DOI:
10.1039/C9RA06771J
(Paper)
RSC Adv., 2019,
9, 30707-30714
Engineering an enhanced thrombin-based GLP-1 analog with long-lasting glucose-lowering and efficient weight reduction
Received
27th August 2019
, Accepted 20th September 2019
First published on 27th September 2019
Abstract
Peptides are considered as potent therapeutic drugs primarily due to the exquisite potency and selectivity to targets. However, the development and clinical application of peptide drugs were severely limited by the poor in vivo lifespans. Here, we designed an improved small albumin-binding polypeptide that can associate with human serum albumin (HSA) and liberate the bioactive peptide. Using glucagon-like peptide-1 (GLP-1) as a model, two new long-lasting GLP-1 analogs (termed XTS1 and XTS2) containing an albumin-binding domain, a protease-cleavable linker and a mutated GLP-1(A8Aib) were designed to demonstrate the sustained release of GLP-1 due to the plasma thrombin (TBN) digestion. Two XTS peptides were prepared of high purity (>99%) and accurate molecular weight determined by reversed high-performance liquid chromatography and mass spectrometry, respectively. In vitro measurements of surface plasmon resonance indicated that XTS1 associate with serum albumins of all species with higher affinity compared with XTS2. Metabolic stability of XTS1 in vitro in human plasma was also better than that of XTS2. Protease cleavage assay results of XTS peptides demonstrated the controlled-release of transient GLP-1 from the XTS1 and XTS2 mixture after thrombin-catalyzed hydrolysis. Then the intraperitoneal glucose tolerance test (IPGTT) showed that the glucose-lowering efficacies of XTS1 were in a dosage-dependent manner within the range of 0.1–0.9 mg kg−1. In addition, XTS1 showed similar hypoglycemic intensity and significantly longer action duration compared to Liraglutide in both multiple IPGTTs and hypoglycemic duration test. Apparently extended plasma half-lives of ∼2.3 and ∼3.5 days were observed after a single subcutaneous administration of XTS1 (0.9 mg kg−1) in rats and cynomolgus monkeys, respectively. Furthermore, twice-weekly subcutaneously dosed XTS1 in db/db mice achieved long-term beneficial effects on body weight, hemoglobin A1C (HbA1C) lowering and the function of pancreatic beta cells. These studies support that XTS1 exerts potential as a therapeutic drug for the treatment of T2DM.
Introduction
Peptides are considered as effective therapeutic agents because of their low immunogenicity, small molecular weight and outstanding specificity.1–3 Especially for the therapeutics based on the hormone peptides which deliver superior therapeutic benefits while displaying exquisite specificity for receptors.4–6 Therefore, there are numerous peptides that have already been used for the development of drugs.7,8 However, the application prospects and actual therapeutic efficacies of peptide drugs are severely limited by the short duration of action in vivo, primarily due to enzyme hydrolysis and the rapid glomerular filtration.9,10
Extending the in vivo exposure of the peptide therapeutic has focused on enlarging molecular size by fusing peptides to molecular scaffolds, such as large natural proteins including albumin11,12 and Fc,13,14 or to a certain domain which can associate with albumin, showing potential for extending the lifespans of peptides.15–18 HSA was widely used as the in vivo molecule transporter due to its wide tissue distribution, long half-life (∼19 days) resulting from neonatal Fc receptor mediated cell recycling and large molecular size. Thereby the peptide drugs, association tightly with serum albumin, will have longer in vivo half-lives compared with the unbound ones.19,20
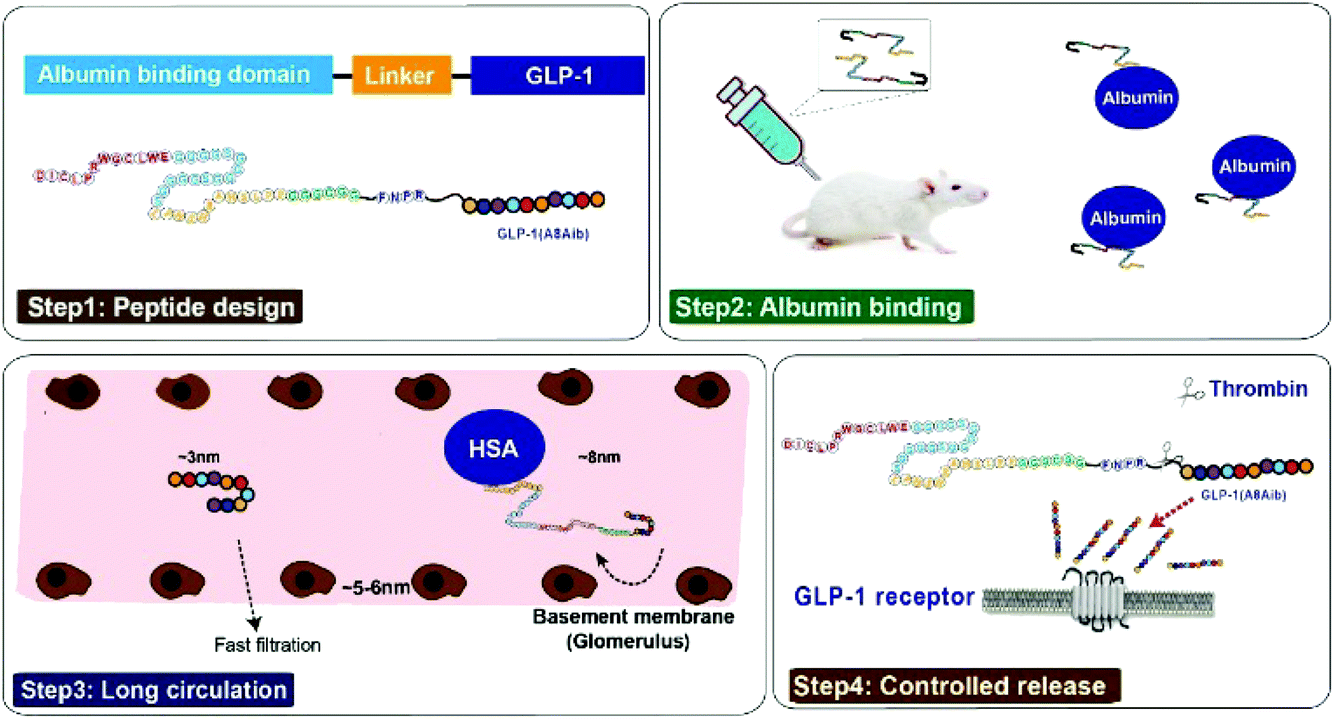
Based on the previous research,21 our strategy for half-life extension was focused on the fusion of peptide drugs with albumin binding peptides that display high affinity for albumins. But the reported results indicated that these fusion peptides, containing only single albumin binding domain, exert only about an affinity of ∼1 μM for HSA, and the sequence of released natural GLP-1 includes DPP-IV digestion site, both of which leading to a short in vivo half-life (no more than 12 hours).22,23 In order to develop the antidiabetic agents with longer duration of action, we chose two previously reported albumin binding peptides with highest affinity for HSA in a tandem array, and remove the DPP-IV digestion site by replacing the 8th alanine with 2-aminoisobutyric acid, a non-natural amino acid with outstanding proteolytic stability, to construct new pro-drugs. When our newly design fusion peptides were subcutaneously administrated into the blood circulation, they will rapidly bind to the albumin which can protect them from elimination action from kidney and proteolytic degradation.21–23 The protease-cleavable linker will be consequentially scissile to TBN hydrolysis and release the mutated GLP-1(A8Aib) (Scheme 1).
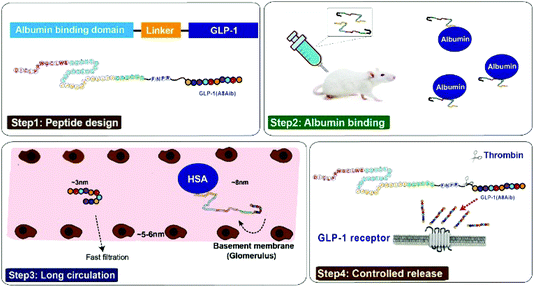 |
| | Scheme 1 Structure and the prolonged persistence mechanism of XTS peptides in vivo. | |
In the present study, we aimed to prepare and demonstrate the XTS peptides showing high efficiency on the glucose-stabilizing and diabetes-protective. Both the preliminary pharmacodynamics and pharmacokinetics characteristics of XTS peptides were also carefully studied in db/db mice and cynomolgus monkeys, respectively.
Experimental
Materials and animals
Synthetic Liraglutide was obtained by GL Biochem Technology Co. (Shanghai, China) which use a traditional solid phase method. Albumins of different species were purchased from Abcam (Shanghai, China). 6–8 weeks old db/db mice were obtained from Cavins Biotechnology Co., Ltd. (Changzhou, China). Male cynomolgus monkeys (Macaca fascicularis) were obtained from Xishan Zhongke Drug Research and Development Co., Ltd. (Suzhou, China) and housed in a conventional facility room. All the animals were acclimated to the controlled ambient conditions of 12 h light/dark cycle and free access to water. Animal care and use committee of the WuXi AppTec. Laboratories (Wuxi, China) approved the animal experiments and the approval codes was ACU19-122 and ACU18-271.
Design, preparation and identification of XTS1 peptide
Design strategy of our peptides was the N-terminal extension of GLP-1(A8Aib) with 39 amino acid sequence of albumin binding domain (Table 1) via a cleavable linker. All the peptides using in this research were synthesized using a prelude automated peptide synthesizer (Protein Technology Inc., America) according to the standard solid phase peptide synthetic manual.24 XTS1/2 were subjected to high performance liquid chromatography (HPLC) on a C18 column (Agilent, America, 4.6 × 250 mm, 5 μm) and detection wavelength was 280 nm. Mass spectrometry analysis of XTS1/2 was performed on a MALDI-TOF-MS AXIMA CFR (Kratos Analytical, Japan).
Table 1 SPR measurements. SPR binding results for XTS1 interaction with serum albumin of different species
| Parameters |
Mouse |
Rat |
Monkey |
Human |
| XTS1 |
k
a (M−1 s−1) |
2.04 × 104 |
3.25 × 104 |
4.18 × 104 |
7.46 × 104 |
|
k
d (s−1) |
6.36 × 10−2 |
4.72 × 10−2 |
9.41 × 10−2 |
1.05 × 10−2 |
|
K
D (M) |
3.13 × 10−6 |
1.45 × 10−6 |
2.25 × 10−7 |
1.41 × 10−7 |
| XTS2 |
k
a (M−1 s−1) |
1.04 × 104 |
2.54 × 104 |
2.27 × 104 |
3.58 × 104 |
|
k
d (s−1) |
8.51 × 10−2 |
5.55 × 10−2 |
7.24 × 10−2 |
2.54 × 10−2 |
|
K
D (M) |
8.11 × 10−6 |
2.18 × 10−6 |
3.18 × 10−7 |
7.09 × 10−7 |
Albumin binding affinity measurements
Characterization of the binding of the synthesized peptides to albumins were conducted by surface plasmon resonance measurements using BIAcore T200 system (General Electric Company, America) for evaluating XTS1′ and XTS2′ affinity for albumins of mouse, rat, monkey and human. Operational details were conducted following the manufacturer's instructions. We fixed the different albumins to the CM5 chips in a fixed level of ∼100 RU and then the XTS1 and XTS2 peptides were furtherly diluted to six gradients (0, 0.11, 0.33, 1, 3, 9, 27 nM) and then flow through the chips with different albumins at a flow rate of 30 μL min−1 under a model of binding (300 s) and dissociating (300 s) using HBS-EP+ as assay buffer at 25 °C. The data fitting procedures were briefly introduced in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 binding model after the double deductions.
:1 binding model after the double deductions.
Protease cleavage assay
XTS peptides samples with or without human serum albumin were dissolved in 0.1 mL PBS buffer (pH 7.4) at a final concentration of 1 mg L−1. Aliquot of human thrombin (0.4 U mL−1) was added to the mixture and then the reaction mixture was fractionated into ten equal parts and incubated at 37 °C for 72 hours in dark. One sample at a time was taken out at 0, 2, 4, 8, 16, 24, 48 and 72 hours, and the reaction was stopped by adding 0.01 mL 0.1% TFA, followed by analysis ELISA and mass spectrometry method.
Plasma stability assay
XTS1 and XTS2 were dissolved in human plasma with or without albumin at a final concentration of 1 mg L−1. The mixture were further incubated at 37 °C for 72 hours. At 0, 1, 2, 4, 8, 16, 24, 48 and 72 h, the collected samples were furtherly analyzed by the above mentioned methods.
Intraperitoneal injection glucose tolerance test
A modified IPGTT was conducted in the db/db mice after overnight fasting. The mice received a single subcutaneous injection of placebo, Liraglutide (0.1 mg kg−1), three dosages of XTS1 (0.1, 0.3 and 0.9 mg kg−1). Animals were administrated with glucose (2.0 g kg−1) after the intraperitoneal injection of Liraglutide or XTS1. Meanwhile, the blood glucose levels (BGLs) were measured using a glucometer at 0, 0.25, 0.5, 1, 2 h after each administration of glucose. Multiple IPGTTs were furtherly conducted at an interval of 30 hours after first administration of saline, Liraglutide (0.1 mg kg−1), three dosages of XTS1 (0.3 and 0.9 mg kg−1). Area under the curves of each IPGTT were also calculated to evaluate the anti-hyperglycemic effects.
Hypoglycemic efficacies in non-fasted db/db mice
Male 4–6 weeks old db/db mice were subcutaneously injected of saline, Liraglutide (0.1 mg kg−1), XTS1 (0.1, 0.3 and 0.9 mg per kg body wt). The blood samples were collected from animal's tail veins at various time-points (0, 0.5, 1, 2, 4, 8, 24, 48, 72 h) and detected using a glucometer.
Pharmacokinetic studies in SD rats and cynomolgus monkeys
SD rats and cynomolgus monkeys were subcutaneously administrated with XTS1 at a dose of 0.3 mg kg−1 and 0.9 mg kg−1. Blood samples were collected from the right forelimb veins pre-dose and at 0.08, 0.5, 1, 2, 4, 8, 24 h post-dose and on days 2, 3, 4, 5, 6, and 7. Plasma samples were separate and quickly frozen by liquid nitrogen. XTS1 concentration in plasma were measured using a liquid chromatography tandem mass spectrometry of which the detection range was 0.1 to 50 μg mL−1.
Chronic in vivo studies
Forty male db/db mice (6–8 weeks old) with average body weight and fasted BGL were assigned to four groups. Animals in each group were once daily subcutaneous (s.c.) injected with saline, Liraglutide (0.1 mg kg−1), XTS1 (0.3 and 0.9 mg per kg body wt) for consecutive 8 weeks. Body weight and the consumption of water and food were measured daily. IPGTT tests without administration of either XTS1 or Liraglutide were conducted at week 1 and 8. HbA1c value and other blood biochemical indexes, including total cholesterol (TC), triglyceride (TG), high-density lipoprotein (HDL), low-density lipoprotein (LDL), alanine aminotransferase (ALT), aspartate aminotransferase (AST) were detected by using automatic biochemical analyzer after the experimental period. After the 8 week treatment, pancreatic tissues from diabetic mice were carefully isolated and processed according to the commonly used procedures, and furtherly stained with hematoxylin–eosin (H&E) according the previous reported method.25
Results
Design, preparation and identification of XTS1 peptides
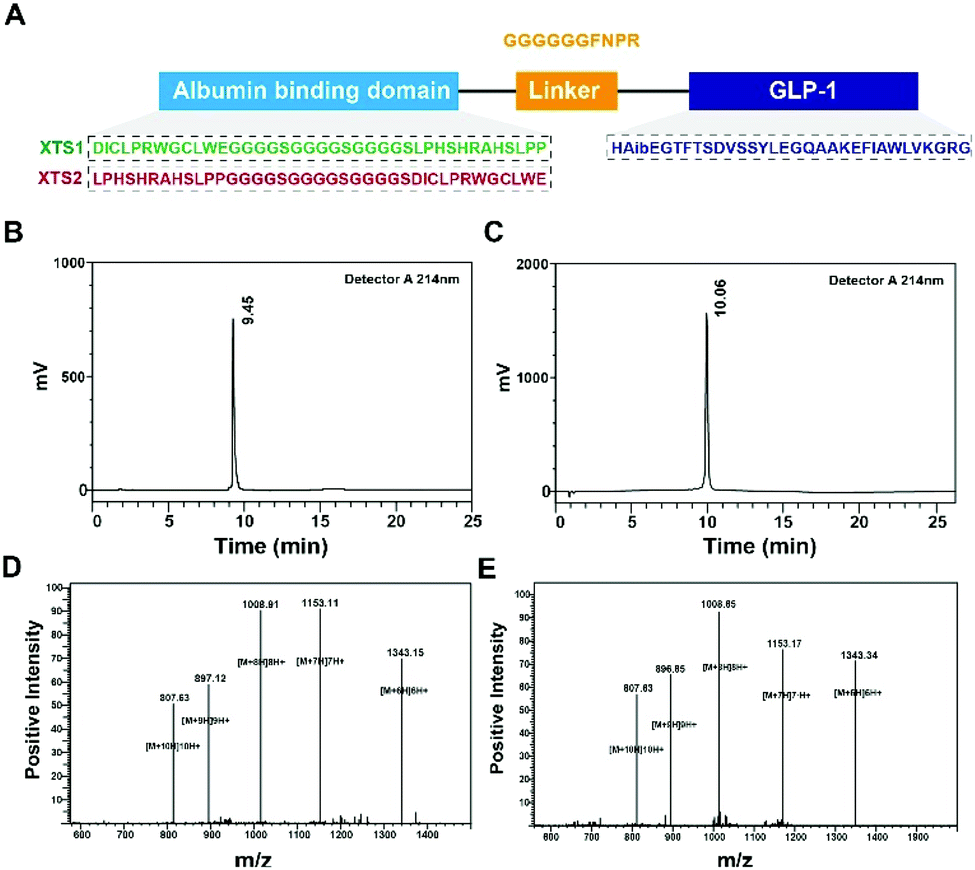
Few albumin binding domains were exploitable that either selected from peptide libraries including synthetic and natural peptide library or exist naturally.19,26,27 In order to extend the medical application of the existing ABDs, more construction modes of ABD-based molecules should be tried. According to the previous research,21 nine 12-mer ABD peptides were identified to exhibit high affinity for HSA. We selected and attached two domains (SEQ1: DIOCLPRWGCLWE and SEQ2: LPHSHRAHSLPP) with higher HSA binding affinity using a (G4S)3 linker to generate two fully new ABD sequences (Fig. 1A). We furtherly fused the two domains to the C-terminal of GLP-1 using a sequence containing flexible linker (GGGGGG) and TBN-cleavable sites (FNPR) to ensure the enough space for binding to GLP-1 receptor (GLP-1R) and controlled release of GLP-1. We also removed the DPP-IV digestion site of natural GLP-1 by replacing the 8th alanine with 2-aminoisobutyric acid, a non-natural amino acid with outstanding proteolytic stability. Therefore, it was expected that the introduction of two ABDs and mutation of GLP-1 would be beneficial for its more long-lasting in vivo glucose-lowering compared with previous reported GLP-1 fusion peptides. As a result, two fusion peptides (termed XTS1 and XTS2) due to the different position of SEQ1 or SEQ2 were successfully constructed (Fig. 1A).
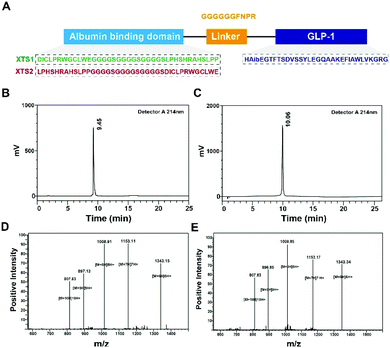 |
| | Fig. 1 Identification of XTS1 peptides. Schematic illustration of peptide design (A); HPLC chromatogram of purified XTS1 (B) and XTS2 (C); mass spectra analysis of (D) XTS1: 8064.77 Da and (E) XTS2: 8065.19 Da. | |
Both two peptides were prepared using a solid phase synthesizing method15,24 and further identified by mass spectrometry after HPLC purification. The peptide purity were both over 99% (Fig. 1B and C) and exact molecular weight (Fig. 1D and E) which were consistent with the theoretical value (XTS1: 7415.17 Da; XTS2: 7415.45 Da).
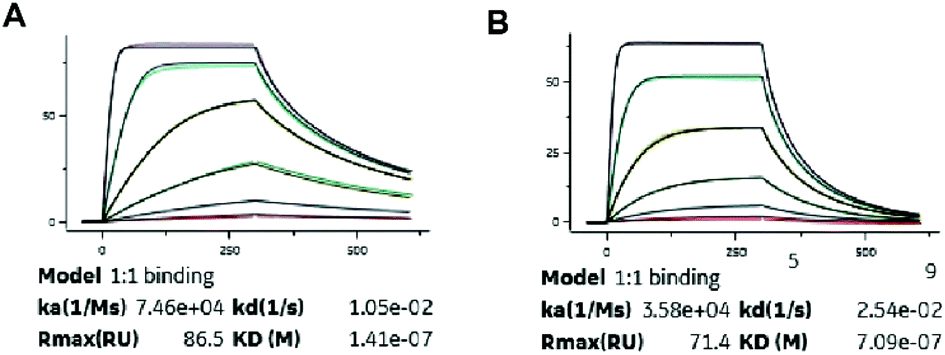
Albumin binding affinity
Binding constants of XTS1 and XTS2 for human and others species, containing association rate constant ka (M−1 s−1), dissociation rate constant kd (s−1) and overall affinity constant KD (M), were presented in Fig. 2 and Table 1, respectively. These results showed that XTS1 associate with all species albumins with higher affinity than XTS2, especially in human (1.41 × 10−7 M vs. 7.09 × 10−7 M). In addition, affinity of both two XTS1 peptides for monkey serum were obvious higher than mouse, rat and similar to human, indicating that pharmacokinetics parameters of XTS1 or XTS2 peptides in monkey were more closely to that in human compared with other species. Compared to the previous albumin binding affinity results,21 stringing SEQ1 and SEQ2 exert almost ten-fold higher affinity for HSA than the single SEQ1 or SEQ2 (1.41 × 10−7 M vs. 1.12 × 10−6 M or 1.67 × 10−6 M).
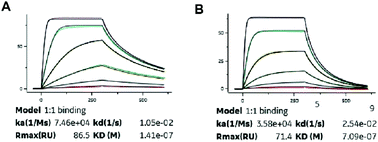 |
| | Fig. 2 SPR measurements of binding affinity of (A) XTS1 and (B) XTS2 for human serum albumin. | |
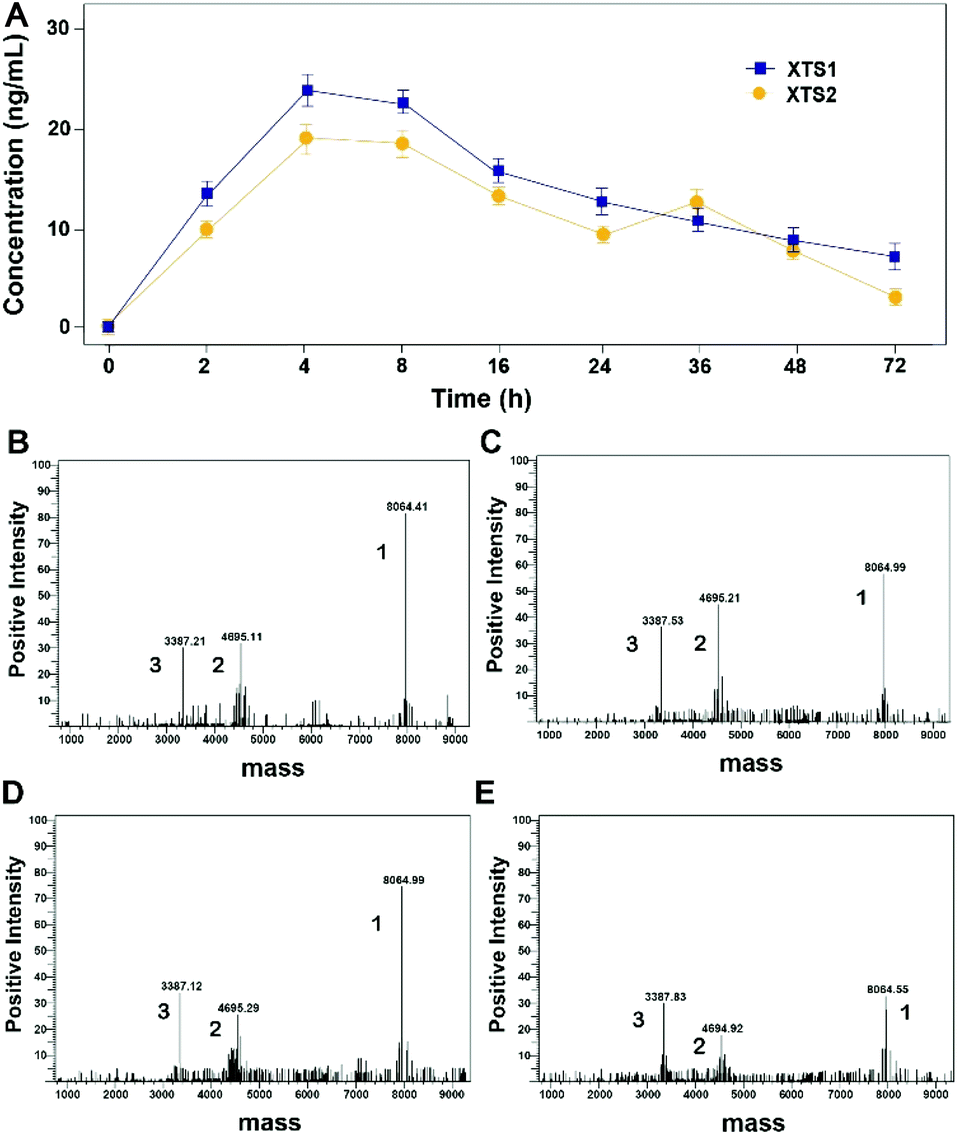
Protease cleavage assay of XTS peptides
As evidenced in Fig. 3, transient GLP-1 peptide can be released from XTS1 and XTS2 via plasma TBN. The amount of released GLP-1 from the XTS1 and XTS2 with TBN-cleavable linkers varies as a function of time during proteolysis, monitored with antibody specifically against GLP-1 and the concentration of released GLP-1 were showed in Fig. 3A. In vitro, the transient existence of three forms of products including intact XTS1 or XTS2, GLP-1(A8Aib), and ABD-GGGGGGFNPR in the TBN-catalyzed reactions were also confirmed by mass spectrometry (Fig. 3B).
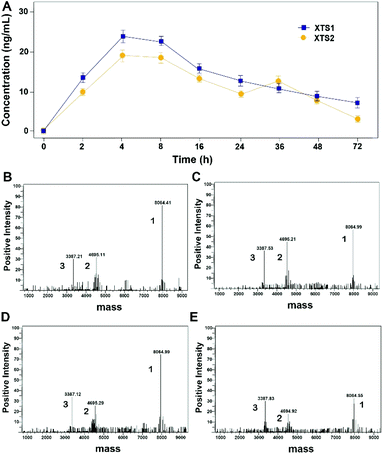 |
| | Fig. 3 Slowly-releasing of GLP-1 from designed XTS fusion peptides in vitro. (A) The transient released GLP-1 concentration–time curve. The mass spectrometric profiles also shows for the hydrolysis of XTS1 at 2 h (B) and 72 h (C) or XTS2 at 2 h (D) and 72 h (E) by TBN. Peak 1, 2 and 3 refer to XTS polypeptide, ABD + linker and GLP-1(A8Aib), respectively. | |
Plasma stability of XTS peptides
The in vitro plasma stability test of XTS1 and XTS2 with or without HSA were furtherly performed. As is showed in Fig. 4, XTS1 and XTS2 had an approximate half-life of 8.1 and 8.6 h, respectively, in human plasma without albumin at 37 °C. As expected, XTS1 exert a nearly 5-fold longer half-life (t1/2 = 36.4 h) in human plasma with albumin which longer than the XTS2 (t1/2 = 25.1 h).
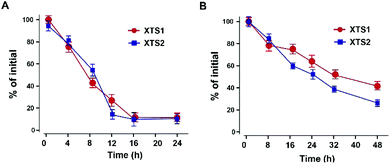 |
| | Fig. 4 Degradation profiles of XTS1 and XTS2 under incubation in human plasma without (A) or with (B) serum albumin. All data are expressed as mean ± SD (n = 5). | |
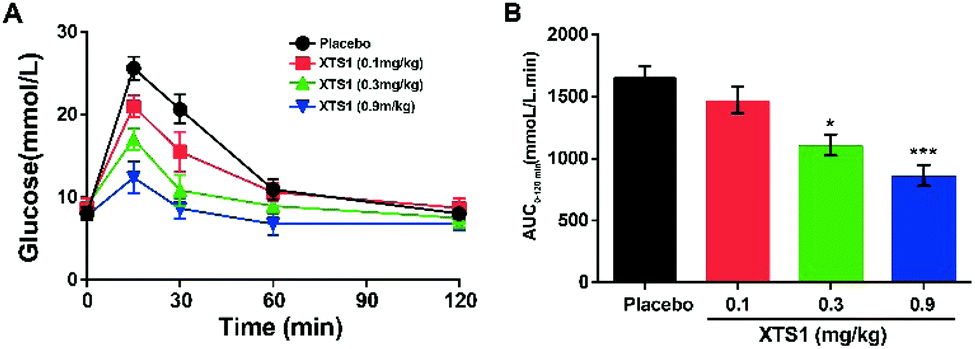
Dose dependency of XTS1 peptides on glucose-lowering in fasted db/db mice
As is described in Fig. 5A and B, IPGTT was conducted after a single subcutaneously administration of XTS1 peptides (0.1, 0.3, 0.9 mg kg−1). All the three dosages of XTS1 exert significantly hypoglycemic efficacies with a lowering percentage of 48.5% for 0.9 mg kg−1 dosage, 35.1% for 0.3 mg kg−1 dosage, and 13.5% for 0.1 mg kg−1 dosage, respectively, compared with the placebo group (Fig. 5B). These results indicate that the glucose-lowering ability of XTS1 was in clearly dosage-dependent way within the range of 0.1–0.9 mg kg−1.
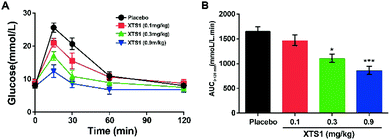 |
| | Fig. 5 Dose-dependence of XTS1 peptide lowering BGL in diabetic mice. (A) IPGTT time-course average BGLs of diabetic mice with placebo, XTS1 (0.1, 0.3, 0.9 mg kg−1) in fasted db/db mice. (B). Areas under curve (AUC) of the BGLs within 0–120 min after treatment. P < 0.05, 0.02, 0.001 using one-way ANOVA (*, **, ***) vs. placebo. All data are expressed as mean ± SD (n = 8). | |
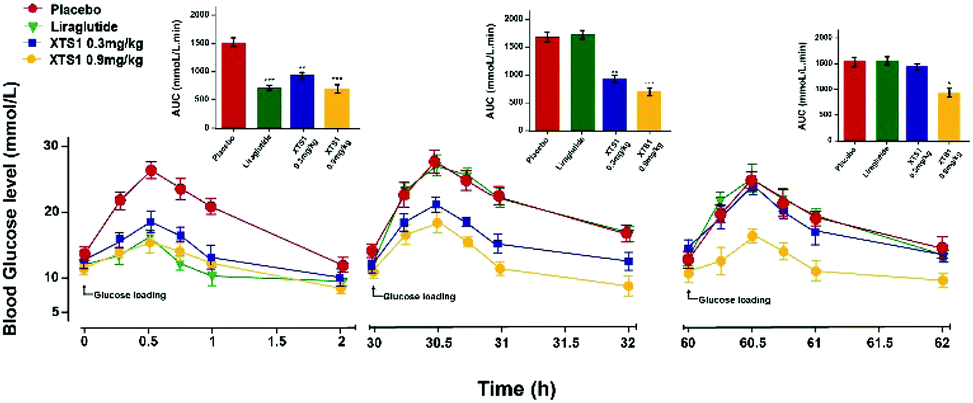
Sustain glucose-stabilizing capabilities of XTS1 peptide in fasted db/db mice
The sustained glucose-stabilizing abilities of XTS1 to lower the blood glucose levels (BGLs) in fasted db/db mice were evaluated by performing a method of modified multiple IPGTTs. The mice received a single intraperitoneal injection of saline, Liraglutide (0.1 mg kg−1), XTS1 (0.3, 0.9 mg kg−1) half an hour before first glucose challenge. The BGLs of saline treated group rapidly reached peak at half an hour and then slowly got back to the base line level within 2 hours after each round administration of glucose (Fig. 6). While the glucose-stabilizing capabilities of XTS1 at both two dose (0.3, 0.9 mg kg−1) were obviously better than that by saline due to the insulinotropic bioactivities. During the IPGTT (30–32 h), the BGLs of XTS1 treated mice were slightly higher than before but remained significantly lower than the control or Liraglutide. In third IPGTT (60–62 h), 0.9 mg kg−1 XTS1 still could significantly lower the BGLs compared with saline. At 60 hours, both two dosages of XTS1 still exert glucose-lowering effects with a glucose-lowering percentage of 40.2% for 0.9 mg kg−1 dosage and 8.2% for 0.3 mg kg−1, respectively, compared with saline-treated ones, while the Liraglutide was almost ineffective at 30 hours (Fig. 6).
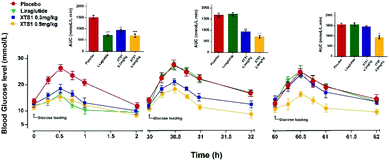 |
| | Fig. 6 Glucose-stabilizing capabilities of XTS1 were studied using a modified multiple IPGTTs in male diabetic mice. Multiple IPGTTs results time-course average BGLs and AUC values of db/db mice with saline, Liraglutide (0.1 mg kg−1) and XTS1 (0.3, 0.9 mg kg−1) for 60 h. P < 0.05, 0.02, 0.001 using one-way ANOVA (*, **, ***) vs. placebo. All data are expressed as mean ± SD (n = 8). | |
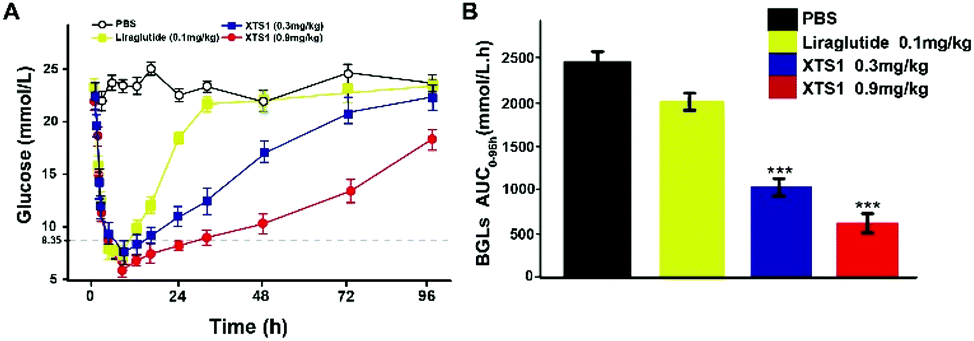
Hypoglycemic duration test
The long-term anti-hyperglycemic activity of XTS1 was further investigated by accessing its hypoglycemic duration in non faceted db/db mice. Using Liraglutide (0.1 mg kg−1) as positive control, two doses of XTS1 (0.3 mg kg−1 and 0.9 mg kg−1) were employed. After the subcutaneous administration, the BGLs of XTS1 groups (0.3 mg kg−1 and 0.9 mg kg−1) both decreased rapidly to the normoglycemic level (less than 16.8 mmol L−1) within 1 h (Fig. 7A). As evidenced by the data shown in Fig. 7A, the lowest glucose levels reached in XTS1 treated group were similar to Liraglutide treated mice, but increased relatively slower. The sustaining time of which the BGLs were under 8.35 mmol L−1 was considered as the hypoglycemic duration. The BGLs in placebo treated mice maintained a hyperglycemic state during the whole experiment and the treatment of XTS1 at two dose (0.3 mg kg−1 and 0.9 mg kg−1) normalized the BGLs for 16.5 h and 33.1 h, respectively, which significant longer than Liraglutide. The glucose-lowering effect of XTS1 at two doses (0.3 mg kg−1 and 0.9 mg kg−1) maintained at least 48 h and 96 h, respectively, which was significant better than Liraglutide (0.1 mg kg−1) in vivo. Meanwhile, the AUC0–96 h values of XTS1 (0.3 mg kg−1 and 0.9 mg kg−1) treated groups exert hypoglycemic effects with a glucose-lowering percentage of 71.3% for 0.9 mg kg−1 and 59.8% for 0.3 mg kg−1, respectively, compared with saline treated ones, while the Liraglutide was only 18.9% (Fig. 7B).
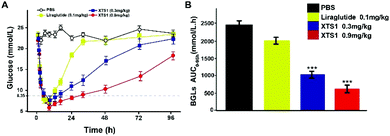 |
| | Fig. 7 Hypoglycemic stabilities of XTS1 as shown by the duration of hypoglycaemia in non-fasted diabetic mice. (A) Anti-hyperglycemic efficacies of Liraglutide (0.1 mg kg−1) and XTS1 (0.3, 0.9 mg kg−1) in T2DM model mice pretreated with each sample for 96 h. (B) Calculated AUC0–96 h values of each group BGLs. P < 0.05, 0.02, 0.001 using one-way ANOVA (*, **, ***) vs. placebo. All data are expressed as mean ± SD (n = 8). | |
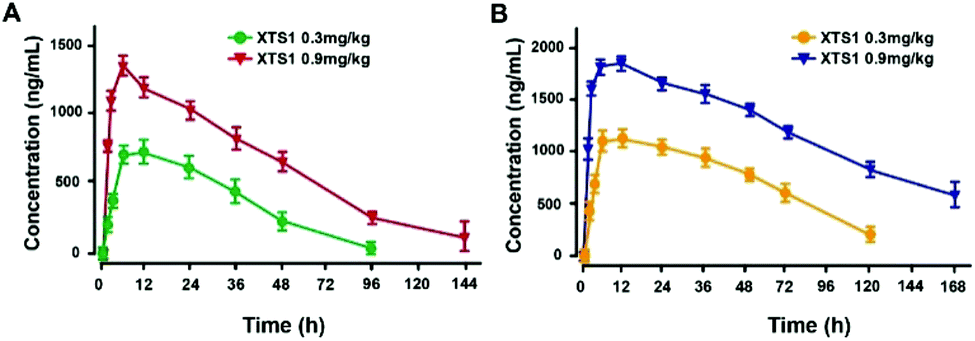
Pharmacokinetics of XTS1 in SD rats and cynomolgus monkeys
Pharmacokinetics parameters of XTS1 in SD rats and cynomolgus monkeys after a single subcutaneous injection were summarized (Fig. 8). The t1/2 of XTS1 after a single dose of 0.3 mg kg−1 or 0.9 mg kg−1 were approximately 33.0 h and 54.8 h in SD rats, respectively, while t1/2 of XTS1 at two doses were approximately 43.1 h and 82.4 h in cynomolgus monkeys, respectively.
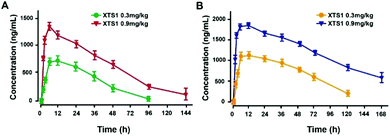 |
| | Fig. 8 Pharmacokinetics of XTS1 in SD rats (A) and cynomolgus monkeys (B). All data are expressed as mean ± SD (n = 4) after treatment. P < 0.05, 0.02, 0.001 using one-way ANOVA (*, **, ***) vs. placebo. All data are expressed as mean ± SD (n = 8). | |
Chronic in vivo study of XTS1 in diabetic mice
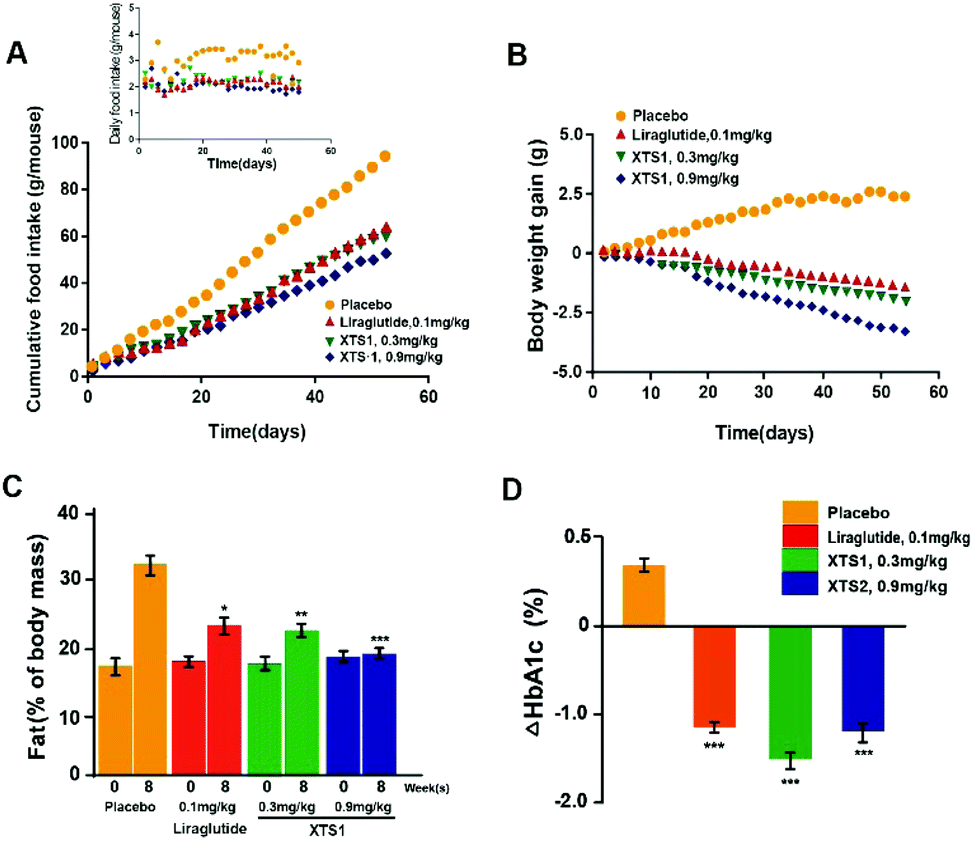
During the 8 weeks treatment course, both food intake and body weight were measured once in two days. Fat% of body mass and HAb1c value were detected at pre-dose and 9 weeks. As is showed in Fig. 9A and B, both food intake and body weight gain of XTS1 at the dose of 0.9 mg kg−1 treatment group showed obvious reduction compared with the placebo treated ones. There was relatively obvious dose reaction relationship between body weight gain or food intake and two dosages of XTS1 (0.3 mg kg−1 and 0.9 mg kg−1) depending on the above results. After the 8 weeks treatment cycle, the % fat and % HbA1c value of both doses of XTS1 treated groups were decreased remarkably compared with placebo treated ones (Fig. 9C and D). Interestingly, treatment of 0.3 mg kg−1 XTS1 exert a better HbA1c reducing effect than not only 0.1 mg kg−1 Liraglutide but also 0.9 mg kg−1 XTS1 on db/db mice.
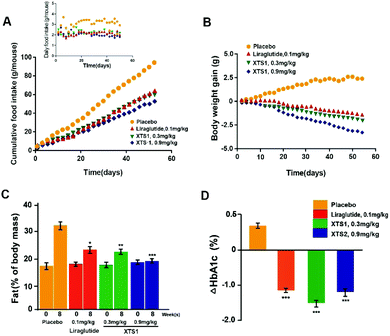 |
| | Fig. 9 Chronic in vivo studies on male db/db mice. Effects of long-term administration of XTS1 peptide on (A) food intake, (B) bodyweight gain, (C) fat% of body mass and (D) HbA1c value in male db/db mice. P < 0.05, 0.02, 0.001 using one-way ANOVA (*, **, ***) vs. placebo. All data are expressed as mean ± SD (n = 8). | |
After 8 week treatment, blood biochemical indexes were detected and listed in Table 2. There is a significant decline in serum TC, TG and LDL-C level in XTS1 group. Furthermore, enzyme activities of ALT and AST were detected to evaluate hepatocyte damage and results showed a significant reduction in both ALT and AST in XTS1 treated mice compared with saline treated ones.
Table 2 Effects of 8 week treatment of XTS1 on db/db mice' blood biochemical indexes
| Parameters |
Placebo |
Liraglutide |
XTS1 (0.3 mg kg−1) |
XTS1 (0.9 mg kg−1) |
| Triglyceride (mmol L−1) |
1.5 ± 0.2 |
1.1 ± 0.2 |
0.8 ± 0.2* |
0.9 ± 0.1* |
| Cholesterol (mmol L−1) |
5.1 ± 0.4 |
4.1 ± 0.6 |
3.8 ± 0.2* |
3.1 ± 0.4* |
| HDL-cholesterol (mmol L−1) |
2.9 ± 0.4 |
2.6 ± 0.1 |
2.5 ± 0.3 |
2.9 ± 0.5 |
| LDL-cholesterol (mmol L−1) |
1.2 ± 0.1 |
0.8 ± 0.1 |
0.9 ± 0.1** |
0.5 ± 0.1** |
| ALT (IU L−1) |
62.7 ± 3.5 |
45.1 ± 5.1 |
50.5 ± 3.4** |
42 ± 3.8*** |
| AST (IU L−1) |
161 ± 13.4 |
112 ± 9.9 |
125 ± 10.5** |
107 ± 7.1** |
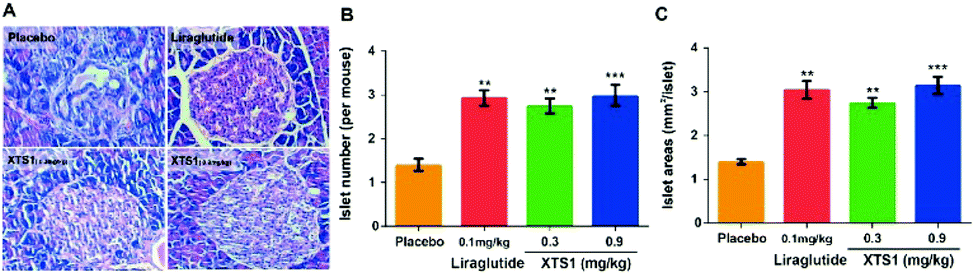
Finally, H&E staining were conducted to evaluate the chronic physiological effects of XTS1 at two doses (0.1 mg kg−1 and 0.9 mg kg−1) on the db/db mice. The pancreas paraffin sections which were stained with H&E and then made to observe the pancreas morphology (Fig. 10A). As is showed in Fig. 10B and C, larger areas and higher numbers of islet were obviously observed in the pancreases from XTS1 treated mice compared with that in the placebo treated group.
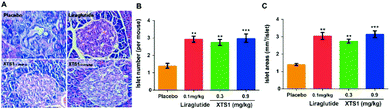 |
| | Fig. 10 Effects of XTS1 on pancreatic islets in db/db mice. (A) Representative images of histologic samples. (B) The number of pancreatic islets in each group. (C) The area of pancreatic islets in each group. P < 0.02, 0.001 using one-way ANOVA (**, ***) vs. placebo. All data are expressed as mean ± SD (n = 8). | |
Discussion
The current research describes the design and characterization of two novel long-acting GLP-1 analogs with prolonged duration of action in plasma circulation. The newly designed fusion peptides (termed XTS1 and XTS2) were comprised of an ABD, a linker with a cleavage site of TBN and a GLP-1(A8Aib). Compared with the previous reports,21 we particularly enhanced the albumin-binding ability of XTS peptides by stringing two albumin binding peptides, and the 8th alanine was changed to 2-aminoisobutyric acid, a non-natural amino acid with outstanding resistance to DPP-IV, to improve the in vivo stability of the released GLP-1.15
In this study, we prepared XTS peptides using a traditional solid phase synthesizing method and the obtained peptides were further identified using HPLC and mass spectrometry. As is showed in Fig. 1B–E, purity of both two peptides were over 99% and exact molecular weight were consistent with the theoretical values.
Both two peptides exerted extremely high affinity for human serum albumin. XTS1 was found with a higher binding affinity for albumin from various species than XTS2 (Fig. 2A, B and Table 1). In order to prove that the transient GLP-1 peptide can be released from designed albumin binding peptide via plasma proteases, we conducted a protease cleavage assay in vitro. Results were showed in Fig. 3A, both two peptides containing a TBN-cleavable linker generated GLP-1 peptide through TBN thrombin-meditated hydrolysis.28–31 Using an ELISA method which was specific for detecting the intact GLP-1, we obtained the transient concentration of the released GLP-1 from XTS1 and XTS2, respectively (Fig. 3A). The mass spectrometric profiles also shows for the hydrolysis of XTS1 (Fig. 3B, 2 hours; Fig. 3C, 72 hours) or XTS2 (Fig. 3D, 2 hours; Fig. 3E, 72 hours) in vitro by TBN. In this study, XTS1 not only displayed the higher albumin-binding affinity, but also possessed the better plasma stability than XTS2, both of which were important for the achievement of prolonged in vivo duration of action. Therefore, the XTS1 was selected to furtherly evaluate its in vivo anti-diabetic activity.
Based on the high affinities for albumin and the presence of enhanced stability in plasma, it is expected that XTS1 will exert long-lasting duration of action in vivo. Furtherly, we study the dose dependency of glucose-lowering effect of XTS1 in diabetic db/db mice by using an IPGTT method (Fig. 5). Compared with vehicle control group, all three doses of XTS1 peptide exert significant glucose-lowering effect and with a clear dose dependency ranging from 0.1 to 0.9 mg kg−1. The long lasting glucose-stabilizing effects XTS1 were confirmed by multiple IPGTTs (Fig. 6) and hypoglycemic duration test (Fig. 7) in db/db mice. The prolonged hypoglycemic duration of XTS1 in diabetic mice probably because of the enzymic metabolism and decreased renal clearance resulting from its strong binding affinity to albumin. Importantly, the hypoglycemia did not occur in the XTS1 treated diabetic mice during the period of multiple IPGTTs and hypoglycemic duration test which mainly because the XTS1 exert its hypoglycemic action by controlled-release of GLP-1(A8Aib), which was widely proved that lower the hyperglycemia in a mode of glucose-dependent way.7,32–34 It's worth mentioning that Liraglutide was tested at a lower dose (0.1 mg kg−1) than XTS1 (0.3 and 0.9 mg kg−1) which was referred to the reported papers35,36 and the clinical trials, and we believe that 0.1 mg kg−1 is a right dose for Liraglutide, as a positive agent, in diabetic mice without obvious risks. Further pharmacokinetics tests of XTS1 in rats and cynomolgus monkeys also showed obviously extended in vivo half-life compared with previous reports37–39 (Fig. 8). Liraglutide, as a GLP-1 analogue which binds to albumin contributes to an in vivo half-life of ∼8 h, is approved for once-daily diabetes treatment. The next generation of GLP-1R agonists are mainly aimed to a once weekly administration. As a result, some agents, such as albiglutide, dulaglutide and semaglutide, were approved as the once-weekly GLP-1 analogues. In this study, the t1/2 of XTS1 after a single dose of 0.3 mg kg−1 or 0.9 mg kg−1 were approximately 33.0 h and 54.8 h in SD rats, respectively, while t1/2 of XTS1 at two doses were approximately 43.1 h and 82.4 h in cynomolgus monkeys, respectively. Interestingly, t1/2 of GLP-1 released from XTS1 (82.4 h, 0.9 mg kg−1) in monkey was relatively higher than semaglutide (54 h, 1 mg kg−1) after a single s.c. injection, which gives us a lot of confidence.
Furthermore, long-term beneficial effects on XTS1 treated db/db mice were achieved, such as reduction on food intake, body weight gain, HbA1c value, fat% of body mass and protective effects on pancreas β-cells (Fig. 9 and 10).
Conclusions
In conclusion, we have shown the design and fusion of a GLP-1 analog to an albumin binding domain comprising of two sequential albumin-binding peptides to obtain the long-acting GLP-1 analogs with significantly prolonged hypoglycemic ability. We believe that the XTS1 peptide is a promising agent deserving further investigation to treat obesity patients with diabetes. Furthermore, this sustained-release strategy may also be applicable for the development of other long-acting peptide drugs rather than only applied to GLP-1 for the prevention of type 2 diabetes.
Author contributions
Design and analysis of ideas, W. W., H. P. and A. S.; methodology, Y. X. and Y. C.; resources, W. L.; writing—original draft preparation, A. S.; writing—review and editing, W. W.; methodology, experimental operation, Z. L. and J. Z.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
We indeed appreciate the linguistic assistance from Mogoedit during the writing and revising this manuscript. This work was supported by the Medical Science and Technology Development Program of Yancheng City (YK2016074, YK2017120, YK2017121).
Notes and references
- W. Danho, J. Swistok, W. Khan, X. J. Chu, A. Cheung, D. Fry, H. Sun, G. Kurylko, L. Rumennik, J. Cefalu, G. Cefalu and P. Nunn, Adv. Exp. Med. Biol., 2009, 611, 467–469 CrossRef CAS.
- G. Pasut, A. Guiotto and F. Veronese, Expert Opin. Ther. Pat., 2004, 14, 859–894 CrossRef CAS.
- X. H. Zhou and A. L. W. PO, Int. J. Pharm., 1991, 75, 97–115 CrossRef CAS.
- T. Kawada, T. Sekiguchi, T. Sakai, M. Aoyama and H. Satake, Zool. Sci., 2010, 27, 134–153 CrossRef CAS PubMed.
- M. J. Atkinson, H. Jüppner, B. Niepel, M. Casaretto, H. Zahn and R.-D. Hesch, J. Immunoassay, 2006, 3, 31–51 Search PubMed.
- D. M. Irwin, BioScience, 2005, 55, 583–591 CrossRef.
- D. D. Mafong and R. R. Henry, Curr. Atheroscler. Rep., 2008, 10, 55–60 CrossRef CAS.
- I. Antal-Zimanyi, M. A. Bruce, K. L. Leboulluec, L. G. Iben, G. K. Mattson, R. T. McGovern, J. B. Hogan, C. L. Leahy, S. C. Flowers, J. A. Stanley, A. A. Ortiz and G. S. Poindexter, Eur. J. Pharmacol., 2008, 590, 224–232 CrossRef CAS.
- L. Diao and B. Meibohm, Clin. Pharmacokinet., 2013, 52, 855–868 CrossRef CAS.
- B. Meibohm and H. Zhou, J. Clin. Pharmacol., 2012, 52, 54S–62S CrossRef.
- H. Chen, G. Wang, L. Lang, O. Jacobson, D. O. Kiesewetter, Y. Liu, Y. Ma, X. Zhang, H. Wu, L. Zhu, G. Niu and X. Chen, Theranostics, 2016, 6, 243–253 CrossRef CAS.
- L. L. Baggio, Q. Huang, T. J. Brown and D. J. Drucker, Diabetes, 2004, 53, 2492–2500 CrossRef CAS.
- C. Li, M. Yang, X. Wang, H. Zhang, C. Yao, S. Sun, Q. Liu, H. Pan, S. Liu, Y. Huan, S. Li, J. Cao, X. Wang, Y. Guo, N. Guo, S. Jing, C. Zhang and Z. Shen, Biochem. Pharmacol., 2018, 150, 46–53 CrossRef CAS.
- A. Ashkenazi and S. M. Chamow, Curr. Opin. Immunol., 1997, 9, 195–200 CrossRef CAS.
- J. Lau, P. Bloch, L. Schaffer, I. Pettersson, J. Spetzler, J. Kofoed, K. Madsen, L. B. Knudsen, J. McGuire, D. B. Steensgaard, H. M. Strauss, D. X. Gram, S. M. Knudsen, F. S. Nielsen, P. Thygesen, S. Reedtz-Runge and T. Kruse, J. Med. Chem., 2015, 58, 7370–7380 CrossRef CAS.
-
S. Lear, Z. Amso and W. Shen, in Method Enzymol, ed. A. K. Shukla, Academic Press, 2019, vol. 622, pp. 183–200 Search PubMed.
- T. Coskun, K. W. Sloop, C. Loghin, J. Alsina-Fernandez, S. Urva, K. B. Bokvist, X. Cui, D. A. Briere, O. Cabrera, W. C. Roell, U. Kuchibhotla, J. S. Moyers, C. T. Benson, R. E. Gimeno, D. A. D'Alessio and A. Haupt, Mol. Metab., 2018, 18, 3–14 CrossRef CAS.
- O. E. Levy, C. M. Jodka, S. S. Ren, L. Mamedova, A. Sharma, M. Samant, L. J. D'Souza, C. J. Soares, D. R. Yuskin, L. J. Jin, D. G. Parkes, K. Tatarkiewicz and S. S. Ghosh, PLoS One, 2014, 9, e87704 CrossRef.
- D. Mark S, Z. Min, Y. G. Meng, K. Miryam, K. Daniel, C. Dan and D. Lisa A, J. Biol. Chem., 2002, 277, 35035–35043 CrossRef.
- P. Kurtzhals, S. Havelund, I. Jonassen and J. Markussen, J. Pharm. Sci., 1997, 86, 1365–1368 CrossRef CAS.
- H. Li, Y. Ma, Y. Chen, Y. Sang, T. Zhou, M. Qiu, X. Huang, C. Zhou and Z. Su, Angew. Chem., 2010, 49, 4930–4933 CrossRef CAS.
- X. Zhong, S. Yang, T. Liu, S. Ji, J. Hu and H. Li, Eur. J. Med. Chem., 2018, 150, 841–850 CrossRef CAS.
- Y. Sang, T. Zhou, H. Li, Y. Ran, D. Jiang, F. Zheng, Y. Chen, C. Wang and X. Zou, Chin. Sci. Bull., 2013, 58, 2447–2453 CrossRef CAS.
- Y. F. Varela, M. Vanegas Murcia and M. E. Patarroyo, Molecules, 2018, 23(11), 2877 CrossRef.
- J. Han, Y. Wang, Q. Meng, G. Li, F. Huang, S. Wu, Y. Fei, F. Zhou and J. Fu, Eur. J. Med. Chem., 2017, 132, 81–89 CrossRef CAS.
- D. Muller, A. Karle, B. Meissburger, I. Hofig, R. Stork and R. E. Kontermann, J. Biol. Chem., 2007, 282, 12650–12660 CrossRef.
- B. Gissel, M. R. Jensen, K. Gregorius, H. I. Elsner, I. Svendsen and D. S. Mouritsen, J. Pept. Sci., 1995, 1, 217–226 CrossRef CAS.
- Y. Ma, S. Zhao, S. Shen, S. Fang, Z. Ye, Z. Shi and A. Hong, Sci. Rep., 2015, 5, 13595 CrossRef.
- M. Gallwitz, M. Enoksson, M. Thorpe and L. Hellman, PLoS One, 2012, 7, e31756 CrossRef CAS.
- K. Borensztajn, M. F. Bijlsma, P. H. Reitsma, M. P. Peppelenbosch and C. A. Spek, Thromb. Res., 2009, 124, 219–225 CrossRef CAS.
-
L. M. Callewaert Nico, US Pat., US20110191913, 2011.
- J. Han, Y. Wang, Q. Meng, G. Li, F. Huang, S. Wu, Y. Fei, F. Zhou and J. Fu, Eur. J. Med. Chem., 2017, 132, 81–89 CrossRef CAS.
- S. Madsbad, Lancet, 2009, 373, 438–439 CrossRef.
- B. D. Green, V. A. Gault, F. P. O'Harte and P. R. Flatt, Curr. Pharm. Des., 2004, 10, 3651–3662 CrossRef CAS.
- L. B. Knudsen and J. Lau, Front. Endocrinol., 2019, 10, 155 CrossRef.
- R. Pastor and J. A. Tur, Curr. Pharm. Des., 2019, 25, 1783–1790 CrossRef CAS.
- R. M. Goldenberg and O. Steen, Can. J. Diabetes, 2019, 43, 136–145 CrossRef.
- A. J. Kugler and M. L. Thiman, Diabetes, Metab. Syndr. Obes.: Targets Ther., 2018, 11, 187–197 CrossRef CAS.
- M. S. Rendell, Expert Opin. Drug Saf., 2017, 16, 1089–1097 CrossRef CAS.
Footnote |
| † These authors are regarded as co-first authors. |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *c and
Anquan
Shang
*c and
Anquan
Shang