
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePalladium/phosphorus-functionalized porous organic polymer with tunable surface wettability for water-mediated Suzuki–Miyaura coupling reaction†
Yizhu Lei *a,
Zaifei Chena and
Guangxing Li*b
*a,
Zaifei Chena and
Guangxing Li*b
aSchool of Chemistry and Materials Engineering, Liupanshui Normal University, Liupanshui, Guizhou 553004, PR China. E-mail: yzleiabc@126.com
bJingchu University of Technology, Jingmen, Hubei 448000, PR China. E-mail: ligxabc@163.com
First published on 11th November 2019
Abstract
A series of phosphorus-functionalized porous organic polymers supported palladium catalysts with tunable surface wettability were successfully prepared using an easy copolymerization and successive immobilization method. The obtained polymers were carefully characterized by many physicochemical methods. Characterization results suggested that the prepared materials featured hierarchically porous structures, high pore volumes, tunable surface wettability and strong electron-donating ability towards palladium species. We demonstrated the use of these solid catalysts for water-mediated Suzuki–Miyaura coupling reactions. It was found that the surface wettability of the prepared catalysts has an important influence on their catalytic activities. The optimal catalyst, which has excellent amphipathicity and relatively high phosphorus concentration, displayed superior catalytic activity compared to the other catalysts. Under ambient conditions, a variety of aryl chlorides can be efficiently transformed to biaryls in high yields. Moreover, the catalyst could be easily recovered and reused at least six times.
Introduction
The increasing environmental concerns about harmful solvent waste has led to a considerable interest of performing organic transformations in water medium.1,2 As nature's chosen medium, water not only offers the advantages of nontoxicity, nonflammability, ecological friendliness and low price,1,2 but also accelerates some reactions' rates through the hydrophobic or “on water” effects.3 On the other hand, heterogeneous switching of homogeneous catalysis is considered as an important strategy for realizing environmentally benign transformations.4 Therefore, there is good reason to believe that developing highly efficient solid catalysts for the aqueous and heterogeneous switch of homogeneous organic reactions would be an ideal pathway for green and sustainable organic synthesis.Triphenylphosphine (PPh3) and its derivatives are one kind of the most important organic ligands that have been widely used in homogeneous transition-metal catalysis.5 However, the air-sensitivity and thermal instability of phosphorus ligands at high temperature make the recycling/recovery of their metal complexes difficult, and thus, restricting their more extensive applications.6 Heterogenization of a homogeneous metal complex by immobilizing the metal complex onto a solid support with the covalent bond has been expected to address these problems,7 where organic polymers8 and porous silicas9 are the most widely used catalyst supports. Porous silicas feature high surface area and stable porous structure, however, their inert chemical nature limits their post-surface chemical modification.10 Conventional polymers feature easy chemical modification, nevertheless, the polymer-anchored molecular catalysts often suffer from poor stability, inhomogeneously distributed active sites, and low efficiency in mass transport.11
Porous organic polymers (POPs), which feature high surface areas, and designable chemical structure, have attracted tremendous research interest as new catalyst supports recently due to their potential to combine the best features of homogeneous and heterogeneous catalysts.12 In recent years, a number of POPs containing PPh3 ligand and its derivatives, have been successfully prepared and applied as the catalyst supports for immobilizing transition metal complexes.5b,6,13 Due to the strong interaction between transition metals and phosphine ligands, transition metals supported on phosphine functionalized POPs usually exhibited long-term reusability, excellent leaching resistant ability, and high catalytic activities.6,13,14 Some of them even outperformed the activities of their homogeneous analogues.6a,15 However, performing an organic reaction in water over a solid catalyst inevitably suffers from poor mass transfer efficiency of the hydrophobic organic substances, and thus, leading to low catalytic efficiency.16 To address this problem, surface of the solid catalyst could be designed to be amphiphilic.17 Nevertheless, the overwhelming majority of POPs, as well as those phosphine functionalized POPs, were mainly composed of hydrophobic aromatic networks. The hydrophobic properties lead to their poor dispersion in water and thus restricted their applications in water.18
Catalytic performance of a heterogeneous catalytic process is usually affected by the following two parameters: (i) structure of the active sites, (ii) the adsorption, desorption, and surface transfer of the reactants and products.19 It is well known that the catalyst wettability plays an important role in manipulation of the adsorption, desorption and surface transfer behaviour of the reactants and products.20 Therefore, designing heterogeneous catalysts with suitable wettability could improve the catalytic performances.19–21 Recently, several investigations about the importance of POPs' wettability in catalysis have been reported,21,22 however, studies of POPs with tunable wettability and their catalytic performances in aqueous-phase catalysis have rarely been explored.
In this study, a series of phosphorus-functionalized porous organic polymers (PTVP-MBA) supported palladium catalysts were successfully prepared by a free-radical cross-linked copolymerization of tris(4-vinylphenyl)phosphine (TVP) and N,N′-methylenebisacrylamide (MBA), and successive immobilization of palladium species method. By simply varying the MBA/TVP mass ratio, the palladium catalysts with varied surface wettability from hydrophobic to hydrophilic could be systematically obtained. The obtained palladium catalysts were tested in Suzuki–Miyaura coupling reactions between aryl chlorides and arylboronic acids. At room temperature, the optimal catalyst exhibited excellent catalytic performance in water.
Results and discussion
Synthesis and characterization of the catalysts
Porous PTVP-MBA-x polymers (x refers to the mass ratio of TVP to the total monomers) were directly synthesized through a solvothermal, free-radical copolymerization reaction of TVP and MBA (Scheme 1). For adjusting the wettability of PTVP-MBA-x polymers, PTVP-MBA-x polymers with different phosphine contents (PTVP-MBA-x, x = 0.2, 0.3, 0.4, 0.5, 0.6, 0.8) were synthesized by varying the mass ratio of tris(4-vinylphenyl)phosphine monomer. For comparison, TVP homopolymer (POL-PPh3, x = 1.0) was also synthesized. | ||
| Scheme 1 Synthetic scheme for the PTVP-MBA-x polymers. | ||
The porous properties of PTVP-MBA-x and POL-PPh3 were measured by nitrogen adsorption–desorption isotherm measurements at 77 K. As shown in Fig. 1, all the samples displayed the combined features of type I and type IV curves. The two obvious steep steps of nitrogen gas uptake in the P/P0 < 0.01 and 0.80 < P/P0 < 1.0 regions indicated the presence of micropores and macropores, while the hysteresis loops at P/P0 in the range of 0.7–1.0 reflected the presence of mesopores. Consistently, pore size distribution curves (inset) based on the NLDFT calculation method also confirmed the presence of hierarchically porous structure. Such a hierarchical pore structure has been found to be beneficial for heterogeneous catalysts by accelerating the mass transport of reactants and products.23 Specific surface areas and total pore volumes of the samples were also listed in Table 1. The results indicated that BET surface areas of the samples decreased from 1146 m2 g−1 to 647 m2 g−1 with the mass ratio (x) of TVP varied from 1.0 to 0.5. Further decreasing the mass ratio (x) of TVP below 0.5, BET surface areas of the samples (PTVP-MBA-x, x = 0.4, 0.3, 0.2) maintained at about 650 m2 g−1. Meanwhile, with TVP mass ratio (x) varied from 1.0 to 0.2, total pore volumes of the samples decreased previously from a maximum value (2.41 cm3 g−1, POL-PPh3) to a minimum value (0.93 cm3 g−1, x = 0.6) and then increased later to a moderate value (1.68 cm3 g−1, x = 0.2).
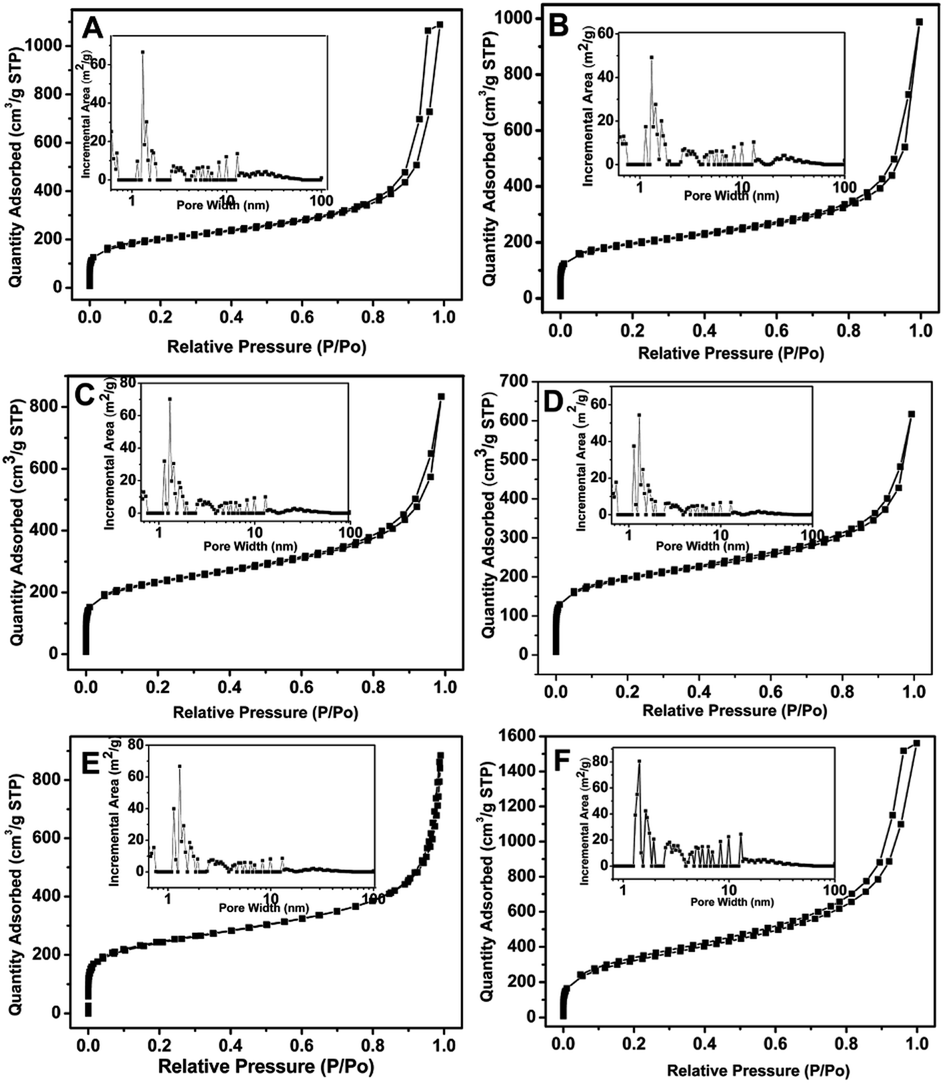 | ||
| Fig. 1 N2 absorption–desorption isotherms and pore size distribution curves (inset) of PTVP-MBA-x samples at 77 K. (A) PTVP-MBA-0.2, (B) PTVP-MBA-0.3, (C) PTVP-MBA-0.4, (D) PTVP-MBA-0.5, (E) PTVP-MBA-0.6, (F) PTVP-MBA-0.8. | ||
Selected SEM images of three representative samples (PTVP-MBA-x, x = 0.2, 0.4, 0.6) were shown in Fig. 2A–C. These images showed that the copolymers had the similar sponge-like morphologies, which consisted of loosely packed and irregular-shape nanoparticles. SEM image of PdII@PTVP-MBA-0.4 in Fig. 2D showed that the sponge-like morphology remained unchanged after coordination with Pd(OAc)2. TEM image of PTVP-MBA-0.4 in Fig. S1 (ESI†) further confirmed the loosely packed and sponge-like morphology.
 | ||
| Fig. 2 SEM images for: (A) PTVP-MBA-0.2; (B) PTVP-MBA-0.4; (C) PTVP-MBA-0.6 and (D) PdII@PTVP-MBA-0.4. | ||
Fig. 3 showed the FT-IR spectra of the monomers and representative polymers. Peaks at 2930 and 2859 cm−1 in associated with the stretching vibration of –CH2 and –CH groups could be clearly observed for all PTVP-MBA-x samples. Peaks at 1661 cm−1 assigned to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond of amide group, were also clearly observed for PTVP-MBA-x samples, indicating the presence of the MBA component. Peaks at 825 cm−1 were related to C–H out-plane flexural vibration of 1,4-substituted benzene ring, indicating the successful introduction of the TVP component. Additionally, Fig. 3a and b showed a stretching vibration band of terminal vinyl group at around 1627 cm−1, and this band was disappeared after polymerization reaction, revealing that the polymerization reactions were finished.
O bond of amide group, were also clearly observed for PTVP-MBA-x samples, indicating the presence of the MBA component. Peaks at 825 cm−1 were related to C–H out-plane flexural vibration of 1,4-substituted benzene ring, indicating the successful introduction of the TVP component. Additionally, Fig. 3a and b showed a stretching vibration band of terminal vinyl group at around 1627 cm−1, and this band was disappeared after polymerization reaction, revealing that the polymerization reactions were finished.
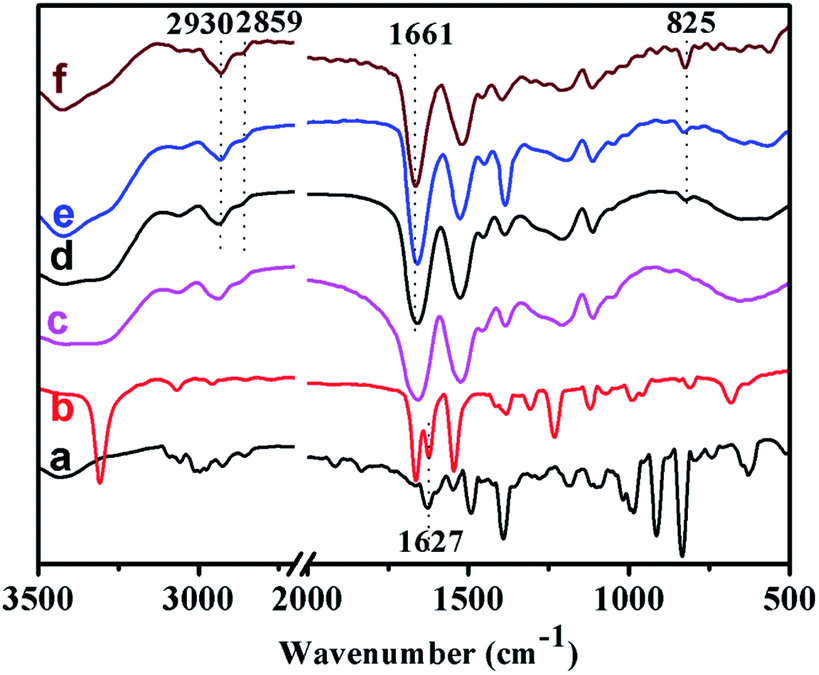 | ||
| Fig. 3 FT-IR spectra of the monomers and polymers: (a) TVP, (b) MBA, (c) POL-PPh3, (d) PTVP-MBA-0.2, (e) PTVP-MBA-0.4, and (f) PTVP-MBA-0.6. | ||
To gain insight into the structural information and coordination states of Pd species, XPS studies have been performed on the PTVP-MBA-0.4 support and three representative PdII@PTVP-MBA-x (x = 0.2, 0.4, 0.6) catalysts. In Fig. S2,† XPS full spectra of PTVP-MBA-0.4 and PdII@P(TSP-MBA)-0.4 validated that C, N, P and O elements were present in the porous polymer and catalysts. As depicted in Fig. 4A, Pd 3d XPS spectra of PdII@PTVP-MBA-x (x = 0.2, 0.4, 0.6) samples revealed that Pd species were present in the Pd(II) state. The binding energies of Pd 3d5/2 were around 337.4 eV for PdII@PTVP-MBA-x (x = 0.2, 0.4, 0.6) samples. These values are lower than that (338.4 eV) of free Pd(OAc)2.24 Simultaneously, the P 2p binding energies of PdII@PTVP-MBA-x (x = 0.2, 0.4, 0.6) samples exhibited higher values than that (132.0 eV) of PTVP-MBA-0.4. These results suggested that there was a strong coordination interaction between Pd species and PPh3 ligand in the frameworks. ICP-AES results (Table S1, ESI†) showed that the palladium contents in PdII@PTVP-MBA-x (x = 0.2, 0.3, 0.4, 0.5, 0.6, 0.8) and PdII@POL-PPh3 catalysts were very close to the nominal amount (2 wt%), suggesting the excellent palladium immobilization abilities of the prepared polymers. In the following catalytic experiments, nominal palladium content (2 wt%) was used for PdII@PTVP-MBA-x (x = 0.2, 0.3, 0.4, 0.5, 0.6, 0.8) and PdII@POL-PPh3 catalysts. Elemental components of the PTVP-MBA-x (x = 0.2, 0.3, 0.4, 0.5, 0.6, 0.8) samples were also examined by elemental analysis (Table S2, ESI†). The results suggested that the C, H, O, N, and P contents of the samples were very close to theoretical values, with relative deviation lower than 5%. To study the element dispersion of PdII@PTVP-MBA-x, SEM-mapping of PdII@PTVP-MBA-0.4 was examined (Fig. S3, ESI†). Obviously, C, N, O, P, and Pd elements were well distributed with high degrees of dispersion. Thermogravimetric analyses (TGA) of PTVP-MBA-x (x = 0.2, 0.4, 0.6) samples (Fig. S4, ESI†) showed that the main weight loss occurred above 300 °C, suggesting that the prepared samples could be stable up to 300 °C. A little weight loss around 4 wt% could be also observed bellow 100 °C; this weight loss could be mainly ascribed to the removal of trapped guest molecules, which is common for porous materials.
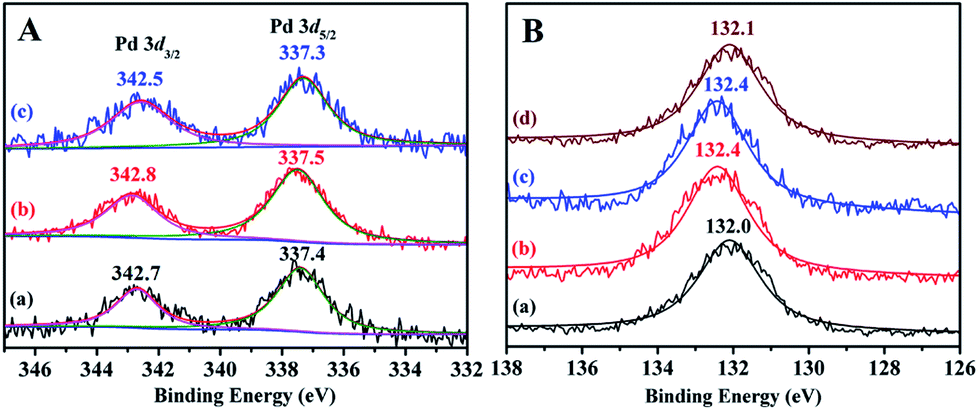 | ||
| Fig. 4 (A) Pd 3d XPS spectra of PdII@PTVP-MBA-0.2 (a), PdII@PTVP-MBA-0.4 (b) and PdII@PTVP-MBA-0.6 (c); (B) P 2p XPS spectra of PTVP-MBA-0.4 (a), PdII@PTVP-MBA-0.2 (b), PdII@PTVP-MBA-0.4 (c) and PdII@PTVP-MBA-0.6 (d). | ||
The solid-state 13C CP/MAS spectra of PTVP-MBA-0.4 and PdII@PTVP-MBA-0.4 were almost identical (Fig. S5, ESI†). The bands at about 175 ppm were assignable to “CO” carbon. The bands ranged from 126 to 149 ppm could be attributed to the aromatic carbons of the samples.13b While the bands ranged from 30 to 43 ppm could be ascribed to the polymerized vinyl groups.15a These results provided additional evidence for the successful synthesis of the copolymer. The solid state 31P CP/MAS spectra of PTVP-MBA-0.4 and PdII@PTVP-MBA-0.4 were shown in Fig. S6 (ESI†). The 31P CP/MAS spectrum of PTVP-MBA-0.4 displayed a main peak at 5.80 ppm and a small peak at 27.52 ppm. The peak at −5.80 ppm could be attributed to phosphorus in a tertiary state.13b,e While the peak at 27.52 ppm was assignable to the oxidation state of phosphorus (PO),13e indicating partial oxidation of tertiary phosphorus occurred during the polymerization reaction. The 31P CP/MAS spectrum of PdII@PTVP-MBA-0.4 displayed two peaks at −6.36 ppm and 28.31 ppm. The peak at −6.36 ppm was assignable to the uncoordinated tertiary phosphorus; while the peak at 28.31 ppm could be ascribed to both the phosphorus atoms coordination with palladium and the oxidation state of phosphorus (PO).13e This downfield shift of the resonance peak confirmed the electron-donating character of phosphine ligands. This result was consistent with XPS analyses.
Wettability control of the solid catalyst is an important and efficient strategy towards high catalytic performance, especially for organic transformations in water.18 The MBA monomer is hydrophilic, while TVP is hydrophobic. Therefore, PTVP-MBA-x samples with different content of MBA should exhibit different wettability properties. To test surface wettability of the obtained catalysts, the water contact angle measurement was conducted. As shown in Fig. 5 and S7,† PdII@POL-PPh3 had the contact angle of nearly 113°, indicating its water-repellent property. With increasement of the content of MBA, the water contact angles for the samples gradually decreased. PdII@PTVP-MBA-0.8 and PdII@PTVP-MBA-0.6 had the contact angles of 59° and 16° for water droplets, respectively. When the mass ratio of MBA in the sample was no less than 0.5, the water droplets were totally absorbed by the porous samples without any residue. Thus, the water contact angles of PdII@PTVP-MBA-x (x = 0.2, 0.3, 0.4, 0.5) were recorded as 0°. These superhydrophilic features should be related to their hydrophilic and porous structures. Contact angle measurements between toluene and PdII@PTVP-MBA-x were also conducted to test their lipophilicity. The results showed that toluene drops were totally absorbed without any residue for all the PdII@PTVP-MBA-x and PdII@POL-PPh3 catalysts. Thus, this copolymerization strategy provides an efficient method for synthesizing amphiphilic catalysts that enables the flexible tuning of their water-wettability at a molecular level.
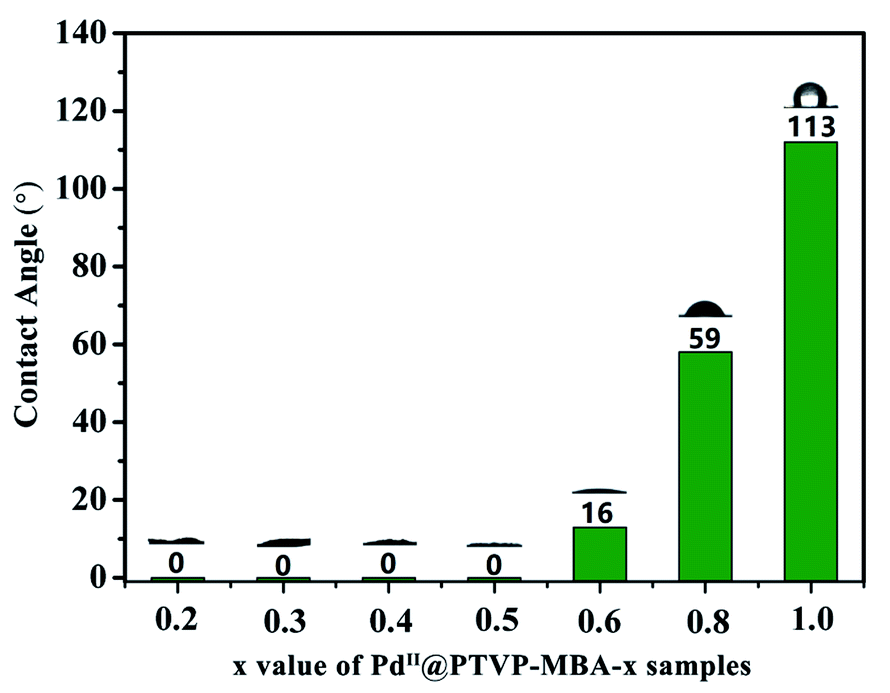 | ||
| Fig. 5 Contact angle measurements of the prepared catalysts for water. | ||
Suzuki–Miyaura coupling reactions of aryl chlorides
With the expected materials in hand, Suzuki–Miyaura coupling of aryl halides, which is a typical coupling reaction, was employed to investigate the catalytic performance of the obtained palladium catalysts.4c,5b,6a,13e To optimize the reaction conditions, coupling reaction of phenylboronic acid with chlorobenzene was adopted for initial studies (Table 2). First the reaction was conducted with PdII@PTVP-MBA-0.5 as a catalyst to screen the bases (entries 1–7). Under the described conditions at 25 °C, K2CO3 afforded the best catalytic performance, and a 75% yield of biphenyl was gained in 8 h (entry 2). Then, catalytic activities of the obtained PdII@PTVP-MBA-x were tested. Interestingly, catalytic activities of PdII@PTVP-MBA-x catalysts changed obviously with the mass ratio (x) of TVP varied from 0.8 to 0.2, and reaching a maximum yield (83%) with PdII@PTVP-MBA-0.4 as a catalyst (entries 9–13). Examination of entry 14 and 15 showed that no homocoupling product was obtained in the absence of chlorobenzene or phenylboronic acid, suggesting that homocoupling reaction of phenylboronic acid or chlorobenzene couldn't occur under the reaction conditions. Thus, the significant difference in yields over different PdII@PTVP-MBA-x catalysts should be mainly attributed to their difference in the surface wettability. PdII@POL-PPh3, which had the highest PPh3 density, however, gave the lowest yield of biphenyl (entry 8). This low catalytic performance could be attributed to its hydrophobic property, which led to its poor dispersion in water and low accessibility to the reactants. With mass ratio of hydrophobic TVP decreased from 1.0 to 0.4, the surface wettability changed from hydrophobic to hydrophilic, and the yields of biphenyl were significantly increased. However, further decreasing the x from 0.4 to 0.2, the yields were decreased. This decrease could be attributed to the low PPh3 concentrations in the PTVP-MBA-0.2 and PTVP-MBA-0.3 networks. The importance of PPh3 ligand was further ascertained by the catalytic performance of Pd/C under the optimal conditions. Pd/C gave no target product, while a 21% yield was gained in the presence of 2.0 mol% PPh3 (entries 16 and 17). For comparison, PdCl2(PPh3)2, a traditional homogeneous catalyst for this coupling reaction, was also tested. However, PdCl2(PPh3)2 gave the target product in trace yield (entry 18); this could be attributed to the poor hydrophilicity and quick formation of palladium black of PdCl2(PPh3)2. The effects of reaction time and catalyst usage were also investigated (entries 19 and 20), and a 98% yield of biphenyl was obtained at 12 h with 1 mol% of PdII@PTVP-MBA-0.4. Thus, an efficient and water-compatible catalyst was obtained with fine-turning the composition of the polymer support.
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Base | T (h) | Yieldb (mol%) |
| a Reaction conditions: Pd 1.0 mol%, chlorobenzene 1.0 mmol, phenylboronic acid 1.5 mmol, base 2.0 mmol, water 3 mL, 25 °C, 1000 rpm.b GC yields.c Without chlorobenzene.d Without phenylboronic acid.e PPh3 2.0 mol%.f Pd 0.5 mol%. | ||||
| 1 | PdII@PTVP-MBA-0.5 | Na2CO3 | 8 | 71 |
| 2 | PdII@PTVP-MBA-0.5 | K2CO3 | 8 | 75 |
| 3 | PdII@PTVP-MBA-0.5 | K3PO4 | 8 | 55 |
| 4 | PdII@PTVP-MBA-0.5 | DBU | 8 | 21 |
| 5 | PdII@PTVP-MBA-0.5 | Et3N | 8 | 14 |
| 6 | PdII@PTVP-MBA-0.5 | NaOH | 8 | 23 |
| 7 | PdII@PTVP-MBA-0.5 | NaHCO3 | 8 | 11 |
| 8 | PdII@POL-PPh3 | K2CO3 | 8 | 12 |
| 9 | PdII@PTVP-MBA-0.8 | K2CO3 | 8 | 29 |
| 10 | PdII@PTVP-MBA-0.6 | K2CO3 | 8 | 57 |
| 11 | PdII@PTVP-MBA-0.4 | K2CO3 | 8 | 88 |
| 12 | PdII@PTVP-MBA-0.3 | K2CO3 | 8 | 81 |
| 13 | PdII@PTVP-MBA-0.2 | K2CO3 | 8 | 72 |
| 14c | PdII@PTVP-MBA-0.4 | K2CO3 | 8 | — |
| 15d | PdII@PTVP-MBA-0.4 | K2CO3 | 8 | — |
| 16 | Pd/C | K2CO3 | 15 | — |
| 17e | Pd/C | K2CO3 | 15 | 21 |
| 18 | PdCl2(PPh3)2 | K2CO3 | 12 | <3 |
| 19 | PdII@PTVP-MBA-0.4 | K2CO3 | 12 | 98 |
| 20f | PdII@PTVP-MBA-0.4 | K2CO3 | 12 | 82 |
To investigate the scope of PdII@PTVP-MBA-0.4 catalyst in Suzuki–Miyaura coupling reaction, aryl chlorides and arylboronic acids bearing different functional groups were tested (Table 3). The results showed that aryl chlorides bearing electron-donating groups such as methyl, methoxy, and amino, gave the biaryls as the sole product in high yields (entries 1–3), but longer reaction times were needed for the latter two substrates. This slower rate of reaction might be due to the strong electron-donating effect of the substituent groups, which resulted in stronger strength of C–Cl bond and lower rate of oxidative addition step.25 Conversely, 4-chlorophenol, although bearing an electron-donating hydroxy group, gave the corresponding biaryl in 99% after only 10 h. This higher reaction rate might be due to the good solubility of 4-chlorophenol in water. Under ambient conditions, aryl chlorides bearing electron-withdrawing groups at the para position also participated in the reactions readily, affording the corresponding biaryls in moderate to excellent yields within 16 h (entries 5–8). Sterically hindered substrates such as 3-nitrochlorobenzene and 2-nitrochlorobenzene also gave good yields of biaryls, but a prolonged reaction time or elevated reaction temperature was needed (entries 9–11). The same protocol was also successfully extended to several para-substituted arylboronic acids, and high yields of desired biaryls were obtained (entries 12–15). Symmetrical biaryls, 4,4′-dimethyl and 4,4′-dicarbaldehyde biphenyls, could be gained in high yields (entries 16 and 17). Rather surprisingly, although most of aryl chlorides in Table 3 are either solids or poorly soluble in water, good yields of biaryls could be gained despite the fact that dissolution could limit reaction efficiency. Thus, this study offers an active and heterogeneous catalyst for the Suzuki–Miyaura coupling of aryl chlorides and arylboronic acids in water.
|
||||
|---|---|---|---|---|
| Entry | R1 | R2 | Time (h) | Yieldb (mol%) |
| a Reaction conditions: PdII@PTVP-MBA-0.4 (Pd 1.0 mol%), aryl chlorides 1.0 mmol, arylboronic acids 1.5 mmol, K2CO3 2.0 mmol, water 3 mL, 25 °C, 1000 rpm.b Isolated yields.c Reaction temperature 50 °C. | ||||
| 1 | 4-CH3 | H | 12 | 93 |
| 2 | 4-OCH3 | H | 14 | 94 |
| 3 | 4-NH2 | H | 15 | 87 |
| 4 | 4-OH | H | 10 | 99 |
| 5 | 4-COCH3 | H | 12 | 96 |
| 6 | 4-F | H | 12 | 96 |
| 7 | 4-Ph | H | 16 | 81 |
| 8 | 4-NO2 | H | 12 | 97 |
| 9 | 3-NO2 | H | 12 | 92 |
| 10 | 2-NO2 | H | 24 | 78 |
| 11 | 2-NO2 | H | 10 | 98c |
| 12 | 4-OCH3 | 4-CH3 | 12 | 92 |
| 13 | 4-OCH3 | 4-C2H5 | 12 | 97 |
| 14 | 4-OCH3 | 4-CF3 | 12 | 91 |
| 15 | 4-OCH3 | 4-CN | 12 | 95 |
| 16 | 4-CH3 | 4-CH3 | 12 | 94 |
| 17 | 4-CHO | 4-CHO | 11 | 95 |
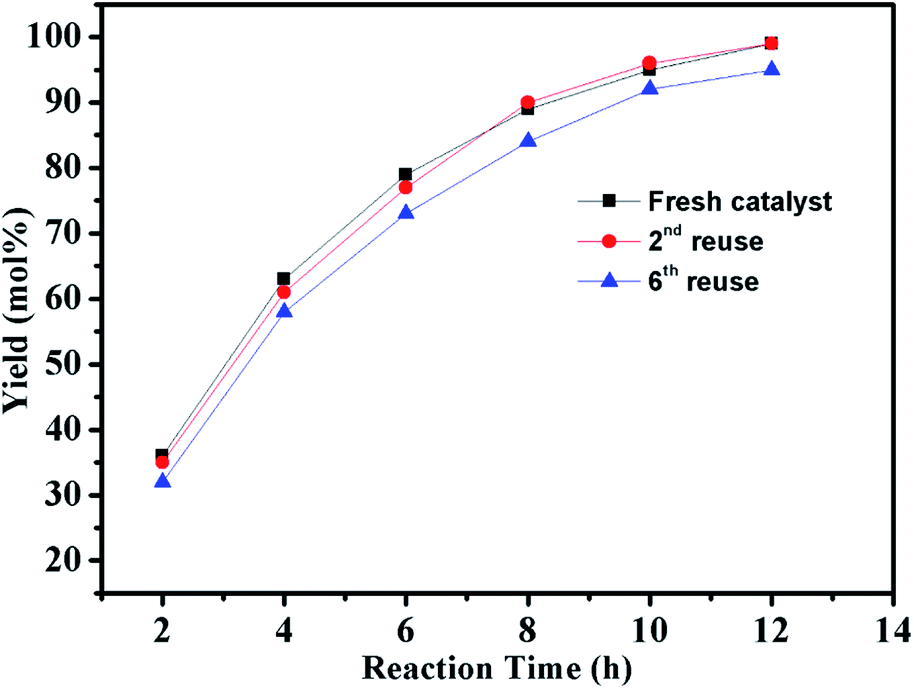
In addition to catalytic activity, reusability and metal leaching of the catalyst are also important factors in the heterogeneous catalytic systems for commercial applications. The reaction of chlorobenzene with phenylboronic acid was selected to evaluate the recycling capacity of PdII@PTVP-MBA-0.4. After each cycle, the yield of desired biaryl was analysed by GC, and the Pd content in the filtrate was analysed by ICP-AES. As shown in Fig. 6A, PdII@PTVP-MBA-0.4 exhibited excellent reusability and could be effectively reused at least six times. Moreover, the Pd leaching in the filtrate was undetectable (<10 ppb). The morphology and composition of the recovered catalyst was studied by TEM and XPS. Fig. 6B showed that the Pd nanoparticles with a mean diameter of about 2.5 nm were observed after the first catalytic run (reuse number, 0). Interestingly, no obvious aggregation of Pd nanoparticles occurred up to the 6th recycle (Fig. 6C). Fig. 6D showed that Pd(0) and Pd(II) species coexisted in the recovered catalyst. For the recovered catalyst after reusing 0 and 6 times, the ratios of Pd0/PdII were 1.7 and 2.1, respectively (estimated by the proportion of relative peak areas). To investigate further the activity of the recovered catalyst, reaction kinetics were also studied over the catalyst recovered in the 2nd and 6th recycle. As shown in Fig. 7, the yields of biphenyl in the 2nd and 6th recycle were similar to those of the fresh PdII@PTVP-MBA-0.4 catalyst. These results suggest that the in situ formed palladium nanoparticles during the recycles are also highly active for Suzuki–Miyaura coupling reaction, and the catalyst could be recycled at least 6 times without significant loss of activity.
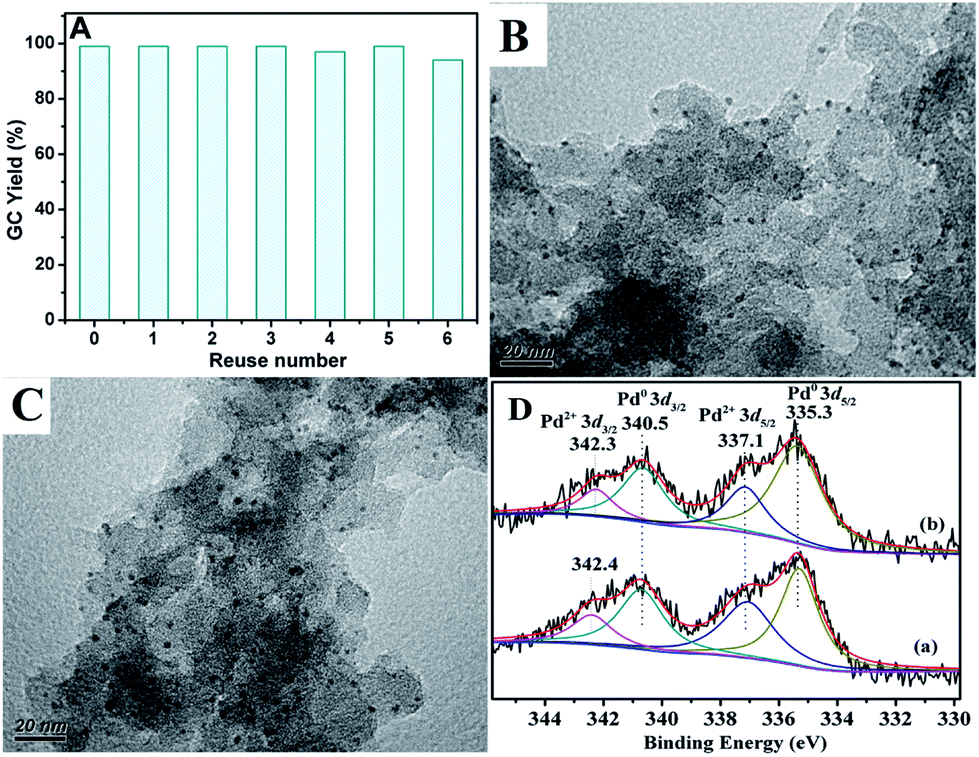 | ||
| Fig. 6 (A) Reuse of the PdII@PTVP-MBA-0.4 for the Suzuki cross-couplings of chlorobenzene with phenylboronic acid. The reaction conditions were the same with that of entry 19 in Table 2; (B) TEM image for the recycling PdII@PTVP-MBA-0.4 after the first run; (C) TEM image for the recovered PdII@PTVP-MBA-0.4 after the 6th recycle; (D) Pd 3d XPS spectra of recycling PdII@PTVP-MBA-0.4: (a) after the first catalytic run, (b) after the 6th recycle. | ||
 | ||
| Fig. 7 Reaction kinetics of the catalyst. | ||
Conclusions
In conclusion, porous organic polymers supported palladium catalysts with tunable surface wettability were successfully prepared via an easy copolymerization and successive immobilization method. The activity test in aqueous Suzuki–Miyaura coupling reaction suggested that both the surface wettability and phosphorus concentration of the catalyst have important influence on the catalytic activity. The catalyst, which featured excellent amphipathicity and relatively high phosphorus concentration, displayed the highest catalytic activity for water-mediated Suzuki–Miyaura coupling of aryl chlorides. At ambient conditions, a variety of aryl chlorides can be efficiently transformed to biaryls in high yields. Additionally, the catalyst can be easily recovered and reused several times without significant loss of catalytic activity. Thus, this protocol not only provides an efficient heterogeneous catalyst for Suzuki–Miyaura coupling reaction, but also some clues to prepare highly efficient heterogeneous catalysts for aqueous organic reactions.Experimental
Materials
N,N′-Methylenebisacrylamide (99%) and palladium diacetate (99%) were purchased from Energy Chemical and used as received. 2,2′-Azobis(2-methylpropionitrile) (AIBN) was got from Aladdin Chemistry Co., Ltd. (Shanghai, China). Tris(4-vinylphenyl)phosphine was prepared according to our previous report.6d Aryl chlorides and arylboronic acids were purchased from commercial suppliers and used without further purification.Synthesis of PTVP-MBA
Porous PTVP-MBA-x polymers (PTVP-MBA-x), where x refers to the mass ratio of TVP to total monomers, were prepared by a hydrothermal and free-radical cross-linked copolymerization route, at 100 °C for 24 h using dimethylformamide (DMF) as the solvent and AIBN as the initiator. As a typical run, TVP (0.8 g), MBA (1.2 g), DMF (20 mL) and AIBN (0.25 wt%, 50 mg) were added into in a 100 mL Teflon autoclave. After replacement of the air in the autoclave with nitrogen, stirred at room temperature for 2 h, the autoclave was heated statically in an oven at 100 °C for 24 h. After cooling to room temperature, the resulting monolith solid was extracted with ethanol and dried under vacuum (60 °C). The obtained white polymer was designated as PTVP-MBA-0.4. Varying the mass ratio of TVP and keeping the same total mass (2.0 g) of two monomers, other PTVP-MBA-x polymers were prepared as the same synthetic procedures of PTVP-MBA-0.4.Synthesis of PdII@PTVP-MBA-x
To prepare 2.0 wt% PdII@PTVP-MBA-x, 42.6 mg of Pd(OAc)2 was first dissolved in 50 mL of acetone, then 1.0 g of PTVP-MBA-x was added to the solution. The mixture was stirred at room temperature for 24 h. The resultant solid was separated by centrifugation, washed with acetone for three times, and followed by a Soxlet-extraction for 24 h with acetone. The obtained solid was dried under vacuum (60 °C) for 12 h to give PdII@PTVP-MBA-x as a light-yellow powder.Synthesis of POL-PPh3 and PdII@POL-PPh3
TVP homopolymer (POP-PPh3) was synthesized by the reported method.13b PdII@POL-PPh3 was prepared via the same procedures as PdII@PTVP-MBA-0.4.Suzuki–Miyaura cross coupling reaction
The catalytic reaction was performed in a 10 mL reaction tube. Generally, PdII@PTVP-MBA-x (53.2 mg, 1.0 mol% Pd) was added to a mixture of arylboronic acid (1.5 mmol), aryl chloride (1.0 mmol) and base (2.0 mmol) in deionized water (3.0 mL). After stirring at 25 °C for the appropriate time, the reaction mixture was treated with ethyl acetate (4 × 5 mL). The catalyst was filtered and washed with ethyl acetate, and the organic fractions combined, dried over anhydrous MgSO4. For the model reaction, the obtained liquid was analyzed by flame ionization (FID) gas chromatography using N,N-dimethylacetamide as an internal standard. For the investigation of substrate scope, the combined liquid was concentrated under reduced pressure, and the residues were purified by thin-layer chromatography (TLC) using petroleum ether as eluting solvent. Purity of products was checked by 1H NMR and yields were based on aryl chlorides. For the recycling tests of the catalyst, the recovered catalyst was washed with methanol and ethyl acetate, dried under vacuum at 40 °C for 6 h, and then reused in a new catalytic recycle.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial support by the National Natural Science Foundation of China (21763017); the Science and Technology Fund Project of Guizhou Province (qian ke he ji chu [2018]1414 and [2016]1133); the Scientific and Technological Innovation Platform of Liupanshui (52020-2018-03-02 and 52020-2017-02-02); the Academician Workstation of Liupanshui Normal University (qiankehepingtairencai [2019]5604 hao); and the Fund of Liupanshui Normal University (LPSSYZDXK201602).References
- (a) T. Kitanosono, K. Masuda, P. Xu and S. Kobayashi, Chem. Rev., 2018, 118, 679–746 CrossRef CAS; (b) M. A. Nasseri, S. A. Alavi, M. Kazemnejadi and A. Allahresani, RSC Adv., 2019, 9, 20749–20759 RSC; (c) Y. Dong, Y. Q. Chen, J. J. Jv, Y. Li, W. H. Li and Y. B. Dong, RSC Adv., 2019, 9, 21671–21678 RSC.
- (a) R. Breslow, A Fifty-Year Perspective on Chemistry in Water, in Organic Reactions in Water, ed. U. M. Lindström, Blackwell, Oxford, UK, 2007, pp. 1–28 Search PubMed; (b) Z. Chen and H. Ren, The Applications of Water as Reagents in Organic Synthesis, in Solvents as Reagents in Organic Synthesis, ed. X.-F. Wu, Wiley-VCH, New York, 2017, pp. 1–44 Search PubMed.
- (a) S. Narayan, J. Muldoon, M. G. Finn, V. V. Fokin, H. C. Kolb and K. B. Sharpless, Angew. Chem., Int. Ed., 2005, 44, 3275–3279 CrossRef CAS; (b) A. Chanda and V. V. Fokin, Chem. Rev., 2009, 109, 725–748 CrossRef CAS.
- (a) H. Xu, J. Gao and D. Jiang, Nat. Chem., 2015, 7, 905 CrossRef CAS; (b) S. Das, P. Heasman, T. Ben and S. Qiu, Chem. Rev., 2016, 117, 1515–1563 CrossRef; (c) C. A. Wang, K. Nie, G. D. Song, Y. W. Li and Y. F. Han, RSC Adv., 2019, 9, 8239–8245 RSC; (d) Y. Dong, J. J. Jv, Y. Li, W. H. Li, Y. Q. Chen, Q. Sun, J. P. Ma and Y. B. Dong, RSC Adv., 2019, 9, 20266–20272 RSC.
- (a) J. F. Hartwig, Organotransition Metal Chemistry: From Bonding to Catalysis, University Science Books, Sausalito, 2010, pp. 33–41 Search PubMed; (b) Z. Guan, B. Li, G. Hai, X. Yang, T. Li and B. Tan, RSC Adv., 2014, 4, 36437–36443 RSC.
- (a) B. Li, Z. Guan, W. Wang, X. Yang, J. Hu, B. Tan and T. Li, Adv. Mater., 2012, 24, 3390–3395 CrossRef CAS; (b) Y. Lei, L. Wu, X. Zhang, H. Mei, Y. Gu and G. Li, J. Mol. Catal. A: Chem., 2015, 398, 164–169 CrossRef CAS; (c) Z. Jia, K. Wang, B. Tan and Y. Gu, Adv. Synth. Catal., 2017, 359, 78–88 CrossRef CAS; (d) Y. Wan, F. Song, T. Ye, G. Li, D. Liu and Y. Lei, Appl. Organomet. Chem., 2019, 33, e4714 CrossRef.
- (a) D. Wang and D. Astruc, Chem. Rev., 2014, 114, 6949–6985 CrossRef CAS; (b) R. Zhong, A. C. Lindhorst, F. J. Groche and F. E. Kühn, Chem. Rev., 2017, 117, 1970–2058 CrossRef CAS; (c) S. Fukuzumi, Y.-M. Lee and W. Nam, ChemCatChem, 2018, 10, 1686–1702 CrossRef CAS.
- (a) M. Gholinejad, M. Afrasi, N. Nikfarjam and C. Nájera, Appl. Catal., A, 2018, 563, 185–195 CrossRef CAS; (b) S. Pan, S. Yan, T. Osako and Y. Uozumi, ACS Sustainable Chem. Eng., 2017, 5, 10722–10734 CrossRef CAS.
- (a) Z. Guan, J. Hu, Y. Gu, H. Zhang, G. Li and T. Li, Green Chem., 2012, 14, 1964–1970 RSC; (b) M. Zhang, R. Ettelaie, T. Yan, S. Zhang, F. Cheng, B. P. Binks and H. Yang, J. Am. Chem. Soc., 2017, 139, 17387–17396 CrossRef CAS.
- Q. Sun, Z. Dai, X. Meng and F.-S. Xiao, Chem. Soc. Rev., 2015, 44, 6018–6034 RSC.
- (a) C. A. Wang, Z. K. Zhang, T. Yue, Y. L. Sun, L. Wang, W. D. Wang, Y. Zhang, C. Liu and W. Wang, Chem.–Eur. J., 2012, 18, 6718–6723 CrossRef CAS; (b) S. Wang, K. Song, C. Zhang, Y. Shu, T. Li and B. Tan, J. Mater. Chem. A, 2017, 5, 1509–1515 RSC.
- (a) R. Dawson, A. I. Cooper and D. J. Adams, Prog. Polym. Sci., 2012, 37, 530–563 CrossRef CAS; (b) Q. Sun, Z. Dai, X. Meng, L. Wang and F. S. Xiao, ACS Catal., 2015, 5, 4556–4567 CrossRef CAS; (c) Y. Xu, S. Jin, H. Xu, A. Nagai and D. Jiang, Chem. Soc. Rev., 2013, 42, 8012–8031 RSC; (d) C. A. Wang, Y. F. Han, Y. W. Li, K. Nie, X. L. Cheng and J. P. Zhang, RSC Adv., 2016, 6, 34866–34871 RSC.
- (a) Q. Zhang, S. Zhang and S. Li, Macromolecules, 2012, 45, 2981–2988 CrossRef CAS; (b) Q. Sun, M. Jiang, Z. Shen, Y. Jin, S. Pan, L. Wang, X. Meng, W. Chen, Y. Ding, J. Li and F.-S. Xiao, Chem. Commun., 2014, 50, 11844–11847 RSC; (c) Z. Yang, B. Yu, H. Zhang, Y. Zhao, Y. Chen, Z. Ma, G. Ji, X. Gao, B. Han and Z. Liu, ACS Catal., 2016, 6, 1268–1273 CrossRef CAS; (d) C. Li, L. Yan, L. Lu, K. Xiong, W. Wang, M. Jiang, J. Liu, X. Song, Z. Zhan, Z. Jiang and Y. Ding, Green Chem., 2016, 18, 2995–3005 RSC; (e) Z. Ding, C. Li, J. Chen, J. Zeng, H. Tang, Y. Ding and Z. Zhan, Adv. Synth. Catal., 2017, 359, 2280–2287 CrossRef CAS; (f) T. Iwai, T. Harada, H. Shimada, K. Asano and M. Sawamura, ACS Catal., 2017, 7, 1681–1692 CrossRef CAS.
- S. Doherty, J. G. Knight, T. Backhouse, A. Bradford, F. Saunders, R. A. Bourne, T. W. Chamberlain, R. Stones, A. Clayton and K. Lovelock, Catal. Sci. Technol., 2018, 8, 1454–1467 RSC.
- (a) T. V. Khamatnurova, M. Johnson, D. Santana, H. S. Bazzi and D. E. Bergbreiter, Top. Catal., 2014, 57, 1438–1444 CrossRef CAS; (b) Q. Sun, Z. Dai, X. Liu, N. Sheng, F. Deng, X. Meng and F.-S. Xiao, J. Am. Chem. Soc., 2015, 137, 5204–5209 CrossRef CAS; (c) X. Wang, S. Min, S. K. Das, W. Fan, K.-W. Huang and Z. Lai, J. Catal., 2017, 355, 101–109 CrossRef CAS.
- (a) B. Lai, F. Mei and Y. Gu, Chem.–Asian J., 2018, 13, 2529–2542 CrossRef CAS; (b) J. O. Weston, H. Miyamura, T. Yasukawa, D. Sutarma, C. A. Baker, P. K. Singh, M. Bravo-Sanchez, N. Sano, P. J. Cumpson, Y. Ryabenkova, S. Kobayashi and M. Conte, Catal. Sci. Technol., 2017, 7, 3985–3998 RSC; (c) J. Miguélez, H. Miyamura and S. Kobayashi, Adv. Synth. Catal., 2017, 359, 2897–2900 CrossRef.
- (a) Y. Gu, C. Ogawa, J. Kobayashi, Y. Mori and S. Kobayashi, Angew. Chem., Int. Ed., 2006, 45, 7217–7220 CrossRef CAS; (b) J. Li, P. Huo, J. Zheng, X. Zhou and W. Liu, RSC Adv., 2018, 8, 24231–24235 RSC; (c) G. Shen, T. Osako, M. Nagaosa and Y. Uozumi, J. Org. Chem., 2018, 83, 7380–7387 CrossRef CAS PubMed; (d) T. M. Yazdely, M. Ghorbanloo and H. Hosseini-Monfared, New J. Chem., 2019, 43, 11926–11933 RSC.
- (a) R. Dawson, A. Laybourn, R. Clowes, Y. Z. Khimyak, D. J. Adams and A. I. Cooper, Macromolecules, 2009, 42, 8809–8816 CrossRef CAS; (b) D. Mullangi, S. Nandi, S. Shalini, S. Sreedhala, C. P. Vinod and R. Vaidhyanathan, Sci. Rep., 2015, 5, 10876 CrossRef; (c) Z. Lv, Q. Sun, X. Meng and F. S. Xiao, J. Mater. Chem. A, 2013, 1, 8630–8635 RSC; (d) E. R. Rangel, E. M. Maya, F. Sánchez, J. G. de La Campa and M. Iglesias, Green Chem., 2015, 17, 466–473 RSC; (e) Y. Lei, Y. Wan, G. Li, X.-Y. Zhou, Y. Gu, J. Feng and R. Wang, Mater. Chem. Front., 2017, 1, 1541–1549 RSC; (f) Y. Lei, G. Lan, D. Zhu, R. Wang, X.-Y. Zhou and G. Li, Appl. Organomet. Chem., 2018, 32, e4421 CrossRef.
- (a) S. Navalon, A. Dhakshinamoorthy, M. Alvaro and H. Garcia, Chem. Rev., 2014, 114, 6179–6212 CrossRef CAS PubMed; (b) L. Wang and F. S. Xiao, Sci. China Mater., 2018, 61, 1137–1142 CrossRef CAS; (c) Y. Wu, J. Feng, H. Gao, X. Feng and L. Jiang, Adv. Mater., 2019, 31, 1800718 CrossRef.
- (a) Y.-Z. Chen, G. Cai, Y. Wang, Q. Xu, S.-H. Yu and H.-L. Jiang, Green Chem., 2018, 18, 1212–1217 RSC; (b) Q. Sun, M. Chen, B. Aguila, N. Nguyen and S. Ma, Faraday Discuss., 2017, 201, 317–326 RSC; (c) X. Chen, P. Qian, T. Zhang, Z. Xu, C. Fang, X. Xu, W. Chen, P. Wu, Y. Shen, S. Li, J. Wu, B. Zheng, W. Zhang and F. Huo, Chem. Commun., 2018, 54, 3936–3939 RSC.
- (a) Y. Zhang, J. Pan, Y. Shen, W. Shi, C. Liu and L. Yu, ACS Sustainable Chem. Eng., 2015, 3, 871–879 CrossRef CAS; (b) G. Huang, Q. Yang, Q. Xu, S.-H. Yu and H.-L. Jiang, Angew. Chem., Int. Ed., 2016, 55, 7379–7383 CrossRef CAS; (c) Q. Sun, H. He, W. Y. Gao, B. Aguila, L. Wojtas, Z. Dai, J. Li, Y.-S. Chen, F.-S. Xiao and S. Ma, Nat. Commun., 2016, 7, 13300 CrossRef CAS PubMed.
- (a) Y. Zhang, J. Pan, Y. Chen, W. Shi, Y. Yan and L. Yu, Chem. Eng. J., 2016, 283, 956–970 CrossRef CAS; (b) J. Wei, L. Zou and J. Li, New J. Chem., 2016, 40, 4775–4780 RSC; (c) H. Gao, J. Pan, D. Han, Y. Zhang, W. Shi, J. Zeng, Y. Peng and Y. Yan, J. Mater. Chem. A, 2015, 3, 13507–13518 RSC; (d) F. Liu, L. Wang, Q. Sun, L. Zhu, X. Meng and F.-S. Xiao, J. Am. Chem. Soc., 2012, 134, 16948–16950 CrossRef CAS; (e) L. Wang, H. Wang, F. Liu, A. Zheng, J. Zhang, Q. Sun, J. P. Lewis, L. Zhu, X. Meng and F.-S. Xiao, ChemSusChem, 2014, 7, 402–406 CrossRef CAS.
- T. Yokoi and T. Tatsumi, Hierarchically Porous Materials in Catalysis, in Hierarchically Structured Porous Materials from Nanoscience to Catalysis, Separation, Optics, Energy, Life Science, ed. B.-L. Su, C. Sanchez and X.-Y. Yang, Wiley-VCH, Weinheim, 2011, pp. 481–515 Search PubMed.
- S. Y. Ding, J. Gao, Q. Wang, Y. Zhang, W. G. Song, C. Y. Su and W. Wang, J. Am. Chem. Soc., 2011, 133, 19816–19822 CrossRef CAS.
- (a) J. P. Stambuli, R. Kuwano and J. F. Hartwig, Angew. Chem., Int. Ed., 2002, 41, 4746–4748 CrossRef CAS; (b) R. B. Bedford, C. S. Cazin and D. Holder, Coord. Chem. Rev., 2004, 248, 2283–2321 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra06680b |
| This journal is © The Royal Society of Chemistry 2019 |