
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTailor-made block copolymers of L-, D- and rac-lactides and ε-caprolactone via one-pot sequential ring opening polymerization by pyridylamidozinc(II) catalysts†
Ilaria
D'Auria
 a,
Massimo Christian
D'Alterio
b,
Consiglia
Tedesco
a and
Claudio
Pellecchia
*a
a,
Massimo Christian
D'Alterio
b,
Consiglia
Tedesco
a and
Claudio
Pellecchia
*a
aDipartimento di Chimica e Biologia “A. Zambelli”, Università Degli Studi di Salerno, Via Giovanni Paolo II 132, 84084 Fisciano, SA, Italy. E-mail: cpellecchia@unisa.it
bDipartimento di Scienze Chimiche, Università “Federico II” di Napoli, Via Cintia 21, 80126 Napoli, Italy
First published on 14th October 2019
Abstract
Three-coordinated Zn(II) complexes bearing sterically encumbered bidentate monoanionic [N,N−] pyridylamido ligands efficiently catalyze the ring opening polymerization of lactide (LA) and ε-caprolactone (CL). Owing to the polymerization controlled nature and high rate, precise stereodiblock poly(LLA-b-DLA) with different block lengths can be easily produced by one-pot sequential monomer addition at room temperature in short reaction times. NMR, SEC and DSC analyses confirm the production of highly isotactic diblock copolymers which crystallize in the high melting stereocomplex phase. Stereo-triblock and tetrablock copolymers of L-LA, D-LA and rac-LA have been synthesized similarly. Finally, a diblock poly(CL-b-LA) has been easily obtained by sequential addition of ε-caprolactone and lactide under mild conditions.
Introduction
Aliphatic polyesters are among the most desirable targets of a sustainable polymer industry in a circular economy.1 Indeed, there is a clear and urgent need for biodegradable polymeric materials for non-durable or disposable applications, as replacements for conventional commodity plastics, i.e. polyolefins and poly(ethylene terephthalate), whose ‘fake degradation’ is behind the problem of microplastic pollution in water.2With a market of roughly 300 ktons per year which is growing steadily, poly(lactic acid) (PLA) is by far the most important polymer in this class from the application standpoint.3,4 PLA is produced via ring opening polymerization (ROP) of lactide, the dimeric lactone of lactic acid, totally derived from annually renewable resources (e.g. starch obtained from corn or sugar beet). The only ROP catalyst used industrially to produce PLA is currently the rather simple Sn(II) 2-ethylhexanoate (Sn(Oct)2).3–5 This cheap and moderately active compound affords rather poor stereocontrol in polymerization and suffers from the occurrence of transesterification processes broadening the polymer dispersity. On the other hand, since the degradation of PLA yields lactic acid, which is easily metabolized in the human body, PLA has several sophisticated biomedical applications, such as resorbable medical implants that require long strength retention, and microspheres and microcapsules of low-molecular-weight for drug delivery systems.5,6 Thus, the cytotoxicity of Sn residues in PLA is a serious matter of concern, and it is expected that, as soon as catalysts with a convenient cost/performance balance are identified, Sn(Oct)2 will ultimately be abandoned. Although hundreds of catalysts for the ROP of lactide and lactones have been reported in the last two decades, catalysts combining non-toxicity, high activity and well controlled polymerization are rare.3,4 Examples of catalysts based on biologically benign metals include heteroleptic complexes of Mg(II) and Zn(II) of the type LM-X, with L = β-diiminate,7–11 trispyrazolylborate,12 N-heterocyclic carbene13,14 or phenolate-based15–21 ligands, and X = alkyl, alkoxy, dialkylamido.
An appealing target for ROP catalysts is the ability to afford “tailor-made” aliphatic polyesters block copolymers.22E.g., since PLLA and PDLA co-crystallize as a stereocomplex phase showing improved properties with respect to the homochiral PLA's,23 the production of stereo-diblock and stereo-multiblock PLA's by stereoselective catalysts has been extensively investigated.24–27 Diblock copolymers of LA and ε-caprolactone (CL) have been also studied since they may combine the complementary features of the two homopolymers.28–31 Moreover, ABA triblock copolymers, comprising e.g. two isotactic crystalline PLLA segments as the A-end blocks and a soft, low Tg amorphous polyester as the B-midblock, have been extensively studied as attractive degradable and sustainable alternatives to industrially relevant SBS thermoplastic elastomers.22,32–36 The synthetic strategy for the production of such ABA block copolyesters usually involves the preparation of an α,ω-dihydroxy capped macroinitiator of the B-midblock followed by ROP of L-lactide (or D-lactide or rac-lactide).22,23 PLLA–PDLA diblocks, PCL–PLA diblocks and ABA triblock copolyesters were also produced by sequential addition of the monomers using living catalysts, although very long reaction times (many hours or days) were required.28–31,37,38 Only recently, Kol et al. reported extremely active Mg catalysts bearing monoanionic tetradentate pyridyldiaminophenolate ligands for the rapid production of stereoblock PLA's and diblock and multiblock PCL–PLA copolymers by one-pot sequential addition of the monomers.39–41
Following our studies concerning pyridylamido or pyridylimino metal complexes as catalysts for the polymerization of olefins,42–46 and the ring opening polymerization of cyclic esters,47–49 here we report heteroleptic Zn(II) pyridylamido complexes (see Scheme 1) functioning as very active catalysts for the ROP of L-, D- and rac-lactides and ε-caprolactone. The controlled nature and high rate of the polymerization allowed the synthesis of well-defined diblock copolymers PLLA-b-PDLA or PCL-b-PLLA, stereotriblock copolymers, such as PLLA-b-PDLA-b-PLLA, PLLA-b-PDLLA-b-PDLA, PCL-b-PLLA-b-PDLA, and stereotetrablock PLLA-b-PDLA-b-PLLA-b-PDLA, by one-pot sequential additions of the monomers under mild conditions and short reaction times.
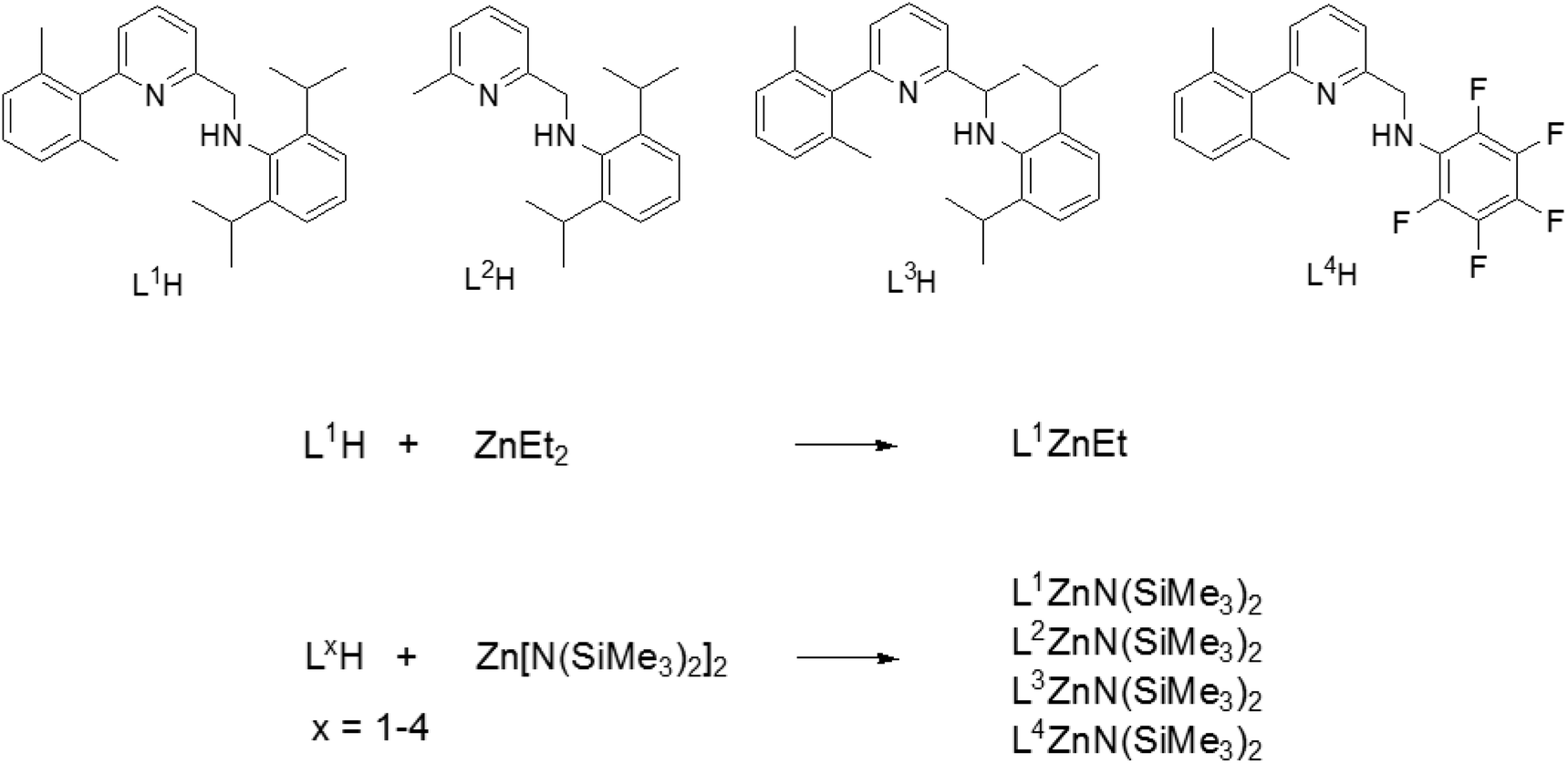 | ||
| Scheme 1 Pyridylamino ligands used for the synthesis of the Zn(II) complexes. | ||
Experimental section
General information
All manipulations involving air and/or moisture-sensitive compounds were performed under a nitrogen atmosphere in a Braun Labmaster glovebox or using Schlenk techniques. Glassware used were dried in an oven at 120 °C overnight and exposed to vacuum–nitrogen cycles. All solvents were dried as follows: toluene was refluxed over metallic sodium, dichloromethane was refluxed over CaH2, benzene, hexane and tetrahydrofuran were refluxed over sodium–benzophenone; they were distilled under nitrogen before use. Deuterated solvents were purchased from Aldrich and stored in the glovebox over 3 Å molecular sieves before use. Lactide was purified by crystallization from dry toluene. ε-Caprolactone was dried with CaH2 for 24 h at room temperature and then distilled under reduced pressure. All other chemicals were commercially available and used as received unless otherwise stated.NMR spectra were recorded using either a Bruker Advance 300, or 400 or a Bruker 600 MHz Ascend 3 HD spectrometer. Chemical shifts (δ) are expressed as parts per million and coupling constants (J) in hertz. 1H NMR spectra are referenced using the residual solvent peak at δ 7.26 for CDCl3, δ 5.32 for CD2Cl2 and δ 7.16 for C6D6. 13C NMR spectra are referenced using the residual solvent peak at δ 77.22 for CDCl3, δ 53.84 for CD2Cl2 and δ 128.06 for C6D6.
MALDI-TOF-MS spectra of polymer samples were acquired using a Bruker solariX XR Fourier transform ion cyclotron resonance mass spectrometer equipped with a 7 T refrigerated actively-shielded superconducting magnet. The samples were ionized in positive ion mode using the MALDI ion source. The mass range was set to m/z 150–9000. The laser power was 15% and 15 laser shots were used for each scan. The mass spectra were calibrated externally using a mix of peptide clusters in MALDI ionization positive ion mode. A linear calibration was applied. The samples were dissolved in THF (1 mg mL−1). trans-2-[3-(4-tert-Butylphenyl)-2-methyl 2-propenylidene]malononitrile (DCTB) was used as the matrix with potassium trifluoroacetate (KTFA) as cationizing agent. The matrix was dissolved in THF at a concentration of 10 mg mL−1 and ∼2 mg of KTFA was added to the solution. The matrix and polymer solutions were mixed together in a 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio and this mixture was spotted on the MALDI plate and left to dry. After evaporation of the solvent, the MALDI target was introduced into the spectrometer.
:1 ratio and this mixture was spotted on the MALDI plate and left to dry. After evaporation of the solvent, the MALDI target was introduced into the spectrometer.
The molecular weights (Mn and Mw) and the dispersities (Mw/Mn) of polymer samples were measured by size exclusion chromatography (SEC) at 30 °C, using THF as solvent, an eluent flow rate of 1 mL min−1, and narrow polystyrene standards as reference. The measurements were performed on a Waters 1525 binary system equipped with a Waters 2414 RI detector using four Styragel columns (range 1000–1000000 Å).
Differential scanning calorimetry (DSC) analysis was performed on a TA Q2000 (TA Instruments) according to the following program: equilibrate at −40 °C; ramp 10.00 °C min−1 to 220 °C; ramp 10.00 °C min−1 to −40.00 °C; ramp 10.00 °C min−1 to 220 °C. The instrument was calibrated for temperature and enthalpy by a high purity indium (156.60 °C, 28.45 J g−1) standard. DSC analyses were carried out on films obtained by casting from 1 wt% dichloromethane solutions at room temperature.
Synthesis and characterization
L1H–L4H and the zinc complexes LxZnN(SiMe3)2 (x = 1–4) were synthesized as previously reported.44,50Homopolymerizations of lactide and ε-caprolactone
In a typical polymerization run, a magnetically stirred reactor vessel was charged with a solution of monomer in the appropriate solvent, to which the alcohol was eventually added, and then 10 μmol of pyridylamidozinc complex. The alcohol used was either isopropanol (used in the runs performed at room temperature, also because the resonances of the resulting end groups are more clearly detected in the NMR spectra), or less volatile benzyl alcohol (used in the runs performed at higher temperature). The reaction mixture was stirred for the prescribed time at the desired temperature. After a specified time, an aliquot of the crude material was sampled and quenched in wet CDCl3. The sample was subjected to monomer conversion determination, which was monitored by integration of monomer versus polymer in 1H-NMR spectrum. The reaction was terminated by exposing to air and the volatiles were removed under vacuum. The crude product was dissolved in CH2Cl2 and precipitated in methanol. The obtained polymer was collected by filtration and further dried in a vacuum oven at 60 °C for 16 h.General procedure for block copolymerizations
In a glove box, to a CH2Cl2 solution of the first monomer were added iPrOH and L2ZnN(SiMe3)2; the reaction mixture was magnetically stirred at room temperature, and the following monomer(s) were sequentially added, maintaining the necessary delay (20–30 min) between each addition. An aliquot of the crude product was sampled and quenched in wet CDCl3 to determine the conversion of each monomer by 1H-NMR. The reaction was terminated by exposing to air and the work up of the reaction mixtures were carried out as described above.Random copolymerization of L-lactide and ε-caprolactone
A magnetically stirred reactor vessel was charged with L-LA, ε-CL, benzyl alcohol and pyridylamidozinc complex. The reaction mixture was thermostated at 130 °C and stirred for 10 minutes. Then the reaction mixture was cooled at room temperature, dissolved in CH2Cl2 and precipitated in methanol. The precipitated polymer was recovered as described above.Kinetic experiments
In a glovebox, a magnetically stirred reactor vessel was charged with a solution of L-LA in dichloromethane (3.5 mL, 0.6 M), a solution of iPrOH (0.5 mL, 0.02 M) and then a solution of L2ZnN(SiMe3)2 (1 mL, 2 mM) and then the reaction mixture was immediately stirred. After time intervals of 3 min each, small amounts of the polymerization mixture were transferred in different vials and taken out of the glovebox; each aliquot was withdrawn and quenched with wet CDCl3; the residual amount was quenched by adding an excess of wet n-hexane. The fractions were analyzed by 1H NMR in CDCl3 to determine the monomer conversion.X-ray crystallography
Yellow prismatic single crystals of compounds L1ZnEt and L3ZnN(SiMe3)2 suitable for X-ray diffraction analysis were obtained by slowly cooling supersaturated hexane solutions. Details of the X-ray crystallographic analysis are reported in the ESI.†Results and discussion
Synthesis and characterization of the complexes
The pyridylamino proligands LxH (x = 1–4, see Scheme 1) were synthesized according to previously reported procedures.44 We initially targeted the synthesis of ethyl Zn complexes: complex L1ZnEt was cleanly obtained in 85% yield from the reaction of equimolar amounts of L1H and ZnEt2 in dichloromethane at 25 °C. The complex was characterized by NMR analysis, confirming a heteroleptic structure (see ESI and Fig. S1 and S2†). Following the initial screening of the performance of L1ZnEt in the ROP of rac-lactide (vide infra), we pursued the synthesis of possibly more reactive silylamido Zn derivatives. Thus, complexes L1ZnN(SiMe3)2, L2ZnN(SiMe3)2, L3ZnN(SiMe3)2, and L4ZnN(SiMe3)2 were synthesized by allowing to react LxH and Zn[N(SiMe3)2]2 as previously reported.50 Single crystals suitable for X-ray diffraction studies of L1ZnEt and L3ZnN(SiMe3)2 were obtained by crystallization from hexane solutions at −20 °C. The X-ray molecular structures are shown in Fig. S3 and S4,† respectively. Selected bond distances and angles are listed in Table S2.† In both compounds the zinc atom is three-coordinated, adopting a slightly distorted trigonal planar geometry. The main geometrical features are commented in the ESI.†Ring opening polymerization of lactide
We initially tested L1ZnEt, bearing a bulky 2,6-dimethylphenyl substituent in the ortho-pyridine moiety and a 2,6-diisopropylphenyl substituent at the amido N, in the ROP of rac-lactide under different conditions (see Table 1). Reaction with 200 equiv. of monomer in dichloromethane at 25 °C for 2 h did not afford any polymer (run 1); also, after addition of 1 equiv. of isopropanol L1ZnEt was inactive under the same conditions (run 2). Only under more drastic conditions L1ZnEt was able to promote the polymerization, converting almost quantitatively 200 equiv. of rac-LA at 80 °C in toluene in 2 h (run 3), and 500 equiv. of rac-LA in the melt without solvent at 130 °C in 10 min (run 4). Monomer conversion was monitored by 1H-NMR in C7D8 at 80 °C until ca. 90% monomer consumption. Plot of ln([L-LA]0/[L-LA]t) (Fig. 1) versus time is linear, indicating that the polymerization process is first-order in monomer concentration and showing an induction time. The latter is reasonably related to the well-known slow reactivity of Zn-ethyl bond toward monomer insertion7–11 and to the generation of more reactive species by in situ reaction with protic impurities. The apparent kinetic constant is (1.87 ± 0.01) × 10−4 s−1 and it correlates well with the activity observed in polymerization run 3.| Run | Catalyst | Monomer | [ROH]0/[Zn]0 | Solvent | T [°C] | Time [min] | Convb [%] | M n,GPC c [kDa] | M n,theo d [kDa] | M w/Mnc |
|---|---|---|---|---|---|---|---|---|---|---|
| a Excepting where differently specified, reactions were performed in 2 mL of solvent, [Zn]0 = 5 mM, with [LA]0/[Zn]0 = 200; ROH = isopropanol. b Conversion of monomer as determined by 1H NMR spectral data. c Experimental Mn and Mw/Mn values determined by SEC in THF against polystyrene standards, using the correction factor 0.58. d Calculated Mn of PLA (in g mol−1) = 144.14 × ([LA]/[Zn]) × conversion of LA. e [LA]0/[Zn]0 = 500. f [LA]0/[Zn]0 = 1000. g ROH = benzyl alcohol. h [LA]0/[Zn]0 = 800. | ||||||||||
| 1 | L1ZnEt | rac-LA | 0 | CH2Cl2 | 25 | 120 | 0 | |||
| 2 | L1ZnEt | rac-LA | 1 | CH2Cl2 | 25 | 120 | 0 | |||
| 3 | L1ZnEt | rac-LA | 0 | Toluene | 80 | 120 | 98 | 19.5 | 28.2 | 1.38 |
| 4e | L1ZnEt | L-LA | 0 | — | 130 | <10 | 92 | 54.1 | 66.3 | 1.31 |
| 5 | L1ZnN(SiMe3)2 | rac-LA | 0 | CH2Cl2 | 25 | 120 | 25 | 26.4 | 7.2 | 1.50 |
| 6 | L1ZnN(SiMe3)2 | rac-LA | 1 | CH2Cl2 | 25 | 11 | >99 | 27.0 | 28.8 | 1.32 |
| 7 | L2ZnN(SiMe3)2 | rac-LA | 1 | CH2Cl2 | 25 | 10 | 97 | 28.2 | 28.0 | 1.25 |
| 8 | L2ZnN(SiMe3)2 | D-LA | 1 | CH2Cl2 | 25 | 10 | 97 | 32.8 | 28.0 | 1.08 |
| 9 | L2ZnN(SiMe3)2 | L-LA | 1 | CH2Cl2 | 25 | 10 | 97 | 35.4 | 28.0 | 1.21 |
| 10 | L3ZnN(SiMe3)2 | rac-LA | 1 | CH2Cl2 | 25 | 10 | 94 | 30.2 | 27.1 | 1.21 |
| 11 | L4ZnN(SiMe3)2 | L-LA | 1 | CH2Cl2 | 25 | 10 | 97 | 25.1 | 28.0 | 1.08 |
| 12 | L2ZnN(SiMe3)2 | rac-LA | 1 | THF | 25 | 60 | 83 | 17.8 | 24.0 | 1.24 |
| 13 | L3ZnN(SiMe3)2 | rac-LA | 1 | THF | 25 | 60 | 97 | 22.0 | 28.0 | 1.35 |
| 14 | L3ZnN(SiMe3)2 | rac-LA | 1 | Toluene | 25 | 30 | 99 | 19.8 | 28.5 | 1.46 |
| 15e | L3ZnN(SiMe3)2 | L-LA | 1g | Toluene | 50 | 20 | 99 | 45.0 | 71.3 | 1.30 |
| 16f | L3ZnN(SiMe3)2 | L-LA | 1g | Toluene | 50 | 20 | 90 | 22.3 | 129.7 | 1.29 |
| 17e | L2ZnN(SiMe3)2 | L-LA | 0 | — | 130 | 10 | 99 | 28.9 | 71.3 | 1.37 |
| 18e | L2ZnN(SiMe3)2 | L-LA | 1g | — | 130 | 10 | 96 | 32.1 | 69.1 | 1.34 |
| 19h | L2ZnN(SiMe3)2 | L-LA | 0 | — | 130 | 30 | 99 | 43.8 | 114.1 | 1.39 |
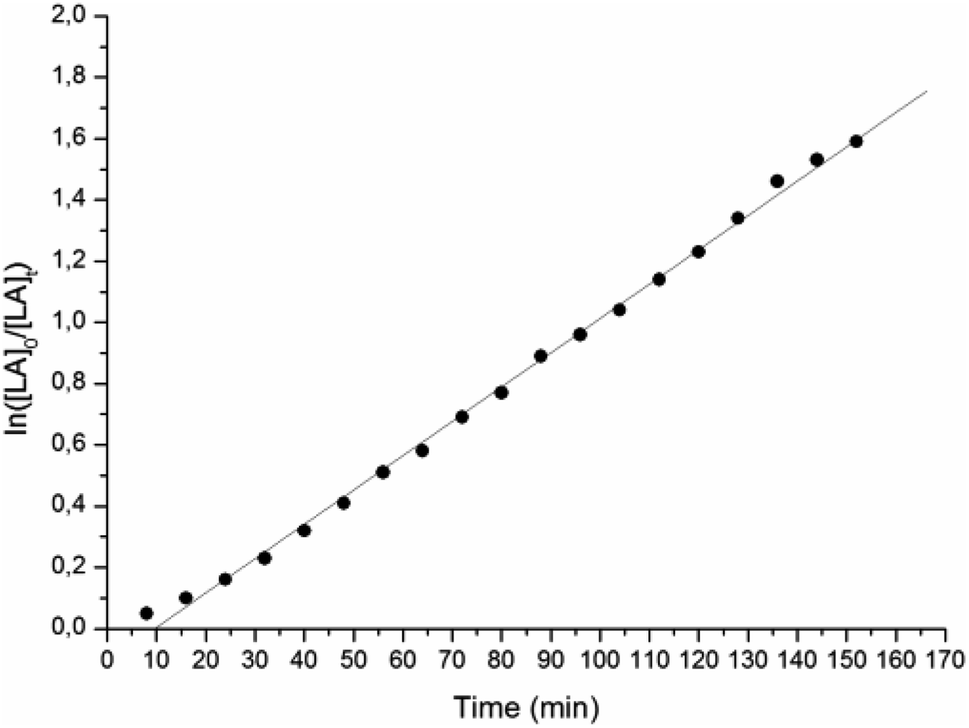 | ||
| Fig. 1 Pseudo-first-order kinetic plot for ROP of L-LA promoted by L1ZnEt: [Zn] = 0.01 M; [L-LA]/[Zn] = 100; T = 80 °C; toluene-d8 as solvent. | ||
Since, as just mentioned, the reactivity of Zn-alkyl bonds vs. monomer insertion,7–11 as well as alcoholysis21 can be slow, we turned to test more reactive Zn-amido complexes. In fact, complex L1ZnN(SiMe3)2, bearing the same ligand, gave 25% monomer conversion when tested under the same mild conditions of run 1, and total conversion in 11 min when 1 equiv. of isopropanol was added (see runs 5 and 6, respectively). The increase of the polymerization rate after the addition of an alcohol is a feature frequently observed in the literature8,18, and it is ascribed to the faster initiation promoted by alkoxy groups with respect to amido moieties.
Complex L2ZnN(SiMe3)2, maintaining the same bulky substituent at the amido N, but having a less bulky Me substituent at the pyridine moiety, gave 97% conversion of rac-LA in 10 min under the same conditions of run 6 (see run 7). Under these latter conditions, L3ZnN(SiMe3)2, maintaining the same bulky substituents of L1ZnN(SiMe3)2, but bearing a methyl substituent on the carbon bridging the amido and the pyridine moieties, was only slightly slower (see run 10). Finally, L4ZnN(SiMe3)2, having a bulky 2,6-dimethylphenyl substituent at the ortho-pyridine moiety and an electron withdrawing pentafluorophenyl substituent at the amido N, performed similarly to L2ZnN(SiMe3)2 (see run 11).
A kinetic study for the polymerization of L-LA by L2ZnN(SiMe3)2 and 1 equiv. of iPrOH was performed by NMR analysis of aliquots of the reaction mixture taken over 3 min intervals. Plot of ln([L-LA]0/[L-LA]t) versus time shows a first-order dependence on the concentration of lactide, with an apparent propagation rate constant of (2.92 ± 0.01) × 10−4 s−1 at 25 °C (see Fig. 2).
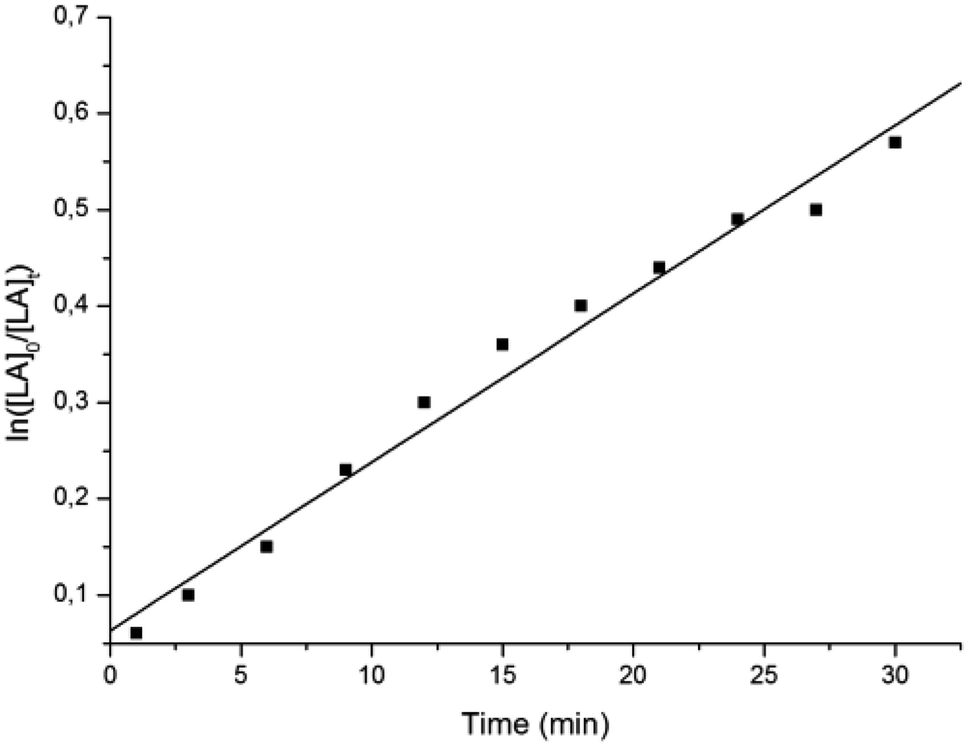 | ||
| Fig. 2 Pseudo-first-order kinetic plot for ROP of L-LA promoted by L2ZnN(SiMe3)2: [Zn] = 2 mM; [L-LA]/[Zn]/[iPrOH] = 200:1:1; T = 25 °C; solvent CH2Cl2. | ||
In summary, the activities of the new Zn complexes in the ROP of rac-LA are poorly sensitive to the ligand structure variations, being in all cases in the range of the best performing Zn catalysts reported in the literature.8,15,21
Polymerization of either L-LA or D-LA by L2ZnN(SiMe3)2 resulted in similar performance to that obtained for rac-LA. (cf. runs 7–9). Homonuclear decoupled 1H NMR spectra of the polymers obtained from rac-LA in runs 3 and 5–7 indicated that atactic PLA's were produced, while highly isotactic PLLA and PDLA were produced from enantiopure monomers (see runs 8 and 9), as expected if no racemization occurs in the reaction. Since there are literature reports of solvent effects on the stereoselectivity in the ROP of rac-LA,51,52 we tested L2ZnN(SiMe3)2 and L3ZnN(SiMe3)2 in THF: while we observed a slower polymerization rate, reasonably owing to the more coordinating solvent, we did not observe any effect on the polymer stereochemistry (see runs 12 and 13). We also tested L3ZnN(SiMe3)2 in toluene: while at 25 °C the polymerization rate was slower than in methylene chloride (200 equiv. of rac-LA were converted in 30 min, see run 14), possibly also owing to monomer poor solubility, at 50 °C L3ZnN(SiMe3)2 converted 500 equivalents of L-LA in 20 min (run 15), while using 1000 equivalents resulted in 90% conversion in the same time (run 16).
Some polymerization tests were also performed at 130 °C without solvent in melt monomer (runs 17–19): L2ZnN(SiMe3)2 converted almost quantitatively 500 equivalents of L-LA in 10 min both without alcohol and in the presence of 1 equiv. of benzyl alcohol, and 800 equivalents in 30 min without alcohol.
SEC analysis of the polymer samples showed a good agreement between the measured and the theoretical molecular weights (see Table 1), indicating a well-controlled polymerization and narrow polydispersities for the runs performed in solution, especially when 1 equiv. of alcohol was added: in fact, using a 200:1:1 monomer:Zn:alcohol ratio, Mn of ca. 30 kDa were measured, as expected (see runs 6–11).
In contrast, polymerizations in neat melt monomer at 130 °C appear to be less controlled: although L1ZnEt converted almost 500 equiv. of LA without added alcohol yielding a Mn of 54 kDa (close to the calculated one, see run 4), L2ZnN(SiMe3)2 afforded significantly lower measured Mn's than theoretical ones, both in the presence and in the absence of alcohol (see runs 17–19).
MALDI-ToF MS analysis of a low MW polymer sample prepared in methylene chloride at 25 °C in the presence of 1 equiv. of iPrOH (see Fig. S6†) showed a major population of linear oligomers of structure H–[OCH(CH3)CO]n–OCH(CH3)2 with peaks separated by 144 Da. On the contrary, MS analysis of a sample produced in the melt at 130 °C (run 18) showed that the low-molecular weight fraction (which is the only fraction that MALDI-ToF can measure) mainly consists of macrocyclic PLA: the formation of the latter has been ascribed53 to backbiting intramolecular transesterification reactions, as confirmed by the presence of odd-membered oligomers, with peaks separated by 72 Da (see Fig. S8†). However, the 1H NMR spectrum (Fig. S7†) of the same sample displays the resonance at δ 4.35 ppm for HOCHMe end groups: the molecular weight calculated from the ratio between the resonance of the methine protons and that of the mentioned end groups is close to the Mn measured by SEC, suggesting that backbiting is only a minor reaction pathway leading to the formation of a little fraction of macrocyclic oligomers.
Ring opening polymerization of ε-caprolactone
Subsequently, the four LxZnN(SiMe3)2 complexes (x = 1–4) were tested in the ROP of ε-caprolactone under the conditions (25 °C, [ε-CL]/[Zn]/[iPrOH] = 200/1/1, 10 min) which had allowed full conversion of LA. At variance with the results of LA polymerizations, the performances of the four catalysts were significantly different (see Table 2): while the complexes bearing the bulkier L1 and L3 ligands afford high monomer conversions, the complex bearing the less bulky L2 was less active. For the latter complex, a polymerization run was performed in toluene at 100 °C with 400 equiv. of CL, resulting in almost complete conversion in 7 min, addressing the effect of the reaction temperature (see run 22, Table 2).| Run | Catalyst | Convb [%] | M n,GPC c [kDa] | M n,theo d [kDa] | M w/Mnc [kDa] |
|---|---|---|---|---|---|
| a Polymerization runs were performed in CH2Cl2 [2 mL] at 25 °C employing 10 μmmol of catalyst with [ε-CL]/[Zn]/[iPrOH] = 200/1/1, time 10 min. b Conversion of ε-CL as determined by 1H NMR spectral data. c Experimental Mn [in g mol−1] and Mw/Mn values determined by SEC in THF against polystyrene standards, using the correction factor 0.56. d Calculated Mn of PCL (in g mol−1) = 114.14 × ([CL]/[Zn]) × conversion of CL. e Polymerization run was performed in toluene at 100 °C with [ε-CL]/[Zn]/[iPrOH] = 400/1/1, time 7 min. f Polymerization time 60 min. | |||||
| 20 | L1ZnN(SiMe3)2 | 94 | 30.6 | 21.5 | 1.11 |
| 21 | L2ZnN(SiMe3)2 | 35 | 11.4 | 8.0 | 1.13 |
| 22e | L2ZnN(SiMe3)2 | 99 | 39.3 | 45.2 | 1.32 |
| 23 | L3ZnN(SiMe3)2 | 81 | 28.2 | 18.5 | 1.16 |
| 24 | L4ZnN(SiMe3)2 | 2 | — | — | — |
| 25 | L4ZnN(SiMe3)2 | 83f | 17.5 | 19.0 | 1.03 |
Finally, the complex bearing the less bulky and electron-withdrawing L4 was only marginally active under mild conditions and short reaction times. However, using a longer reaction time (60 min) 80% monomer conversion was achieved, pointing to a possible induction period. As a matter of fact, as previously reported,50L4ZnN(SiMe3)2 is involved in a slow Schlenk-type equilibrium between the heteroleptic and the homoleptic bis(chelate) complexes: it is possible that the position of the equilibrium is affected by the coordination ability of the monomer.
Block copolymerization of L-LA, D-LA, rac-LA and CL by sequential monomer addition
In view of the controlled nature and high rate of the polymerization promoted by our new pyridylamido Zn(II) catalysts, we targeted the synthesis of stereoblock copolymers by sequential addition of L-LA, D-LA and rac-LA (see Scheme 2).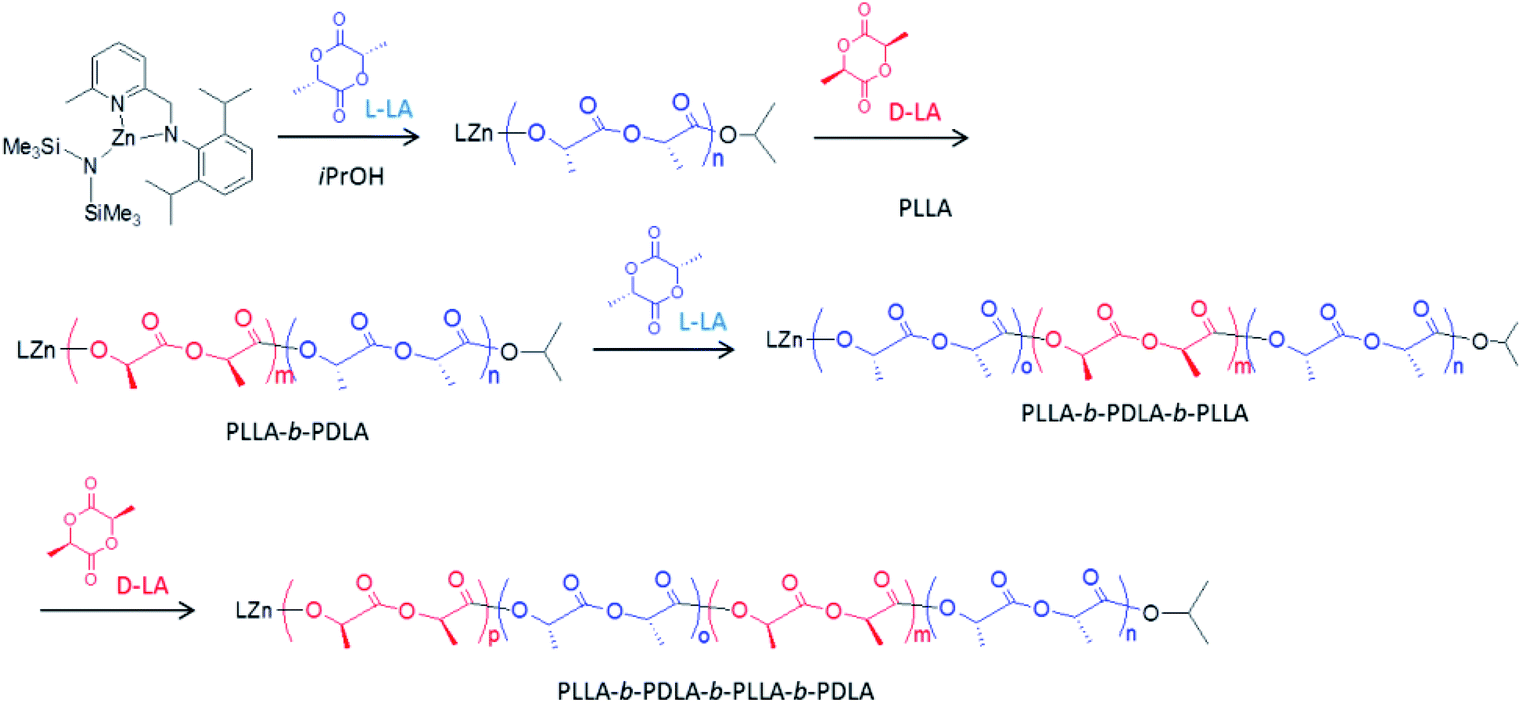 | ||
| Scheme 2 One-pot synthesis of stereoblock PLA's. | ||
Thus, a diblock copolymer was synthesized by allowing to react 10 μmol of L2ZnN(SiMe3)2, 1 equiv. of isopropanol and 100 equiv. of L-LA at 25 °C in CH2Cl2 for 20 min, and then adding 100 equiv. of D-LA continuing the polymerization for further 20 min (see Table 3, run 26). 1H and 13C NMR analysis confirmed the production of a highly isotactic copolymer, according to literature assignments25,39 (Pm > 98%, see Fig. 3 and 4).
| Run | Copolymer (theoretical block length) | Timeb [min] | M n c [kDa] | M w/Mnc | T m d [°C] | ΔHmd [Jg−1] |
|---|---|---|---|---|---|---|
| a Polymerizations performed in CH2Cl2 [2 mL] at 25 °C employing 10 μmol of [L2ZnN(SiMe3)2] and 1 equiv. of iPrOH. Full conversion was confirmed by 1H NMR. b 20–30 min was maintained between each monomer addition, depending on the monomer amount and length of polymer chain. c Experimental Mn [in g mol−1] and Mw/Mn values determined by SEC in THF against polystyrene standards, using the correction factor 0.58 for lactide and 0.56 for caprolactone. d The Tm and ΔHm was the melting temperatures and enthalpies during the first heating under a heating rate of 10 °C min−1. | ||||||
| 26 | PLLA-b-PDLA [100-b-100] | 40 | 18.8 | 1.20 | 209 | 58.1 |
| 27 | PLLA-b-PDLA [200-b-200] | 60 | 41.1 | 1.16 | 215 | 51.3 |
| 28 | PLLA-b-PDLA-b-PLLA [100-b-100-b-100] | 60 | 36.8 | 1.11 | 199 | 43.4 |
| 29 | PLLA-b-PDLLA-b-PDLA [100-b-100-b-100] | 60 | 34.4 | 1.20 | 205 | 42.3 |
| 30 | PLLA-b-PDLA-b-PLLA-b-PDLA [100-b-100-b-100-b-100] | 80 | 37.8 | 1.22 | 191 | 15.8 |
| 31 | PCL-b-PLLA [100-b-100] | 40 | 27.5 | 1.39 | 56[PCL] | 19.6[PCL] |
| 175[PLA] | 33.3[PLA] | |||||
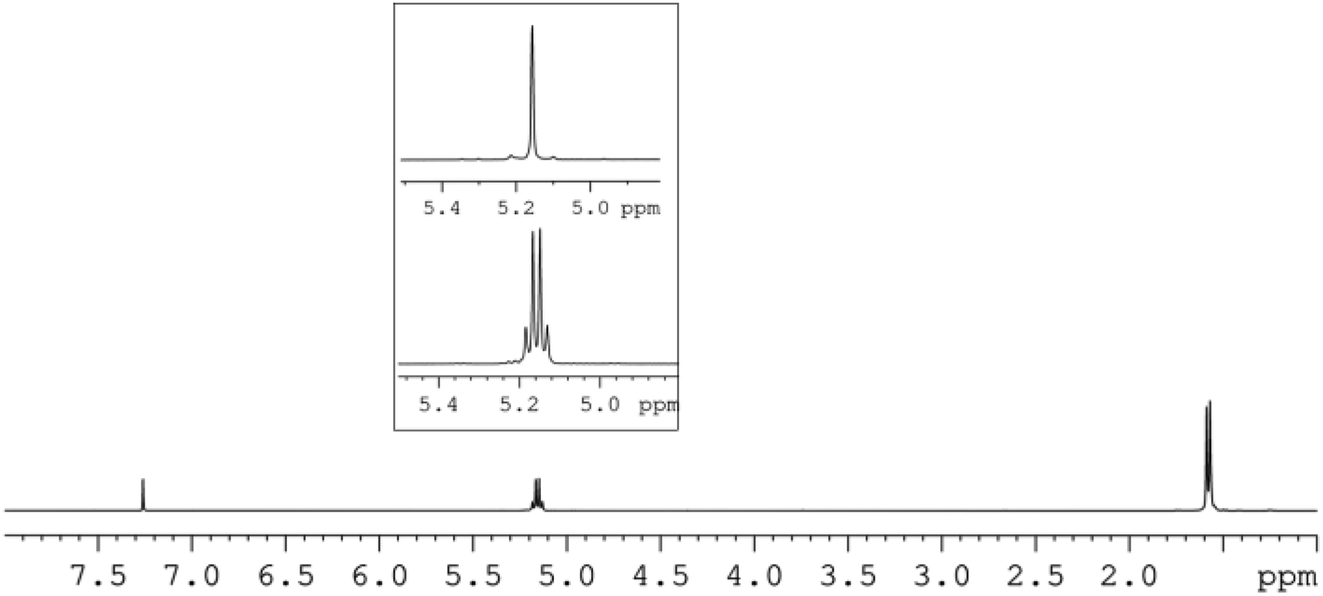 | ||
| Fig. 3 1H-NMR and HD-NMR (square) (CDCl3, 600 MHz) of L100-b-D100 diblock PLA copolymer sample (run 26, Table 3). | ||
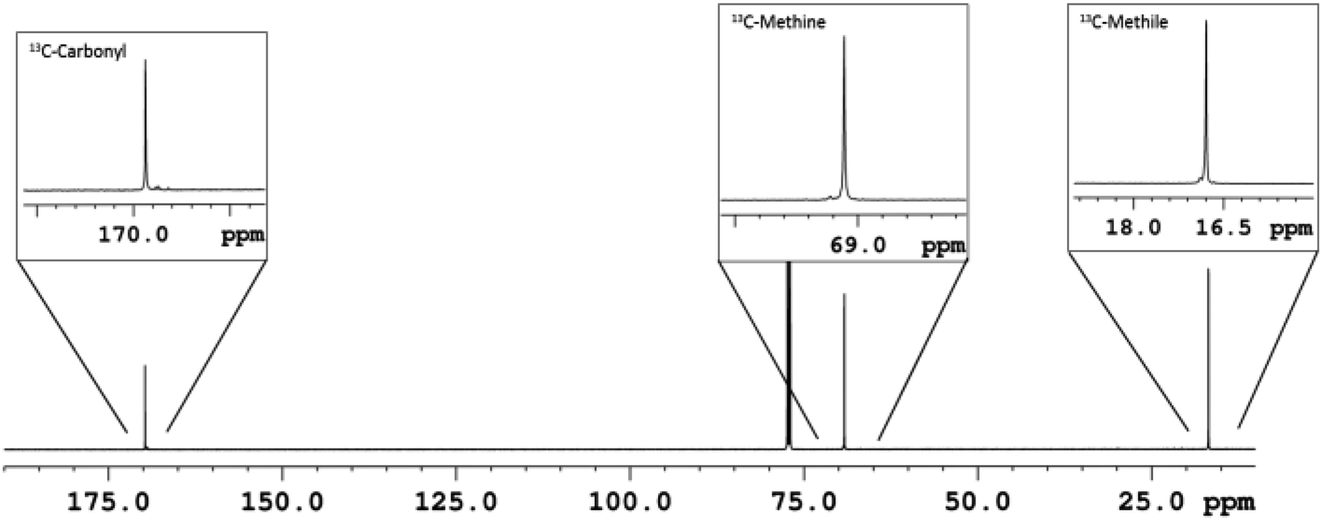 | ||
| Fig. 4 13C-NMR (CDCl3, 150 MHz) of L100-b-D100 diblock PLA copolymer sample (run 26, Table 3). | ||
SEC analysis performed after the formation of each block confirmed the synthesis of a diblock copolymer (see Fig. 5), although, as noticed by a reviewer, some tailing effects suggest contamination by some low-MW minor fractions, possibly due to the presence of protic impurities in the reaction medium.
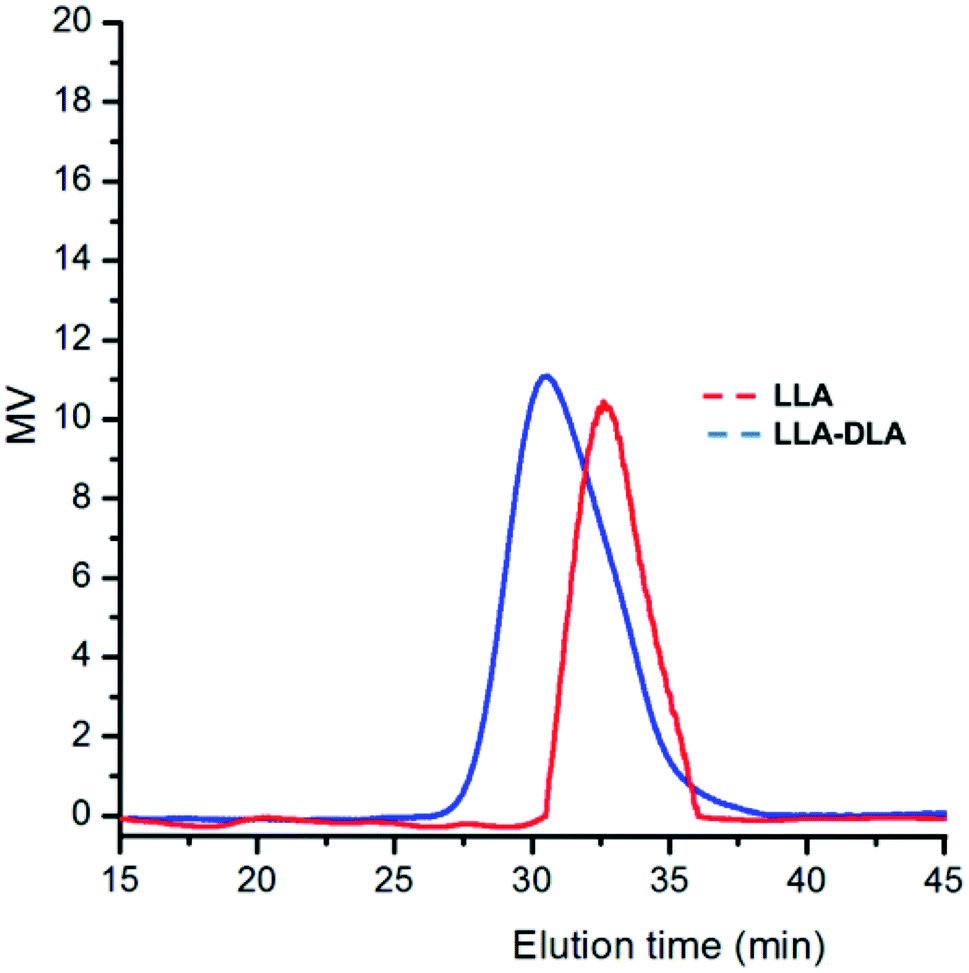 | ||
| Fig. 5 SEC profiles for diblock poly(LLA-b-DLA) in run 26 of Table 3. | ||
Finally, DSC analysis indicated that the stereodiblock copolymer crystallizes in the stereocomplex form, with a Tm = 209 °C in the first heating, followed by recrystallization and a Tm = 202 °C in the second heating run (see Fig. S10–S11†).
A diblock poly(LLA-b-DLA) having longer L-LA and D-LA sequences was synthesised similarly by sequential addition of 200 equivalents of the monomers over a 1 h total reaction time (Table 3, run 27).
A triblock poly(LLA-b-DLA-b-LLA) was prepared similarly (Table 3, run 28) by sequential additions of 100 equiv. of the proper monomer in each step over 20 min-each intervals, in order to ensure full conversion. NMR analysis again showed a stereoregular blocks composition, SEC analysis confirmed the increase of MW over each monomer addition (see Fig. S15†), and DSC analysis confirmed the production of a stereocomplex phase (Tm = 199 °C). The lower melting temperature and melting enthalpy observed with respect to the diblock copolymer can be ascribed to the inequivalent amounts of the sequences of L-LA and D-LA, the latter being one half of the former.
A triblock poly(LLA-b-DLLA-b-DLA), containing two crystalline end blocks of isotactic PLA's with opposite chirality and an amorphous midblock of atactic PLA, was also prepared (Table 3, run 29) by a similar procedure.38 A stereocomplex phase of the two crystalline end blocks was evidenced by DSC analysis (Tm = 205 °C). Interestingly, the melting temperature of this sample is higher than that of the previous one, probably because here there are equivalent amounts of the sequences of L-LA and D-LA, spanned by an amorphous block of rac-lactide. NMR analysis confirms the presence of both the isotactic and the atactic blocks (see Fig. S16†).
A stereotetrablock PLLA-b-PDLA-b-PLLA-b-PDLA was also prepared with the same procedure (Table 3, run 30): also in this case the stereocomplex phase was produced (Tm = 191 °C). The lower melting temperature and melting enthalpy are ascribed to the fact that the stereoregular block integrity was only partially preserved, as indicated by NMR analysis (Pm = 0.9), owing to incomplete monomer conversion during the subsequent steps.
Block copolymerization of LA and CL was also explored. Thus, a run was performed using L2ZnN(SiMe3)2, 1 equiv. of iPrOH and 100 equiv. of CL at 25 °C: full conversion was achieved after 20 min and then 100 equiv. of L-LA were added and the reaction was prolonged for additional 20 min, affording a diblock PCL–PLA copolymer (see run 31 of Table 3). NMR, DSC and SEC analyses confirmed the formation of the diblock PCL–PLA copolymer (see Fig. S21–S24†).
We also explored the feasibility of the reversed sequence addition of the two monomers: however, after the conversion of 200 equiv. of LA under the same reaction conditions, the added CL was not polymerized at all. Accordingly, a polymerization performed under similar conditions, but adding the two monomers at the beginning of the run resulted in the production of PLA homopolymer, with CL remaining unreacted. The importance of the order of monomer addition was addressed in an early work on the matter28 and the difficulty to achieve polymerization of CL after LA was observed frequently,18,37–41 although we30 and Ma et al.52 have reported catalysts able to produce diblock PLA–PCL copolymers via the “PLA first” route. No CL polymerization was observed in the presence of LA even in toluene at 50 °C, while a LA–CL copolymer was obtained at 130 °C in neat monomers: 1H and 13C NMR analysis of the latter sample indicated that a pseudorandom copolymer was produced owing to extensive transesterifications as indicated by the presence of single lactic acid monomer units (13C resonance of the carbonyl at δ = 171 ppm (see Fig. S25 and S26†)).
Conclusions
We have introduced a family of three-coordinated Zn(II) complexes bearing chelating pyridylamido ligands with variable steric and electronic features: these complexes have been tested for the ROP of lactide and ε-caprolactone, resulting in fast and controlled polymerizations. The latter performances allowed the synthesis of precise stereodiblock, stereotriblock and stereotetrablock copolymers of L-LA, D-LA and rac-LA by one-pot sequential addition of the monomers under mild conditions and in short reaction times. In the same way, diblock copolymers of ε-caprolactone and lactide were produced. The reported complexes are the first catalysts based on non-toxic and environmentally benign Zn complexes able to produce efficiently “tailor-made” block copolymers by the one-pot sequential addition strategy.37–41 Future work will be focused on the physical properties of new polymeric materials produced by this strategy, with a particular attention to the synthesis of ABA triblock copolymers behaving as biodegradable thermoplastic elastomers.22,32–36Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by Fondazione Cariplo (Milano, Italy), Project “APOLLO”, CUP B42F17000080005 and by the University of Salerno (FARB). The authors acknowledge technical assistance by Dr Patrizia Oliva for NMR analyses, Dr Patrizia Iannece for MS analyses, and Dr Mariagrazia Napoli for SEC measurements.References
- See e.g. D. K. Schneiderman and M. A. Hillmyer, Macromolecules, 2017, 50, 3733–3749 CrossRef CAS.
- For a recent review of the matter, see, e.g. S. Karbalaei, P. Hanachi, T. R. Walker and M. Cole, Environ. Sci. Pollut. Res., 2018, 25, 36046–36063 CrossRef CAS.
- See e.g. X. Zhang, M. Fevre, O. J. Jones and R. M. Waymouth, Chem. Rev., 2018, 118, 839–885 CrossRef CAS.
- M. J.-L. Tschan, E. Brulé, P. Haquette and C. M. Thomas, Polym. Chem., 2012, 3, 836 RSC.
- S. Saeidlou, M. A. Huneault, H. Li and C. B. Park, Prog. Polym. Sci., 2012, 37, 1657–1677 CrossRef CAS.
- E. Castro-Aguirre, F. Iniguez-Franco, H. Samsudin, X. Fang and R. Auras, Adv. Drug Delivery Rev., 2016, 107, 333–366 CrossRef CAS.
- M. Cheng, A. B. Attygalle, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 1999, 121, 11583–11584 CrossRef CAS.
- B. M. Chamberlain, M. Cheng, D. R. Moore, T. M. Ovitt, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2001, 123, 3229–3238 CrossRef CAS.
- A. Dove, V. Gibson, E. Marshall, A. White and D. Williams, Dalton Trans., 2004, 570–578 RSC.
- N. Ayala, M. H. Chisholm, J. C. Gallucci and C. Krempner, Dalton Trans., 2009, 9237 RSC.
- M. Keram and H. Ma, Appl. Organomet. Chem., 2017, 31, e3893 CrossRef.
- M. H. Chisholm, N. W. Eilerts, J. C. Huffman, S. S. Iyer, M. Pacold and K. Phomphrai, J. Am. Chem. Soc., 2000, 122, 11845–11854 CrossRef CAS.
- C. Fliedel, S. Mameri, S. Dagorne and T. Aviles, Appl. Organomet. Chem., 2014, 28, 504–511 CrossRef CAS.
- C. Fliedel, D. Vila-Vicosa, M. J. Calhorda, S. Dagorne and T. Aviles, ChemCatChem, 2014, 6, 1357–1367 CrossRef CAS.
- C. K. Williams, L. E. Breyfogle, S. K. Choi, W. Nam, V. G. Young Jr, M. A. Hillmyer and W. B. Tolman, J. Am. Chem. Soc., 2003, 125, 11350–11359 CrossRef CAS.
- V. Poirier, T. Roisnel, J.-F. Carpentier and Y. Sarazin, Dalton Trans., 2009, 9820–9827 RSC.
- L. Wang and H. Ma, Macromolecules, 2010, 43, 6535–6537 CrossRef CAS.
- A. Pilone, M. Lamberti, M. Mazzeo, S. Milione and C. Pellecchia, Dalton Trans., 2013, 42, 13036–13047 RSC.
- C. Fliedel, V. Rosa, F. M. Alves, A. M. Martins, T. Aviles and S. Dagorne, Dalton Trans., 2015, 44, 12376–12387 RSC.
- A. Pilone, N. De Maio, K. Press, V. Venditto, D. Pappalardo, M. Mazzeo, C. Pellecchia, M. Kol and M. Lamberti, Dalton Trans., 2015, 44, 2157–2165 RSC.
- T. Rosen, Y. Popowski, I. Goldberg and M. Kol, Chem.–Eur. J., 2016, 22, 11533–11536 CrossRef CAS.
- M. A. Hillmyer and W. B. Tolman, Acc. Chem. Res., 2014, 47, 2390–2396 CrossRef CAS.
- Y. Ikada, K. Jamshidi, H. Tsuji and S. H. Hyon, Macromolecules, 1987, 20, 904–906 CrossRef CAS.
- N. Spassky, M. Wisniewski, C. Pluta and A. Le Borgne, Macromol. Chem. Phys., 1996, 197, 2627–2637 CrossRef CAS.
- T. M. Ovitt and G. W. Coates, J. Am. Chem. Soc., 2002, 124, 1316–1326 CrossRef CAS.
- Z. Zhong, P. J. Dijkstra and J. Feijen, J. Am. Chem. Soc., 2003, 125, 11291–11298 CrossRef CAS PubMed.
- N. Nomura, R. Ishii, Y. Yamamoto and T. Kondo, Chem.–Eur. J., 2007, 13, 4433–4451 CrossRef CAS.
- Jacobs, P. Dubois, R. Jerome and P. Teyssie, Macromolecules, 1991, 24, 3027–3034 CrossRef CAS.
- A. Amgoune, C. M. Thomas, T. Roisnel and J.-F. Carpentier, Chem.–Eur. J., 2006, 12, 169–179 CrossRef CAS.
- D. Pappalardo, L. Annunziata and C. Pellecchia, Macromolecules, 2009, 42, 6056–6062 CrossRef CAS.
- T. Ebrahimi, D. C. Aluthge, B. O. Patrick, S. G. Hatzikiriakos and P. Mehrkhodavandi, ACS Catal., 2017, 7, 6413–6418 CrossRef CAS.
- M. Ryner and A.-C. Albertsson, Biomacromolecules, 2002, 3, 601–608 CrossRef CAS.
- Z. Zhang, D. W. Grijpma and J. Feijen, Macromol. Chem. Phys., 2004, 205, 867–875 CrossRef CAS.
- J. O. Lin, W. Chen, Z. Shen and J. Ling, Macromolecules, 2013, 46, 7769–7776 CrossRef CAS.
- Y. Nakayama, K. Aihara, H. Yamanishi, H. Fukuoka, R. Tanaka, Z. Cai and T. Shiono, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 489–495 CrossRef CAS.
- K. Schneiderman, E. M. Hill, M. T. Martello and M. A. Hillmyer, Polym. Chem., 2015, 6, 3641–3651 RSC.
- N. Othman, C. Xu, P. Mehrkhodavandi and S. G. Hatzikiriakos, Polymer, 2012, 53, 2442–2452 CrossRef.
- C. Aluthge, C. Xu, N. Othman, N. Noroozi, S. G. Hatzikiriakos and P. Mehrkhodavandi, Macromolecules, 2013, 46, 3965–3974 CrossRef.
- T. Rosen, I. Goldberg, V. Venditto and M. Kol, J. Am. Chem. Soc., 2016, 138, 12041–12044 CrossRef CAS.
- T. Rosen, I. Goldberg, W. Navarra, V. Venditto and M. Kol, Chem. Sci., 2017, 8, 5476–5481 RSC.
- T. Rosen, I. Goldberg, W. Navarra, V. Venditto and M. Kol, Angew. Chem., 2018, 130, 7309–7313 CrossRef.
- L. Annunziata, D. Pappalardo, C. Tedesco and C. Pellecchia, Macromolecules, 2009, 42, 5572–5578 CrossRef CAS.
- G. Li, M. Lamberti, S. D'Amora and C. Pellecchia, Macromolecules, 2010, 43, 8887–8891 CrossRef CAS.
- L. Annunziata, S. Pragliola, D. Pappalardo, C. Tedesco and C. Pellecchia, Macromolecules, 2011, 44, 1934–1941 CrossRef CAS.
- G. Li, C. Zuccaccia, C. Tedesco, I. D'Auria, A. Macchioni and C. Pellecchia, Chem.–Eur. J., 2014, 20, 232–244 CrossRef CAS PubMed.
- I. D'Auria, S. Milione, T. Caruso, G. Balducci and C. Pellecchia, Polym. Chem., 2017, 8, 6443–6454 RSC.
- G. Li, M. Lamberti, M. Mazzeo, D. Pappalardo, G. Roviello and C. Pellecchia, Organometallics, 2012, 31, 1180–1188 CrossRef CAS.
- G. Li, M. Lamberti, D. Pappalardo and C. Pellecchia, Macromolecules, 2012, 45, 8614–8620 CrossRef CAS.
- I. D'Auria, C. Tedesco, M. Mazzeo and C. Pellecchia, Dalton Trans., 2017, 46, 12217–12225 RSC.
- I. D'Auria, M. C. D'Alterio, G. Talarico and C. Pellecchia, Macromolecules, 2018, 51, 9871–9877 CrossRef.
- M. Crisholm, K. Choojun, J. Gallucci and P. Wambua, Chem. Sci., 2012, 3, 3445–3457 RSC.
- M. Keram and H. Ma, Appl. Organomet. Chem., 2017, 31, e3893 CrossRef.
- See e.g. F. Bonnet, F. Stoffelbach, G. Fontained and S. Bourbigot, RSC Adv., 2015, 5, 31303–31310 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1916723 and 1916724. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra07133d |
| This journal is © The Royal Society of Chemistry 2019 |