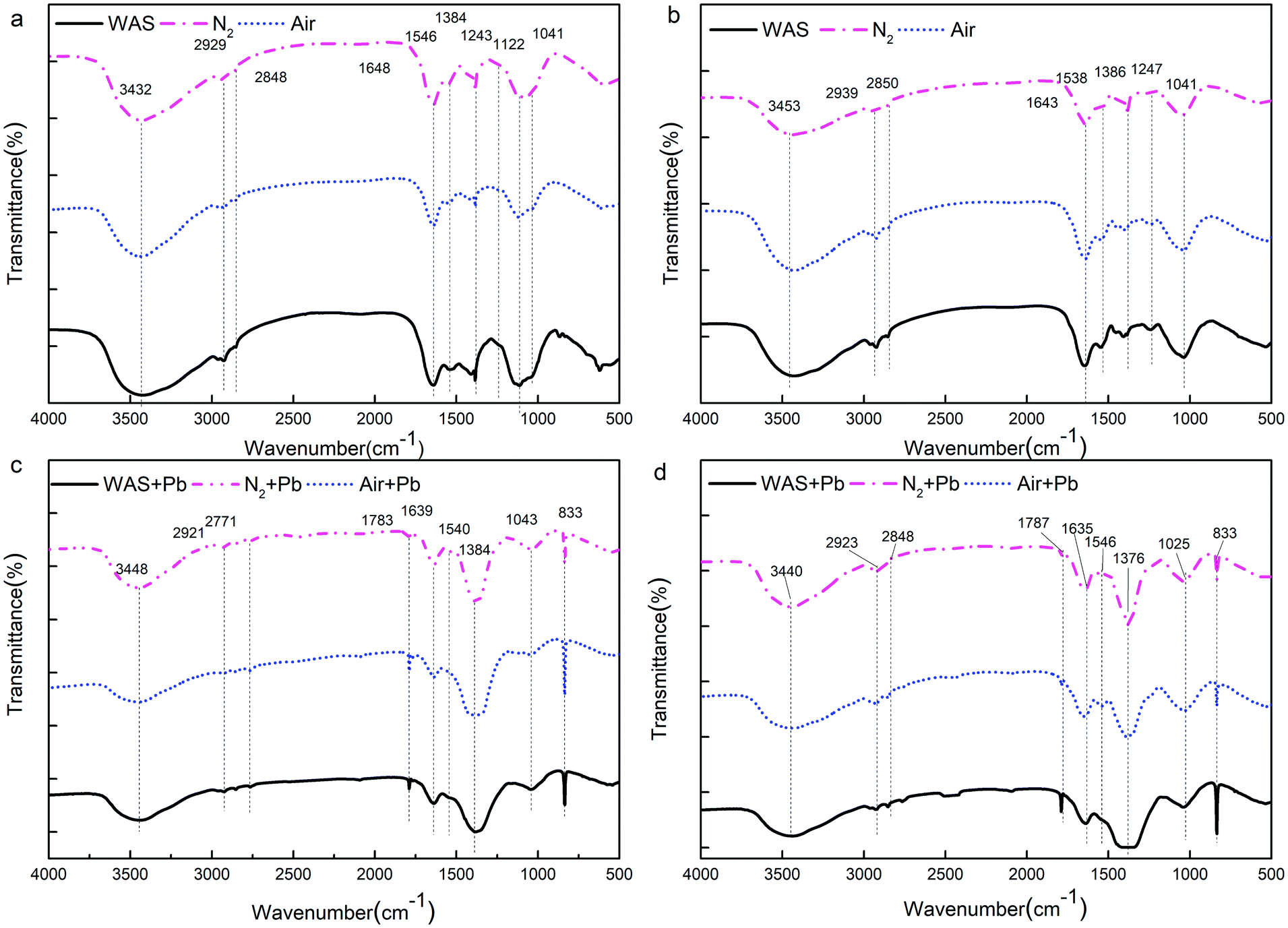 | ||
| Fig. 5 FTIR spectra of the extracted LB-EPS (a), TB-EPS (b) LB-EPS after the adsorption of Pb2+ (c) and TB-EPS after the adsorption of Pb2+ (d) from WAS by nitrogen or air aeration. | ||
Fig. 5c and d show the functional group region in the FTIR spectra of the Pb2+-adsorbed LB-EPS and TB-EPS obtained from the raw WAS, nitrogen or air-aerated sludge, respectively; for both the LB-EPS and the TB-EPS, the intensity of the peak at around 3401 cm−1 became weak, indicating that –OH and –NH2 were involved in the adsorption process. The decrease and even disappearance of the peaks between 2950 and 2920 cm−1 after the adsorption of Pb2+ indicated that the –CH2– asymmetric stretching vibration was a consequence of adsorption. The absorption bands at around 1639 cm−1 are characteristic for the stretching vibration of –COOH. The apparent shift in –COOH after the adsorption of Pb2+ indicates that the hydrogen ions existing in the carboxyl groups are replaced by Pb2+. Thus, an ion exchange between the functional groups and the metal ions should take place during the adsorption process. The decline in the C–N stretching vibration and N–H deformation vibration of Amide II in the proteins at 1540 cm−1 meant they participated in the adsorption process. The intensity of the adsorption peak at 1386 cm−1 dramatically increased after the binding of Pb2+, indicating that the stretching vibration of CO and the deformation vibration of –OH were involved in the adsorption process. The disappearance of the peak at 1247 cm−1 indicated that the stretching vibration of C–O was also involved in the adsorption process. The results showed that the functional groups in the EPS responsible for the adsorption of Pb2+ mainly consisted of –OH, –NH2, –CH2–, –COOH, C–N, N–H, CO, CC, C–O, and C–O–C. Previous studies have mentioned that proteins and carbohydrates are the main extracellular compounds that have functional groups such as carboxyl (–COOH), hydroxyl (–OH), and amide (N–H), which are primarily involved in metal ion binding.
 | ||
| Fig. 6 SEM images of WAS (a) before and (b) after being aerated by nitrogen for 4 h. The EPS extracted from WAS aerated by nitrogen (c) before and (d) after the adsorption of Pb2+. | ||
Fig. 6c shows the SEM images of the EPS obtained from the nitrogen aeration before the adsorption of Pb2+. Original EPS shows a meshy structure with small branches, which are longer, finer and denser. Fig. 6d shows the SEM image of the Pb2+-adsorbed EPS obtained from nitrogen aeration. More large flaky substances and a rough surface topography appeared after the adsorption of Pb2+. This indicated that Pb2+ was attached to the edge of the EPS and present as small particles, which further deposited on the surface of the EPS by surface precipitation. This finding was similar to that of the EPS extracted from the WAS subjected to air sparging.3,32,33
4. Conclusions
Herein, nitrogen aeration was disclosed to significantly improve the contents of PS in the S-EPS, LB-EPS and TB-EPS as well as the PN content in the S-EPS and LB-EPS extracted from the WAS. The PN content in the TB-EPS slightly increased. During nitrogen aeration, the compositions and fractions of the EPS gradually increased and were different from those obtained after air aeration. The changes in the fluorescence intensities of the peaks I and II for the S-EPS, LB-EPS and TB-EPS were basically consistent with the variations in their concentrations. Especially, nitrogen aeration could dramatically improve the contents of the protein-like substances in the S-EPS and LB-EPS as compared to air aeration. The Pb2+ reclamation rates of the LB-EPS extracted from the nitrogen-aerated WAS were much higher than those in the case of the air-aerated WAS. The FTIR results further indicated that nitrogen aeration could improve the contents of the functional groups, primarily –OH, –COOH and –NH2, which were responsible for the binding of heavy metals, in both the LB-EPS and TB-EPS. The SEM analyses verified that the nitrogen scour contributed to the release of the EPS from the sludge flocs into the solution, and the Pb2+ reclamation was achieved by the attachment of Pb2+ onto the edge of the EPS followed by surface precipitation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was supported by the National Key R&D Program of China (No. 2017YFC0403400), the Foundation of State Key Laboratory of Pollution Control and Resource Reuse (Tongji University, China) (No. PCRRE16019), the Fundamental Research Funds for the Central Universities, the National Natural Science Foundation of China (No. 51678422 and No. 51378368), and the higher school innovative engineering plan (111 Project), and sponsored by the K. C. Wong Magna Fund of Ningbo University.References
- Y.-K. Kim, J.-H. Bae, B.-K. Oh, W. Hong Lee and J.-W. Choi, Bioresour. Technol., 2002, 82, 157–164 CrossRef CAS.
- G.-P. Sheng, H.-Q. Yu and X.-Y. Li, Biotechnol. Adv., 2010, 28, 882–894 CrossRef CAS.
- W. W. Li and H. Q. Yu, Bioresour. Technol., 2014, 160, 15–23 CrossRef CAS.
- G. Guibaud, S. Comte, F. Bordas and M. Baudu, Process Biochem., 2005, 40, 661–668 CrossRef CAS.
- D. Wei, M. Li, X. Wang, F. Han, L. Li, J. Guo, L. Ai, L. Fang, L. Liu and B. Du, J. Hazard. Mater., 2016, 301, 407–415 CrossRef CAS.
- Z. Zhang, Y. Zhou, J. Zhang and S. Xia, Environ. Sci. Pollut. Res. Int., 2014, 21, 2132–2140 CrossRef CAS.
- Z. Zhang, J. Zhang, J. Zhao and S. Xia, Environ. Sci. Pollut. Res. Int., 2015, 22, 1812–1818 CrossRef CAS.
- D. T. Sponza, Process Biochem., 2002, 37, 983–998 CrossRef CAS.
- B. Durmaz and F. Sanin, Water Sci. Technol., 2001, 44, 221–229 CrossRef CAS.
- X. Jia, H. Furumai and H. H. Fang, Water Res., 1996, 30, 1439–1444 CrossRef CAS.
- G.-P. Sheng, H.-Q. Yu and Z.-B. Yue, Appl. Microbiol. Biotechnol., 2005, 69, 216–222 CrossRef CAS.
- Y. Zhou, S. Xia, J. Zhang, Z. Zhang and S. W. Hermanowicz, Desalin. Water Treat., 2016, 57, 9343–9353 CrossRef CAS.
- Y. Zhou, Z. Zhang, J. Zhang and S. Xia, J. Environ. Sci., 2016, 45, 248–256 CrossRef.
- Y. Liu, W. Lv, Z. Zhang and S. Xia, RSC Adv., 2018, 8, 32172–32177 RSC.
- Z. Zhang, Y. Zhou, J. Zhang, S. Xia and S. W. Hermanowicz, Chem. Eng. J., 2016, 299, 177–183 CrossRef CAS.
- S. Xia, Y. Zhou, E. Eustance and Z. Zhang, Sci. Rep., 2017, 7, 13491 CrossRef.
- Y. Zhou, J. Zhang, Z. Zhang, C. Zhou, Y. S. Lai and S. Xia, Chem. Eng. J., 2017, 320, 494–500 CrossRef CAS.
- V. L. Pachapur, S. J. Sarma, S. K. Brar, Y. Le Bihan, G. Buelna and C. R. Soccol, Int. J. Hydrogen Energy, 2015, 40, 8669–8676 CrossRef CAS.
- B. Fr, T. Griebe and P. Nielsen, Appl. Microbiol. Biotechnol., 1995, 43, 755–761 CrossRef.
- N. Li, D. Wei, S. Wang, L. Hu, W. Xu, B. Du and Q. Wei, J. Colloid Interface Sci., 2017, 490, 754–761 CrossRef CAS PubMed.
- W. Hou, Z. Ma, L. Sun, M. Han, J. Lu, Z. Li, O. A. Mohamad and G. Wei, J. Hazard. Mater., 2013, 261, 614–620 CrossRef CAS.
- H. Diao, X. Li, J. Gu, H. Shi and Z. Xie, Process Biochem., 2004, 39, 1421–1426 CrossRef CAS.
- Z. Wang, Z. Wu and S. Tang, Water Res., 2009, 43, 1533–1540 CrossRef CAS.
- W. Chen, P. Westerhoff, J. A. Leenheer and K. Booksh, Environ. Sci. Technol., 2003, 37, 5701–5710 CrossRef CAS.
- Y. Zhou, S. Xia, J. Zhang, B. T. Nguyen and Z. Zhang, Chem. Eng. J., 2017, 308, 1098–1104 CrossRef CAS.
- M. C. Veiga, M. K. Jain, W. Wu, R. I. Hollingsworth and J. G. Zeikus, Appl. Environ. Microbiol., 1997, 63, 403–407 CAS.
- G. Guibaud, N. Tixier, A. Bouju and M. Baudu, Chemosphere, 2003, 52, 1701–1710 CrossRef CAS.
- Y. Zhou, S. Xia, Z. Zhang, J. Zhang and S. W. Hermanowicz, J. Environ. Eng., 2016, 142, 04016032 CrossRef.
- M. Iqbal, A. Saeed and S. I. Zafar, J. Hazard. Mater., 2009, 164, 161–171 CrossRef CAS.
- G. Guibaud, S. Comte, F. Bordas, S. Dupuy and M. Baudu, Chemosphere, 2005, 59, 629–638 CrossRef CAS.
- L. Wei, Y. Li, D. R. Noguera, N. Zhao, Y. Song, J. Ding, Q. Zhao and F. Cui, J. Hazard. Mater., 2017, 321, 473–483 CrossRef CAS.
- Z. Zhang, P. Wang, J. Zhang and S. Xia, Environ. Sci. Pollut. Res. Int., 2014, 21, 10823–10829 CrossRef CAS.
- W. Liu, J. Zhang, Y. Jin, X. Zhao and Z. Cai, J. Environ. Chem. Eng., 2015, 3, 1223–1232 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2019 |