
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOne-step scalable synthesis of honeycomb-like g-C3N4 with broad sub-bandgap absorption for superior visible-light-driven photocatalytic hydrogen evolution
Shanshan
Li
,
Chun
Hu
 ,
Yannan
Peng
and
Zhihong
Chen
*
,
Yannan
Peng
and
Zhihong
Chen
*
Key Laboratory for Water Quality and Conservation of the Pearl River Delta, Ministry of Education, Institute of Environmental Research at Greater Bay, Guangzhou University, Guangzhou 510006, China. E-mail: chenzhihong@gzhu.edu.cn
First published on 14th October 2019
Abstract
Integration of a nanostructure design with a sub-bandgap has shown great promise in enhancing the photocatalytic H2 production activity of g-C3N4via facilitating the separation of photogenerated charges while simultaneously increasing the active sites and light harvesting ability. However, the development of a synthetic route to generate an ordered g-C3N4 structure with remarkable sub-bandgap absorption via a scalable and economic approach is challenging. Herein, we report the preparation of a honeycomb-like structured g-C3N4 with broad oxygen sub-bandgap absorption (denoted as HOCN) via a scalable one-pot copolymerization process using oxamide as the modelling agent and oxygen doping source. The morphology and sub-bandgap position can be tailored by controlling the oxamide to dicyandiamide ratio. All HOCN samples exhibit remarkable enhancement of photocatalytic H2 production activity due to the synergistic effect between the sub-bandgap and honeycomb structure, which results in strong light absorption extending up to 1000 nm, fast separation of photogenerated charge carriers, and rich photocatalytic reaction sites. In particular, HOCN4 exhibits a remarkable photocatalytic H2 production rate of 1140 μmol h−1 g−1 under visible light irradiation (>420 nm), which is more than 13.9 times faster than the production rate of pristine g-C3N4. Moreover, even under longer wavelength light irradiation (i.e., >500 and >800 nm), HOCN4 still exhibits a high H2 production rate of 477 and 91 μmol h−1 g−1, respectively. In addition, HOCN4 possesses an apparent quantum yield (AQY) of 4.32% at 420 nm and 0.12% at 800 nm. These results confirm that the proposed synthesis strategy allow for scalable production of g-C3N4 with an ordered nanostructure via electronic modulation, which is beneficial for its practical application in photocatalytic H2 production.
Introduction
With the increasing energy crisis and environmental pollution, hydrogen energy, a promising renewable alternative to traditional fossil fuels, has attracted much attention, in particular hydrogen production from photocatalytic water splitting.1 As a metal-free polymer semiconductor with high thermal and chemical stability, and beneficial electronic structure, graphitic carbon nitride (g-C3N4) has drawn considerable research interest for photocatalytic H2 production from water splitting since its discovery.2,3 However, the photocatalytic efficiency of pristine g-C3N4 is substantially suppressed by fast photogenerated electron–hole pair recombination, limited photocatalytic activity and insufficient response to visible light irradiation. During the past decade, various strategies have been developed to enhance the photocatalytic H2 production activity of g-C3N4 including nanostructure design,4–10 doping with heteroatoms,11–14 copolymerization,15,16 crystal control,17 creating a sub-bandgap18–21 and constructing heterojunctions.22–28 However, a single strategy approach cannot simultaneously overcome all the intrinsic shortcomings of g-C3N4. Therefore, our approach involves the incorporation of these strategies to further optimize the photocatalytic H2 production activity.Recently, integration of nanostructure design with a sub-bandgap has provided an efficient route for facilitating the separation of photogenerated electron–hole pairs while simultaneously increasing the number of active sites as well as the light harvesting of g-C3N4.29–37 Additionally, a synergistic effect between nanostructure design and sub-bandgap creation endowed the obtained g-C3N4 with remarkably improved photocatalytic H2 production activity. Tao et al. reported g-C3N4 nanoflakes with a phosphorus sub-bandgap that was prepared by post-calcination treatment of buck g-C3N4 with a phosphorus sub-bandgap and exhibited a strong sub-bandgap absorption over the entire visible region, enlarged surface area and improved charge separation and transfer efficiency. These properties led to remarkably enhanced visible-light photocatalytic H2 production.36 Yi et al. reported a combinative strategy involving hydrothermal pre-treatment and calcination to synthesize mesoporous g-C3N4 with an oxygen and sulfur sub-bandgap for outstanding photocatalytic hydrogen evolution.37 Apparently, the formation of an ordered structure along with the creation of a sub-bandgap is an effective route to further increase the photocatalytic activity of g-C3N4. However, an additional post- or pre-treatment procedure and salt addition are time consuming, expensive and complex operation processes that allow for facile heteroatom introduction, which severely limits the scalable synthesis of g-C3N4 with an ordered nanostructure and sub-bandgap modulation. To the best of our knowledge, the development of a scalable and simple approach for the synthesis of g-C3N4 with an ordered structure and sub-bandgap is challenge.
Haung et al. reported that the replacement of O atoms for lattice N atoms via the formation of C–O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds generated an acceptor level below the bottom of the conduction band, resulting in an oxygen sub-bandgap between the valence band and oxygen acceptor.38 Inspired by previous studies, honeycomb-like g-C3N4 with an oxygen sub-bandgap (denoted as HOCN) was fabricated using a scalable one-pot copolymerization method using oxamide as a modelling agent and oxygen doping source. The morphology and sub-bandgap position can be tuned by controlling the oxamide to dicyandiamide ratio. The HOCN samples exhibit a strong light absorption region up to 1000 nm due to the narrowed sub-bandgap, which is composed of a valence band (VB) and oxygen doping states. The HOCNs exhibit a honeycomb-like structure with a high surface area of 185.6 m2 g−1, leading to efficient charge transfer and separation. Therefore, all HOCN samples exhibit enhanced photocatalytic H2 evolution rates under visible light irradiation (λ > 420 nm). In particular, HOCN4 produced a remarkable enhanced photocatalytic H2 production rate of 1140 μmol h−1 g−1 under visible light irradiation, which is much higher than that of pure g-C3N4 (CN, 82 μmol h−1 g−1). Further, HOCN4 exhibits a high H2 production rate of 477 and 91 μmol h−1 g−1 under light illumination with longer wavelengths (i.e., λ > 500 nm and λ > 800 nm, respectively).
C bonds generated an acceptor level below the bottom of the conduction band, resulting in an oxygen sub-bandgap between the valence band and oxygen acceptor.38 Inspired by previous studies, honeycomb-like g-C3N4 with an oxygen sub-bandgap (denoted as HOCN) was fabricated using a scalable one-pot copolymerization method using oxamide as a modelling agent and oxygen doping source. The morphology and sub-bandgap position can be tuned by controlling the oxamide to dicyandiamide ratio. The HOCN samples exhibit a strong light absorption region up to 1000 nm due to the narrowed sub-bandgap, which is composed of a valence band (VB) and oxygen doping states. The HOCNs exhibit a honeycomb-like structure with a high surface area of 185.6 m2 g−1, leading to efficient charge transfer and separation. Therefore, all HOCN samples exhibit enhanced photocatalytic H2 evolution rates under visible light irradiation (λ > 420 nm). In particular, HOCN4 produced a remarkable enhanced photocatalytic H2 production rate of 1140 μmol h−1 g−1 under visible light irradiation, which is much higher than that of pure g-C3N4 (CN, 82 μmol h−1 g−1). Further, HOCN4 exhibits a high H2 production rate of 477 and 91 μmol h−1 g−1 under light illumination with longer wavelengths (i.e., λ > 500 nm and λ > 800 nm, respectively).
Experimental section
Synthesis
The two precursors, which consisted of oxamide (OA, Sinopharm Chemical Reagent Co., Ltd., 98%) and dicyandiamide (DCDA, Sinopharm Chemical Reagent Co., Ltd., 99%), were of analytic grade and used without further purification. The melting point of oxamide is 300 °C, and oxamide will gradually volatilize above this temperature. Therefore, a pre-polymerization temperature and time for dicyandiamide and oxamide of 300 °C and 1 hour, respectively, were chosen. The detailed preparation method was as follows: dicyandiamide (2 g) and a certain amount of oxamide were uniformly mixed and thoroughly ground in a mortar. The mixture was placed into a muffle furnace and heated to 300 °C for 1 hour using a heating rate of 1.7 °C min−1 followed by heating to 520 °C for 2 hours with a heating rate of 5 °C min−1. After cooling to room temperature, the samples were ground and collected. The samples with oxamide to dicyandiamide molar ratios of 1%, 2%, 3%, 4% and 5% are referred to as HOCN1, HOCN2, HOCN3, HOCN4 and HOCN5, respectively. In comparison, pristine carbon nitride (CN) was synthesized using same thermal polymerization method with dicyandiamide (2 g) as the precursor.Characterization
The chemical state of the compound was analysed by X-ray photoelectron spectroscopy (XPS) using 150 W Al Kα X-ray irradiation (Thermo ESCALAB 250). The microstructures were analysed using scanning electron microscopy (SEM, HITACHI S-4800) and transmission electron microscopy (TEM, JEM-2100 JEOL, Japan). Direct measurements of the UV-vis diffuse reflectance spectra (DRS) of the samples were performed with a UV-vis spectrophotometer (Hitachi UH4150). The phase structures of the as-prepared samples were characterized by X-ray diffraction (XRD) on a Bruker instrument with Cu Kα radiation. The Fourier-transform infrared (FTIR) spectra were recorded on a Thermo spectrometer in a frequency range of 4000–500 cm−1. The specific surface areas of the samples were determined by BET analysis of nitrogen adsorption–desorption experiments (Micromeritics ASAP 2460). The fluorescence (FL) spectra were recorded on a Hitachi F-7000 fluorescence spectrophotometer. The electron paramagnetic resonance (EPR) spectra were obtained at room temperature using a Bruker A300-10/12 EPR spectrometer.Photocatalytic hydrogen evolution test
The photocatalytic hydrogen production experiment under visible light irradiation was conducted in a Pyrex top–irradiation reaction vessel. A 300 W xenon lamp (CEL-HXF300, perfectlight, Beijing) was used as the light source. A 420 nm cutoff filter was chosen as the visible light source. The catalyst was mixed with a H2PtCl6 aqueous solution for ultraviolet radiation treatment to provide supporting platinum on the catalyst as an auxiliary catalyst. Then, 50 mg of the photocatalyst was dispersed in 100 mL of ultrapure water along with a sacrificial agent (i.e., 10 mL of triethanolamine). Nitrogen was pumped into the reactor for 30 min to remove the dissolved oxygen prior to the experiment. During the entire reaction process, the temperature of the reaction system was maintained at 5 °C by the flow of cooling water. 1 mL of gas was collected and injected for gas chromatography analysis (Techcomp GC7900, equipped with a TCD and 5 Å molecular sieve columns). The AQY was calculated according to the following formula: AQY (%) = number of evolved H2 molecules × 2 × 100/number of incident photons.Results and discussion
The photocatalytic performance of the CN and HOCN samples with the addition of different amounts of oxamide was evaluated based on the photocatalytic evolution rate (HER) under visible light irradiation (λ > 420 nm) with 1 wt% Pt as the cocatalyst, which was photo-deposited prior to photocatalytic hydrogen evolution test. The results are shown in Fig. 1. The HOCN samples exhibit enhanced photocatalytic H2 evolution rates that are 4.5–13.9 times higher than that of pristine carbon nitride (82 μmol h−1 g−1), suggesting that the introduction of oxamide in the thermal polymerization process of dicyandiamide is a feasible method for improving the photocatalytic activity of CN. In particular, the HER of HOCN4 was 1140 μmol h−1 g−1, which is approximately 13.9 times that of pristine g-C3N4. Notably, a decrease in the HER rate was observed when the oxamide to dicyandiamide molar ratio exceeded 4%, which may be due to the increase in oxamide that would produce recombination sites on the surface of g-C3N4. Moreover, to gain additional insight into the photocatalytic response in the long wavelength region, H2 production experiments were carried out using a cutoff filter that allows for λ > 500 and > 800 nm. As shown in Fig. 1B, under irradiation with long wavelengths of light (i.e., > 500 and > 800 nm), HOCN4 exhibited a high H2 production rate of 477 and 91 μmol h−1 g−1, respectively. However, pristine CN did not exhibit any photocatalytic H2 production activity at these longer wavelengths. The apparent quantum yield (AQY) of H2 production was consistent with the characteristic absorption of HOCN4. As shown in Fig. 1C, HOCN4 possesses a high AQY of 4.32%, 0.69% and 0.12% at 420, 600 and 800 nm, respectively. In contrast, CN exhibits no AQY at 600 and 800 nm due to the lack of light absorption at these wavelengths. These results confirm that the introduction of oxamide in the synthesis process is a feasible method for enhancing the photocatalytic activity of CN, especially in the long wavelength region. The photocatalytic stability and reusability of the optimal HOCN4 photocatalyst was evaluated by four consecutive tests, and the entire reactor was evacuated every 4 h, as shown in Fig. 1D. During the four cycles, the HER rate was rather stable and centred at approximately 1100 μmol h−1 g−1, indicating that introduction of oxamide does not affect the stability of CN.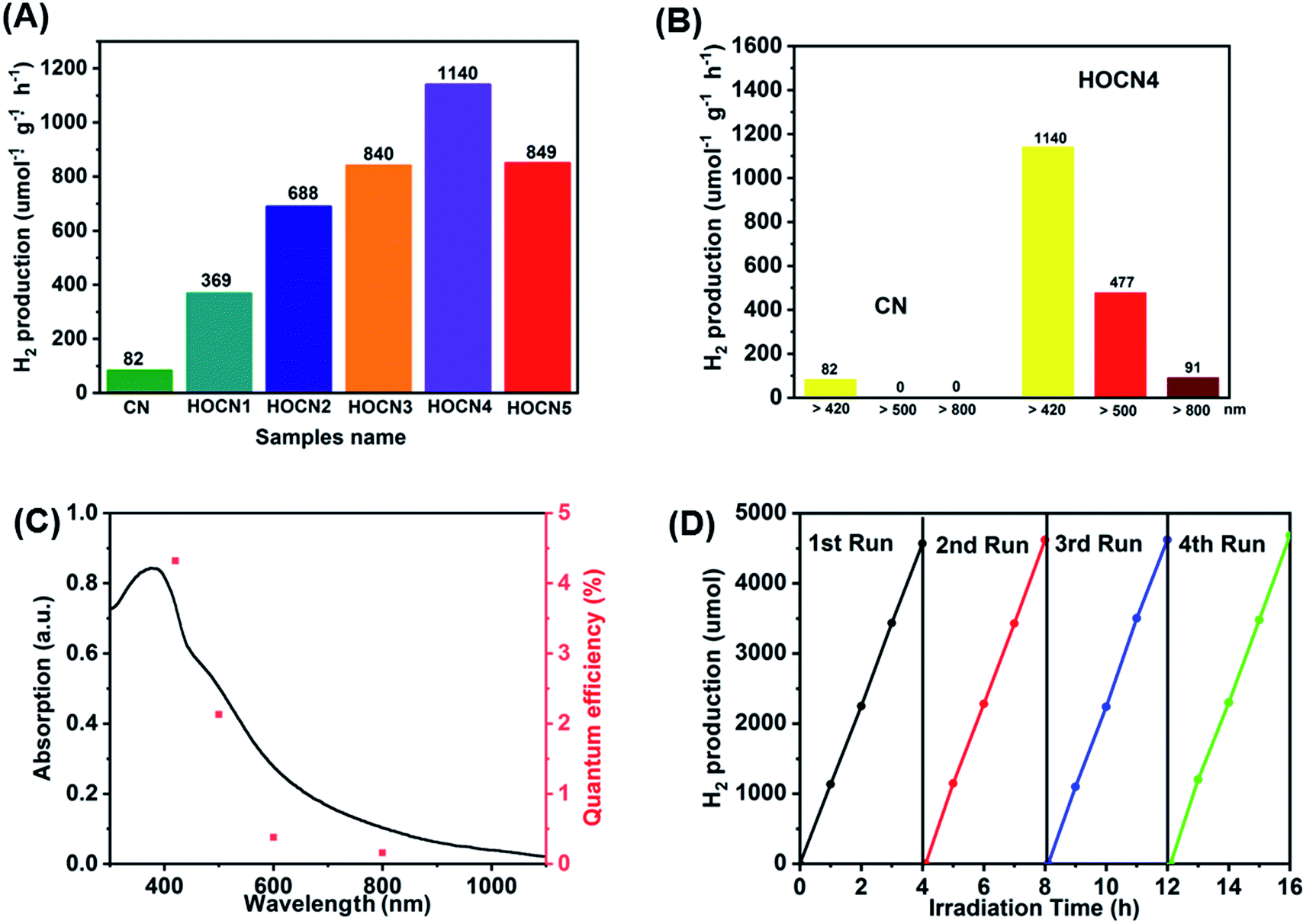 | ||
| Fig. 1 (A) Photocatalytic H2 evolution under visible light irradiation (λ > 420 nm) of CN and HOCNX (X = 1–5) samples with 1wt% Pt as cocatalyst (photo-deposited prior to photocatalytic hydrogen evolution test); (B) Photocatalytic H2 evolution under different light irradiation (λ > 420 nm; λ > 500 nm; λ > 800 nm) of CN and HOCN4; (C) UV-vis diffuse-reflectance spectrum and wavelength-dependent AQY of HOCN4. (D) The stability test for four cycling H2 evolution of HOCN4 under visible light irradiation. | ||
The formation of a honeycomb-like structure was observed and confirmed via SEM and TEM analyses. As shown in Fig. 2A and B, in the absence of oxamide, CN exhibits the typical morphology with aggregated and stacked layers. However, with the addition of oxamide, HOCN exhibits a honeycomb-like morphology, which is accompanied by the formation of many irregular pores (Fig. 2C and D). Further observation of the TEM images indicates that HOCN consists of dense thick nanosheets with irregular pore sizes on its surface (Fig. 2E). However, the surface of CN is smooth with no nanopores (Fig. 2F). The specific surface area of CN and HOCN was further investigated based on their nitrogen adsorption and desorption isotherms. As shown in Fig. 2G, the BET surface area of HOCN4 is approximately 185.8 m2 g−1, which is approximately 12.8 times higher than that of pristine CN (14.6 m2 g−1). As shown in the inset of Fig. 2G, the pore size of HOCN4 is 60–110 nm and 5–20 nm, which is consistent with the diameter observed from the TEM analysis. Only small mesopores (5–15 nm) were observed in the pristine CN samples. These results suggest that the additional oxamide results in the formation of CN with a honeycomb structure.
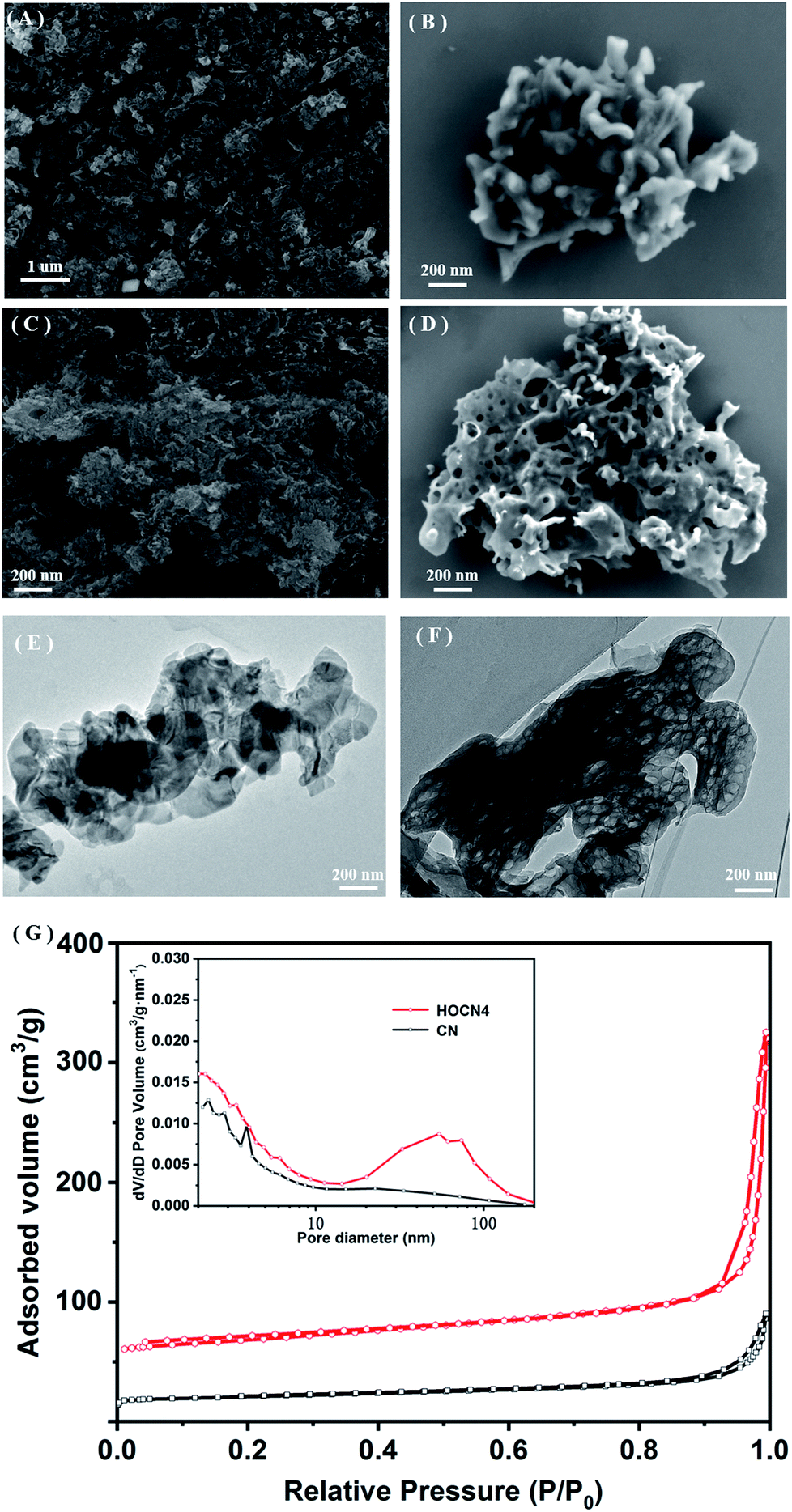 | ||
| Fig. 2 Low and high-magnification SEM image of (A and B) CN, (C and D) HOCN4; TEM image of (E) CN and (F) HOCN4; (G) N2 adsorption–desorption isotherms and the corresponding pore-size distribution curves (inset) of CN and HOCN4. | ||
The crystallinity of the as-prepared CN and HOCN4 were characterized by XRD, as shown in Fig. 3. The HOCN4 samples possess XRD patterns that are similar to CN due to the graphitic-like layered structure. The (002) peak was indexed to the strong interplanar stacking peak of the aromatic systems at 27.1°, confirming the sheet-like structure.39 The minor peak at 13.2° corresponds to the (100) diffraction peak, arising from the in-plane graphene structure. In comparison to that of CN, the characteristic (100) and (002) peaks of the HOCN4 samples were weaker and broader due to the formation of a honeycomb structure.
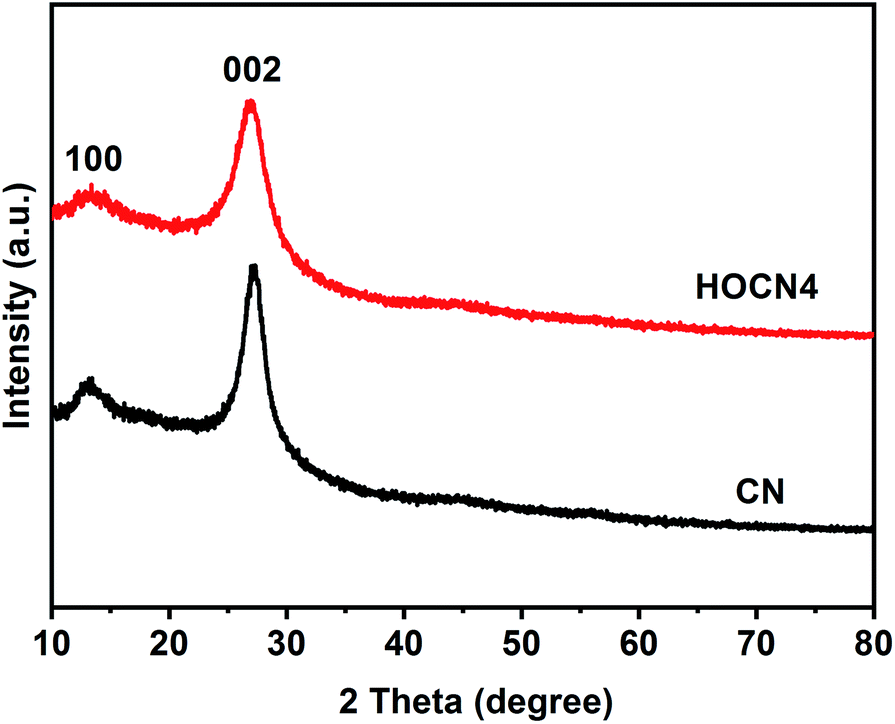 | ||
| Fig. 3 XRD patterns of CN and HOCN4. | ||
As shown in Fig. 4, the Fourier transform infrared (FTIR) spectra for CN and HOCN4 contained several bands in the 1250–1700 cm−1 region for the typical stretching vibrations of C–N heterocycles as well as a characteristic peak at 809 cm−1 corresponding to the tri-s-triazine units. It is important to note that HOCN4 exhibits –OH groups at 2900–3300 cm−1 and C–O stretching vibrations at 1130 and 1206 cm−1. However, the peak at 980 cm−1, which corresponds to the N–O groups, was not observed, implying that the O inset into the carbon nitride framework may occur via replacement of the N atom in the C–NC bonds of the carbon nitride framework.40 To further investigate the location of the O atoms in the carbon nitride framework, X-ray photoelectron spectroscopy (XPS) of CN and HOCN was carried out, and the results are shown in Fig. 5. The results can be fitted to two peaks at 531.6 eV and 532.8 eV, corresponding to C–O and surface-adsorbed water, respectively. The peak of the C–O bond for HOCN is more intense than that for CN. In the C 1s high-resolution XPS spectra, the peak at 284.6 eV is due to graphitic C–C bonds, and the peaks at 286.5 eV and 288.2 eV are due to C–N bonds and hybridized C–CN, respectively. The intensity of the peaks at 284.6 eV and 288.7 eV, which correspond to C–C and C–O bonds, in the C 1s high-resolution spectra is much stronger than those for CN, demonstrating that more C–C and C–O bonds exist in HOCN. In the N 1s high-resolution XPS spectra, no signals corresponding to N–O species were observed for the CN and HOCN samples, suggesting that O is not bound to N. The FTIR and XPS results indicate that the O atoms are incorporated into the framework of CN by replacing the coordinated N atoms (i.e., C–NC).
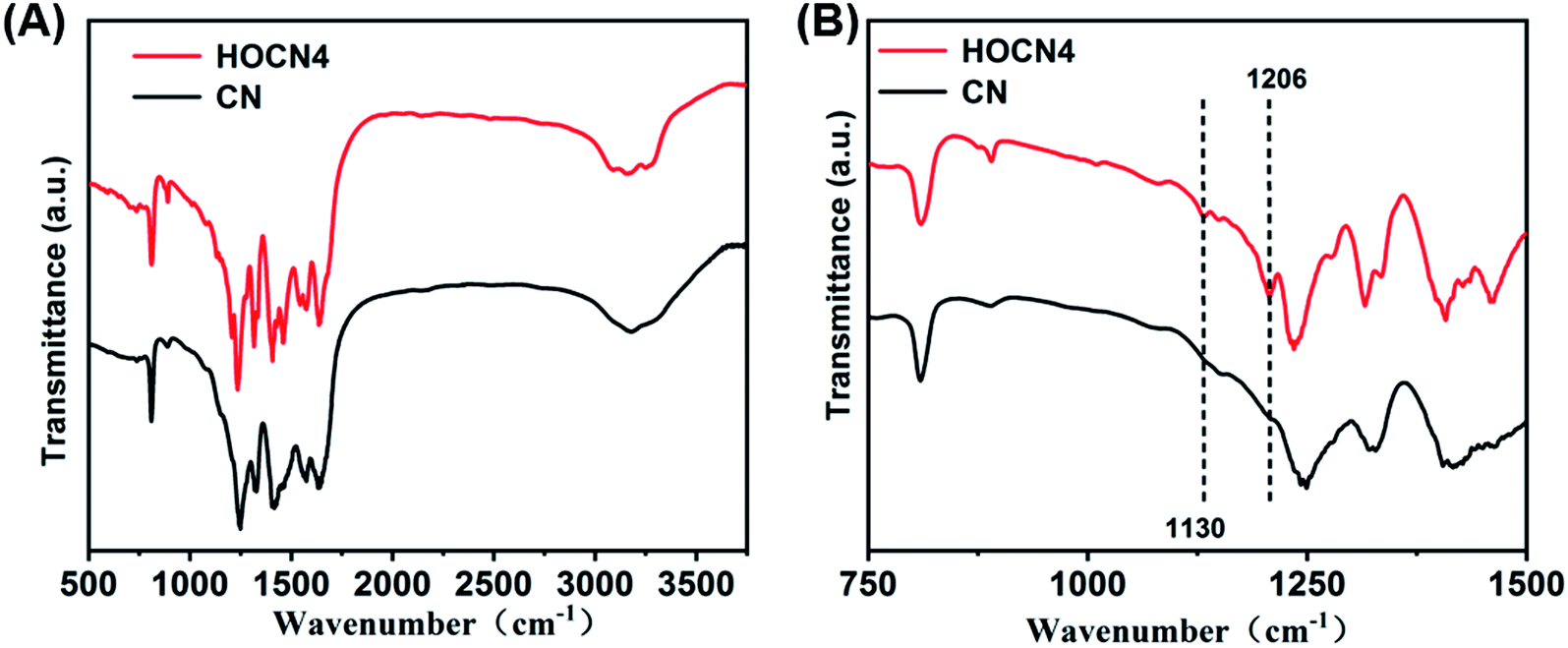 | ||
| Fig. 4 FT-IR spectra of CN and HOCN4. | ||
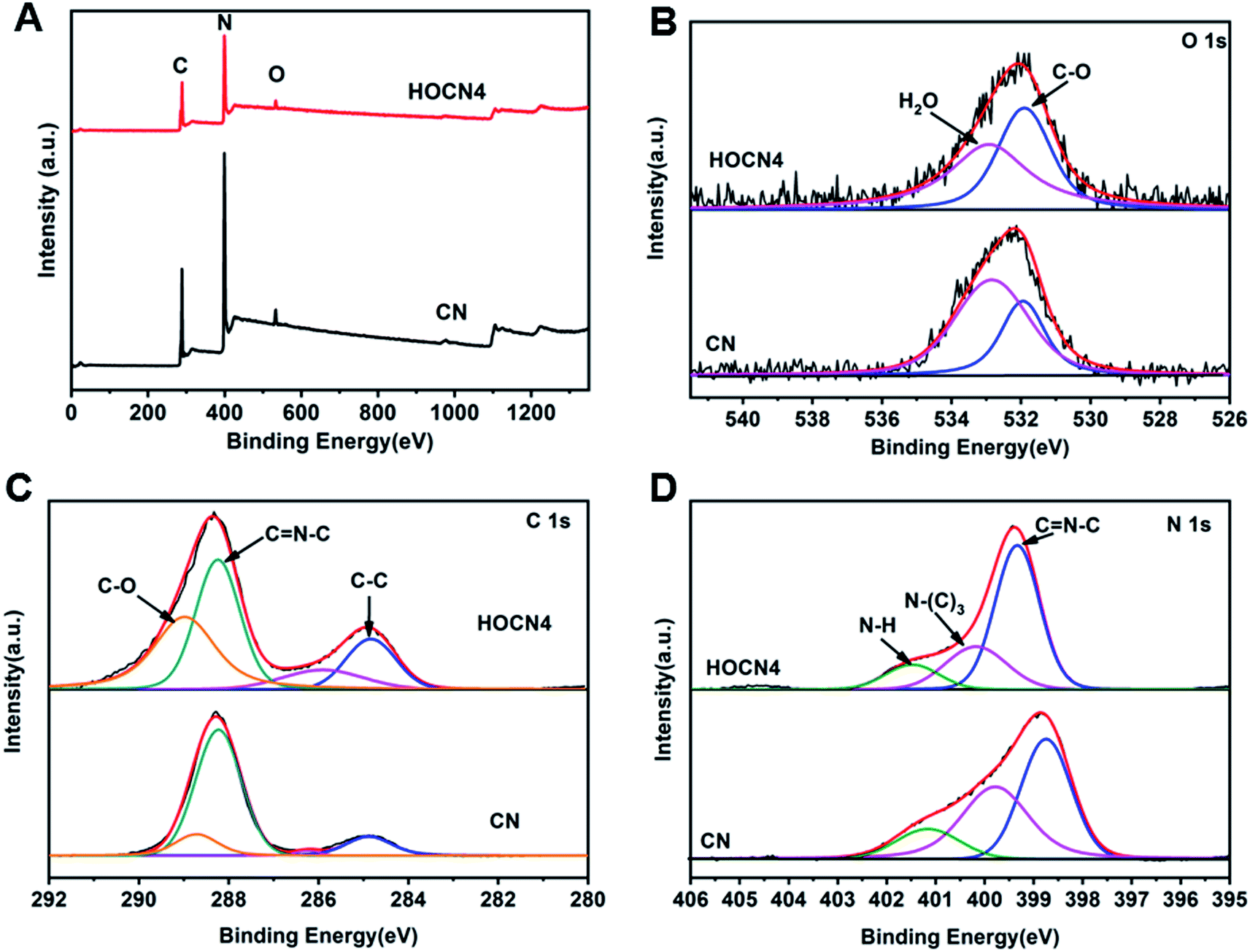 | ||
| Fig. 5 (A) Full XPS spectra of CN and HOCN4; (B) high-resolution XPS spectra of O; (C) high-resolution XPS spectra of C 1s; (D) high-resolution XPS spectra of N 1s. | ||
Based on these results, the possible reaction for the thermal polymerization of oxamide and dicyandiamide as well as the role of oxamide in the formation of the honeycomb structure are shown in Scheme 1. At a medium–high temperature, the carbon atoms of the OA nucleophilically attack the amino groups of DCDA by releasing ammonia to form a similar structure to that of melem (prepolymer 1) followed by polymerization with dicyandiamide to form prepolymer 2. Simultaneously, the polymerization between two dicyandiamides occurs to form melem.16,41 At a high temperature, further polymerization between prepolymer 2 and melem occurs, leading to the introduction of a hetero oxygen atom into the CN framework. Simultaneously, some of prepolymer 2 would decompose into H2O and NH3 and CO2 gases at a high temperature, and then, these gases could expand the packing layers, resulting in the formation of pores in the CN bulk to afford the honeycomb structure. As a result, an oxygen co-doped carbon nitride with a honeycomb structure was obtained.
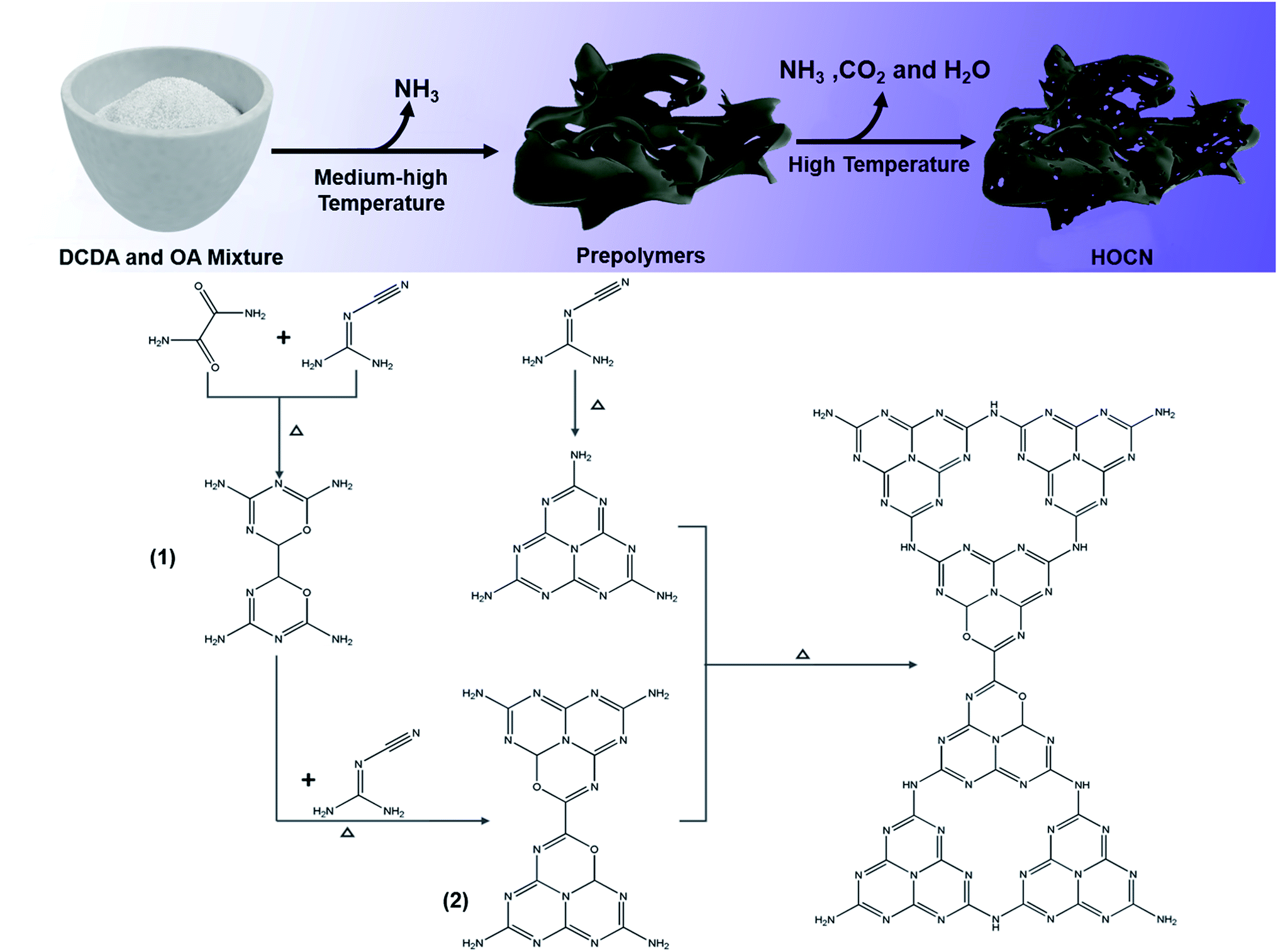 | ||
| Scheme 1 The possible reaction and role of oxamide for the formation HOCN. | ||
The light absorption properties of CN and HOCN were investigated using UV-vis diffuse reflectance spectroscopy (DRS). As shown in Fig. 6A, the light absorption edge of CN was located at approximately 442 nm. Comparatively, HOCN exhibits a light absorption edge at approximately 476 nm with a strong absorption tail (Urbach tail) over the entire visible light region, and the absorption tail edge gradually red shifts to longer wavelengths with increasing oxamide addition, which is beneficial for enhancing the utilization of solar light. As reported, the Urbach tail arises from the sub-bandgap, which originates from the VB to doping states and is located within the band gap of CN.24 The band gap (Eg) of CN and HOCN4 can be determined by fitting the Tauc plots. CN exhibits only one characteristic band gap of 2.80 eV. However, HOCN4 exhibits a characteristic band gap (Eg) of 2.6 eV with a sub-band gap (ES) of 2.13 eV (Fig. 6B). To estimate the band position and structure of CN and HOCN4, the valence band (VB) XPS spectra were recorded (Fig. 6C), and the VB positions for CN and HOCN4 were 1.70 and 1.51 eV, respectively. The conduct band (CB) and doping states (Ed) could be determined as follows: Eg = ECB − EVB, ES = Ed − EVB. The ECB of CN was calculated to be −1.10 V, and the ECB and Ed of HOCN4 was calculated to be at −1.09 and −0.63 V vs. NHE, respectively (Fig. 6D). Both the ECB and Ed of HOCN4 are more negative than the water reduction level, indicating that the photogenerated electron on the CB and doping states can thermodynamically reduce H2O to H2.
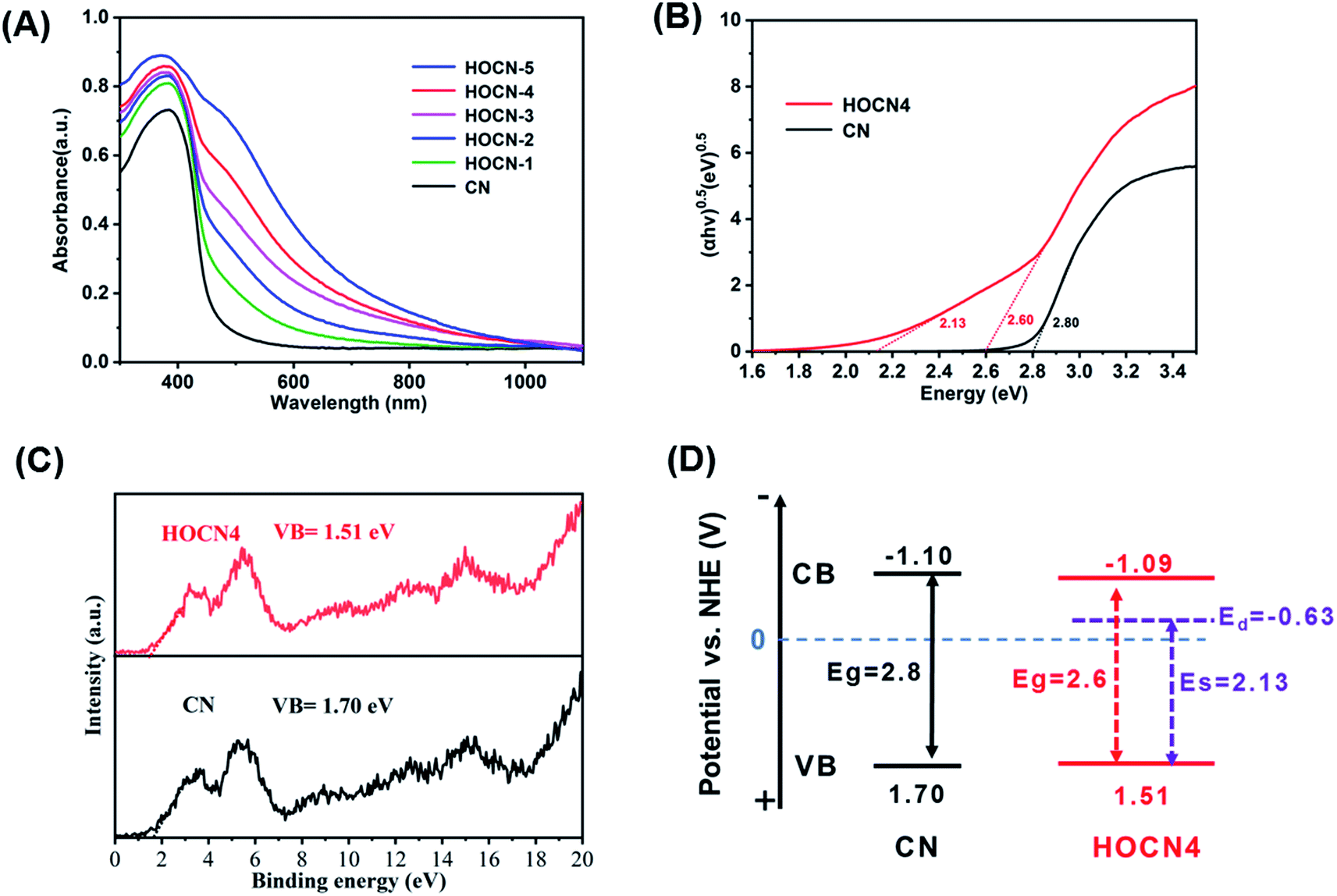 | ||
| Fig. 6 (A) DRS spectra image of CN and HOCNX (X = 1–5) samples; (B) plots of (αhν) 1/2 vs. photon energy of CN and HOCN4; (C) XPS-VB patterns of CN and HOCN4; (D) the corresponding band position of CN and HOCN4. | ||
To further confirm the beneficial role of the sub-band gap and honeycomb-like structure for facilitating separation of the photogenerated electron–hole pairs, electron paramagnetic resonance (EPR) spectroscopy, photoluminescence spectroscopy (PL), photoresponse current and electrochemical impedance spectroscopy (EIS) were performed, and the results are shown in Fig. 7. As shown in Fig. 7A, HOCN4 exhibits a much higher EPR signal intensity than CN both under dark and light irradiation. Fig. 7B shows the PL spectrum of CN and HOCN4. CN exhibits a high fluorescence emission peak at approximately 458 nm, corresponding to band-to-band recombination of photogenerated electron–hole pairs. However, the intensity of this emission peak for HOCN4 decreased, suggesting that the loss of photogenerated electron–hole pairs due to band-to-band recombination is effectively prevented. Moreover, in comparison to CN, the PL signal of HOCN4 was red shifted due to narrowing of the band gap as well as the existence of the sub-band gap, which is consistent with the DRS results. HOCN4 exhibits a much higher photoresponse current and smaller diameter than CN (Fig. 7C and D), implying much easier generation of electron–hole pairs and efficient electron transport at the interfaces. Based on these results, the sub-band gap and honeycomb-like structure efficiently facilitated the generation of electron–hole pairs and inhibited their recombination in CN, which is advantageous for photocatalytic water reduction. In addition, when the oxamide to dicyandiamide molar ratio exceeds 4% (HOCN5 samples), a higher PL signal intensity, lower photocurrent and larger diameter were observed, corresponding to a higher recombination probability for the photogenerated charge carriers in HOCN5. Therefore, the excess oxamide results in the formation of recombination sites and a decreased HER rate.
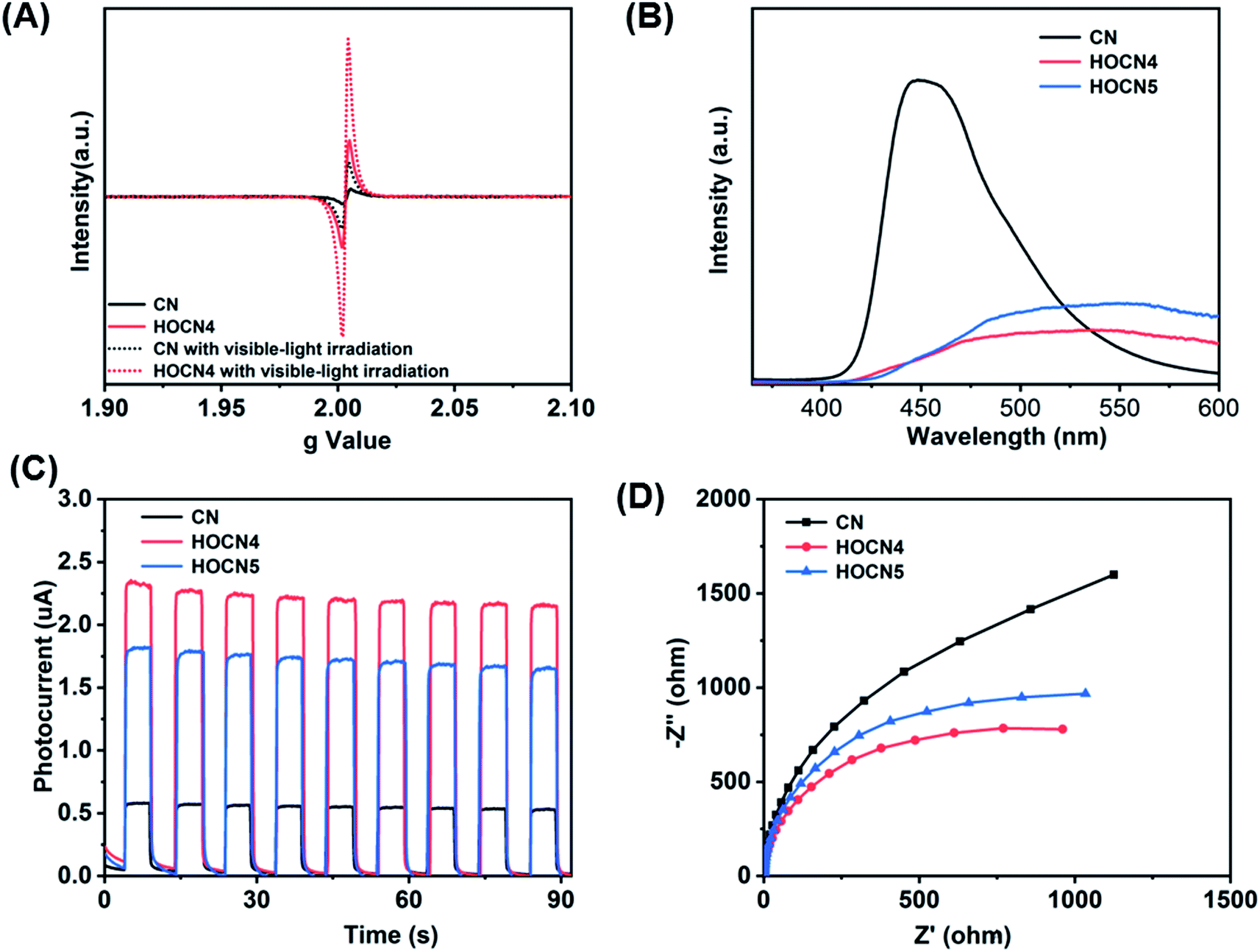 | ||
| Fig. 7 EPR spectra of CN and HOCN4 (A), PL spectra (B), I–T (C) and Nyquist (D) plots of CN, HOCN4 and HOCN5 under visible light. | ||
Based on these results, the proposed photocatalytic H2 evolution mechanism is shown in Scheme 2. Engineering of textural and electronic structures can enable synergetic tuning and optimization of photocatalytic water reducing reactions, resulting in maximum enhancement of the photocatalytic H2 production performance. The three most important features are as follows: (1) the oxygen sub-bandgap contributes to enhanced visible light absorption, (2) the oxygen acceptor states further expedite the transfer of photogenerated charge carriers, and (3) the honeycomb structure increases the number of available active sites and results in more efficient visible light absorbance due to the reflection effect. First, pristine CN can only absorb visible light with wavelengths less than 450 nm through an excited bandgap. However, the HOCN samples can generate electron–hole pairs due to the bandgap as well as the excited oxygen sub-bandgap. Therefore, this material can utilize both short and long wavelength visible light to generate photoelectrons from the VB to the CB as well as oxygen acceptor states while leaving the photogenerated holes in the VB. Second, because the CB position is more negative than that of the oxygen acceptor states, the photoelectrons on the CB of HOCN would transfer to the oxygen acceptor states, which is beneficial for separation of photogenerated electron–hole pairs. Finally, the photoelectrons transferred to the oxygen acceptor and the original photogenerated electrons on the oxygen acceptor states will shift to Pt and reduce H2O to H2. In addition, the photo-holes on the valence band of HOCN will react with sacrificial TEOA.
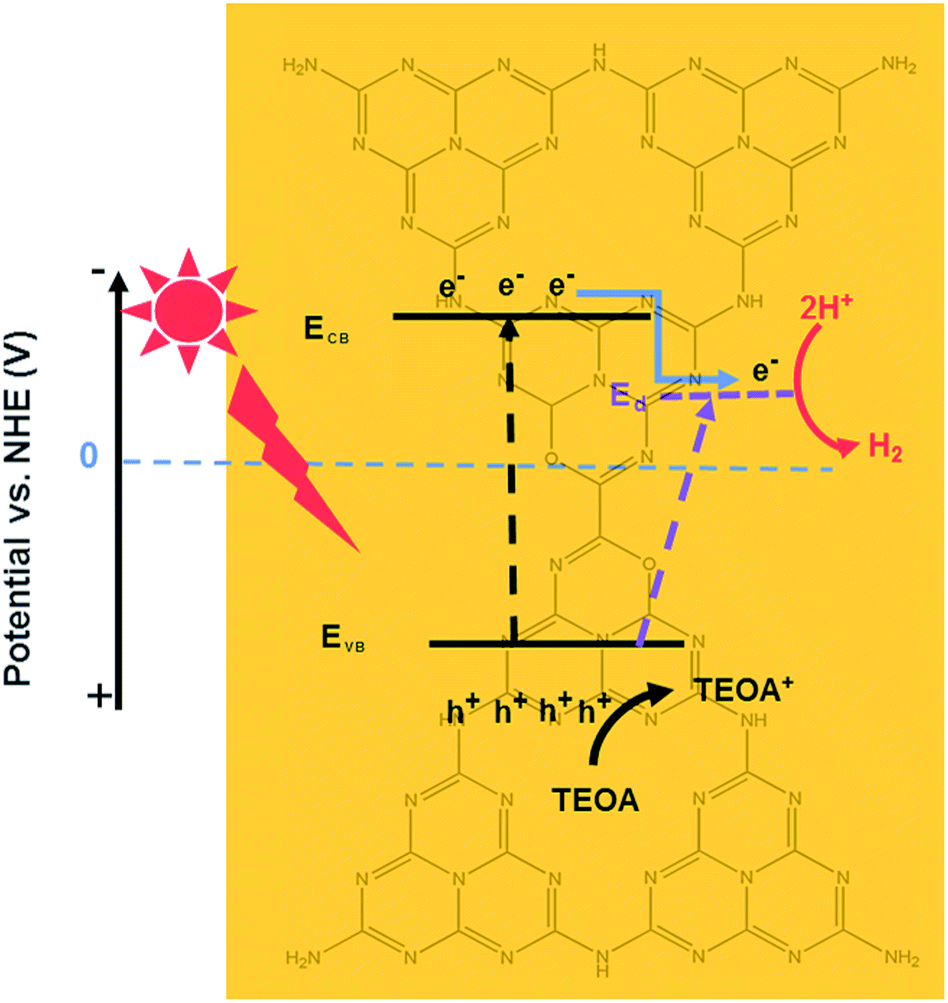 | ||
| Scheme 2 The proposed photocatalytic H2 evolution mechanism of HOCN under light irradiation. | ||
Conclusions
In summary, a honeycomb-like graphitic carbon nitride with a sub-band gap and efficient charge separation was prepared by simple introduction of oxamide into the thermal condensation process of dicyandiamide. In comparison to pristine g-C3N4, all HOCN samples exhibit a higher photocatalytic H2 production due to three advantageous properties including (a) a larger surface area that leads to more photocatalytic sites, (b) better optical absorption, and (c) higher separation efficiency of the photogenerated electron–hole pairs that originate from the sub-band gap structure. This study provides a guide for designing controllable nanostructures with broad light absorption for high performance g-C3N4 photocatalysts using simple co-thermal condensation.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the National Key Research and Development Plan (2016YFA0203200) and the Science Starting Foundation of Guangzhou University (290020316).References
- Z. Wang, C. Li and K. Domen, Chem. Soc. Rev., 2019, 48(7), 2109–2125 RSC.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2008, 8, 76 CrossRef.
- G. F. Liao, Y. Gong, L. Zhang, H. Y. Gao, G. J. Yang and B. Z. Fang, Energy Environ. Sci., 2019, 12(7), 2080–2147 RSC.
- S. Sun and S. Liang, Nanoscale, 2017, 9, 10544–10578 RSC.
- Z. Wang, W. Guan, Y. Sun, F. Dong, Y. Zhou and W.-K. Ho, Nanoscale, 2015, 7(6), 2471–2479 RSC.
- F. Yang, D. Liu, Y. Li, L. Cheng and J. Ye, Appl. Catal., B, 2019, 240, 64–71 CrossRef CAS.
- G. G. Zhang, A. Savateev, Y. B. Zhao, L. N. Li and M. Antonietti, J. Mater. Chem. A, 2017, 5(25), 12723–12728 RSC.
- C. X. Zhao, Z. P. Chen, J. S. Xu, Q. Q. Liu, H. Xu, H. Tang, G. S. Li, Y. Jiang, F. Q. Qu, Z. X. Lin and X. F. Yang, Appl. Catal., B, 2019, 256, 117867 CrossRef CAS.
- Y. Jiang, F. Q. Qu, L. Tian, X. F. Yang, Z. Y. Zou and Z. X. Lin, Appl. Surf. Sci., 2019, 487, 59–67 CrossRef CAS.
- H. Tang, R. Wang, C. X. Zhao, Z. P. Chen, X. F. Yang, D. Bukhvalovb, Z. X. Lin and Q. Liu, Chem. Eng. J., 2019, 374, 1064–1075 CrossRef CAS.
- L. Jiang, X. Yuan, Y. Pan, J. Liang, G. Zeng, Z. Wu and H. Wang, Appl. Catal., B, 2017, 217, 388–406 CrossRef CAS.
- F. Wei, Y. Liu, H. Zhao, X. Ren, J. Liu, T. Hasan, L. Chen, Y. Li and B.-L. Su, Nanoscale, 2018, 10(9), 4515–4522 RSC.
- Y.-Y. Li, B.-X. Zhou, H.-W. Zhang, S.-F. Ma, W.-Q. Huang, W. Peng, W. Hu and G.-F. Huang, Nanoscale, 2019, 11(14), 6876–6885 RSC.
- Q. Q. Liu, J. Y. Shen, X. H. Yu, X. F. Yang, W. Liu, J. Yang, H. Tang, H. Xu, H. M. Li, Y. Y. Li and J. S. Xue, Appl. Catal., B, 2019, 248, 84–94 CrossRef CAS.
- J. S. Zhang, X. F. Chen, K. Takanabe, K. Maeda, K. Domen, J. D. Epping, X. Z. Fu, M. Antonietti and X. C. Wang, Angew. Chem., Int. Ed., 2010, 49(2), 441–444 CrossRef CAS.
- Z. H. Chen, P. Sun, B. Fan, Q. Liu, Z. G. Zhang and X. M. Fang, Appl. Catal., B, 2015, 170, 10–16 CrossRef.
- L. H. Lin, Z. Y. Yu and X. C. Wang, Angew. Chem., Int. Ed., 2019, 58(19), 6164–6175 CrossRef CAS.
- C. Y. Liu, H. W. Huang, W. Cui, F. Dong and Y. H. Zhang, Appl. Catal., B, 2018, 230, 115–124 CrossRef CAS.
- Y. Yu, W. Yan, W. Y. Gao, P. Li, X. F. Wang, S. M. Wu, W. G. Song and K. J. Ding, J. Mater. Chem. A, 2017, 5(33), 17199–17203 RSC.
- W. Yan, Y. Yu, H. H. Zou, X. F. Wang, P. Li, W. Y. Gao, J. Z. Wang, S. M. Wu and K. Ding, Sol. RRL, 2018, 2(7), 1800058–1800066 CrossRef.
- J. S. Cheng, Z. Hu, K. L. Lv, X. F. Wu, Q. Li, Y. H. Li, X. F. Li and J. Sun, Appl. Catal., B, 2018, 232, 330–339 CrossRef CAS.
- D. Huang, X. Yan, M. Yan, G. Zeng, C. Zhou, J. Wan, M. Cheng and W. Xue, ACS Appl. Mater. Interfaces, 2018, 10(25), 21035–21055 CrossRef CAS.
- Y. J. Yuan, Z. K. Shen, S. T. Wu, Y. B. Su, L. Pei, Z. J. Ji, M. Y. Ding, W. F. Bai, Y. F. Chen, Z. T. Yu and Z. G. Zou, Appl. Catal., B, 2019, 246, 120–128 CrossRef CAS.
- Y. J. Yuan, Y. Yang, Z. J. Li, D. Q. Chen, S. T. Wu, G. L. Fang, W. F. Bai, M. Y. Ding, L. X. Yang, D. P. Cao, Z. T. Yu and Z. G. Zou, ACS Appl. Energy Mater., 2018, 1, 1400–1407 CrossRef CAS.
- X. F. Yang, L. Tian, X. L. Zhao, H. Tang, Q. Q. Liu and G. S. Li, Appl. Catal., B, 2019, 244, 240–249 CrossRef CAS.
- L. Tian, X. F. Yang, Q. Q. Liu, F. Q. Qu and H. Tang, Appl. Surf. Sci., 2018, 455, 403–409 CrossRef CAS.
- L. Tian, X. F. Yang, X. K. Cui, Q. Q. Liu and H. Tang, Appl. Surf. Sci., 2019, 463, 9–17 CrossRef CAS.
- W. Liu, J. Shen, Q. Q. Liu, X. F. Yang and H. Tang, Appl. Surf. Sci., 2018, 462, 822–830 CrossRef CAS.
- G. Zhang, G. Li, Z.-A. Lan, L. Lin, A. Savateev, T. Heil, S. Zafeiratos, X. Wang and M. Antonietti, Angew. Chem., Int. Ed., 2017, 56(43), 13445–13449 CrossRef CAS PubMed.
- D. M. Ruan, S. Kim, M. Fujitsuka and T. Majima, Appl. Catal., B, 2018, 238, 638–646 CrossRef CAS.
- D. Zhang, Y. L. Guo and Z. K. Zhao, Appl. Catal., B, 2018, 226, 1–9 CrossRef CAS.
- J. Wu, N. Li, H.-B. Fang, X. Li, Y.-Z. Zheng and X. Tao, Chem. Eng. J., 2019, 358, 20–29 CrossRef CAS.
- Y. Wang, F. Silveri, M. K. Bayazit, Q. Ruan, Y. M. Li, J. J. Xie, C. R. A. Catlow and J. W. Tang, Adv. Energy Mater., 2018, 8(24), 1801084–1801094 CrossRef.
- N. Tian, Y. H. Zhang, X. W. Li, K. Xiao, X. Du, F. Dong, G. I. N. Waterhouse, T. R. Zhang and H. W. Huang, Nano Energy, 2017, 38, 72–81 CrossRef CAS.
- H. Huang, K. Xiao, N. Tian, F. Dong, T. Zhang, X. Du and Y. Zhang, J. Mater. Chem. A, 2017, 5(33), 17452–17463 RSC.
- H. B. Fang, X. H. Zhang, J. J. Wu, N. Li, Y. Z. Zheng and X. Tao, Appl. Catal., B, 2018, 225, 397–405 CrossRef CAS.
- Z. Chen, T. Fan, M. Shao, X. Yu, Q. Wu, J. Li, W. Fang and X. Yi, Appl. Catal., B, 2019, 242, 40–50 CrossRef CAS.
- C. Liu, H. Huang, W. Cui, F. Dong and Y. Zhang, Appl. Catal., B, 2018, 230, 115–124 CrossRef CAS.
- Z. F. Chen, S. C. Lu, Q. L. Wu, F. He, N. Q. Zhao, C. N. A. He and C. S. Shi, Nanoscale, 2018, 10(6), 3008–3013 RSC.
- Z. Song, Z. Li, L. Lin, Y. Zhang, T. Lin, L. Chen, Z. Cai, S. Lin, L. Guo, F. Fu and X. Wang, Nanoscale, 2017, 9(45), 17737–17742 RSC.
- Y. Kofuji, S. Ohkita, Y. Shiraishi, H. Sakamoto, S. Tanaka, S. Ichikawa and T. Hirai, ACS Catal., 2016, 6(10), 7021–7029 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2019 |