DOI:
10.1039/C9RA07224A
(Paper)
RSC Adv., 2019,
9, 34136-34143
An advanced and efficient Co3O4/C nanocomposite for the oxygen evolution reaction in alkaline media†
Received
9th September 2019
, Accepted 9th October 2019
First published on 23rd October 2019
Abstract
The design of efficient nonprecious catalysts for the hydrogen evolution reaction (HER) or the oxygen evolution reaction (OER) is a necessary, but very challenging task to uplift the water-based economy. In this study, we developed a facile approach to produce porous carbon from the dehydration of sucrose and use it for the preparation of nanocomposites with cobalt oxide (Co3O4). The nanocomposites were studied by the powder X-ray diffraction and scanning electron microscopy techniques, and they exhibited the cubic phase of cobalt oxide and porous structure of carbon. The nanocomposites showed significant OER activity in alkaline media, and the current densities of 10 and 20 mA cm−2 could be obtained at 1.49 and 1.51 V versus reversible hydrogen electrode (RHE), respectively. The impedance study confirms favorable OER activity on the surface of the prepared nanocomposites. The nanocomposite is cost-effective and can be capitalized in various energy storage technologies.
1. Introduction
To strengthen the renewable energy reservoirs, electrochemical water splitting is a promising technology and can boost the efficiency of rechargeable metal–air batteries, fuel cells and other energy storage devices.1–6 To date, the state-of-art electrocatalysts for the dissociation of water into oxygen are IrO2 or RuO2.1,7 However, the high cost and scarcity of these precious catalysts hinder their application on a large scale. Extensive efforts have been made towards the design of nonprecious, active, and stable OER catalysts using earth-abundant materials, including cobalt phosphate, perovskite oxides and transition metal oxides/layer double hydroxides, for the OER.8–14 Moreover, these electrocatalysts are highly stable and exhibit excellent activity in different electrolytes. In the recent past, numerous studies have been reported on the use of earth-abundant metal oxides, including cobalt oxide (Co3O4),15 nickel oxide,16 and manganese oxide,17 as active materials for the OER. Among these materials, cobalt oxide has been found to be highly efficient and stable.18
Co3O4 nanostructures combined with conducting carbon materials, such as carbon nanotubes, graphene and mesoporous carbon, are widely used as supporting materials due to their high conductivity and high specific surface area.19–22 Regardless of this revolutionary advancement, new functional electrocatalysts with low overpotential and high activity are still demanded. The fabrication of carbon nanotubes and graphene is a very expensive and complicated process; therefore, facile approaches are needed to produce carbon materials for the design of efficient composite materials for the production of hydrogen and oxygen gases on a large scale. However, there is no study related to the dehydration of common sugar into carbon and its coupling with cobalt oxide nanostructures. The use of porous carbon materials obtained from the dehydration of sucrose for the coupling of cobalt oxide nanostructures in the design of a functional catalyst is presented in this study. The enhanced performance is mainly attributed to the high density of active edges, which facilitate the OER process at low potential as compared to the pristine carbon and cobalt oxide materials. The improved OER activity is due to the synergetic effect produced between the carbon and cobalt oxide nanostructures due to the quantum confinement effect.
Herein, we present a simple strategy for the design of a new functional nanocomposite based on the combination of cobalt oxide nanostructures with porous carbon materials. The nanocomposite was successfully characterized by various analytical techniques such as X-ray diffraction, Fourier transform infrared spectroscopy, scanning electron microscopy and energy dispersive spectroscopy. The presented nanocomposites showed excellent OER activity at 1.49 V vs. RHE to achieve the current density of 10 mA cm−2 in a 1 M KOH electrolyte. The impedance study revealed fast OER kinetics over the proposed nanocomposites.
2. Experimental
Cobalt nitrate hexahydrate, urea, sulfuric acid, sucrose sugar, potassium hydroxide and 99.5% ethanol were used without any further purification. The synthesis of cobalt oxide and carbon composite was carried out in three steps. Step 1: the carbon material was obtained by the dehydration of sucrose in the presence of an 8 M concentrated sulfuric acid solution. The carbon material was washed several times with deionized water followed by ethanol treatment. The carbon material was dried overnight at 60 °C. Step 2: the deposition of cobalt hydroxide nanostructures on porous carbon was carried out by a hydrothermal method. An equimolar solution of cobalt nitrate hexahydrate and urea was prepared in 100 mL of deionized water in four separate beakers. Then, successive addition of porous carbon with different weights, such as 100 mg, 200 mg, 300 mg, and 400 mg, which were labeled as samples 1, 2, 3, and 4, respectively, was carried out. The precursor solution was tightly covered with an aluminum foil and kept in an electric oven at 95 °C for 5 hours. After the completion of growth, the cobalt hydroxide nanostructures were obtained by filtration and washed with deionized water. The samples were dried at room temperature. Step 3: the calcination of all the samples was carried out at 500 °C for three hours, and then, these samples were ready for application in the catalysis process. In a similar way, the pristine cobalt oxide nanostructures were synthesized without the addition of carbon materials.
The morphology of the as-prepared nanostructured materials was investigated by the JEOL scanning electron microscope at the accelerating voltage of 20 kV. The crystalline and phase purity was explored by the Philips powder diffractogram at room temperature. Fourier transform IR technique was used to confirm the interaction between cobalt oxide and porous carbon material. Energy dispersive spectroscopy was used to monitor the elemental composition of composite catalysts. The electrochemical experiments were performed in a 1 M KOH aqueous solution. The catalyst ink was prepared by mixing 15 mg of the prepared composite in 2.5 mL of deionized water and 1 mL of glutaraldehyde as a binder polymer. The geometric area of the glassy carbon electrode was 3 mm. The drop cast method was used to modify the surface of the glassy carbon electrode using 10 μliter of each catalyst ink. The modified electrode was dried in a preheated oven at 60 °C. The platinum wire and silver–silver chloride (Ag/AgCl) saturated with the KCl electrolyte were used as the counter and reference electrodes, respectively. The cyclic and linear sweep voltammetry were used as the primary modes for the investigation of the OER activity. Electrochemical impedance spectroscopy was carried out in the frequency range from 1000 kHz to 0.1 Hz at the amplitude of 10 mV and onset potentials of 0.5 V in a 1 M KOH electrolyte.
All the potentials reported throughout the manuscript were converted to RHE through the Nernst equation as follows:
| | |
ERHE = EAg/AgCl + 0.059pH + E0Ag/AgCl.
| (1) |
The current density was calculated by dividing the current with the geometric area of the glassy carbon electrode.
3. Results and discussion
3.1. Structural characterization of the as-obtained Co3O4-based electrocatalysts
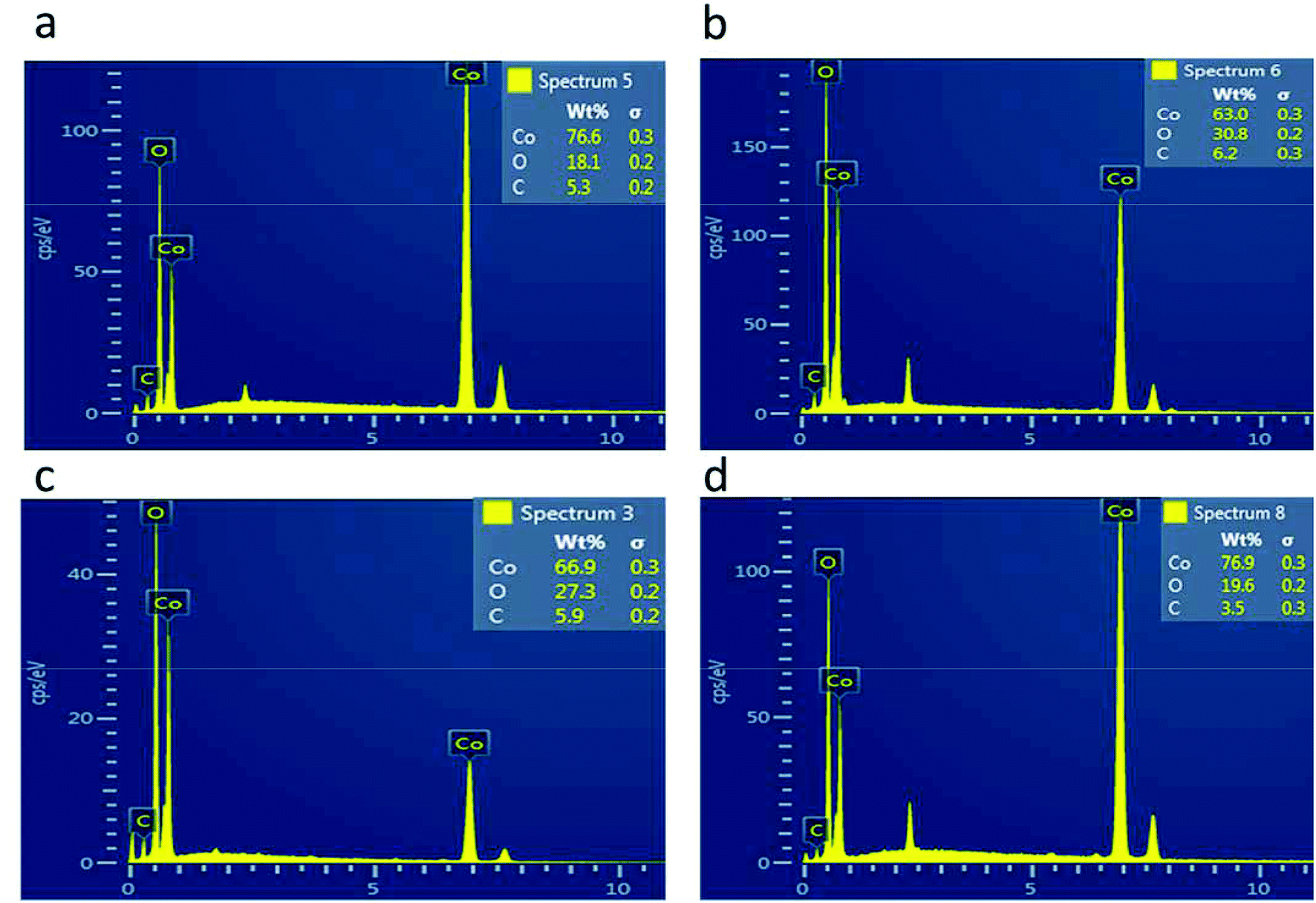
The morphology of the as-prepared samples was investigated by scanning electron microscopy, as shown in Fig. 1. Fig. 1(a) shows the morphology of the pristine cobalt oxide sample that exhibits the morphology of a bundle of nanowires. The morphology of the carbon material before the deposition of cobalt oxide is shown in Fig. 1(b), which is a porous structure. The sharp edges are found for the cobalt oxide nanostructures on porous carbon, as depicted in Fig. 1(c–f). The variation in the carbon content did not alter the morphology of the cobalt oxide nanostructures. The Co3O4/C composite structure showed a large number of active sites for catalyzing the water splitting reaction. The chemical composition of the as-prepared Co3O4/C composite materials was analyzed by energy dispersive spectroscopy, and the results indicated the presence of Co, O and C as the main elements, as shown in Fig. 2. No other impurities were found, and the EDS results were consistent with those of the XRD analysis.
 |
| | Fig. 1 SEM images: (a) pristine cobalt oxide, (b) pure carbon, (c) sample 1, (d) sample 2, (e) sample 3, and (f) sample 4. | |
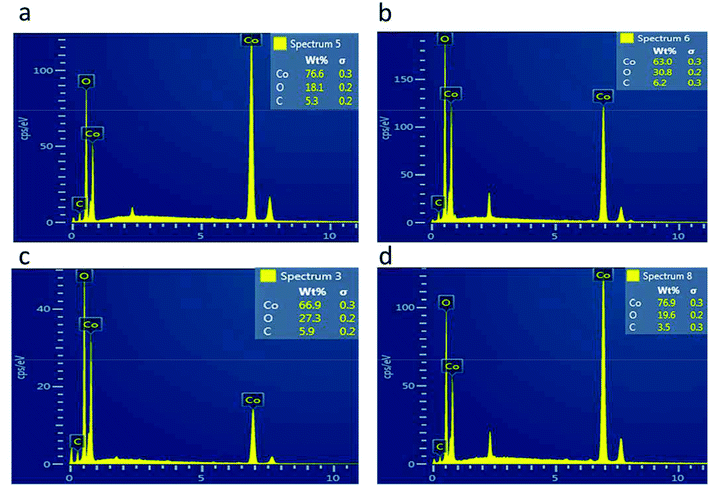 |
| | Fig. 2 EDS spectra: (a) sample 1, (b) sample 2, (c) sample 3, and (d) sample 4. | |
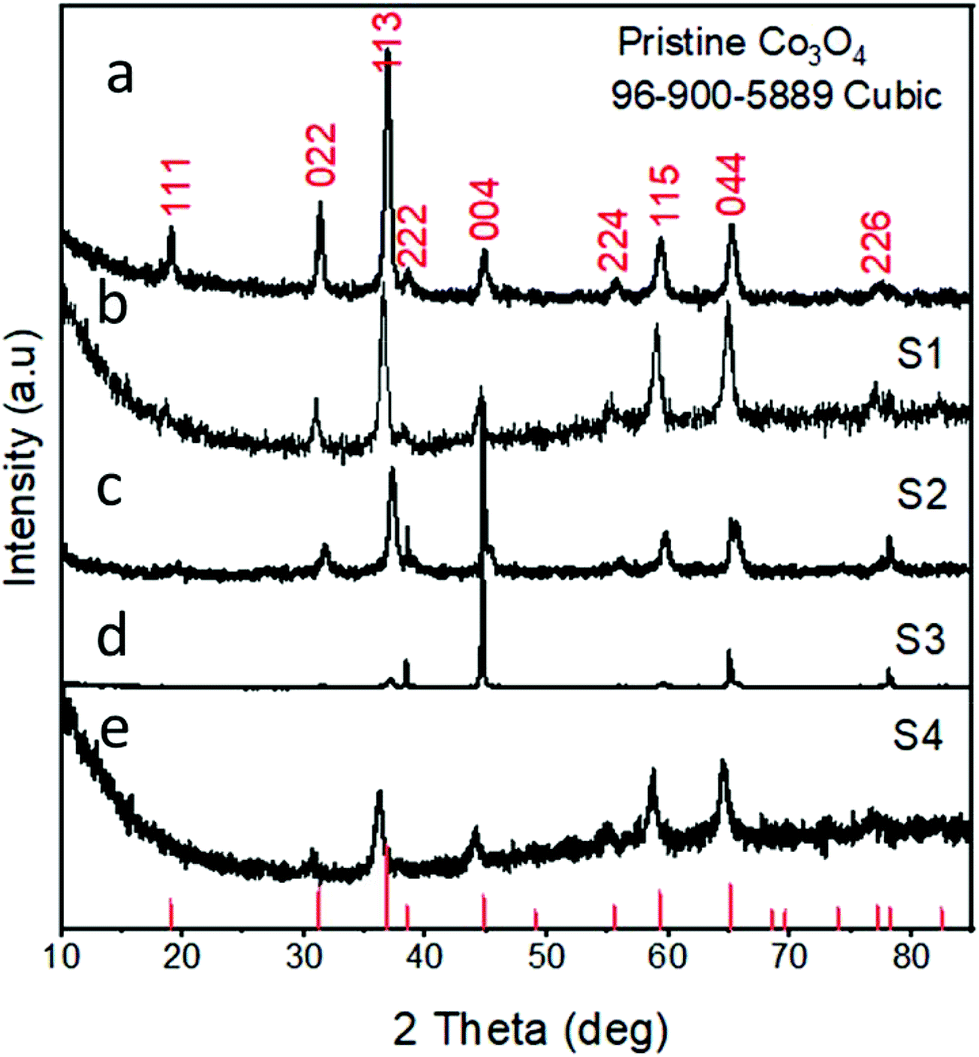
Fig. 3 shows the XRD diffraction patterns of the as-prepared Co3O4 samples. Fig. 3(a) shows the reflection peaks for pristine Co3O4, and this confirms the cubic phase of Co3O4. All the diffraction patterns corresponded to those of Co3O4 and fully agreed with the standard JCPDS card no: 96-900-5889. The XRD study was also carried out for the various Co3O4/C composite samples, which also confirmed the cubic phase of cobalt oxide, as shown in Fig. 3(b–e). All the diffraction patterns are also in good agreement with the standard JCPDS card no: 96-900-8589. No other phases or impurities were detected by XRD. The XRD patterns for the pure carbon material are shown in S1,† and the sample exhibits an amorphous phase.
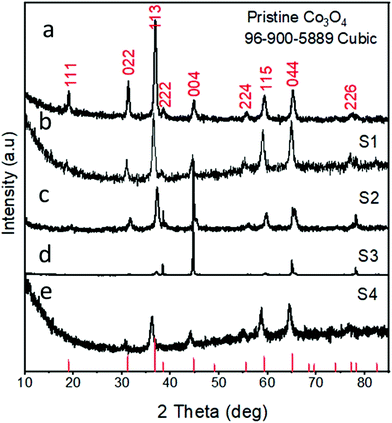 |
| | Fig. 3 XRD spectra: (a) sample 1, (b) sample 2, (c) sample 3, (d) sample 4, and (e) pristine Co3O4. | |
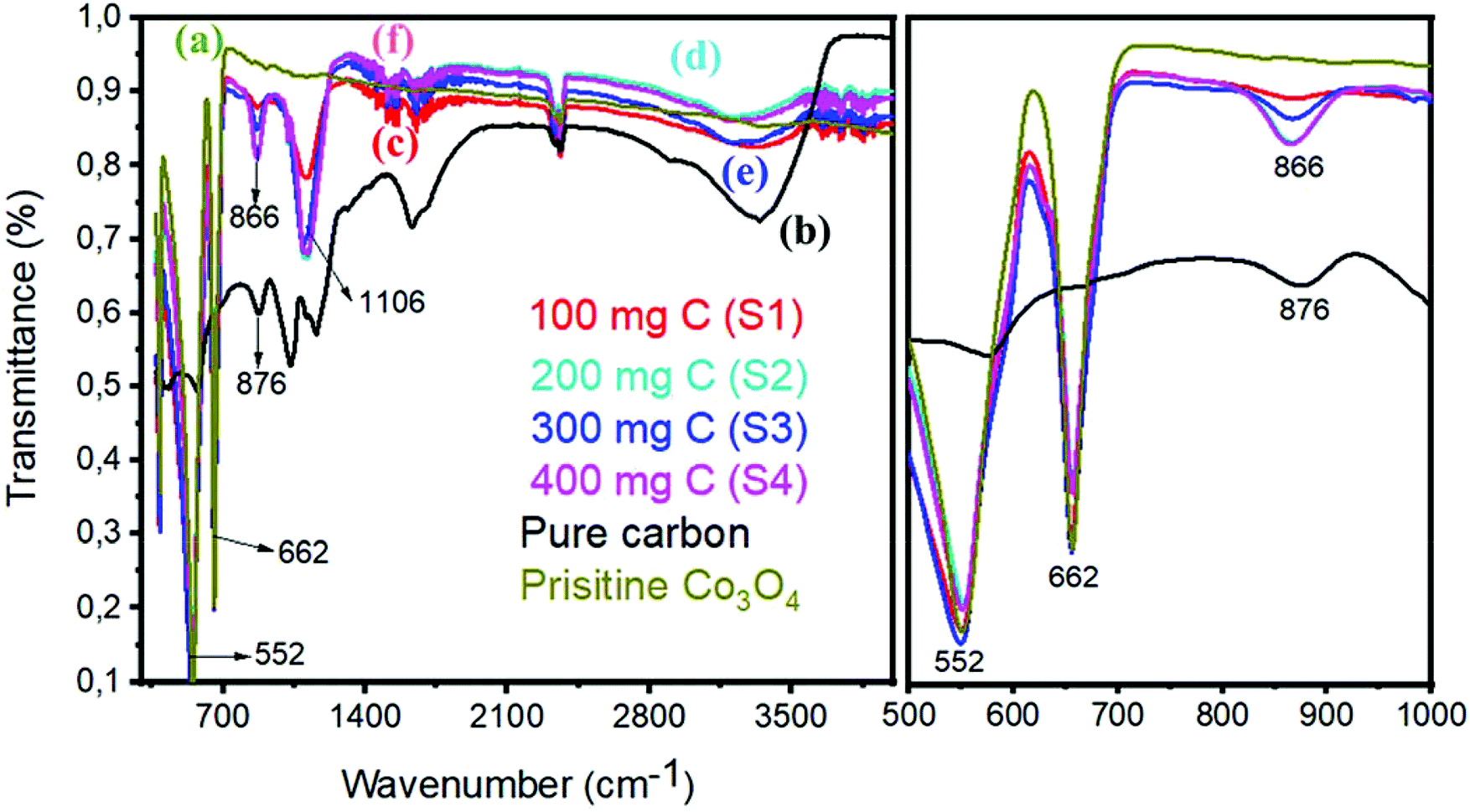
Fig. 4 shows the FTIR spectrum of pristine Co3O4 and Co3O4/C composite samples. The right part of Fig. 4 indicates the magnified region from 500 to 1000 cm−1 of the FTIR spectrum. The FTIR study shows that the spectrum of the Co3O4/C composite does not exhibit any new absorption peak, and this validates that the structure of cobalt oxide and carbon is not damaged. The sharp peaks at 552 cm−1 and 662 cm−1 of pristine cobalt oxide are assigned to the Co–O stretching vibration modes. Similar stretching vibrations are noticed in the composite samples, confirming the strong interaction between Co3O4 and carbon material. The bands at 1649 cm−1 of pristine Co3O4 are indexed to the vibration of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C. The composite samples also have CC of Co3O4/C and a large difference, suggesting the electronic interaction between CC and Co–O. The absorption bands in the region of 1000–1500 cm−1 are due to the O– symmetric and asymmetric stretching and the C–O stretching vibrations, and these features indicate that the adsorption of H2O and CO2 is unavoidable as the sample preparation was conducted in open air. In the case of pure carbon, the C–C, CC, C–O, and C–H stretching vibrations are prominent. We believe that the bonding features between Co3O4 and carbon material are due to the in situ growth of cobalt oxide.
C. The composite samples also have CC of Co3O4/C and a large difference, suggesting the electronic interaction between CC and Co–O. The absorption bands in the region of 1000–1500 cm−1 are due to the O– symmetric and asymmetric stretching and the C–O stretching vibrations, and these features indicate that the adsorption of H2O and CO2 is unavoidable as the sample preparation was conducted in open air. In the case of pure carbon, the C–C, CC, C–O, and C–H stretching vibrations are prominent. We believe that the bonding features between Co3O4 and carbon material are due to the in situ growth of cobalt oxide.
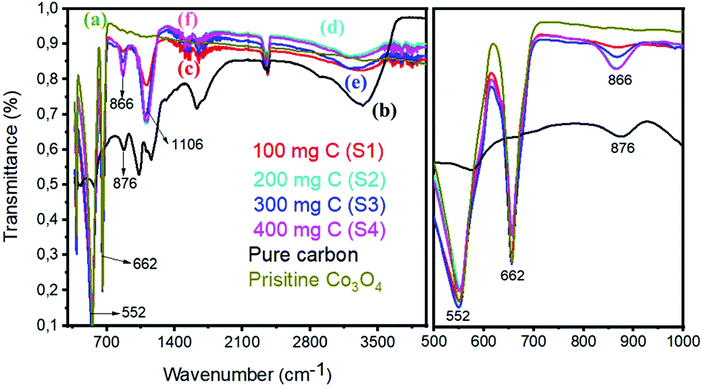 |
| | Fig. 4 FTIR spectra: (a) pristine cobalt oxide, (b) pure carbon, (c) sample 1, (d) sample 2, (e) sample 3, and (f) sample 4. Right hand part indicates the zoom in region from 500–1000 cm−1. | |
3.2. OER activity in alkaline media
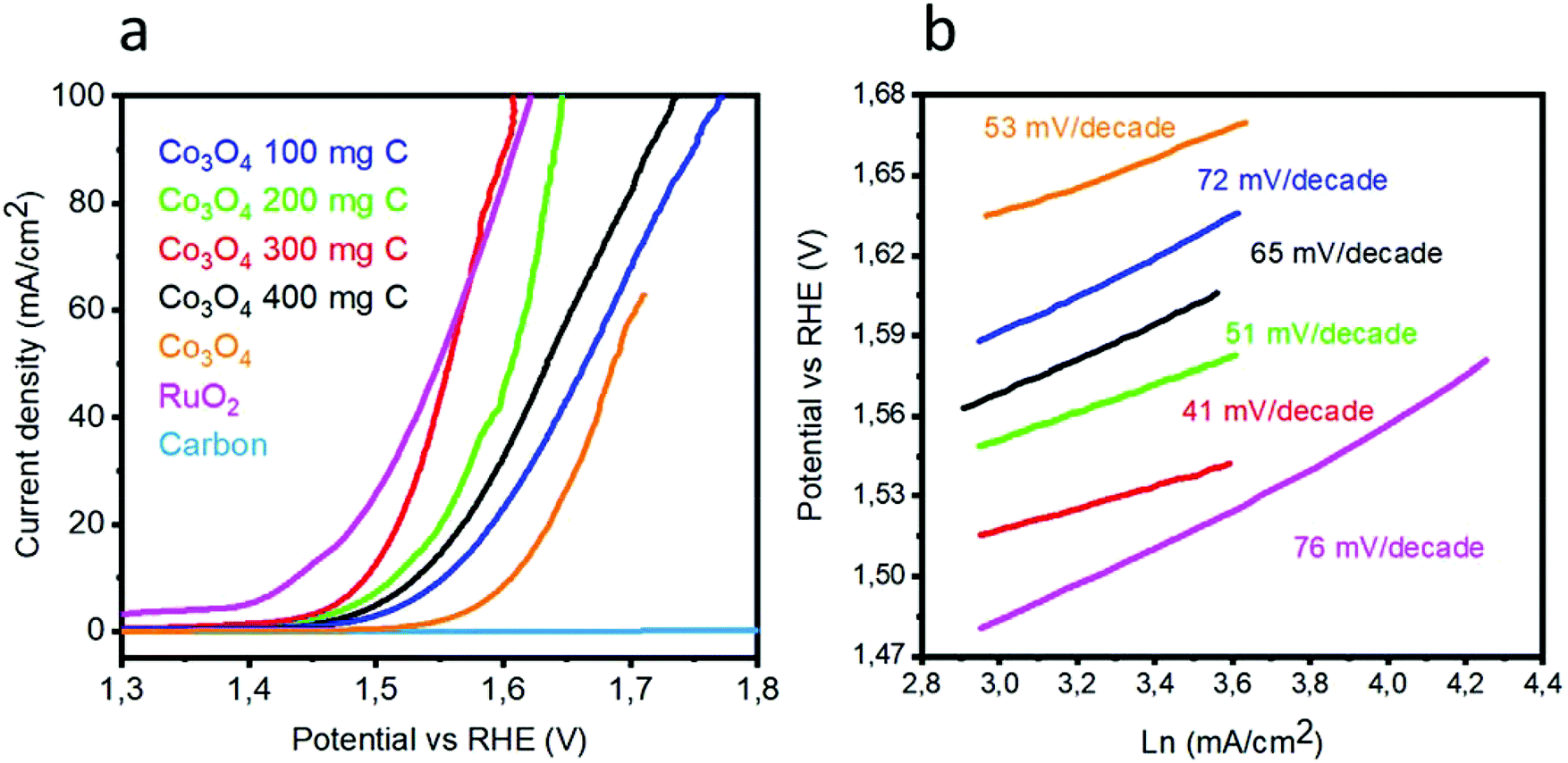
To investigate the OER activity of various catalysts based on the Co3O4/C composite and pristine Co3O4 in a 1 M KOH electrolyte, LSV polarization curves were obtained at the scan rate of 1 mV s−1, as shown in Fig. 5(a). Pristine cobalt oxide showed a slow OER activity at higher onset potential, but the addition of carbon as a supporting material for the deposition of Co3O4 nanostructures not only reduced the onset potential but also resulted in higher current density. The lowest overpotential to achieve a current density of 10 mA cm−2 is 1.49 V vs. RHE for the sample 3. The excellent OER activity of the composite catalysts can be attributed to the high surface area with improved active edges in the presence of the porous carbon material. In addition to the sample 3, the sample 1, 2, and 4 are efficient catalysts for the OER but require slightly higher OER potential. The OER activity is improved with the addition of carbon material. However, after a certain amount, the carbon material facilitates the aggregation of cobalt oxide nanostructures; this decreases the OER performance of the composite sample, as shown for the sample 4. It is believed that in the wet chemistry process, a nanostructured material has more defects, and further, the combination of these defect materials with a porous material can lead to enhanced OER performance, as demonstrated by the sample 3. It can be observed from Fig. 5(a) that peroxidation peaks are assigned to M2+ (M–OH; herein, M is = Co/Ru) and M3+ (M–OOH) oxidation, which can be found in the range from 1.25 to 1.43 V vs. RHE. It is believed that the height of the pre-oxidation peak current or the integrated peak area provides the density of the active edges (M–OOH) available within the metal oxide, and consequently, a favorable OER performance is achievable. The composition of each catalyst is solely responsible for the position of the pre-oxidation peak; thus, it varies from sample to sample.23–25 It is very difficult to know all the information about the OER mechanism, and generally, the OER process on transition metal oxides in alkaline media has been proposed by Li et al.26 and is expressed in following chemical equations:| | |
M*OH + OH− → M*O + H2O + e−
| (3) |
| | |
M*O + OH− → M*OOH + e−
| (4) |
| | |
M*OOH + OH− → M*O2 + H2O + e−
| (5) |
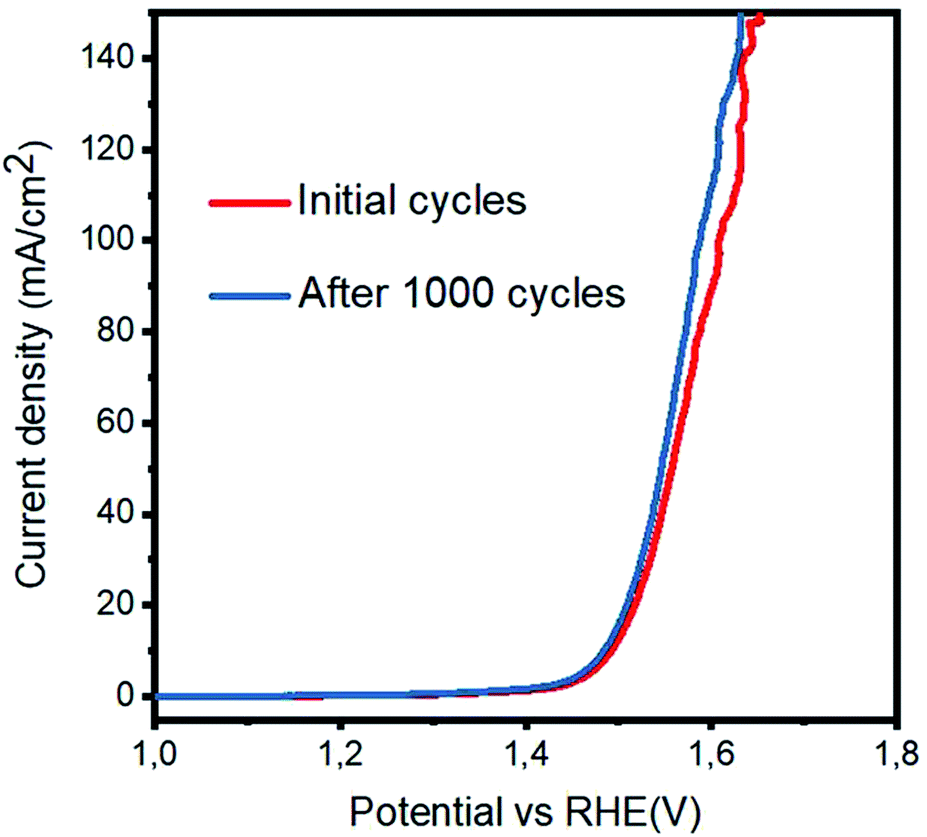
Herein, M* indicates the active edges on cobalt oxide, and the step (4) is considered as the rate-determining step; moreover, the Volmer, Heyrovsky and Tafel reactions correspond to the Tafel slope values of 120, 40 and 30 mV dec−1, respectively.27,28 The sample 3 has a lower Tafel slope value of 41 mV dec−1 than RuO2, which suggests that the OER kinetics has a more favorable environment on the sample 3 and it follows the Volmer–Heyrovsky reaction mechanism, as shown in Fig. 5(b). Based on the polarization curves, it can be said that only metal oxide is responsible for the OER kinetics, and porous carbon only enhances the performance of cobalt oxide by providing more surface for the growth of cobalt oxide nanocrystals, which may be involved in the adsorption of OH− on the surface and its further conversion into oxygen gas. In addition to this, the Tafel slopes of RuO2, the samples 1, 2, and 4 and pristine Co3O4 were found to be 76, 51, 72, 65, and 53 mV dec−1, respectively. Based on the available literature and to the best of our knowledge, the OER results are encouraging and ensure the possible configuration of the electrolyzer for efficient water splitting devices. These observations of superior OER results are mainly attributed to the high surface area provided by the carbon material during the deposition of the Co3O4 nanostructures. It is safe to say that the composite materials may have enhanced conductivity, synergetic effect produced from the active sites of Co3O4 and more surface area of the carbon material for the favorable OER activity. The durability and stability of the sample 3 was investigated through cyclic voltammetry for the OER, as shown in Fig. 6. After 1000 CV runs at 100 mV s−1, no significant loss in the catalytic activity was found, but a negligible loss of potential was visible at higher current densities.
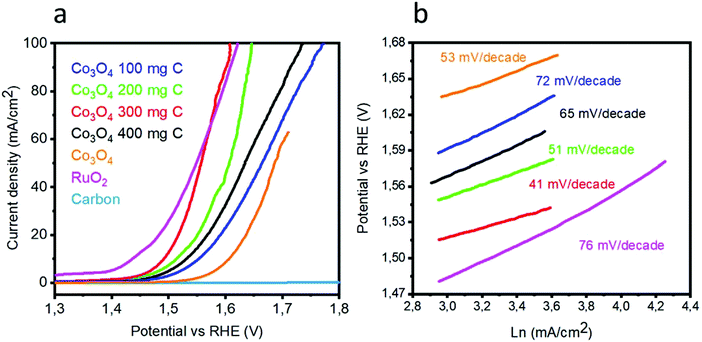 |
| | Fig. 5 (a) Linear sweep voltammetry of pristine cobalt oxide, pure carbon, sample 1, sample 2, sample 3, and sample 4, RuO2 at the scan rate 5 mV s−1 in 1 M KOH, (b) corresponding Tafel plots. | |
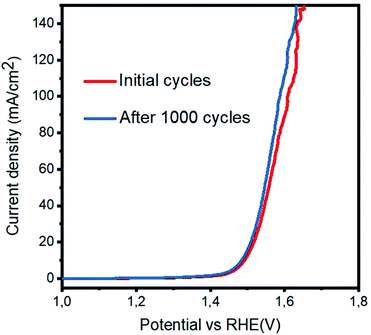 |
| | Fig. 6 Durability experiment via cyclic voltammetry at the scan rate of 20 mV s−1 after 2000 cycles in 1 M KOH. | |
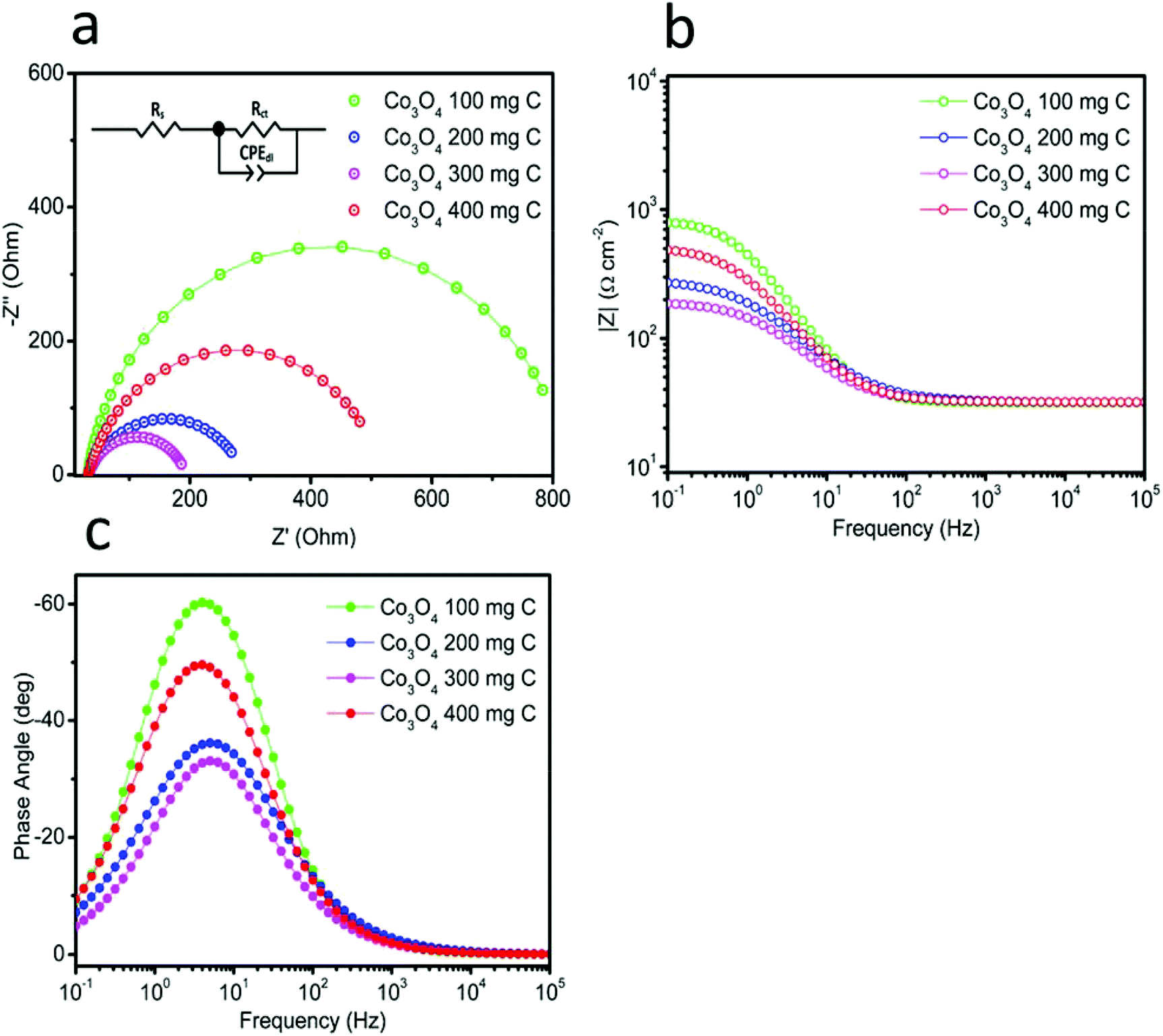
Catalytic activity is strongly associated with the charge transfer resistance. Therefore, electrochemical impedance spectroscopy was carried out at high, intermediate and low frequencies to understand the kinetics of charge transfer and the recombination mechanisms at the interfaces of the electrode and electrolyte. Fig. 7(a) shows the Nyquist plots obtained for the catalysts in the frequency range from 0.1 Hz to 1 MHz under an applied potential of 1.45 V vs. RHE. The same data is also plotted in the Bode format, as shown in Fig. 7(b) and (c). This impedance plots can be explained with the help of an equivalent circuit model, as depicted in inset of Fig. 7(a). The circuit fitting values are provided in Table 1. Rs corresponds to the solution resistance, which is nearly similar for all the electrocatalysts. The diameter of the semicircle corresponds to the charge transfer resistance (Rct). The quick charge transfer process and faster kinetics of the reaction are due to the lower Rct values. The obtained Rct values for Co3O4 100 mg C, Co3O4 200 mg C, Co3O4 300 mg C and Co3O4 400 mg C are 792, 258, 162 and 486 Ω, respectively. The result shows that Co3O4 300 mg C corresponds to a faster reaction rate than the other electrocatalysts. Single peak is observed in the Bode plots with the corresponding relaxation being assigned to the charge transfer process. The maximum oscillation frequency (fmax) of the impedance semicircle of Co3O4 300 mg C is less than that of the other electrocatalysts, thus the corresponding electron recombination lifetime (τn = 1/2πfmax) increased Co3O4 300 mg C can be attributed to favorable adsorption on the working electrode.29–31 The EIS observations revealed favorable OER activity on the composite samples. The performance of the produced catalyst was compared with that of the reported efficient catalysts, as presented in Table 2, and the produced catalyst was superior in terms of the Tafel slope and overpotential.
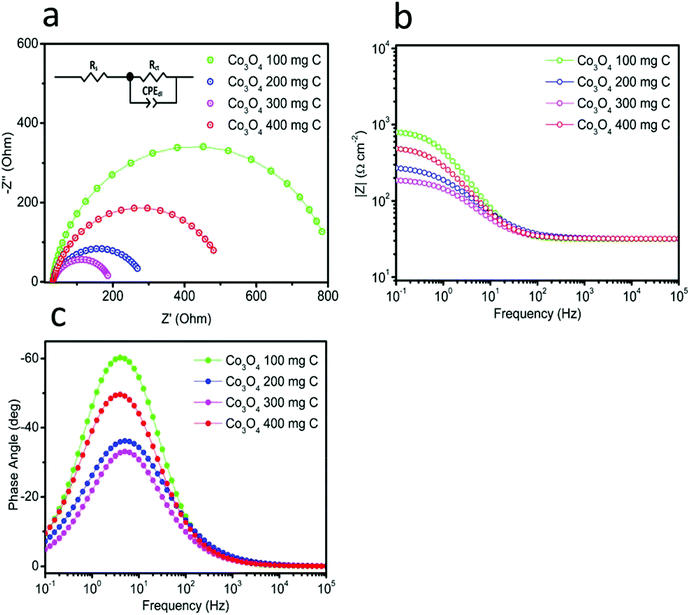 |
| | Fig. 7 Electrochemical impedance spectroscopy in the frequency range from 100 kHz to 0.1 Hz in 1 M KOH at the amplitude of 10 mV: (a) Nyquist plots, inset showing the fitted equivalent circuit model, and (b and c) Bode plots. | |
Table 1 The fitted circuit values
| |
Rs Ω |
Rct Ω |
CPEdl mF |
| 100 |
31.56 |
792.30 |
0.30 |
| 200 |
31.82 |
258.20 |
0.54 |
| 300 |
31.78 |
162.00 |
0.83 |
| 400 |
31.88 |
486.30 |
0.45 |
Table 2 Comparison of the presented overall water splitting results of the electrocatalyst (sample 3) with those of the recently reported studies
| Catalyst |
Overpotential mV |
Tafel slope mV dec−1 |
Reference |
| CoCo LDH |
393 |
59 |
Nat. Commun., 2014, 5, 4477 |
| CoOx@CN |
∼385 |
|
J. Am. Chem. Soc., 2015, 137, 2688 |
| Co–P films |
345 |
47 |
Angew. Chem. Int. Ed., 2015, 54, 6251 |
| Co3O4/NrmGO |
310 |
67 |
Nat. Mater., 2011, 10, 780 |
| NixCo3−xO |
∼370 |
59–64 |
Adv. Mater., 2010, 22, 1926 |
| N-G-CoO |
340 |
71 |
Energy Environ. Sci., 2014, 7, 609 |
| Co3O4/C |
260 |
41 |
This work |
4. Conclusions
In summary, herein, simple Co3O4/C composite-based catalysts were designed by the dehydration of common sucrose followed by the wet chemical method for the in situ growth of the Co3O4 nanostructures. The composite material has shown excellent OER activity, which can be assigned to high surface area, large number of active centers, synergetic effect and rich defect chemistry within the catalyst structure. The simple integration of interface between Co3O4 and the carbon material could facilitate the flow of water molecules for dissociation and consequently support the adsorption of intermediate species. Particularly, the OER could be conducted at low overpotentials by achieving the 10 mA cm−2 current density at 1.49 V vs. RHE. The lowest possible Tafel slopes are achieved for the nonprecious catalysts, which highly indicate the capitalization of composite materials for practical applications. The proposed strategy of designing the catalysts is very simple and cost-effective and can be capitalized on a large scale to produce oxygen.
Conflicts of interest
Authors declare no conflict of interest in this research work.
References
- M. G. Walter, L. Emily, J. R. Warren, S. W. McKone, S. W. Boettcher, Q. M. Elizabeth, A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS.
- H. B. Gray, J. Nat. Gas Chem., 2009, 1, 7 CrossRef CAS.
- D. G. Nocera, Chem. Res., 2012, 45, 767–776 CrossRef CAS.
- M. S. Dresselhaus and I. L. Thomas, J. Nat., 2001, 414, 332–337 CrossRef CAS.
- C. Liu, J. Tang, H. M. Chen, B. Liu and P. Yang, Nano Lett., 2013, 13, 2989–2992 CrossRef CAS PubMed.
- H. Wang, H. W. Lee, Y. Deng, Z. Lu, P. C. Hsu, Y. Liu, D. Lin and Y. Cui, J. Nat. Gas Chem., 2015, 23, 7261 Search PubMed.
- Y. Lee, J. Suntivich, K. J. May, E. E. Perry and Y. Shao-Horn, J. Phys. Chem. Lett., 2012, 3, 399–404 CrossRef CAS.
- M. W. Kanan and D. G. Nocera, Science, 2008, 321, 1072–1075 CrossRef CAS PubMed.
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-Horn, Science, 2011, 334, 1383–1385 CrossRef CAS.
- M. Gong, Y. Li, H. Wang, Y. Liang, J. Z. Wu, J. Zhou, J. Wang, T. Regier, F. Wei and H. Dai, J. Am. Chem. Soc., 2013, 135, 8452–8455 CrossRef CAS.
- S. Cobo, J. Heidkamp, P. A. Jacques, J. Fize, V. Fourmond, L. Guetaz, B. Jousselme, V. Ivanova, H. Dau, S. Palacin, M. Fontecave M and V. A. Artero Janus, Nat. Mater., 2012, 11, 802–807 CrossRef CAS.
- F. Song and X. Hu, Nat. Commun., 2014, 5, 4477 CrossRef CAS.
- T. Maiyalagan, K. A. Jarvis, S. Therese, P. J. Ferreira and A. Manthiram, Nat. Commun., 2014, 5, 3949 CrossRef CAS.
- R. D. Smith, M. S. Prévot, R. D. Fagan, Z. Zhang, P. A. Sedach, M. K. Siu, S. Trudel and C. P. Berlinguette, Science, 2013, 340, 60–63 CrossRef CAS.
- N. H. Chou, P. N. Ross, A. T. Bell and T. D. Tilley, ChemSusChem, 2011, 4, 1566–1569 CrossRef CAS PubMed.
- M. Dinca, Y. Surendranath and D. G. Nocera, Science, 2010, 107, 10337–10341 CAS.
- F. Jiao and H. Frei, Environ. Sci., 2010, 3, 1018–1027 CAS.
- J. A. Koza, Z. He, A. S. Miller and J. A. Switzer, Chem. Mater., 2012, 24, 3567–3573 CrossRef CAS.
- Y. Y. Mu, H. P. Liang, J. S. Hu, L. Jiang and L. J. Wan, J. Phys. Chem. B, 2005, 109, 22212–22216 CrossRef CAS.
- Q. Liu, J. T. Jin and J. Y. Zhang, ACS Appl. Mater. Interfaces, 2013, 5, 5002–5008 CrossRef CAS.
- Y. Liang, H. Wang, J. Zhou, Y. Li, J. Wang, T. Regier and H. Dai, J. Am. Chem. Soc., 2012, 134, 3517–3523 CrossRef CAS.
- X. Bian, J. Zhu, L. Liao, M. D. Scanlon, P. Ge, C. Ji, H. H. Girault and B. Liu, Electrochem. Commun., 2012, 22, 128–132 CrossRef CAS.
- B. S. Yeo and A. T. Bell, J. Phys. Chem. C, 2012, 116, 8394 CrossRef CAS.
- L. Trotochaud, J. K. Ranney, K. N. Williams and S. W. Boettcher, J. Am. Chem. Soc., 2012, 134, 17253 CrossRef CAS.
- M. W. Louie and A. T. Bell, J. Am. Chem. Soc., 2013, 135, 12329 CrossRef CAS.
- Y. Li, H. X. Li, K. Z. Cao, T. Jin, X. J. Wang, H. M. Sun, J. X. Ning, Y. J. Wang and L. F. Jiao, Ener. Stor. Mate., 2018, 12, 44 Search PubMed.
- H. Su, H. H. Wang, B. Zhang, K. X. Wang, X. H. Lin and J. S. Chen, Nano Energy, 2016, 22, 79 CrossRef CAS.
- H. Y. Jin, J. Wang, D. F. Su, Z. Z. Wei, Z. F. Pang and Y. Wang, J. Am. Chem. Soc., 2015, 137, 2688 CrossRef CAS.
- R. Naeem, M. A. Ehsan, R. Yahya, M. Sohail, H. Khaledi and M. Mazhar, Dalton Trans., 2016, 38, 14928–14939 RSC.
- Y. Liu, H. He, J. Li, W. Li, Y. Yang, Y. Li and Q. Chen, RSC Adv., 2015, 58, 46928–46934 RSC.
- Q. Song, J. Li, L. Wang, Y. Qin, L. Pang and H. Liu, J. Catal., 2019, 370, 176–185 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra07224a |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  b,
Umair Aftab
b,
Umair Aftab