
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHalogenation of graphene triggered by heteroatom doping†
Samson O. Olanreleabc,
Zan Lianab,
Chaowei Siab and
Bo Li *a
*a
aShenyang National Laboratory for Materials Science, Institute of Metal Research, Chinese Academy of Sciences, 72 Wenhua Road, Shenyang 110016, China. E-mail: boli@imr.ac.cn
bSchool of Materials Science and Engineering, University of Science and Technology of China, Shenyang 110016, China
cChemical Sciences Department, Mountain Top University, Km 12 Lagos-Ibadan Expressway, Ogun State, Nigeria
First published on 18th November 2019
Abstract
Halogenation is one of the most important ways to tailor the properties of graphene. We demonstrate for the first time that boron and nitrogen doping can effectively tune the interactions between halogen diatomic molecules and graphene from first principles calculations. Boron and nitrogen doping disrupt the regular pi-electron pattern and create spin density and orbital polarization. More interesting, nitrogen and boron doping not only significantly increases the binding energies of Cl2, Br2, and I2 but also induces the spontaneous dissociation of F2. The tunable effects from nitrogen and boron doping can adjust the interactions in a wide range. Overall, it is suggested that doping can be a very promising method for the facile halogenation of graphene.
Introduction
Graphene is an extended honeycomb network of sp2-hybridized carbon atoms and the first example of a close-packed two dimensional (2D) crystalline material isolated in nature. Since its discovery in 2004, graphene has been ascribed as a super/wonder material which has been widely studied in various fields owing to its exciting and extraordinary characteristics. In particular, graphene exhibits superior electronic, mechanical, optical, and transport characteristics. These exceptional intrinsic properties have been widely exploited in applications ranging from photocatalysis, electrochemical sensing, energy storage, those involving solar fuels, etc.On the other hand, various graphene derivatives which are obtained from either non-covalent or covalent functionalization have become a new class of materials which not only inherit the extraordinary properties of graphene but also open up the new horizon to further extend the application of graphene.1 One of the effective functionalization methods for graphene is halogenation by the halogen elements including fluorine, chlorine, bromine and iodine.2 It has been reported that fluorographene, i.e. the fully fluorinated graphene was successfully synthesized with C![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :F ratio 1:1.3 However, the other halogen elements which constitute the partially halogenated graphene are experimentally obtained. The covalent bonded halogens significantly modify the intrinsic properties of graphene such as electric conductivity, mechanical strength, optical transparency, carrier mobility and chemical stability. Overall, the halogenation is a very versatile method to tailor the graphene for on-purpose application.
:F ratio 1:1.3 However, the other halogen elements which constitute the partially halogenated graphene are experimentally obtained. The covalent bonded halogens significantly modify the intrinsic properties of graphene such as electric conductivity, mechanical strength, optical transparency, carrier mobility and chemical stability. Overall, the halogenation is a very versatile method to tailor the graphene for on-purpose application.
The interaction between halogen molecule and the graphene is of vital importance to effective halogenation of graphene. Although the synthesis routes might be different for various halogenated graphene, the halogen source is mainly from halogen diatomic molecule atmosphere. Therefore, the strength and nature of interactions between halogen molecules and graphene arouse the intensive interests from computational point view. Rudenko et al. have investigated adsorption of fluorine, chlorine, bromine, and iodine diatomic molecules on graphene using density functional theory calculations.4 This study reveals that van der Waals correction plays a crucial role in the estimation of binding strength of halogen molecule. In-plane orientation of the molecules has been found to be more stable than the orientation perpendicular to the graphene layer. Sahin et al. perform the first principle DFT and molecular dynamics on the Cl adatom adsorption and diffusion on graphene.5 They found single Cl atom is in ionic bonding with graphene and the diffusion of Cl adatom is quite facile without noticeable barrier. Moreover, they also predict the stable structure of fully chlorinated graphene which Cl has alternatively bonding at two sides of graphene. Yaya et al. study the adsorption of bromine diatomic molecule on graphene and graphite.6 They found that the most stable configuration of Br2 is very different for graphene and graphite respectively with much stronger binding energy on graphene. Moreover, the charge redistribution causes a dipole in adsorbed Br2 on graphene. Tristant et al. used van der Waals-corrected DFT to study iodine atom and molecule adsorption on graphene.7 They also conclude the iodine molecule will not spontaneously dissociate with contact of graphene. On the other hand, the adsorption of halogens can significantly modify the electronic structure of graphene including openness of band gap and Dirac point shift etc.8
One common feature found from these computational studies is that the nature of bonding between halogen diatomic molecule and graphene is intermolecular interaction which is generally around 400 meV or below, and this interaction does not really help to bind halogen molecules and subsequently undermine the formation of halogenated graphene derivatives. Therefore, an effective strategy which can adjust the interactions will be useful to the development of novel halogenated graphene. In this work, doping method by nitrogen and boron is investigated to tune the adsorption of halogen diatomic molecule on graphene. The nitrogen or boron doping for graphene have been witnessed the increasing applications in various fields such as electrochemistry, heterogeneous catalysis etc.9 Due to the similar radius with carbon, nitrogen and boron can be substituted into graphene lattice. Moreover, nitrogen and boron with one more and less valence electrons compared with carbon, resembles p/n doping and these doping types induce significant changes in the electronic structure of graphene. As our previous work indicate that doping can adjust the adsorption of small molecules on nanostructured carbon materials, therefore nitrogen and boron doping are explored for the adsorption of halogen diatomic molecules on graphene.10 Hence, the studies on the doping method for halogen molecule adsorption on graphene not only bring forth new ways to adjust the interactions between halogen molecule and graphene but also deepen the understandings of doping effect.
Computational methods
In this study, the electronic and structural properties of halogen-doped and undoped graphene systems were calculated using spin-polarized density functional theory (DFT) as implemented in Vienna ab initio simulation package (VASP).11,12 For valence electrons, a plane-wave basis set was adopted with an energy cutoff of 500 eV and the ionic cores were described with the projector augmented-wave (PAW) method.13,14 The Perdew–Burke–Ernzerhof (PBE) functional was used as the exchange–correlation functional approximation.8 A Monkhorst–Pack k-point mesh15 of 1 × 1 × 3 were used for geometric relaxation of all structures. The vacuum distance was set to be 12 Å in order to eliminate the interactions between layers and their neighbouring layers.Aiming to analyze the interaction between halogen molecules (F2, Cl2, Br2 and I2) and doped/undoped graphene, the binding energy (Eb) was calculated using the following eqn (1):
| Eb = Esys(doped/undoped) − Egrap(doped/undoped) − Emol | (1) |
To accurately describe the effect of weakly dispersive interaction of halogen molecules on doped/undoped graphene surface, we added van der Waals corrections to our DFT calculations using DFT-D3 correction method of Grimme.16 The atoms in molecules method of Bader has been used for the charge analysis.17 The climbing image nudged elastic band (CI-NEB) method18 was used to locate the transition states. The highest image along the MEP is denoted as the transition state (TS). The energy barrier Ea of each elementary reaction was calculated by the energy difference between the transition state and the initial state.
Results and discussion
Firstly, the halogen diatomic molecules adsorptions on pristine graphene are calculated. Six different configurations considered are shown in Fig. 1(a) and their corresponding binding energies are included in Fig. 1(b). The configurations can be categorized into parallel (A–F) or perpendicular (G–I) structures respectively. The adsorption of F2 molecule is significantly bigger than the other halogen molecules. It is also revealed that the binding energies of different configurations of F2 molecule are quite close to each other. The most stable configuration is identified to be perpendicular on the top of carbon. For Cl2, Br2, and I2 molecules, there is an increasing trend of binding energies along these three molecules for parallel configurations (A–F) as shown in Fig. 1(b). Moreover, the charge transfer is quite small for the adsorption of halogen diatomic molecules on pristine graphene except for F2 molecule, as shown in Table S1.† The height of halogen molecules above pristine graphene is shown in Table S2.† The height is almost above 3 Å except fluorine molecule in perpendicular configurations. The bonding distance of adsorbed halogen molecules (Cl2, Br2, and I2) are slightly elongated within 0.1 Å compared with gas phase molecule as shown in Table S3.†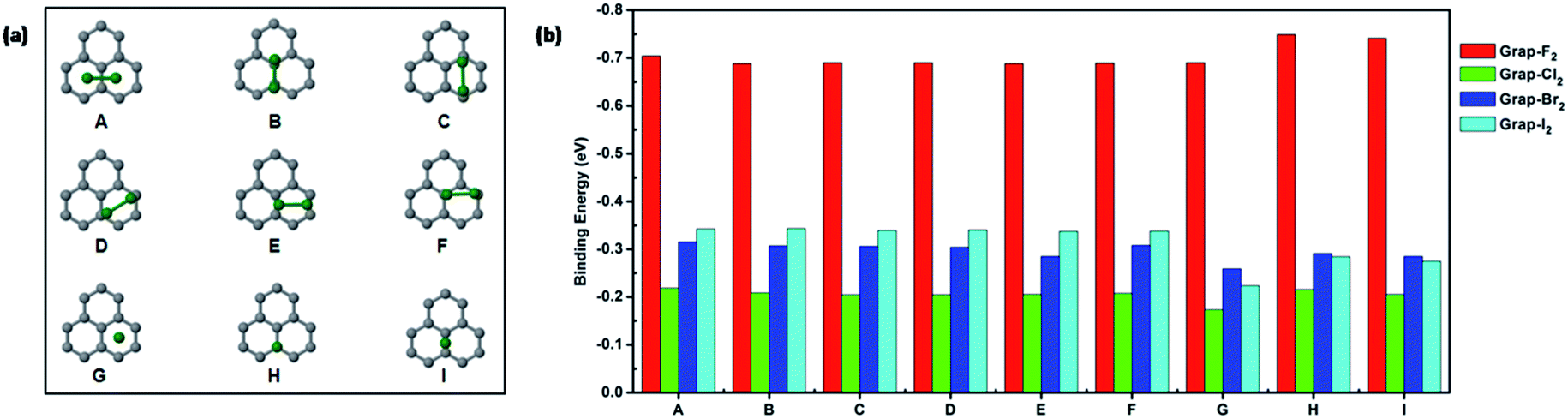 | ||
| Fig. 1 (a) Configurations of halogen molecule adsorption (F2, Cl2, Br2 and I2) on pristine graphene. Gray and green spheres represent carbon and halogen atoms respectively. (b) The adsorption energy (eV) at various binding sites. | ||
For F2, the bond distance of adsorbed molecules exhibit above 0.5 Å elongations than the gas phase molecule. Overall, the interactions between halogen diatomic molecules (Cl2, Br2, and I2) and pristine graphene is weak physisorption. The adsorption of F2 molecule is much stronger than the other halogens due to its strong electronegativity. The predicted adsorption configurations of halogen molecules on pristine graphene are similar to the previous studies. However, the binding strength is different from previous studies.4 To further understand the nature of interaction, non-covalent interaction analysis19 is performed for adsorbed fluorine molecule in configuration (A and I) as shown in Fig. S1.† It is clearly indicated the interactions mainly resides in between fluorine molecule and graphene.
Due to the similar atomic radius, boron and nitrogen can enter graphene lattice and substitute a carbon atom as shown in Fig. S2.† The boron and nitrogen dopants make a major impact on the electronic structure of graphene. From Bader charge analysis, the charges are transferred from three carbons around nitrogen to nitrogen and the opposite trend is found for boron which charges are transferred to surrounding carbons instead of nitrogen becomes negatively charged by 1.33e and boron becomes positively charged by 1.84e respectively. The charge accumulation and depletion around boron and nitrogen dopants are also observed in previous studies.20,21 Moreover, this effect is also revealed from ELF analysis as shown in Fig. 2(a and b). It is observed that the electron depletions around nitrogen dopant and electron accumulation around boron dopant respectively. Moreover, the dopants also disrupt the regular pattern of HOMO orbital of pristine graphene as shown in Fig. 2(c and d). For nitrogen doping, there is a lone pair lobe located on nitrogen dopant and boron forms a delocalized orbital with three neighboring carbons.
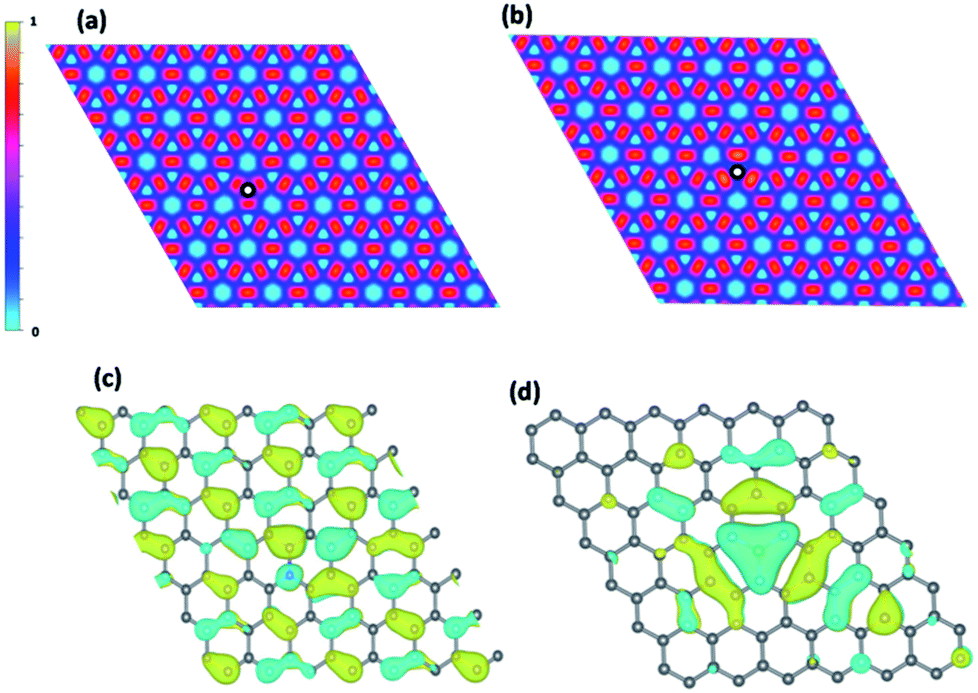 | ||
Fig. 2 ElF analysis (a) nitrogen doping (b) boron doping. The  symbol indicates dopant position. HOMO orbital plot (c) nitrogen doping (d) boron doping. symbol indicates dopant position. HOMO orbital plot (c) nitrogen doping (d) boron doping. | ||
The perturbed electronic structure from doping has a direct influence on the binding energies of halogen diatomic molecules.
The configurations and binding energies of halogen diatomic molecules on boron and nitrogen doped graphene are shown in Fig. 3(a and b). For boron-doped graphene, fluorine molecule adsorption show reduced binding energies for all parallel configurations (A–F) compared with the counterparts on the pristine graphene. For the other halogen diatomic molecules (Cl2, Br2, and I2), different trends are observed in its parallel and perpendicular configurations. The binding energies of the parallel configurations (A–F) do not experience too much change. However, the perpendicular configurations (G and H) witnessed a significant increase in binding energies particularly for Br2 and I2 molecules.
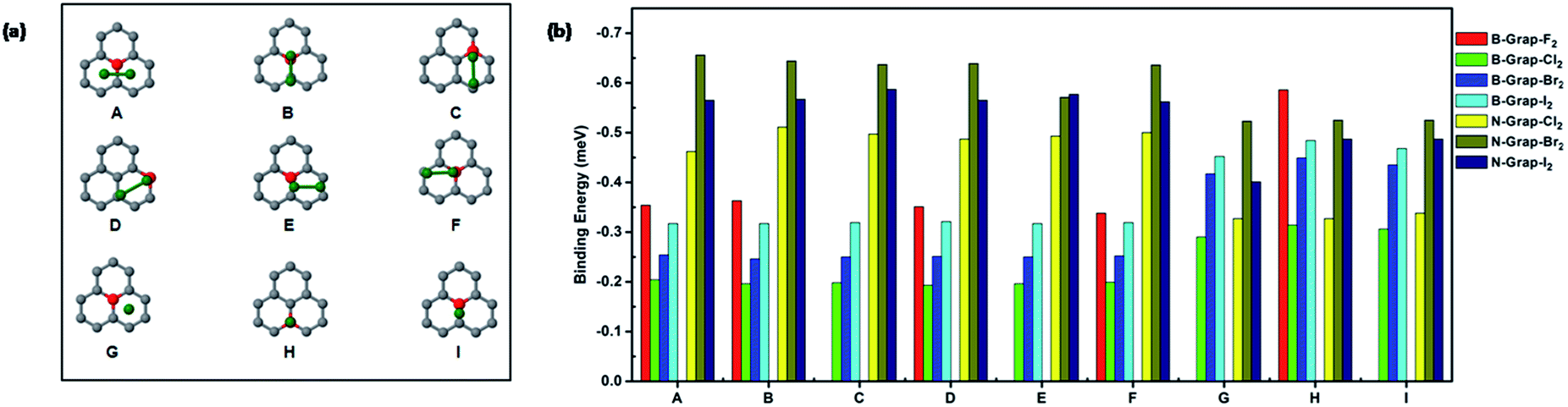 | ||
| Fig. 3 (a) Configurations of halogen molecule adsorption on doped graphene. Gray, green and red spheres represent carbon, halogen and boron or nitrogen atoms respectively. (b) The adsorption energy (eV) at various binding sites. | ||
The increased binding energies also coincided with the decreased distance between adsorbed molecule and graphene as shown in Table S2.† Overall, the boron doping brings modest effects on the binding of halogen diatomic molecules. For nitrogen doping, the discussions firstly are constrained to Cl2, Br2, and I2 molecules.
The binding energy increases are quite obvious for all of three molecules at most configurations. The increasing binding energies corresponds with the more charges obtained by the adsorbed halogen molecules as shown in Table S1† compared with pristine graphene. The increased charge density and polarization of orbital induced by nitrogen doping could be accountable to the increase of binding energies.
The binding of F2 molecule shows different feature from the other halogen molecules. The calculations indicate that the F2 molecule undergoes a spontaneous dissociation on nitrogen doped graphene which does not have a barrier. The initial and final states of dissociations on nitrogen doped graphene are shown in Fig. S3.† After dissociation, two fluorine atoms are binding with two carbons around nitrogen dopant. The bond distance being 1.48 Å is a pure indication that covalent bonding is formed between dissociated fluorine atom and carbon atom on graphene. In contrast, the dissociation of F2 on pristine graphene has a barrier of 0.12 eV. The calculation clearly suggests that nitrogen doping indeed can enhance the halogenation of graphene. On the other hand, boron doping also can induce spontaneous dissociation of F2 molecule as shown in Fig. S3.† Overall, nitrogen doping more significantly increase the adsorption energies of halogen molecules compared with boron doping. Hence, doping can adjust the interactions between halogen molecule and graphene in a wide range. This tunable effect from different dopants is also observed in our previous studies.10 Furthermore, the increases of dopants contents consequently enhance the binding strength of halogen molecule as shown in Fig. S4.†
Conclusions
The first principle calculations clearly indicate that the doping by boron and nitrogen can effectively adjust interactions between halogen diatomic molecule and graphene. Boron and nitrogen dopants can substitute carbon in graphene lattice and induce the significant changes of electronic structure. Furthermore, nitrogen doping can significantly increase the binding strength of halogen molecules (Cl2, Br2, and I2) and arouse the spontaneous dissociation of F2 molecule compared with pristine graphene. Moreover, nitrogen and boron doping exhibit some tunable effects on graphene–halogen interactions. The doping mechanism of graphene by nitrogen and boron is expected to open the opportunities for rational design of halogenated graphene derivatives. In the end, the current work demonstrates that the doping is a valid method for the facile halogenation of graphene and pave the way for the further application of halogenated graphene.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the NSFC (21573255) and Natural Science Foundation of Liaoning Province (20180510014). This work was also supported by the State Key Laboratory of Catalytic Materials and Reaction Engineering (RIPP, SINOPEC). The computations were also supported by the Special Program for Applied Research on Super Computation of the NSFC Guangdong Joint Fund (the second phase) under Grant No. U1501501.Notes and references
- V. Georgakilas, M. Otyepka and A. B. Bourlinos, et al., Functionalization of Graphene: Covalent and Non-Covalent Approaches, Derivatives and Applications, Chem. Rev., 2012, 112(11), 6156–6214 CrossRef CAS PubMed.
- F. e. Karlický, K. Kumara Ramanatha Datta and M. Otyepka, et al., Halogenated Graphenes: Rapidly Growing Family of Graphene Derivatives, ACS Nano, 2013, 7(8), 6434–6464 CrossRef PubMed.
- W. Feng, P. Long and Y. Feng, et al., Two-Dimensional Fluorinated Graphene: Synthesis, Structures, Properties and Applications, Adv. Sci., 2016, 3(7), 1500413 CrossRef PubMed.
- A. N. Rudenko, F. J. Keil and M. I. Katsnelson, et al., Adsorption of diatomic halogen molecules on graphene: A van der Waals density functional study, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82(3), 35427 CrossRef.
- H. Sahin and S. Ciraci, Chlorine Adsorption on Graphene: Chlorographene, J. Phys. Chem. C, 2012, 116(45), 24075–24083 CrossRef CAS.
- A. Yaya, C. P. Ewels and I. Suarez-Martinez, et al., Bromination of graphene and graphite, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83(4), 45411 CrossRef.
- D. Tristant, P. Puech and I. C. Gerber, Theoretical Study of Graphene Doping Mechanism by Iodine Molecules, J. Phys. Chem. C, 2015, 119(21), 12071–12078 CrossRef CAS.
- C. Y. Xu, P. A. Brown and J. Lu, et al., Electronic Properties of Halogen-Adsorbed Graphene, J. Phys. Chem. C, 2015, 119(30), 17271–17277 CrossRef CAS.
- J. Albero and H. Garcia, Doped graphenes in catalysis, J. Mol. Catal. A: Chem., 2015, 408, 296–309 CrossRef CAS.
- S. Ali, T. F. Liu and Z. Lian, et al., The tunable effect of nitrogen and boron dopants on a single walled carbon nanotube support on the catalytic properties of a single gold atom catalyst: a first principles study of CO oxidation, J. Mater. Chem. A, 2017, 5(32), 16653–16662 RSC.
- G. Kresse and J. Furthmüller, Efficiency of ab initio total energy calculations for metals and semiconductors using a plane-wave basis set, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Efficient iterative schemes for total-energy calculations using a plane-wave basis set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- G. Kresse and D. Joubert, From ultrasoft pseudopotentials to the projector augmented wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- P. E. Blöchl, Projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- H. J. Monkhorst and J. D. Pack, Special points for Brillouin-zone integrations, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- G. Henkelman, A. Arnaldsson and H. Jónsson, A fast and robust algorithm for Bader decomposition of charge density, Comput. Mater. Sci., 2006, 36, 354–360 CrossRef.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, A climbing image nudged elastic band method for finding saddle points and minimum energy paths, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
- E. R. Johnson, S. Keinan and P. Mori-Sanchez, et al., Revealing Noncovalent Interactions, J. Am. Chem. Soc., 2010, 132(18), 6498–6506 CrossRef CAS PubMed.
- B. Li and D. Su, First-principles studies of the activation of oxygen molecule and its role in partial oxidation of methane on boron-doped single-walled carbon nanotubes, J. Phys. Chem. C, 2013, 11734, 17485–17492 CrossRef.
- K. Gong, F. Du, Z. Xia, M. Durstock and L. Dai, Nitrogen-Doped Carbon Nanotube Arrays with High Electrocatalytic Activity for Oxygen Reduction, Science, 2009, 323, 760–764 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra06962c |
| This journal is © The Royal Society of Chemistry 2019 |