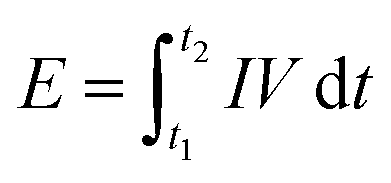DOI:
10.1039/C9RA07157A
(Paper)
RSC Adv., 2019,
9, 37882-37888
Improved pseudocapacitive charge storage in highly ordered mesoporous TiO2/carbon nanocomposites as high-performance Li-ion hybrid supercapacitor anodes†
Received
6th September 2019
, Accepted 6th November 2019
First published on 20th November 2019
Abstract
A Li-ion hybrid supercapacitor (Li-HSCs), an integrated system of a Li-ion battery and a supercapacitor, is an important energy-storage device because of its outstanding energy and power as well as long-term cycle life. In this work, we propose an attractive material (a mesoporous anatase titanium dioxide/carbon hybrid material, m-TiO2-C) as a rapid and stable Li+ storage anode material for Li-HSCs. m-TiO2-C exhibits high specific capacity (∼198 mA h g−1 at 0.05 A g−1) and promising rate performance (∼90 mA h g−1 at 5 A g−1) with stable cyclability, resulting from the well-designed porous structure with nanocrystalline anatase TiO2 and conductive carbon. Thereby, it is demonstrated that a Li-HSC system using a m-TiO2-C anode provides high energy and power (∼63 W h kg−1, and ∼4044 W kg−1).
Introduction
High-power energy-storage devices have been regarded as indispensable systems for medium- and large-scale energy-storage applications such as electric vehicles (EVs) and smart grid technologies. This is because the medium- and large-scale energy storage applications require fast charging and discharging behaviors with long cycle lifetime.1,2 An electric double-layer capacitor (EDLC), which is one of the typical supercapacitors, is the representative energy-storage device that provides highly excellent power abilities (2–5 kW kg−1) with stable cyclability.3–6 However, in spite of its superb abilities, it suffers from low energy performance (∼10 W h kg−1) due to its charge-storage mechanism being based on the physisorption of solvated ions at electrode/electrolyte interfaces.7–9
As its alternative device, Li-ion hybrid supercapacitors (Li-HSCs) composed of Li-ion battery (LIB) electrode materials as anodes and EDLC electrode materials as cathodes in a non-aqueous electrolyte containing a Li salt have been intensively researched and developed in recent years.10–12 This is because that their electrochemical performance delivering both high energy and power abilities with stable cyclability is highly attractive. Such unique characteristics result from asymmetric charge-storage mechanisms that Li+ from the electrolyte inserts to the anode materials (faradaic reaction) and anions such as PF6− and ClO4− are physically adsorbed on the surface of the cathode materials (non-faradaic reaction) during the charge process.10 However, because of the different charge-storage mechanisms between two electrodes, a kinetics imbalance issue is one of the major problems in Li-HSCs, which is should be solved to develop high-performance Li-HSCs.13,14 In other words, the reaction mechanism of the anode materials using ionic diffusion in a crystal framework of electrode materials is much more sluggish than that of cathode materials.15,16
To address this kinetics issue, two kinds of strategies have intensively been considered. One of the effective strategies is to reduce particle size of electrode materials in the nanoscale regime.15 Well-nanosized electrode materials have a variety of merits such as shortened lengths for Li+ diffusion/electron mobility (high-power ability) and enhanced electrode/electrolyte interface area (high capacity).17–19 In addition, reducing particle size of electrode materials in the nanoscale leads to improved pseudocapacitive reactions associated with surface-controlled reactions, which is kinetically not limited by diffusion-controlled reactions.20 Another strategy is to use attractive carbonaceous and Ti-based anode materials such as graphite and titanium dioxide (TiO2).21–24 Particularly, anatase TiO2 is one of the promising anode materials for Li-HSCs, because of its beneficial Li+ insertion/extraction behaviors and a variety of merits. Firstly, it provides theoretical capacity of ∼168 mA h g−1 in the potential range of ∼1.7 V (vs. Li/Li+) with small volume change (<4%) during cycling.21,25 Therefore, compared to the graphite anode working in the potential range of ∼0.2 V (vs. Li/Li+), it is free from Li-plating problem and can provide highly stable cycle performance.26,27 Secondly, Li-HSCs using anatase TiO2 anodes can use a cost-attractive and lightweight Al current collector in the anodic part instead of Cu current collector because alloying reaction between Li+ and Al does not take place in the potential range of 1.0–3.0 V (vs. Li/Li+) for anatase TiO2. In addition, it is non-toxic and one of the highly abundant materials.28,29 However, the drawback of anatase TiO2 is its poor electrical conductivity and relatively low Li+ diffusion rate.28,30,31
Here, to rationally design the anatase TiO2 as anode materials for Li-HSCs, we synthesized mesoporous anatase TiO2/carbon nanocomposite (denote as m-TiO2-C) by using block copolymer assisted simple synthesis method. It is clearly demonstrated in this work that design of the synthesized mesoporous electrode material comprising both nanocrystalline anatase TiO2 and conductive carbon is highly appropriate to solve drawback of the anatase TiO2 and to maximize its electrochemical performance (high capacity and rate capability), which result from improved Li+ diffusion kinetics with rapid electron transport. Furthermore, we proved that the well-designed m-TiO2-C is highly attractive as anode materials for Li-HSCs delivering high energy and power.
Experimental
Synthesis of m-TiO2-C
0.15 g of PEO-b-PS (poly(ethylene oxide)-b-poly(styrene), Mn = 27![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 466 g mol−1) was dissolved in 4 mL tetrahydrofuran (THF). 0.9 mL of titanium isopropoxide (TTIP) with 0.3 mL of 35–37% hydrochloric acid (concentrated HCl) was added dropwise to the block copolymer/THF mixture solution with continuous stirring. After 1 h of stirring, the mixture solution was poured to the glass Petri dish. The solvent was evaporated overnight at 40 °C, and then dried at 100 °C. The as-synthesized transparent film was obtained and then, the collected power was heat-treated in Ar atmosphere at 700 °C for 2 h. Commercial TiO2 purchased from Sigma Aldrich was employed for control group (denoted as com-TiO2).
466 g mol−1) was dissolved in 4 mL tetrahydrofuran (THF). 0.9 mL of titanium isopropoxide (TTIP) with 0.3 mL of 35–37% hydrochloric acid (concentrated HCl) was added dropwise to the block copolymer/THF mixture solution with continuous stirring. After 1 h of stirring, the mixture solution was poured to the glass Petri dish. The solvent was evaporated overnight at 40 °C, and then dried at 100 °C. The as-synthesized transparent film was obtained and then, the collected power was heat-treated in Ar atmosphere at 700 °C for 2 h. Commercial TiO2 purchased from Sigma Aldrich was employed for control group (denoted as com-TiO2).
Materials characterization
Structure and chemical characterization. The structure and morphology of prepared samples were investigated using scanning electron microscopy (SEM; S-4200 field-emission, Hitachi) and high resolution-transmission electron microscopy (HR-TEM; JEOL JEM-2010). Nitrogen adsorption–desorption analysis was conducted with 77 K with Micromeritics Tristar II 3020 system to estimate pore size and specific surface area. To confirm the specific pore morphology, small-angle X-ray scattering (SAXS) patterns was detected using 4C SAXS beamlines at the Pohang Light Source (PLS). To investigate crystalline phase, X-ray diffraction (XRD) pattern was identified by D/max-2500 a diffractometer (Rigaku, Cu-Kα radiation). The carbon content of m-TiO2-C was estimated using thermogravimetric analysis (TGA; NETZSCH STA 449C). Electron energy loss spectroscopy (EELS) analysis was performed to identify containing elements using energy-filtering transmission electron microscopy (EF-TEM, JM-220FS).
Electrochemical measurements. For half and full-cell electrochemical tests, active materials including m-TiO2-C and com-TiO2 (80 wt%) were homogeneously mixed with super-P carbon (10 wt%) and polyvinyledene fluoride (PVDF, 10 wt%) in N-methyl-2-pyrrolidone (NMP). The slurries were pasted on current collector using doctor blade. The prepared electrodes were dried at 60 °C for 6 h and then, 110 °C for 12 h in vacuum oven. Subsequently, the electrodes were roll-pressed. For half-cell test, 2032-type coin cells were fabricated with lithium metal as both counter and reference electrodes in Ar-filled glovebox. The mass loading of active materials used as anode materials were carefully controlled around 1.0 mg cm−2. The electrolyte was 1.0 M of LiPF6 in mixture of ethylene carbonate/dimethyl carbonate (EC/DMC, 1:1 volume ratio, Panaxetec. Co., Korea). The activated carbon electrode used as cathode materials was prepared using commercially available MSP-20 (90 wt%), conductive carbon (5 wt%), and polytetrafluoroethylene (PTFE, 5 wt%). In the half-cell tests, the working voltages for anode and cathode materials were 1.0–3.0 and 3.0–4.5 V (vs. Li/Li+), respectively. In the case of full-cell, Li-HSC was assembled using m-TiO2-C as an anode and MSP-20 as a cathode and the weight ratio of anode and cathode active materials was controlled to 1:3.5 in working voltages of 0–3.0 V. The energy and power of the Li-HSCs was calculated by numerically integrating the galvanostatic discharge profiles using eqn (1) and (2) as follows.32| |
 | (1) |
where I is the constant current (A g−1), V is the working voltage (V), t1 and t2 are the start/end of discharge time (s) of Li-HSCs, respectively, and| |
 | (2) |
where t is the discharge time (s). All electrochemical tests were conducted using the WBCS-3000 battery cycler (WonA Tech, Korea).
Results and discussion
Material characterization
The m-TiO2-C was synthesized using a block copolymer-assisted synthesis method and schematic representation on the synthesis method is shown in Fig. 1. In brief, the Ti precursor (TTIP) and laboratory-made amphiphilic block copolymer (poly(ethylene oxide)-b-poly(styrene), PEO-b-PS) synthesized by atomic transfer radical polymerization (ATRP) were dissolved in tetrahydrofuran (THF). And then, to induce sol–gel reaction of TTIP, concentrated hydrochloric acid (HCl) was slowly added to the TTIP/block copolymer/THF mixture solution. The titanium oxide sol made by hydrolysis selectively interacts with the hydrophilic PEO part of PEO-b-PS via hydrogen bonds. During the evaporation of organic solvent at 40 °C (evaporation-induced self-assembly, ESIA), highly ordered mesostructure is formed by self-organization of titanium oxide sol/block copolymer mixture.33–36 After drying at 100 °C to induce the cross-linkage of titanium oxide sol, the as-synthesized TiO2/block copolymer composite was heat-treated at 700 °C under inert atmosphere (Ar condition). During heat-treatment, as-synthesized TiO2 is converted to crystalline TiO2. In addition, PS part of PEO-b-PS is converted to mechanically stable and conductive carbon (in situ formed carbon in m-TiO2-C). For comparison, commercially available TiO2 (com-TiO2) was employed.
 |
| | Fig. 1 Schematic illustration of the synthesis of m-TiO2-C. | |
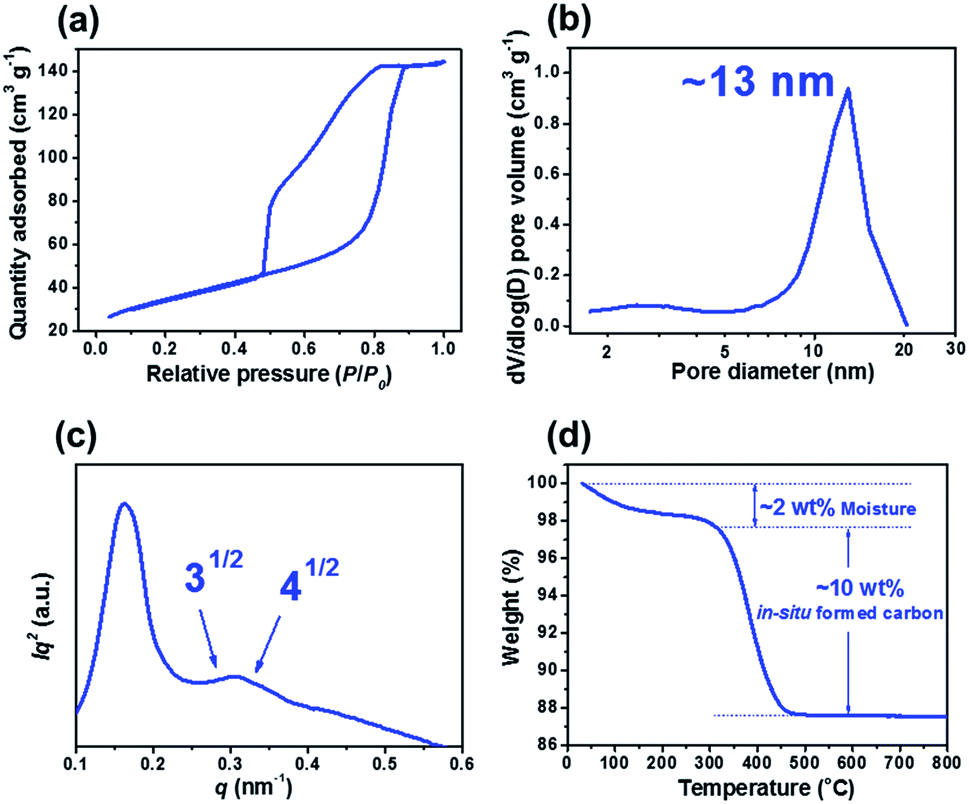
Fig. 2a and b show that X-ray diffraction (XRD) patterns of m-TiO2-C and com-TiO2 are in good agreement with anatase TiO2 (JCPDS no. 21-1272) with no noticeable impurities. Their average crystallite sizes calculated using the Debye–Scherrer equation were ∼14 (m-TiO2-C) and ∼100 (com-TiO2) nm, respectively.37 In addition, high-resolution transmission electron microscopy (HR-TEM, Fig. 2c) image of m-TiO2-C clearly shows the lattice fringes of anatase TiO2 with (101) and (004) spacing of 0.353 and 0.238 nm, respectively, again indicating that the crystal structure of m-TiO2-C well match the anatase TiO2 phase. Mesoporous structure of m-TiO2-C is identified through scanning electron microscopy (SEM) image, N2 adsorption–desorption technique, and small-angle X-ray scattering (SAXS) patterns. SEM image (Fig. 2d) shows highly ordered mesoporous structure with uniform pore size. Compared to m-TiO2-C, com-TiO2 has irregular and larger particle shape/size without porosity (Fig. S1†). In addition, N2 adsorption isotherm (Fig. 3a) of m-TiO2-C corresponds to type IV curve with a sharp adsorption at ∼0.9 P/P0, indicating that uniform mesopores are predominant. The main pore size calculated using the Barret–Joyner–Halenda (BJH) method and specific surface area calculated using the Brunauer–Emmett–Teller (BET) of m-TiO2-C were ∼13 nm and ∼123 m2 g−1 (Fig. 3b), respectively, which is significantly higher than that of com-TiO2 (<2 m2 g−1). The mesoporous structural characterization of m-TiO2-C is further demonstrated by SAXS pattern (Fig. 3c). Scattering peaks of m-TiO2-C with a peak position ratio of 1:31/2:41/2 suggest that hexagonally ordered TiO2 structure with a long-range order is well established.38 To confirm the presence of in situ formed carbon in m-TiO2-C, thermogravimetric analysis (TGA) and electron energy loss spectroscopy (EELS) analysis were employed. TGA result (Fig. 3d) proves that the in situ formed carbon content in the m-TiO2-C is around 10 wt%. Furthermore, EELS analysis image (Fig. 4) directly shows the existence of in situ formed carbon in the m-TiO2-C, representing that Ti, O, and C are uniformly dispersed.
 |
| | Fig. 2 (a) XRD pattern on m-TiO2-C. (b) Comparison of XRD patterns on m-TiO2-C and com-TiO2. (c) HR-TEM image of m-TiO2-C. (d) SEM image of m-TiO2-C. | |
 |
| | Fig. 3 (a) N2 physisorption isotherm and (b) pore size distribution of m-TiO2-C. (c) SAXS pattern of m-TiO2-C. (d) TGA result of m-TiO2-C. | |
 |
| | Fig. 4 EELS mapping images of m-TiO2-C. | |
Electrochemistry
Galvanostatic charge–discharge (de-lithiation and lithiation) test on m-TiO2-C was conducted in the potential range of 1.0–3.0 V (vs. Li/Li+), showing that the m-TiO2-C provides reversible charge capacity of ∼198 mA h g−1 at current of 0.05 A g−1 (Fig. 5a). The typical plateau at a potential of ∼1.7 V (vs. Li/Li+) demonstrates reversible Li+ intercalation into anatase TiO2 lattice, where TiO2 + xLi+ + xe− ↔ LixTiO2 (0 ≤ x ≤ 0.5).22,28,39 Fig. 5b shows the specific capacity of m-TiO2-C is much higher (∼198 mA h g−1) than that of com-TiO2 (∼110 mA h g−1) at current of 0.05 A g−1. In addition, compared to com-TiO2, the m-TiO2-C showed much better rate capability with increasing currents from 0.05 to 5 A g−1, also indicating that capacity retention of m-TiO2-C with increasing currents is significantly outstanding than that of com-TiO2 (Fig. S2a†). Fig. 5c and S2b† shows that the galvanostatic charge–discharge curves of m-TiO2-C with increasing currents are well maintained with small overpotentials (reduced internal resistance) compared to those of com-TiO2. It represents that well-ordered structure with conductive in situ formed carbon is highly beneficial to improve electrochemical performances of anatase TiO2, mainly due to a variety of merits including (i) shortened diffusion lengths of Li+, (ii) superior electron mobility, (iii) easy penetration of electrolyte, (iv) plentiful charge-storage sites, and etc.34,40–42 It should be noted that the m-TiO2-C exhibits better rate capability than other anatase TiO2 anodes previously reported (Fig. S3†) and delivers highly stable cycle stability (capacity retention of ∼94% with ∼100% coulombic efficiency for ∼350 cycles) at the current of 0.5 A g−1 (Fig. 5d and S4†).43–47 In addition, it shows stable long-term cyclability at high current of 3 A g−1 for 1000 cycles (Fig. S4,† capacity retention of ∼97% with ∼100% coulombic efficiency). Because the excellent rate capability and long-term cycle stability are main factors for application to anodes of Li-HSCs, m-TiO2-C developed in this work could be the extremely potential Li-HSC anode material.
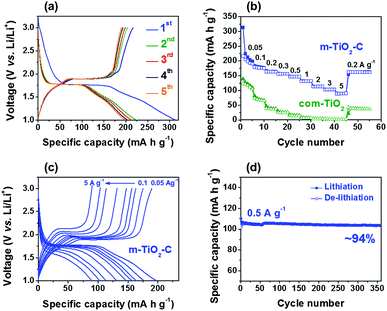 |
| | Fig. 5 (a) Galvanostatic charge–discharge profiles of m-TiO2-C at 0.05 A g−1. (b) Comparison of rate capability of TiO2 electrodes at different currents from 0.05 to 5 A g−1. (c) Galvanostatic charge–discharge profiles of m-TiO2-C at various currents from 0.05 to 5 A g−1. (d) Cycle performance of m-TiO2-C at a current of 0.5 A g−1. | |
To further reveal the reason why m-TiO2-C provides superior electrochemical behaviors, we performed cyclic voltammetry (CV) tests on m-TiO2-C and com-TiO2 conducted at sweep rates from 0.1 to 1.0 mV s−1 in the potential range of 1.0–3.0 V (vs. Li/Li+) as shown in Fig. 6a and S5.† The pair of redox peaks (1.7–2.0 V vs. Li/Li+) of m-TiO2-C and com-TiO2 at sweep rate of 0.1 mV s−1 well match the lithiation and de-lithiation reactions. In previous studies, it is well known that the anatase TiO2 is influenced by the diffusion-controlled process with a two-phase process. Therefore, from i = avb equation (where a (mV s−1)) with CV tests, b value of anatase TiO2 is close to 0.5, indicating that the diffusion-controlled reactions is more dominant than the surface-controlled reaction (b = 1.0).20,48,49 As shown in Fig. 6b and S6,†b value of m-TiO2-C obtained from peak currents of the lithiation process is around 0.62, indicative of that the charge-storage reaction mechanism in m-TiO2-C is influenced by both the surface- and diffusion-controlled reactions (improved pseudocapacitive reaction).49 It is considerably contrast to that of com-TiO2 showing severe deviation with increasing sweep rates from 0.1 to 1.0 mV s−1 because of huge internal resistance. In addition, b values of m-TiO2-C obtained in potential range of 1.2–2.1 V (vs. Li/Li+) are also higher than 0.5 (Fig. S6†).
 |
| | Fig. 6 (a) CV curves of m-TiO2-C at different sweep rates of 0.1–1.0 mV s−1. (b) log(i) vs. log(v) plot of cathodic peak current on m-TiO2-C electrode. | |
Before building Li-HSC system using the m-TiO2-C anode, electrochemical behavior of MSP-20 (commercially available activated carbon) as a cathode material for the Li-HSC was investigated by galvanostatic charge–discharge half-cell test. Fig. S7† shows that reversible specific capacity of the MSP-20 is ∼60 mA h g−1 at a current of 0.05 A g−1 in the voltage range of 3.0–4.5 V (vs. Li/Li+). Considering the specific capacities and working voltages between m-TiO2-C anode and MSP-20 cathode, the Li-HSC system using m-TiO2-C anode and MSP-20 cathode was carefully assembled and its galvanostatic charge–discharge tests were conducted at various currents in the potential range of 0.0–3.0 V (Fig. 7a and S8†). Galvanostatic charge–discharge curves of Li-HSC using m-TiO2-C as anode and MSP-20 as cathode at current rates from 0.01 to 5 A g−1 do not exhibit typical triangular shape, dissimilar to those of conventional symmetric SCs.50 It is again confirmed from CV data at sweep rates of 1–20 mV s−1 (Fig. 7b and c) that the CV profile does not follows a typical rectangular shape of conventional symmetric SCs. The galvanostatic charge–discharge and CV shapes of the Li-HSC are mainly due to combination of the faradaic reaction at the m-TiO2-C anode and the non-faradaic reaction at the MSP-20 cathode.22 The maximum energy and power of the Li-HSC calculated using eqn (1) and (2) with galvanostatic charge–discharge curves were ∼63 W h kg−1 and ∼4044 W kg−1, respectively. The Ragone plot on the trade of relationship between energy and combination of the faradaic reaction at the m-TiO2-C anode and the non-faradaic reaction at the MSP-20 cathode.22 The maximum energy and power of the Li-HSC calculated using eqn (1) and (2) with galvanostatic charge–discharge curves were ∼63 W h kg−1 and ∼4044 W kg−1, respectively. The Ragone plot on the trade of relationship between energy and power shows that its energy and power is much better than that of other results previously reported (Fig. 7d).43,51–54 Finally, long-term cycle stability of the Li-HSC was investigated. Fig. 7e shows that the cycling stability of the Li-HSC at a current of 0.5 A g−1 is well maintained with ∼100% coulombic efficiency for 1000 cycles. The Ragone plot and cycle performance data imply that m-TiO2-C is highly suitable as the anode material for Li-HSC system.
 |
| | Fig. 7 (a) Galvanostatic charge–discharge profiles of the Li-HSC at different currents from 0.01 to 0.1 A g−1. (b and c) CV curves of the Li-HSCs at different sweep rates of 1–20 mV s−1. (d) Ragone plots compared with previously reported results. (e) Cycle performance of the Li-HSC at a current of 0.5 A g−1. | |
Conclusions
In summary, we reported the block copolymer assisted one-pot synthesis method of m-TiO2-C and its application for high-power anode materials of Li-HSC. The m-TiO2-C delivered high specific capacity (∼198 mA h g−1 at 0.05 A g−1) and rate capability (∼90 mA h g−1 at 5 A g−1) with stable cycle performance. Its electrochemical performance was superior compared to com-TiO2, mainly due to synergistic effects of unique mesostructure and hybridization between anatase TiO2 and in situ formed carbon. Thereby, the Li-HSC system using the m-TiO2-C anode possessed high energy and power abilities (∼63 W h kg−1 and ∼4044 W kg−1) in the potential range of 0.0–3.0 V, implying that the energy-storage device (Li-HSC) can be a promising alternative to conventional symmetric SCs.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT [NRF-2017R1A2B3004648, and NRF-2018M1A2A2061987]. This work was supported by the Korea Institute of Energy Technology Evaluation and Planning (KETEP) and the Ministry of Trade, Industry & Energy (MOTIE) of the Republic of Korea [No. 20174030201600, and No. 20182010600430].
Notes and references
- B. Kang and G. Ceder, Nature, 2009, 458, 190–193 CrossRef CAS PubMed.
- M. H. Hsieh, G. A. Li, W. C. Chang and H. Y. Tuan, J. Mater. Chem. A, 2017, 5, 4114–4121 RSC.
- J. Yan, Q. Wang, T. Wei and Z. Fan, Adv. Energy Mater., 2014, 4, 1300816 CrossRef.
- G. Zhang and X. W. Lou, Adv. Mater., 2013, 25, 976–979 CrossRef CAS PubMed.
- G. F. Chen, Y. Z. Su, P. Y. Kuang, Z. Q. Liu, D. Y. Chen, X. Wu, N. Li and S. Z. Qiao, Chem. - Eur. J., 2015, 21, 4614–4621 CrossRef CAS PubMed.
- S. Ghosh, S. Jeong and S. R. Polaki, Korean J. Chem. Eng., 2018, 35, 1389–1408 CrossRef CAS.
- P. Simon, Y. Gogotsi and B. Dunn, Science, 2014, 343, 1210–1211 CrossRef CAS PubMed.
- D. Pech, M. Brunet, H. Durou, P. Huang, V. Mochalin, Y. Gogotsi, P. L. Taberna and P. Simon, Nat. Nanotechnol., 2010, 5, 651–654 CrossRef CAS PubMed.
- E. M. Jin, H. Lee, H. Jun and S. Jeong, Korean J. Chem. Eng., 2017, 34, 885–891 CrossRef CAS.
- K. Naoi, S. Ishimoto, J. Miyamoto and W. Naoi, Energy Environ. Sci., 2012, 5, 9363–9373 RSC.
- C. M. Lai, T. L. Kao and H. Y. Tuan, J. Power Sources, 2018, 379, 261–269 CrossRef CAS.
- R. Yi, S. Chen, J. Song, M. L. Gordin, A. Manivannan and D. Wang, Adv. Funct. Mater., 2014, 24, 7433–7439 CrossRef CAS.
- E. Lim, C. Jo, H. Kim, M. Kim, Y. Mun, Y. Ye, J. Hwang, K. Ha, K. Roh and J. Lee, ACS Nano, 2015, 9, 7497–7505 CrossRef CAS PubMed.
- E. Lim, W. Lim, C. Jo, J. Chun, M. Kim, K. Roh and J. Lee, J. Mater. Chem. A, 2017, 5, 20969–20977 RSC.
- E. Lim, C. Jo and J. Lee, Nanoscale, 2016, 8, 7827–7833 RSC.
- V. Aravindan, J. Gnanaraj, Y. S. Lee and S. Madhavi, Chem. Rev., 2014, 114, 11619–11635 CrossRef CAS PubMed.
- Z. Wang, L. Zhou and X. W. Lou, Adv. Mater., 2012, 24, 1903–1911 CrossRef CAS PubMed.
- Z. Yang, D. Choi, S. Kerisit, K. M. Rosso, D. Wang, J. Zhang, G. Graff and J. Liu, J. Power Sources, 2009, 192, 588–598 CrossRef CAS.
- A. K. Mondal, K. Kretschmer, Y. Zhao, H. Liu, H. Fan and G. Wang, Microporous Mesoporous Mater., 2017, 246, 72–80 CrossRef CAS.
- J. Wang, J. Polleux, J. Lim and B. Dunn, J. Phys. Chem. C, 2007, 40, 14925–14931 CrossRef.
- D. Deng, M. Kim, J. Y. Lee and J. Cho, Energy Environ. Sci., 2009, 2, 818–837 RSC.
- H. Kim, M. Cho, M. Kim, K. Park, H. Gwon, Y. Lee, K. C. Roh and K. Kang, Adv. Energy Mater., 2013, 3, 1500–1506 CrossRef CAS.
- S. Ghosh, W. D. Yong, E. M. Jin, S. R. Polaki, S. Jeong and H. Jun, Korean J. Chem. Eng., 2019, 36, 312–320 CrossRef CAS.
- W. Lee, S. Jeong, H. Lee, B. Kim, K. An, Y. Park and S. Jung, Korean J. Chem. Eng., 2017, 34, 2993–2998 CrossRef CAS.
- L. Thirugunanam, S. Kaveri, V. Etacheri, S. Ramaprabhu, M. Dutta and V. G. Pol, Mater. Charact., 2017, 131, 64–71 CrossRef CAS.
- Y. Liu and Y. Yang, J. Nanomater., 2016, 2016, 1–15 CAS.
- V. Etacheri, C. N. Hong, J. Tang and V. G. Pol, ACS Appl. Mater. Interfaces, 2018, 10, 4652–4661 CrossRef CAS PubMed.
- J. Shin, D. Samuelis and J. Maier, Adv. Funct. Mater., 2011, 21, 3464–3472 CrossRef CAS.
- Y. Guan, T. Hu, J. Wu, L. Zhao, F. Tian, W. Pan, P. He, W. Qi, F. Li and K. Xu, Korean J. Chem. Eng., 2019, 36, 115–125 CrossRef CAS.
- Y. Qiu, K. Yan, S. Yang, L. Jin, H. Deng and W. Li, ACS Nano, 2010, 4, 6515–6526 CrossRef CAS PubMed.
- J. S. Chen, H. Liu, S. Z. Qiao and X. W. Lou, J. Mater. Chem., 2011, 21, 5687–5692 RSC.
- F. Zhang, T. Zhang, X. Yang, L. Zhang, K. Leng, Y. Huang and Y. Chen, Energy Environ. Sci., 2013, 6, 1623 RSC.
- J. Hwang, C. Jo, M. Kim, J. Chun, E. Lim, S. Kim, S. Jeong, Y. Kim and J. Lee, ACS Nano, 2015, 9, 5299–5309 CrossRef CAS PubMed.
- C. Jo, Y. Kim, J. Hwang, J. Shim, J. Chun and J. Lee, Chem. Mater., 2014, 26, 3508–3514 CrossRef CAS.
- J. Lee, M. C. Orilall, S. C. Warren, M. Kamperman, F. J. DiSalvo and U. Wiesner, Nat. Mater., 2008, 7, 222–228 CrossRef CAS PubMed.
- J. Hwang, J. Kim, E. Ramasamy, W. Choi and J. Lee, Microporous Mesoporous Mater., 2011, 143, 149–156 CrossRef CAS.
- A. D. Krawitz, Introduction to Diffraction in Materials Science and Engineering, Wiley, New York, 2001, p. 168 Search PubMed.
- M. Templin, A. Franck, A. D. Chensne, H. Leist, Y. Zhang, R. Ulrich, V. Schadler and U. Wiesner, Science, 1997, 278, 1795–1798 CrossRef CAS PubMed.
- Y. Guo, Y. Hu and J. Maier, Chem. Commun., 2006, 26, 2783–2785 RSC.
- Y. Ye, C. Jo, I. Jeong and J. Lee, Nanoscale, 2013, 5, 4584–4605 RSC.
- C. Jo, J. Hwang, H. Song, A. H. Dao, Y. Kim, S. Lee, S. Hong, S. Yoon and J. Lee, Adv. Funct. Mater., 2013, 23, 3747–3754 CrossRef CAS.
- E. Kang, Y. Jung, G. Kim, J. Chun, U. Wiesner, A. C. Dillon, J. Kim and J. Lee, Adv. Funct. Mater., 2011, 21, 4349–4357 CrossRef CAS.
- G. Tang, L. Cao, P. Xiao, Y. Zhang and H. Liu, J. Power Sources, 2017, 355, 1–7 CrossRef CAS.
- E. Lim, H. Shim, S. Fleischmann and V. Presser, J. Mater. Chem. A, 2018, 6, 9480 RSC.
- H. Wang, C. Guan, X. Wang and H. J. Fan, Small, 2015, 11, 1470–1477 CrossRef CAS PubMed.
- T. Brousse, R. Marchand, P. Taberna and P. Simon, J. Power Sources, 2006, 158, 571–577 CrossRef CAS.
- V. Aravindan, M. V. Reddy, S. Madnavi, S. G. Mhaisalkar, G. V. Subba Rao and B. V. R. Chowdari, J. Power Sources, 2011, 196, 8850–8854 CrossRef CAS.
- V. Augustyn, J. Come, M. A. Lowe, J. W. Kim, P. L. Taberna, S. H. Tolbert, H. D. Abruna, P. Simon and B. Dunn, Nat. Mater., 2013, 12, 518–522 CrossRef CAS PubMed.
- H. Lindstrom, S. Sodergren, A. Solbrand, H. Rensmo, J. Hjelm, A. Hagfeldt and S. Lindquist, J. Phys. Chem. B, 1997, 101, 7717–7722 CrossRef.
- K. Karthikeyan, S. Amaresh, V. Aravindan, H. Kim, K. S. Kang and Y. S. Lee, J. Mater. Chem. A, 2013, 1, 707–714 RSC.
- H. Jung, N. Venugopal, B. Scrosati and Y. Sun, J. Power Sources, 2013, 221, 266–271 CrossRef CAS.
- Q. Wang, Z. Wen and J. Li, Adv. Funct. Mater., 2006, 16, 2141–2146 CrossRef CAS.
- V. Aravindan, N. Shubha, W. Chui and S. Madhavi, J. Mater. Chem. A, 2013, 1, 6145–6151 RSC.
- Y. Wang, W. Wang and Y. Xia, Electrochim. Acta, 2005, 50, 5641–5646 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra07157a |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  b,
Eunho Lim
b,
Eunho Lim