
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe selective hydrosilylation of norbornadiene-2,5 by monohydrosiloxanes†
Marina A. Gusevaa,
Dmitry A. Alentiev *a,
Evgeniya V. Bermeshevab,
Ilya A. Zamilatskovc and
Maxim V. Bermeshev*a
*a,
Evgeniya V. Bermeshevab,
Ilya A. Zamilatskovc and
Maxim V. Bermeshev*a
aA. V. Topchiev Institute of Petrochemical Synthesis of Russian Academy of Sciences, 29 Leninsky Prospekt, 119991 Moscow, Russia. E-mail: d.alentiev@ips.ac.ru; bmv@ips.ac.ru
bI. M. Sechenov First Moscow State Medical University, 8 Bld. 2 Trubetskaya Str., 119991 Moscow, Russia
cA. N. Frumkin Institute of Physical Chemistry and Electrochemistry of Russian Academy of Sciences, 31 Bld. 4 Leninsky Prospekt, 119071 Moscow, Russia
First published on 16th October 2019
Abstract
A simple one-step approach for the selective synthesis of exo-norbornenes with organosilicon substituents is suggested through the direct hydrosilylation of norbornadiene-2,5 with chlorine-free silanes. Using the example of norbornadiene-2,5 hydrosilylation with pentamethyldisiloxane and 1,1,1,3,5,5,5-heptamethyltrisiloxane, the possibility of obtaining exo-isomers of norbornenes with 100 exo-/endo-selectivity is shown. The investigation of Pt-, Rh-, and Pd-complexes in combination with various ligands as catalysts was performed. The hydrosilylation of norbornadiene-2,5 in the presence of Pt- or Rh-catalysts was not selective and led to a mixture consisting of three isomers (exo-/endo-norbornenes and substituted nortricyclane). In the case of the Pd-salt/ligand catalytic system, the formation of an endo-isomer was not observed at all and only two isomers were formed (exo-norbornene and nortricyclane). The selectivity of exo-norbornene/nortricyclane formation strongly depended on the nature of the ligand in the Pd-catalyst. The best selectivity was revealed when R-MOP was the ligand, while the highest catalytic activity was reached with a dioxalane-containing ligand.
Introduction
The hydrosilylation of alkenes and dienes is a powerful synthetic tool for the production of various organosilicon compounds1–3 and modification of the properties of the material.4,5 Among the low-molecular-weight organosilicon compounds, alkenylsilanes are the most commonly used building blocks in organic synthesis owing to their versatile reactivity. Usually alkenylsilanes are obtained by the hydrosilylation of alkynes6 and dienes.6,7 Nevertheless, this reaction may lead to several isomeric products and therefore the development of regio- and stereoselective approaches is a challenge.The chemistry of substituted cycloalkenes is also fruitful and not only includes different convenient organic modifications but also polymerization transformations. In particular, it is diverse for strained cycles such as cyclopropenes or norbornenes. The combination of versatile organosilicon chemistry combined with the possibilities of cycloalkenes within a molecule can reveal grand perspectives. Recently it has been shown that Si-containing norbornenes are monomers that are in high demand for the design and synthesis of membrane,8–11 microporous12,13 and adhesive14,15 materials. However, although the traditional synthesis with the help of the [4π + 2π]- (Diels–Alder) reaction between cyclopentadiene and vinylsilanes is simple and is performed in onestep, it provides a mixture of exo- and endo-isomers (Scheme 1a). This mixture is, as a rule, enriched by an endo-isomer that is less reactive in polymerizations.16,17 To overcome this disadvantage, the [2σ + 2σ + 2π]-cycloaddition of vinylsilanes with quadricyclane was realized (Scheme 1b).18 This selectively gives the exo-isomer, but only “activated” vinylsilanes (which contain chlorine atoms at silicon) were active in this reaction.19–22 This limits the scope of the reaction. Furthermore, it is also necessary to use an additional step to transform the Si–Cl-bonds into the desired silicon-containing group. A further difficulty in the considered approach is the photochemical synthesis of quadricyclane from norbornadiene-2,5.
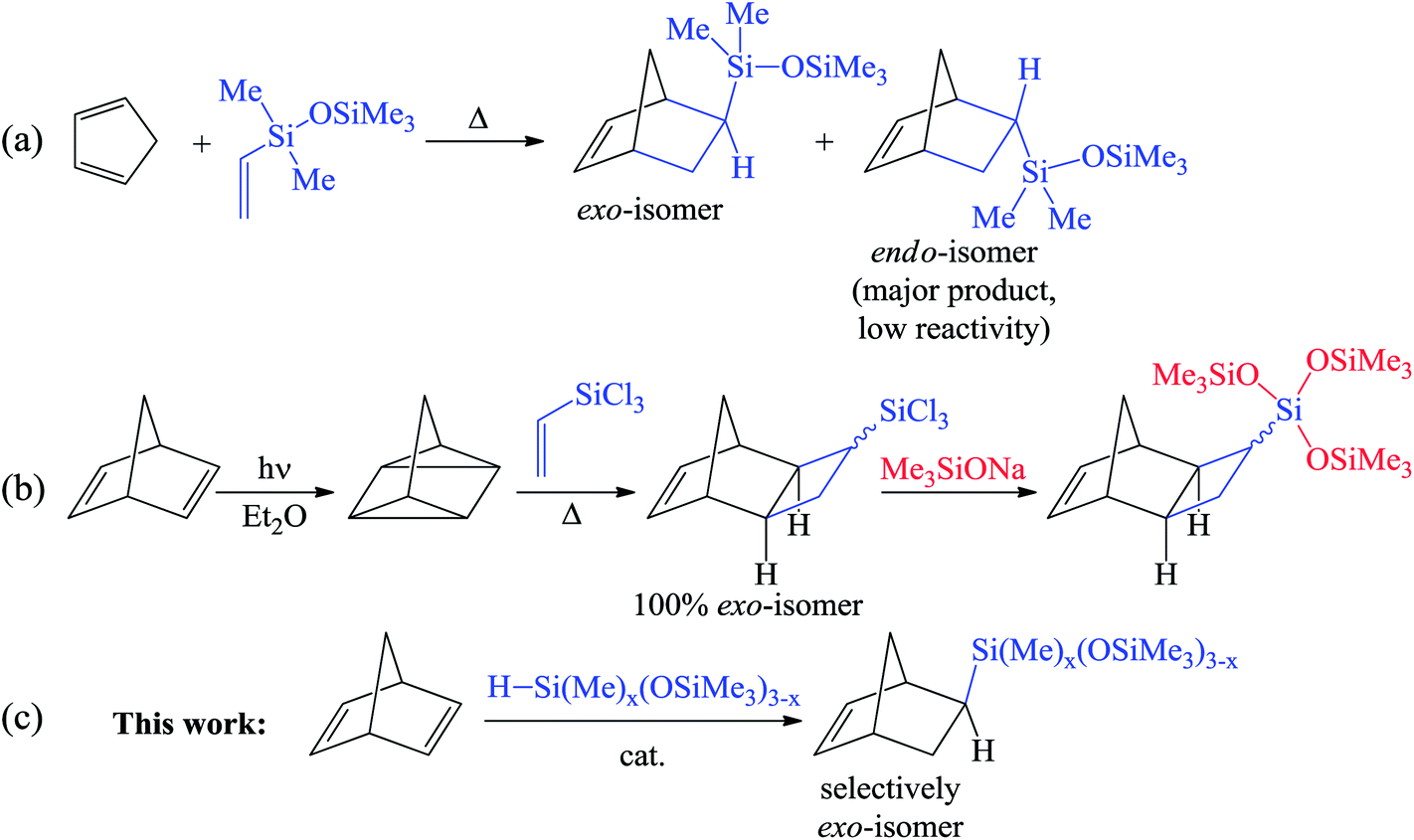 | ||
| Scheme 1 Approaches to the synthesis of Si-containing norbornenes: Diels–Alder reaction (a), [2σ + 2σ + 2π]-cycloaddition (b), hydrosilylation (c). | ||
Therefore, the development of a one-step and selective approach for the synthesis of Si-substituted exo-norbornenes is highly warranted. The incorporation of an oligosiloxanyl-group into a norbornene moiety is of particular interest. These compounds are suitable monomers for the preparation of “comb-shaped” polymers consisting of rigid main chains (polynorbornene main chains) and flexible side groups (oligosiloxanyl-groups). Owing to the dual nature of such polymers, they display an excellent performance in the membrane separation of gaseous hydrocarbons.23,24 An attractive method for the synthesis of Si-substituted exo-norbornenes is the regio- and stereoselective hydrosilylation of norbornadiene-2,5 (NBD) with non-activated monohydrosilanes. This approach might allow the desired products to be obtained in one-step. To date little is known about the selective hydrosilylation of NBD. Concerning the selective synthesis of Si-containing exo-norbornenes, a single example of the hydrosilylation of NBD with only an “activated” silane (trichlorosilane) has been described before.25,26 At the same time, a selective hydrosilylation of NBD with non-activated silanes (e.g. trialkylsilanes or hydrosiloxanes) has still not been realized. Thus, the usual Pt-catalysts or radical initiators result in the formation of a mixture consisting of exo- and endo-isomers and a nortricyclane derivative.27 An attempt has also been previously reported involving a non-activated silane (triethoxysilane) in the reaction with NBD using a Co-catalyst, but it turned out to be unsuccessful.28 One further attempt for the hydrosilylation of NBD with a triethylsilane was performed over a Mo-catalyst.29 It led selectively to an endo-isomer of Si-containing norbornene, which spontaneously underwent the metathesis polymerization under the studied conditions. Here, we report the selective synthesis of exo-norbornenes bearing oligosiloxane-moieties via a direct hydrosilylation of NBD using commercially available silanes. The suggested method gives only the exo-isomer with a minor content of an inert saturated nortricyclane, while the formation of the endo-isomer is not observed at all.
Experimental
Methods and materials
The NMR spectra were recorded on MSL-300 (Bruker, USA) and Varian Inova 500 (Agilent Technologies, USA) spectrometer at 300.13 MHz and 499.8 MHz correspondingly for 1H spectra, 133.3 MHz for 13C spectra and 99.3 MHz for 29Si spectra, using CDCl3 as a solvent. Gas chromatography/mass-spectrometry (GC/MS) analysis was conducted on a gas-chromato-mass-spectrometer “Finnigan MAT 95 XL” (ionization energy of 70 eV, mass range of 20–800, resolution of 1000, source temperature of 200 °C, scan rate of 1 s per mass decade, chromatograph HP 6890+, PDMS DB-5 column 30 m × 0.25 mm, using helium as a carrier gas). The infrared (IR) spectra were registered on an IFS-66 v/s Bruker spectrometer.All hydrosilylation experiments (excluding experiment with R-MONOPHOS in air) were conducted under argon using the standard Schlenk technique.
Toluene was dried over Na, distilled under argon and stored over Na wire. Norbornadiene-2,5, pentamethyldisiloxane and 1,1,1,3,5,5,5-heptamethyltrisiloxane (Aldrich) were dried over Na and distilled at reduced pressure under argon. All ligands and chemicals were purchased from Aldrich and used without additional purification.
Hydrosilylation study
Determination of conversion and product ratios
The conversion of norbornadiene-2,5 was determined from the ratio of the integral intensities of toluene (2.23 ppm) and the norbornadiene-2,5 double bond protons (6.66 ppm) in the NMR spectra. The selectivity of the mono-silicon-substituted norbornene formation was determined from the integral intensity of its double bonds (6.12 and 5.90 ppm). GC/MS was also applied to determine the relative amounts of the products. The exo- and endo-isomers differed according to their 1H NMR spectra. The signal at 0.25–0.30 ppm is assigned to the C(5)H in exo-isomer.Synthesis of exo-silicon-substituted norbornenes
A mixture of norbornadiene-2,5, hydrosilane and the (AllPdCl)2/R-MOP catalyst (see Example 1) was placed in a round-bottom glass ampoule that had been previously evacuated and filled with argon three times. The ampoule was sealed and allowed to remain at 45 °C for 15 or 20 days in the case of PMDS or HMTS respectively. Then, the reaction mixture was evacuated (1 mm Hg, room temperature) for 20 min under stirring to remove volatile compounds. The product (in the mixture with nortricyclane) was isolated by distillation under vacuum (0.1 mm Hg).1H NMR (δ, ppm, CDCl3): 6.12 (m, 1H, C(2,3)H), 5.91 (m, 1H, C(2,3)H), 2.89 (m, 1H, C(1,4)H), 2.77 (m, 1H, C(1,4)H), 1.61–1.54 (m, 1H, C(6)H), 1.17–1.02 (m, 3H, C(6,7)H), 0.31–0.23 (m, 1H, C(5)H), 0.12 to (−)0.02 (m, 15H, Si–CH3).
13C NMR (δ, ppm, CDCl3): 138.28 (C(2,3)), 133.48 (C(2,3)), 46.76 (C(7)), 42.70 (C(1,4)), 42.41 (C(1,4)), 26.60 (C(6)), 26.56 (C(5)), 1.97 (Si–CH3), −0.02 (Si–CH3).
29Si NMR (δ, ppm, CDCl3): 7.49, 7.11.
IR (cm−1): 2360, 2330, 1110 (νSi–O–Si), 790 (νSi(CH3)3).
MS (EI): 240 (M+, 1%); 147 (SiMe2OSiMe3+, 100%); 92 (NBD+, 7%); 73 (SiMe3+, 25%); 66 (CPD+, 55%).
1H NMR (δ, ppm, CDCl3): 6.11 (m, 1H, C(2,3)H), 5.90 (m, 1H, C(2,3)H), 2.87 (m, 1H, C(1,4)H), 2.77 (m, 1H, C(1,4)H), 1.64–1.57 (m, 1H, C(6)H), 1.21–0.96 (m, 3H, C(6,7)H), 0.25–0.19 (m, 1H, C(5)H), 0.09–0.04 (m, 18H, Si–CH3), 0.02 (m, 3H, Si–CH3).
13C NMR (δ, ppm, CDCl3): 138.09 (C(2,3)), 133.60 (C(2,3)), 46.69 (C(7)), 42.68 (C(1,4)), 42.36 (C(1,4)), 26.46 (C(6)), 25.87 (C(5)), 1.87 (Si–CH3), −0.66 (Si–CH3).
29Si NMR (δ, ppm, CDCl3): 6.91, −22.23.
IR (cm−1): 2360, 2330, 1110 (νSi–O–Si), 790 (νSi(CH3)3).
MS (EI): 314 (M+, 1%); 221 (SiMe(OSiMe3)2+, 100%); 73 (SiMe3+, 15%); 66 (CPD+, 12%).
Results and discussion
The hydrosilylation of NBD by monohydrosilanes was systematically investigated in the presence of different catalytic systems based on Pd-, Pt-, and Rh-containing complexes. Commercially available pentamethyldisiloxane (PMDS) and 1,1,1,3,5,5,5-heptamethyltrisiloxane (HMTS) were chosen as hydrosilanes. Depending on the conditions, the reaction may result in different products in various ratios (Scheme 2). In the case of an NBD/silane molar ratio of 1/1 the main product can be a mono-substituted norbornene bearing organosilicon substituent in the exo- or endo-position. Silicon-substituted nortricyclane and disubstituted norbornane could be also formed as co-products. To determine the conditions for the selective hydrosilylation of NBD, we varied the nature of the ligand and catalyst and the reaction temperature. The ratio of products was controlled using 1H NMR spectroscopy and GC/MS.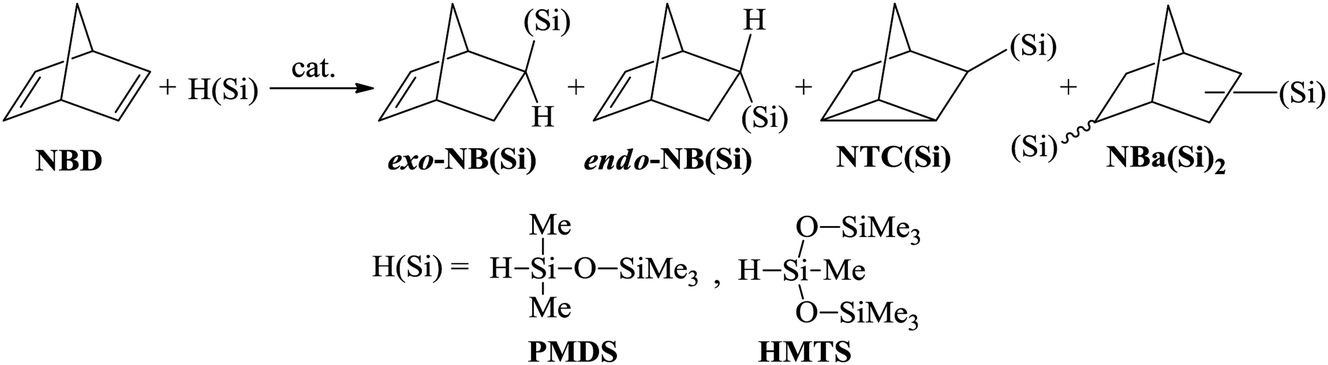 | ||
| Scheme 2 Hydrosilylation of norbornadiene-2,5 by monohydrosilanes and possible products. | ||
Effect of catalyst nature
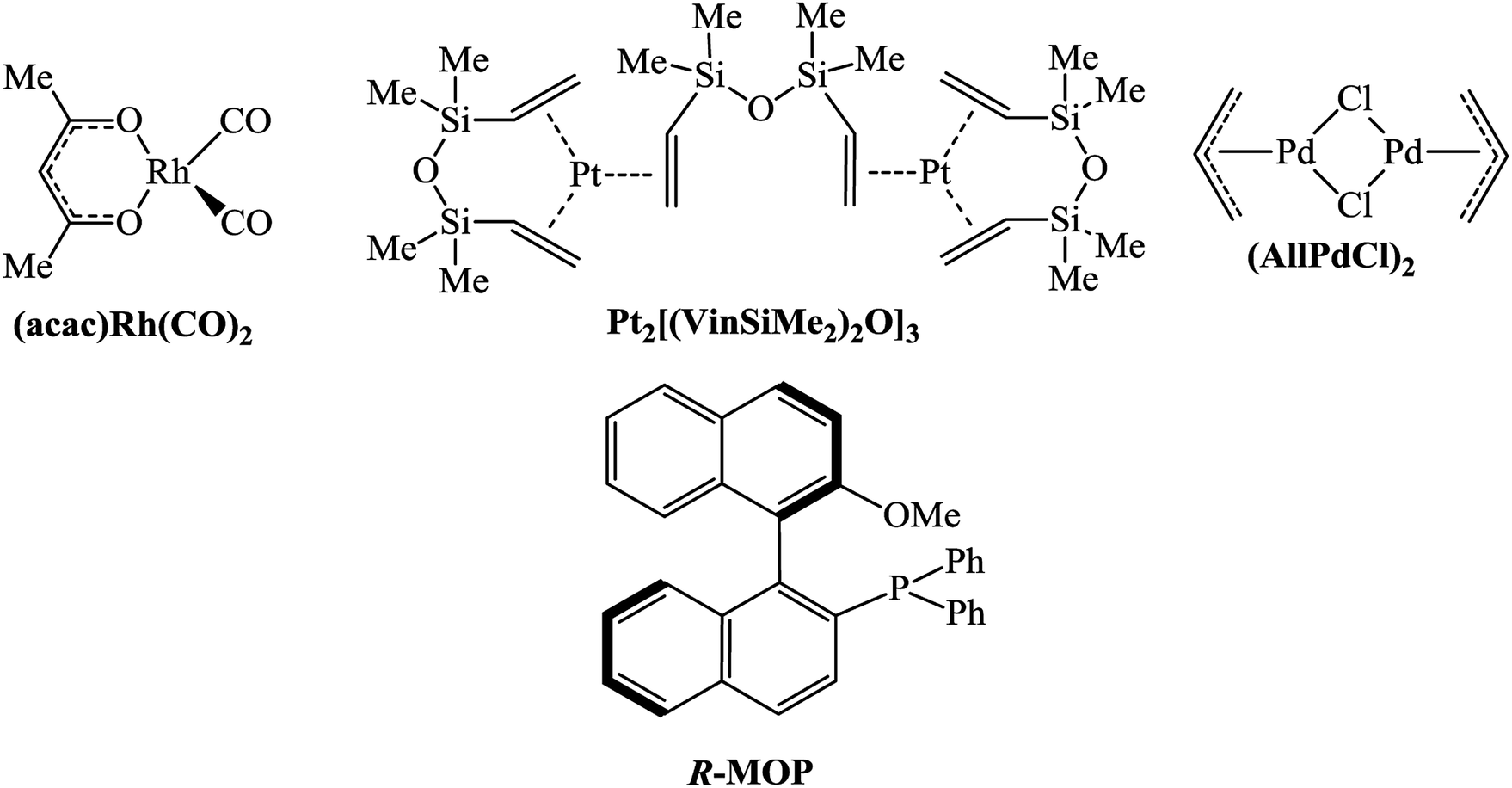
Various compounds of transition metals have previously been proved to be effective catalysts for the hydrosilylation of olefins and unsaturated polymers.1,5,30 The most active of them are Pt salts and its complexes. Catalysts based on other Pt-group metals are slightly less active. A catalyst can exert a strong effect on the selectivity of the hydrosilylation reaction. For example, in the hydrosilylation reaction the Pt-containing systems tolerate any hydrosilane (e.g., HSiClnMe3−n (n = 1–3), HSi(OR)3, and HnSiR4−n (n = 1–3)), while Pd-catalysts are mostly applicable to HSiClnR3−n (n = 2, 3) and Rh-compounds display a good tolerance to HSiR3 (R – alkyl or aryl).31,32 In this work the catalysts based on Rh-, Pd-, and Pt-complexes were compared for the hydrosilylation of NBD using PMDS or HMTS (Scheme 2). All experiments were performed up to the quantitative conversion of NBD in order to compare the selectivities correctly. In the presence of Rh- and Pt-catalysts the hydrosilylation of NBD by PMDS and HMTS proceeded at room temperature (Table 1). However, the reactions were not selective and the formation of three products was detected: exo- and endo-isomer of mono-silyl-substituted norbornene and mono-silyl-substituted nortricyclane. The composition of the products depended on the nature of the catalyst. Thus, in the case of the Rh-catalyst the endo-isomer was the major product and essential amounts of Si-containing nortricyclane were formed. The Pt-catalyst gave the exo-isomer as a major product and the content of nortricyclane in the mixture was significantly lower. Bis-silyl-substituted norbornane formed in trace amounts in all cases and it was easily separated by distillation.| Catalystb | Reaction temperature, °C | Reaction time, h | Ratio of products, % | ||
|---|---|---|---|---|---|
| exo-NB(Si) | endo-NB(Si) | NTC(Si) | |||
| a The reactions were conducted up to the quantitative conversion of NBD. The NBD/silane molar ratio was 1/1.1; Pd-loading was 0.1 mol%, Pt-loading was 0.05 mol%, Rh-loading was 0.05 mol%; molar ratio Pd/phosphine was 1/2; C(NBD) = 2.5 M in the reaction with PMDS and 2.0 M in the reaction with HMTS.b The structures of the catalysts and a ligand.c HMTS was used as a hydrosilane in the reaction. | |||||
| (acac)Rh(CO)2 | 25 | 48 | 33 | 48 | 19 |
| 48 | 32c | 48c | 20c | ||
| Pt2[(VinSiMe2)2O]3 | 25 | 48 | 55 | 37 | 8 |
| 48 | 75c | 24c | 1c | ||
| (AllPdCl)2/PPh3 | 75 | 72 | — | — | — |
| (AllPdCl)2/PCy3 | 75 | 72 | — | — | — |
| (AllPdCl)2/R-MOP | 75 | 48 | 76 | 0 | 24 |
 |
|||||
Simple Pd-based systems such as allylpalladium(II) chloride dimer ((AllPdCl)2) in combination with PPh3 or PCy3 unexpectedly turned out to be inactive for the hydrosilylation of NBD by PMDS even upon heating up to 75 °C. The change in the readily available phosphine by R-MOP in the Pd-catalyst allowed us to perform, for the first time, the selective and direct synthesis of silicon-substituted exo-norbornenes from NBD and chlorine-free silanes. It should be noted that although this catalytic system ((AllPdCl)2/R-MOP) reported previously was applied for the hydrosilylation of alkenes by various silanes,25,33,34 its activity in the hydrosilylation of NBD by inactivated silanes has not yet been studied. In contrast to the Pt- and Rh-containing systems considered above, (AllPdCl)2/R-MOP catalyzed hydrosilylation of NBD only upon heating. Traces of the products were detected at room temperature after a week. The activity of (AllPdCl)2/R-MOP increases with an increase in the temperature (see below). The usage of (AllPdCl)2/R-MOP as a catalyst allows the exo-isomer to be obtained selectively with a small content of nortricyclane and without traces of the endo-isomer (Table 1). This by-product is a saturated compound and will not affect the polymerization of the prepared Si-containing exo-isomers.26
In addition to (AllPdCl)2, Pd(OAc)2 was also estimated as a source of Pd. The activity of the Pd(OAc)2-containing system turned out to be higher at a low temperature than that of the (AllPdCl)2/R-MOP catalyst, but at 75 °C it becomes less active (Table 2). The possible reason for this is that there is less thermal stability for Pd(OAc)2; at higher temperatures it decomposes forming Pd(0) faster than (AllPdCl)2. As it was found in the case of the (AllPdCl)2/R-MOP catalyst, the Pd(OAc)2/R-MOP system also selectively gave the exo-isomer of norbornene without traces of the endo-isomer, while the saturated Si-substituted nortricyclane was a by-product.
| Catalyst | Temperature, °C | Conversion, % | Selectivity, % |
|---|---|---|---|
| a NBD/silane molar ratio was 1/1.1; Pd/R-MOP molar ratio was 1/2; Pd-loading was 0.1 mol%; C(NBD) = 2.5 M; the reaction time was 24 h. | |||
| (AllPdCl)2/R-MOP | 30 | 0 | — |
| 45 | 10 | 92 | |
| 60 | 55 | 86 | |
| 75 | 86 | 76 | |
| Pd(OAc)2/R-MOP | 30 | 0 | — |
| 45 | 24 | 87 | |
| 60 | 60 | 84 | |
| 75 | 70 | 74 | |
Therefore, among the studied Pt-, Rh-, and Pd-catalysts, only the latter exhibited 100% selectivity for exo-/endo-isomers formation and therefore it is promising to vary the composition of the Pd-based catalyst in order to evaluate the influence of the catalyst nature on its activity and selectivity.
Ligand screening
The dramatic difference in the catalytic activity of (AllPdCl)2/R-MOP, (AllPdCl)2/PPh3 and (AllPdCl)2/PCy3 displayed the significant influence of the ligand on the activity of the Pd-system in the hydrosilylation reaction of NBD. Therefore it was interesting to find other ligands which are suitable for the considered reaction. For this study the hydrosilylation of NBD with PMDS in the presence of (AllPdCl)2 was chosen as a model reaction. As R-MOP contains a phosphine group, an anisole moiety and a chiral binaphthyl framework, the screening was performed in three directions. Firstly, it was interesting to consider various phosphines as ligands for hydrosilylation (Fig. 1(I)). The reaction for the hydrosilylation of NBD by PMDS was studied at different temperatures from 25 to 75 °C. In contrast to PCy3 and PPh3, the usage of P(OEt)3 as a ligand led to the hydrosilylation products; the formation of the exo-silicon-substituted norbornene (16%) and nortricyclane (27%) was observed within 9 h at 75 °C. Nevertheless, this catalytic system was much less active and selective than the system containing R-MOP. The catalytic systems with aromatic phosphines (DPEphos, DPPF, Fig. 1(I)) proved to be inactive. The silyl-substituted norbornene and nortricyclane were not detected, even at high temperatures.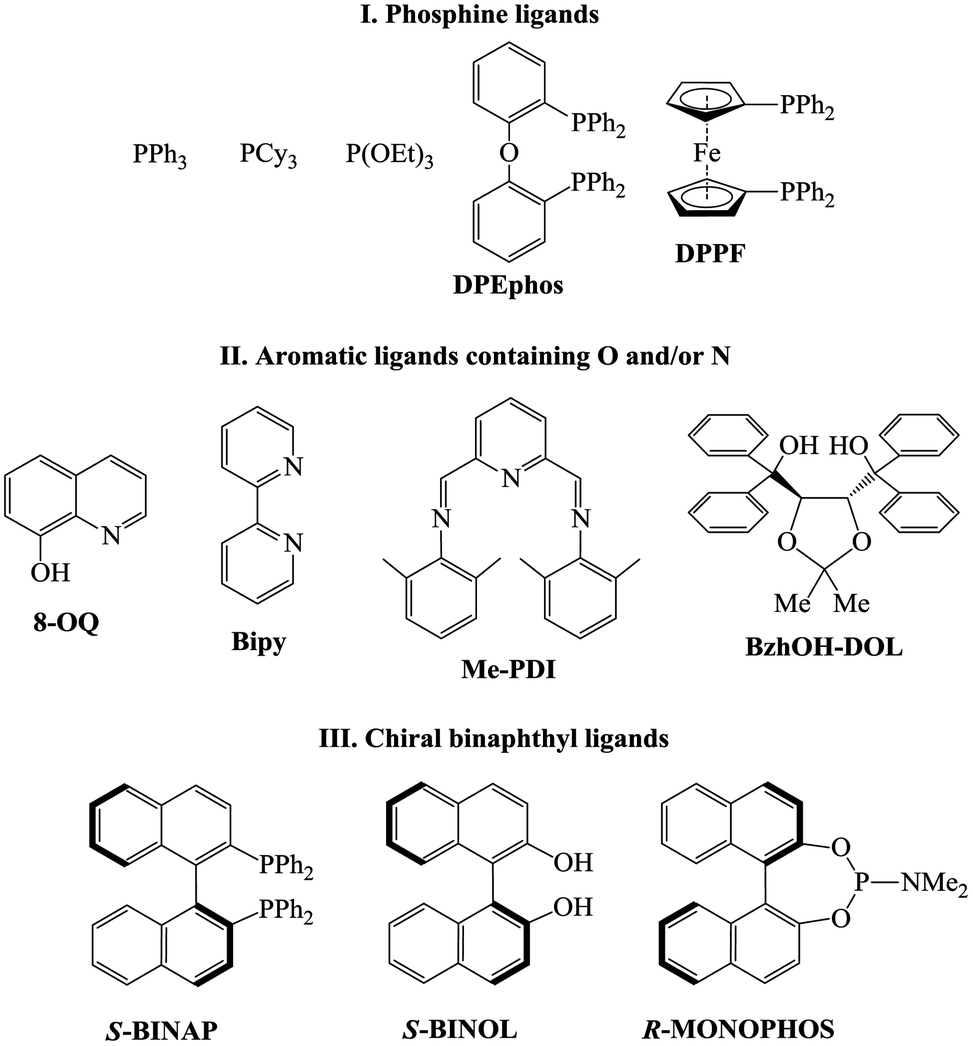 | ||
| Fig. 1 The ligands used for the hydrosilylation of norbornadiene-2,5. | ||
The second group of studied ligands is the aromatic compounds containing oxygen or nitrogen as a donor atom: 8-oxyquinoline, 2,2′-bipyridyl and others (Fig. 1(II)). The catalytic systems based on (AllPdCl)2 in combination with these ligands turned out to be active in the hydrosilylation of NBD by PMDS. In all cases the formation of silicon-substituted norbornene (pure exo-isomer) and nortricyclane was observed. For each ligand the dependence of the NBD conversion and the selectivity of exo-norbornene on time at a constant temperature (75 °C) was determined (Fig. 2). The activity of (AllPdCl)2 with a ligand from this group was found to be greater than that of (AllPdCl)2/R-MOP. The ligand containing a dioxalane fragment (BzhOH-DOL) provided the highest catalytic activity revealing quantitative conversion for several hours. The catalytic systems with ligands containing pyridine fragments (Bipy, Me-PDI, 8-OQ) were less active. One more interesting feature for this group of ligands is the presence of an induction period; during the few first hours, these systems did not exhibit catalytic activity (Fig. 2A). This can be explained by the slower formation of the active catalytic species with such ligands. The selectivity of exo-norbornene formation for these systems was lower (45–55%) than it was for (AllPdCl)2/R-MOP and generally tended to decrease with the increase of NBD conversion. The lowest selectivity was observed in the case of BzhOH-DOL and 8-OQ, the highest was for Bipy. As a result, all considered catalytic systems based on the second group of ligands disclosed a higher activity than that of the system containing R-MOP as a ligand, at the same time the selectivity of exo-norbornene formation was lower.
 | ||
| Fig. 2 Time plot of NBD conversion (a) and exo-norbornene formation selectivity (b) for the hydrosilylation of NBD by PMDS (at a ratio of 1/1.1) at 75 °C in the presence of (AllPdCl)2 with various ligands. | ||
If R-MOP as a ligand provides the highest selectivity, it is interesting to consider the influence of other similar ligands on the selectivity. Therefore, the third group of ligands are ones containing the chiral binaphthyl fragment: S-BINAP, S-BINOL and R-MONOPHOS (Fig. 1(III)). Despite S-BINAP and R-MOP possessing a similar structure, the system with S-BINAP surprisingly turned out to be inactive, even at high temperature (75 °C). This result for S-BINAP, a ligand containing only phosphines centers, is in a good agreement with the above described inactivity of Pd-systems with other phosphines (PPh3 or PCy3). The activities of the (AllPdCl)2 systems with R-MONOPHOS and R-MOP were close, while the (AllPdCl)2/S-BINOL catalyst displayed the highest activity among the ligands from the considered group (Fig. 3B). The higher activity of (AllPdCl)2/S-BINOL also agrees well with the observed highest activity of another system with an OH-containing ligand ((AllPdCl)2/BzhOH-DOL). For the catalysts with S-BINOL and R-MONOPHOS an induction period of 1–2 hours was found that differs with the R-MOP-containing system. The selectivities of the systems with the third group of ligands were moderate. Nevertheless, they also allowed exo-substituted norbornene to be obtained selectively without the endo-isomer (Fig. 3B).
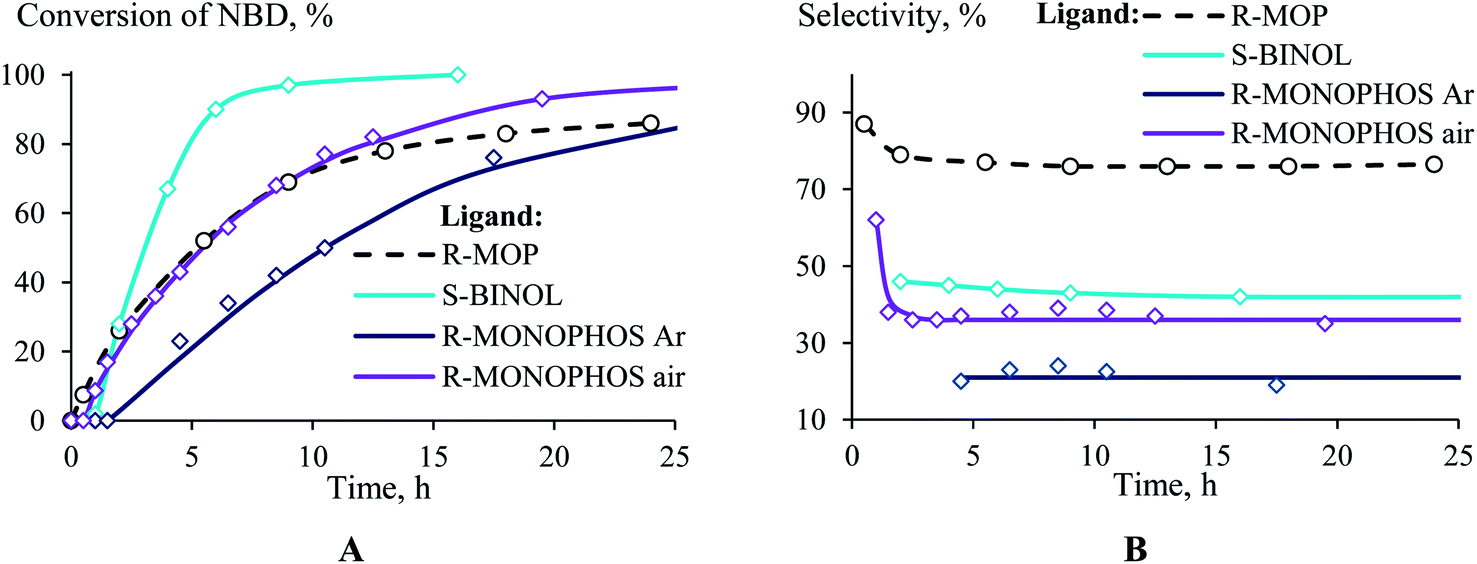 | ||
| Fig. 3 Time plot of the conversion (a) and selectivity (b) for the hydrosilylation of NBD by PMDS (in the ratio of 1/1.1) at 75 °C in the presence of (AllPdCl)2 with various binaphthyl-type ligands. | ||
The system with R-MONOPHOS was tested in an inert atmosphere (like all of the other ligands) and in air. In an inert atmosphere, the system containing R-MONOPHOS exhibited a reduced activity and provided the selective formation of substituted nortricyclane (the selectivity of the exo-norbornene formation was 20%). The conduction of the reaction in air exhibited a surprising effect, consisting of an increase in the conversion and exo-norbornene formation selectivity (Fig. 3). The time plot of the conversion for the R-MONOPHOS-containing system in air is similar to that of R-MOP, while the selectivity of exo-norbornene formation in air is still lower and close to that of the S-BINOL system (35% of the main product). Study of the reasons for the oxygen influence on the activity and selectivity of the catalyst system is now in progress.
The observed strong dependence of the Pd-catalyst activity from the nature of the ligand could be explained by the electron donating properties of the ligand. Thus, PPh3 and PCy3 are stronger electron donors than P(OR)3 and, therefore, they better compensate for the charge on the Pd-atom. The same is true for S-BINAP, containing only PPh2Ar groups. As a result, PAr3 or PAlk3 are not suitable as ligands for the catalysts of the considered reaction. P(OEt)3 and R-MOP contain less electron-donating phosphorous atoms and systems based on them are already active. When P(OEt)3 and R-MOP are replaced by an N-containing ligand, which are weaker electron donors, the activity of the catalytic systems again increases.
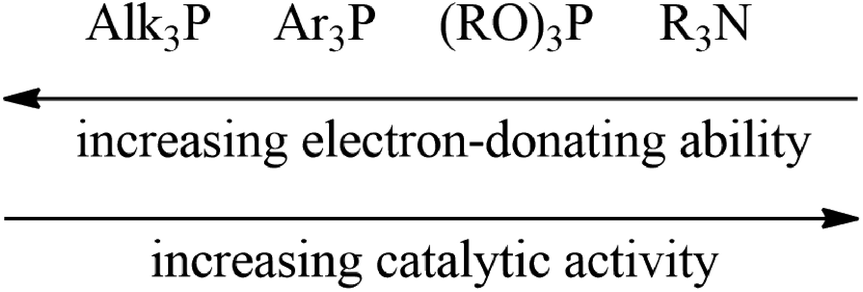
The electron-donating ability of the ligands also affects the selectivity. It seems that systems bearing a non-compensated charge on a transition metal have a greater tendency to give rearranged products (nortricyclane). For example, the system with a ligand from the second group (Fig. 2) has weaker electron donors than R-MOP and the selectivities for these systems are lower. For BZhOH-DOL and R-MONOPHOS a more complex influence can be realized, as well as the steric hindrances of the ligand which can also contribute to the activity and selectivity of the considered systems.
Therefore, among the three studied groups of ligands, the highest conversions of NBD during the hydrosilylation over Pd-based system were achieved with BzhOH-DOL and 8-OQ as a ligand, while the catalyst with the R-MOP ligand displayed a better selectivity at a moderate catalytic activity.
Effect of reaction temperature
As expected, the conversion and selectivity for hydrosilylation were found to depend on the reaction temperature; the former tends to increase with the increase of the temperature, and the latter to decrease (Table 3). At room temperature the reaction did not proceed; at 45 °C the reaction proceeded with low conversion and high selectivity; at 60 °C the conversion increased to a moderate level with the selectivity remaining high; at 75 °C the reaction proceeded with almost quantitative conversion and moderate selectivity.| Temperature, °C | Time, d | Conversion, % | Selectivity, % |
|---|---|---|---|
| a NBD/silane molar ratio was 1/1.1; (AllPdCl)2/R-MOP (1/4 mol. ratio) was the catalyst; Pd-loading was 0.1 mol%; C(NBD) = 2.5 M. | |||
| 25 | 1 | 0 | — |
| 2 | 0 | — | |
| 45 | 1 | 10 | 92 |
| 2 | 37 | 88 | |
| 60 | 1 | 55 | 86 |
| 2 | 77 | 84 | |
| 75 | 1 | 86 | 76 |
| 2 | 99 | 76 | |
Effect of hydrosilane nature
The structure of the hydrosilane was found to influence how the hydrosilylation proceeded. PMDS was more reactive than HMTS (Fig. 4A). This may be due to the difference in the steric hindrances of these silanes; in the case of HMTS, the activation of the Si–H bond as well as the insertion of the silyl-group to a carbon atom are complicated by the presence of two bulky Me3Si-groups. This probably decreases the hydrosilylation rate. Unlike the conversion, the selectivity, in general, did not depend on the nature of the monohydrosilane (PMDS or HTMS). The selectivity of the NBD hydrosilylation by HMTS was lower only at the beginning of the time period than it was when PMDS was used as the silane, but after 2.5 h the selectivities became equal (Fig. 4B).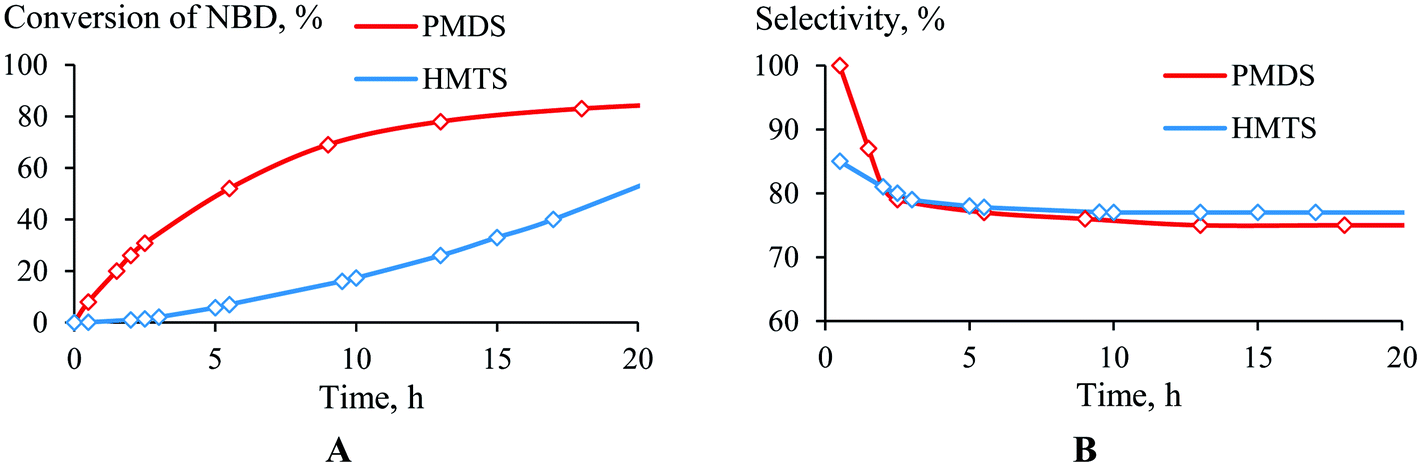 | ||
| Fig. 4 Time plot of the conversion (a) and selectivity (b) for the hydrosilylation of NBD by PMDS or HMTS (in the ratio of 1/1.1) at 75 °C in the presence of (AllPdCl)2/R-MOP. | ||
Proposed mechanism
There are two well-documented mechanistically different pathways for the metal-catalyzed hydrosilylation of alkenes. The first one is the Chalk–Harrod mechanism, it was described for Pt- and Pd-containing catalysts. According to the mechanism, an alkene is inserted into the metal–H-bond. The second one is the modified Chalk–Harrod mechanism, it was proposed for Rh-catalysts. According to this, an alkene is inserted into the metal–Si-bond. In the case of NBD as an alkene, these mechanisms become a little more complex because NBD can coordinate to the metal-center by different sides (Scheme 3). Thus, if the coordination occurs through the exo-face, only the exo-adduct is obtained. The coordination from the endo-face can be realized in two ways. In this case NBD can be considered as a monodentate or bidentate ligand. When NBD binds with a metal via a double bond from the endo-face, the endo-isomer of norbornene is obtained. At the same time, if NBD coordinates to a metal-center by two double bonds from the endo-face, this results in the formation of the nortricyclane derivative.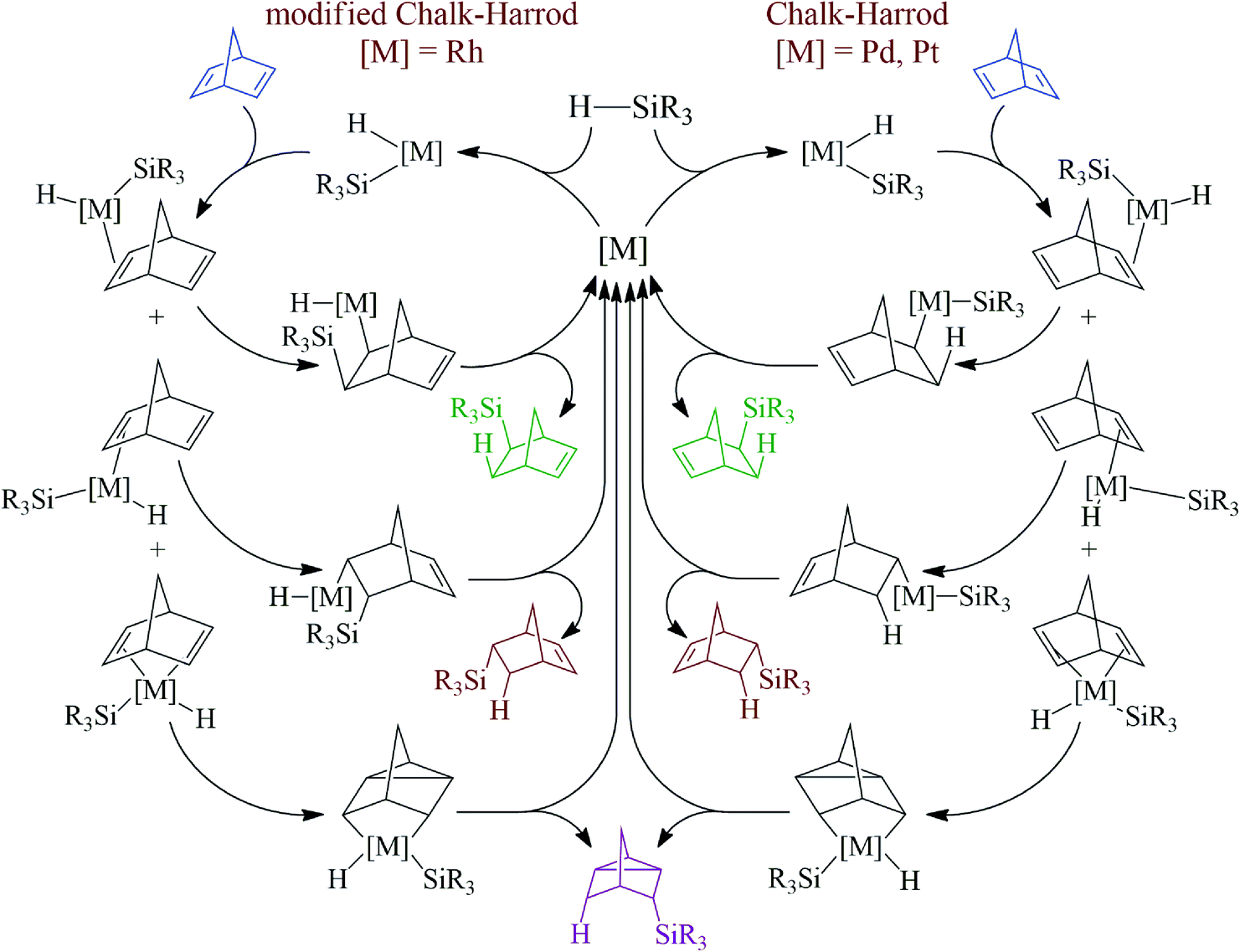 | ||
| Scheme 3 Proposed mechanism for the hydrosilylation of NBD by monohydrosilanes. | ||
An interesting finding was that all of the studied Pd-systems did not give an endo-isomer at all. Only exo-norbornene and nortricyclane were isolated as products of monohydrosilylation. This could be due to the fact that NBD tends to bind with the Pd-atom as a monodentate ligand via an exo-face or as a bidentate ligand via an endo-face, while for the Pt- and Rh-compounds the monodentate coordination of NBD from both the exo- and endo-face seems more preferable than the bidentate coordination of NBD from the endo-face. This is a possible reason for the observed difference in selectivity for the studied catalysts of the hydrosilylation reaction. Analysis of the literature data has also revealed just few Pd-complexes in which a norbornene derivative binds to a Pd-atom as a monodentate ligand from the endo-face35–38 and there are many examples in which the Pd-atom coordinates to a norbornene derivative from the exo-face.39–41
The role of the ligand in the mechanism could appear in certain stages. Firstly, Pd-complexes with less electron-donating ligands probably possess a more appropriate electron configuration to be inserted into the Si–H-bond than the same complexes with more electron-donating ligands. Secondly, the presence of stronger electron-donating ligands could make the complex more stable, which, in turn, complicates the coordination of NBD. Thirdly, similar effects take place on the stage when the π-complex of NBD with Pd transforms to the σ-one. This effect still remains unclear and study of this mechanism is currently in progress.
Conclusions
The direct and selective hydrosilylation of norbornadiene-2,5 by chlorine-free silanes was realized. This opens up a route to a simple, one-step and effective approach for the synthesis of exo-norbornenes bearing organosilicon substituents, which are promising monomers for the design of membranes, adhesives and gas-storage materials. The influence of the nature of the catalyst and the ligand structure on the catalytic activity and selectivity in the hydrosilylation reaction was evaluated. It has been shown that Pd-based catalytic systems provide the selective formation of exo-substituted norbornene without any trace of the endo-isomer, unlike Rh- and Pt-systems which are not selective. The activity and selectivity of the Pd-catalysts strongly depends on the nature of the catalyst. A higher activity was achieved for systems containing weak electron-donating ligands. The most selective Pd-system among the ones studied was found to be (AllPdCl)2/R-MOP. Using the examples of norbornadiene-2,5 hydrosilylation with pentamethyldisiloxane and 1,1,1,3,5,5,5-heptamethyltrisiloxane, the possibility of obtaining exo-isomers of norbornenes with 100% exo-/endo-selectivity was disclosed.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the Russian Science Foundation (grant no. 18-13-00415).References
- M. Zaranek and P. Pawluc, ACS Catal., 2018, 8, 9865–9876 CrossRef CAS.
- X. Du and Z. Huang, ACS Catal., 2017, 7, 1227–1243 CrossRef CAS.
- F. V. Drozdov, G. V. Cherkaev and A. M. Muzafarov, J. Organomet. Chem., 2019, 880, 293–299 CrossRef CAS.
- C. Drohmann, M. Möller, O. B. Gorbatsevich and A. M. Muzafarov, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 741–751 CrossRef CAS.
- R. J. Hofmann, M. Vlatković and F. Wiesbrock, Polymers, 2017, 9, 534 CrossRef.
- C. Wu, W. J. Teo and S. Ge, ACS Catal., 2018, 8, 5896–5900 CrossRef CAS.
- S. E. Parker, J. Börgel and T. Ritter, J. Am. Chem. Soc., 2014, 136, 4857–4860 CrossRef CAS.
- D. A. Alentiev, E. S. Egorova, M. V. Bermeshev, L. E. Starannikova, M. A. Topchiy, A. F. Asachenko, P. S. Gribanov, M. S. Nechaev, Y. P. Yampolskii and E. S. Finkelshtein, J. Mater. Chem. A, 2018, 6, 19393–19408 RSC.
- C. R. Maroon, J. Townsend, K. R. Gmernicki, D. J. Harrigan, B. J. Sundell, J. A. Lawrence, S. M. Mahurin, K. D. Vogiatzis and B. K. Long, Macromolecules, 2019, 52, 1589–1600 CrossRef CAS.
- T. Katsumata, M. Shiotsuki, F. Sanda and T. Masuda, Polymer, 2009, 50, 1389–1394 CrossRef CAS.
- H. Tetsuka, M. Hagiwara and S. Kaita, Polym. J., 2011, 43, 97–100 CrossRef CAS.
- P. Chapala, M. Bermeshev, L. Starannikova, V. Shantarovich, N. Gavrilova, V. Lakhtin, Y. Yampolskii and E. Finkelshtein, Macromol. Chem. Phys., 2017, 218, 1600385 CrossRef.
- P. P. Chapala, M. V. Bermeshev and N. N. Gavrilova, Polym. Sci., Ser. A, 2017, 59, 143–148 CrossRef CAS.
- E. V. Bermesheva, D. A. Alentiev, A. P. Moskalets and M. V. Bermeshev, Polym. Sci., Ser. B, 2019, 61, 314–322 CrossRef CAS.
- N. R. Grove, P. A. Kohl, S. A. Bidstrup-Allen, R. A. Shick, B. L. Goodall and S. Jayaraman, MRS Proceedings, 1997, 476, 3–8 CrossRef CAS.
- M. V. Bermeshev and P. P. Chapala, Prog. Polym. Sci., 2018, 84, 1–46 CrossRef CAS.
- S. D. Tsai and R. A. Register, Macromol. Chem. Phys., 2018, 219, 1800059 CrossRef.
- M. V. Bermeshev and E. S. Finkelshtein, INEOS OPEN, 2018, 1, 39–54 CrossRef , http://ineosopen.org/2018_issue_1.
- P. P. Chapala, M. V. Bermeshev, V. G. Lakhtin, A. M. Genaev, A. N. Tavtorkin and E. S. Finkelshtein, Mendeleev Commun., 2015, 25, 344–345 CrossRef CAS.
- M. Bermeshev, P. Chapala, V. Lakhtin, A. Genaev, M. Filatova, A. Peregudov, K. Utegenov, N. Ustynyuk and E. Finkelshtein, Silicon, 2015, 7, 117–126 CrossRef CAS.
- B. A. Bulgakov, M. V. Bermeshev, D. V. Demchuk, V. G. Lakhtin, A. G. Kazmin and E. S. Finkelshtein, Tetrahedron, 2012, 68, 2166–2171 CrossRef CAS.
- M. Bermeshev, B. Bulgakov, L. Starannikova, G. Dibrov, P. Chapala, D. Demchuk, Y. Yampolskii and E. Finkelshtein, J. Appl. Polym. Sci., 2015, 132, 41395 CrossRef.
- H. Tetsuka, K. Isobe and M. Hagiwara, Polym. J., 2009, 41, 643–649 CrossRef CAS.
- E. Finkelshtein, M. Gringolts, M. Bermeshev, P. Chapala, and Y. Rogan, Polynorbornenes, in Membrane Materials for Gas and Vapor Separation, ed. Y. Yampolskii, and E. Finkelshtein, Wiley, Chichester, 2017, pp. 143–221 Search PubMed.
- Y. Uozumi, L. Sang-Yong and T. Hayashi, Tetrahedron Lett., 1992, 33, 7185–7188 CrossRef CAS.
- D. A. Alentiev, M. V. Bermeshev, L. E. Starannikova, E. V. Bermesheva, V. P. Shantarovich, V. G. Bekeshev, Y. P. Yampolskii and E. S. Finkelshtein, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 1234–1248 CrossRef CAS.
- H. G. Kuivila and C. R. Warner, J. Org. Chem., 1964, 29, 2845–2851 CrossRef CAS.
- A. J. Cornish, M. F. Lappert and T. A. Nile, J. Organomet. Chem., 1977, 136, 73–85 CrossRef CAS.
- M. Stosur and T. Szymańska-Buzar, J. Mol. Catal. A: Chem., 2008, 286, 98–105 CrossRef CAS.
- H. Wen, K. Wang, Y. Zhang, G. Liu and Z. Huang, ACS Catal., 2019, 9, 1612–1618 CrossRef CAS.
- B. Marciniec, Comprehensive Handbook on Hydrosilylation, Pergamon, Oxford, 1992 Search PubMed.
- T. Hayashi, and K. Yamasaki, 10.18 – C–E Bond Formation Through Asymmetric Hydrosilylation of Alkenes, in Comprehensive Organometallic Chemistry III, D. M. P. Mingos, and R. H. Crabtree, ed. Elsevier, Oxford, 2007, pp. 815–838 Search PubMed.
- Y. Uozumi, K. Kitayama and T. Hayashi, Tetrahedron: Asymmetry, 1993, 4, 2419–2422 CrossRef CAS.
- K. Kitayama, H. Tsuji, Y. Uozumi and T. Hayashi, Tetrahedron Lett., 1996, 37, 4169–4172 CrossRef CAS.
- D. Meinhard, F. Hollmann, W. Huhn, U. Thewalt, M. Klinga and B. Rieger, Organometallics, 2004, 23, 5637–5639 CrossRef CAS.
- A. K. Hearley, R. J. Nowack and B. Rieger, Organometallics, 2005, 24, 2755–2763 CrossRef CAS.
- R. P. Hughes and J. Powell, J. Organomet. Chem., 1973, 60, 427–441 CrossRef CAS.
- A. L. Safir and B. M. Novak, Macromolecules, 1995, 28, 5396–5398 CrossRef CAS.
- B. Commarieu and J. P. Claverie, Chem. Sci., 2015, 6, 2172–2181 RSC.
- M. D. Walter, P. S. White and M. Brookhart, Chem. Commun., 2009, 6361–6363 RSC.
- K.-H. Yu, S.-L. Huang, Y.-H. Liu, Y. Wang, S.-T. Liu, Y.-C. Cheng, Y.-F. Lin and J.-T. Chen, Molecules, 2017, 22, 1095 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra06784a |
| This journal is © The Royal Society of Chemistry 2019 |