
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiaminophosphinoboranes: effective reagents for phosphinoboration of CO2†
Natalia Szynkiewicz ,
Anna Ordyszewska,
Jarosław Chojnacki and
Rafał Grubba*
,
Anna Ordyszewska,
Jarosław Chojnacki and
Rafał Grubba*
Department of Inorganic Chemistry, Faculty of Chemistry, Gdańsk University of Technology, G. Narutowicza St. 11/12. PL-80-233, Gdansk, Poland. E-mail: rafal.grubba@pg.edu.pl
First published on 3rd September 2019
Abstract
The monomeric diaminophosphinoboranes readily react with CO2 under mild conditions to cleanly form products of the general formula  in the absence of a catalyst. The isolated products from the CO2-phosphinoboration were fully characterized by NMR spectroscopy, IR spectroscopy, and X-ray diffraction. The mechanism of CO2 phosphinoboration with diaminophosphinoboranes was elucidated by DFT calculations.
in the absence of a catalyst. The isolated products from the CO2-phosphinoboration were fully characterized by NMR spectroscopy, IR spectroscopy, and X-ray diffraction. The mechanism of CO2 phosphinoboration with diaminophosphinoboranes was elucidated by DFT calculations.
Introduction
Increasingly stringent environmental standards are prompting us to look for clean and efficient methods for the synthesis of organic compounds. In recent years, much attention has been focused on reactions employing simple, nonmetallic P,B-based1–6 systems that can activate waste gases such as CO,7,8 CO2,9–13 NOx,10,14 SO2,14,15 or even H2.5,9,16–19 These metal-free reactions not only involve easily accessible and inexpensive gaseous reagents as the feedstock but also enable the straightforward and efficient synthesis of organic compounds through the functionalization of small inorganic molecules.20–26 The resulting products can be directly used as substrates for the synthesis of more complex systems that are difficult or impossible to obtain by other means.The largest portion of reports are those devoted to CO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 11,12,17,22,27 and its incorporation into organic molecules under mild conditions. While highly nucleophilic phosphines form stable adducts with CO2,11 boranes capture CO2 only in the presence of a Lewis base12 that can act as either a stoichiometric4,27 or catalytic23 coreagent or as the site of the ambiphilic molecule. One of the most common systems of this kind is frustrated Lewis pairs (FLPs), involving inter- or intramolecular combinations of sterically encumbered Lewis acids and bases that cannot quench each other.18 In the reaction of P,B-based FLPs with CO2, the synergistic interactions of the P-center with a carbon atom and the B-center with an oxygen atom lead to the formation of P–C and B–O bonds, respectively, giving zwitterionic products with the formula
11,12,17,22,27 and its incorporation into organic molecules under mild conditions. While highly nucleophilic phosphines form stable adducts with CO2,11 boranes capture CO2 only in the presence of a Lewis base12 that can act as either a stoichiometric4,27 or catalytic23 coreagent or as the site of the ambiphilic molecule. One of the most common systems of this kind is frustrated Lewis pairs (FLPs), involving inter- or intramolecular combinations of sterically encumbered Lewis acids and bases that cannot quench each other.18 In the reaction of P,B-based FLPs with CO2, the synergistic interactions of the P-center with a carbon atom and the B-center with an oxygen atom lead to the formation of P–C and B–O bonds, respectively, giving zwitterionic products with the formula  .27 In the vast majority of FLPs, the presence of highly electron-withdrawing substituents to increase the acidity of the B atom is crucial for efficient CO2 fixation. However, it was shown that geminal FLPs, in which the donor and acceptor sites are separated by one atom, can activate CO2 despite the mild Lewis acidity of the boron center.28,29
.27 In the vast majority of FLPs, the presence of highly electron-withdrawing substituents to increase the acidity of the B atom is crucial for efficient CO2 fixation. However, it was shown that geminal FLPs, in which the donor and acceptor sites are separated by one atom, can activate CO2 despite the mild Lewis acidity of the boron center.28,29
These reports inspired us to go one step further and investigate whether systems in which the B atom is directly bound to the phosphorus atom also activate CO2. Hence, we focused on monomeric phosphinoboranes, which are another type of ambiphilic P,B-based molecules.30,31 A recent very comprehensive review31 by Pringle et al. classifies these species containing a single P–B bond with a pyramidal P atom and the general formula R2P–BR2 as borylphosphines30,32,33 and those with a double P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) B bond and trigonal planar P atom and the general formula
B bond and trigonal planar P atom and the general formula  as phosphinoboranes.34–36 Notably, we found that there are a limited number of reports on the reactivity of P–B bond systems towards small molecules. Phosphinoboranes R2PB(C6F5)2 (R = tBu, Cy) exhibit FLP-like reactivity towards H2.34,37 The Westcott group explored the chemistry of phosphinoboronate ester Ph2P–Bpin38 (pin = 1,2-O2C2Me4), which effectively functionalizes a wide variety of heterorganic systems, such as carbonyls,38,39 N-heterocycles,40 aldimines,38,39 carbodiimides41 and isocyanates.41 Very recently Wescott and Stephan tested reactivity of R2PBpin (R = Ph, tBu), R2PBMes2 (R = Ph, tBu), and R2PBcat (R = Ph, tBu, Mes) towards CO2, where the reactions with the first and the second group of phosphinoboranes gave R2PCO2Bpin and R2PCO2BMes respectively, whereas the reaction involving R2PBcat yielded (R2P)2CO and O(Bcat)2.42
as phosphinoboranes.34–36 Notably, we found that there are a limited number of reports on the reactivity of P–B bond systems towards small molecules. Phosphinoboranes R2PB(C6F5)2 (R = tBu, Cy) exhibit FLP-like reactivity towards H2.34,37 The Westcott group explored the chemistry of phosphinoboronate ester Ph2P–Bpin38 (pin = 1,2-O2C2Me4), which effectively functionalizes a wide variety of heterorganic systems, such as carbonyls,38,39 N-heterocycles,40 aldimines,38,39 carbodiimides41 and isocyanates.41 Very recently Wescott and Stephan tested reactivity of R2PBpin (R = Ph, tBu), R2PBMes2 (R = Ph, tBu), and R2PBcat (R = Ph, tBu, Mes) towards CO2, where the reactions with the first and the second group of phosphinoboranes gave R2PCO2Bpin and R2PCO2BMes respectively, whereas the reaction involving R2PBcat yielded (R2P)2CO and O(Bcat)2.42
As a part of our research program on applying P–P bond systems to the activation of small molecules, we recently reported the first example of CO2 diphosphination by unsymmetrical diphosphanes43 in the presence of BPh3. Herein, a weak Lewis acid catalyzes the insertion of CO2 into the P–P bond of the Lewis basic component with the formation of P–C and P–O bonds in a reversible manner (Scheme 1).44
 | ||
| Scheme 1 Diphosphination of CO2 by unsymmetrical diphosphanes.44 | ||
Our studies revealed that the presence of long and polarized P–P bonds is crucial for the activation of CO2 by diphosphanes. Herein, we decided to apply this synthetic approach to trivalent phosphorus and boron compounds with direct P–B bonds.
Results and discussion
To this end, we designed and synthesized a series of new phosphinoboranes 1–3, which are structural analogs of unsymmetrical diphosphanes where one P atom has been replaced by a B atom (Scheme 2). The first step of the synthesis was the reaction of BBr3 with a four-fold excess of iPr2NH in petroleum ether with the elimination of an ammonium salt and formation of the bromo(diamino)borane. Then, the obtained product was used in the equimolar reaction with the corresponding lithium phosphide (RR′PLi) in toluene at −50 °C. Diaminophosphinoboranes 1–3 were isolated by removal of the LiBr followed by evaporation of the solvent, giving analytically pure products in high yields (83–88%).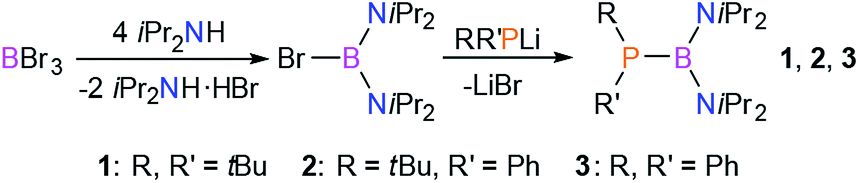 | ||
| Scheme 2 Synthesis of diaminophosphinoboranes 1–3. | ||
The 31P{1H} NMR spectra of 1, 2 and 3 show broad singlets at −8.0 ppm, −26.2 ppm and −36.1 ppm, respectively. The upfield resonances in the 31P{1H} NMR spectra of 1–3 indicate a lack of significant P–B π-interaction within these molecules. The 11B NMR spectra of 1–3 each exhibit only one broad singlet (39.5 ppm (1); 40.4 ppm (2); and 38.8 ppm (3)) at values typical for trivalent  species.30,31 The broadness of the signals in the 31P{1H} and 11B spectra can be explained by the quadrupolar nature of boron. Moreover, the 1JP–B coupling is not visible likely because of the small value of this coupling constant and mentioned broadness of the signals. The crystallization from petroleum ether at −20 °C resulted in colorless crystals of 2 and 3. The X-ray structures of 2 and 3 are presented in Fig. 1. The most characteristic structural features of 2 and 3 are the planar geometry of the N and B atoms with the sum of the angles being close to 360°, the pyramidal geometry of the P atom (sum 323.8° (2); 316.95 (3)) and the very long B–P distance (1.983(2) Å (2); 1.984(1) Å (3)). The B–P bond lengths in 2 and 3 are among the longest distances reported for phosphinoboranes,30,31 and they are even slightly longer than the sum of the single covalent bond radii of B and P (1.96 Å).45
species.30,31 The broadness of the signals in the 31P{1H} and 11B spectra can be explained by the quadrupolar nature of boron. Moreover, the 1JP–B coupling is not visible likely because of the small value of this coupling constant and mentioned broadness of the signals. The crystallization from petroleum ether at −20 °C resulted in colorless crystals of 2 and 3. The X-ray structures of 2 and 3 are presented in Fig. 1. The most characteristic structural features of 2 and 3 are the planar geometry of the N and B atoms with the sum of the angles being close to 360°, the pyramidal geometry of the P atom (sum 323.8° (2); 316.95 (3)) and the very long B–P distance (1.983(2) Å (2); 1.984(1) Å (3)). The B–P bond lengths in 2 and 3 are among the longest distances reported for phosphinoboranes,30,31 and they are even slightly longer than the sum of the single covalent bond radii of B and P (1.96 Å).45
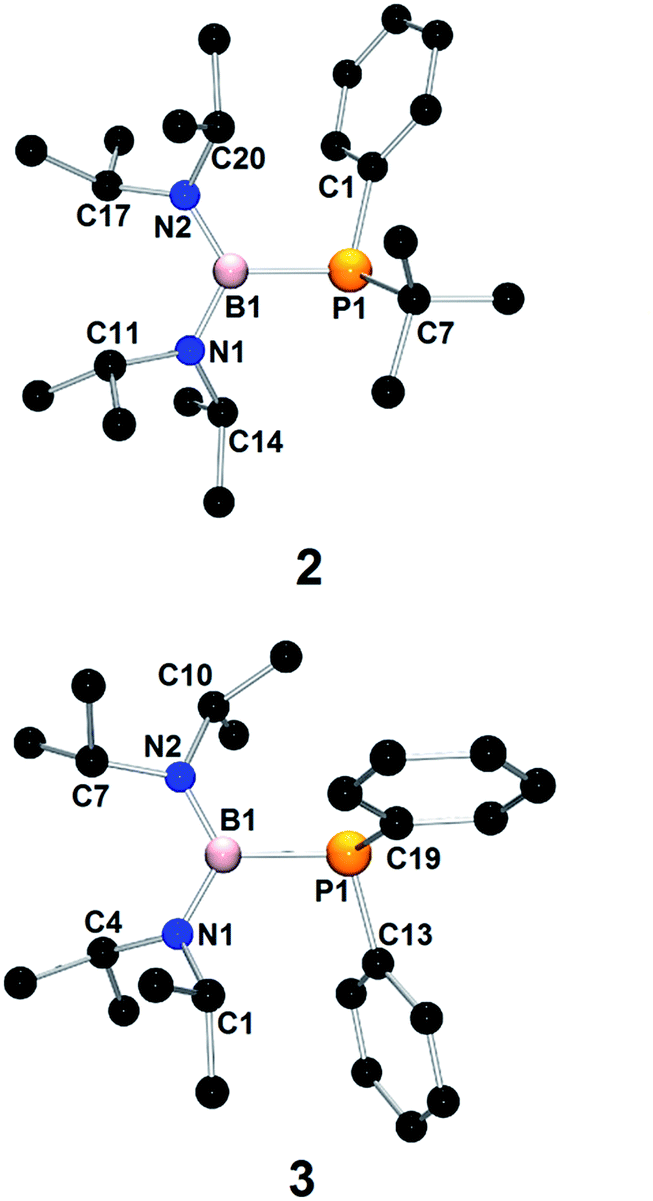 | ||
| Fig. 1 Molecular structures of 2 and 3. | ||
Moreover, the shortening of the B–N bonds in 2 and 3 to B–N distances in the range of 1.425(1)–1.442(3) Å (sum of the single covalent bond radii of B and N: 1.56 Å; sum of the double covalent bond radii for B and N: 1.38 Å) was observed.45,46 This feature, together with the planar geometry of the N atom, indicates significant B–N π-bonding. NBO analysis of these species provided further insight into the electronic structures of 1–3. The strong interaction between the lone pairs on the N atoms and the formally empty p-orbital of the B atom resulting from the formation of B–N π-bonds is clearly visible. Otherwise, donor–acceptor interactions between the P and B centers are very weak. Hence, the lone pair on the P atom is accessible for reactions with electrophilic compounds. The nucleophilic and electrophilic properties of 1–3 were studied by analysis of the condensed Fukui functions. In general, 1–3 have strong nucleophilic character with the nucleophilicity centered at the P atom. The nucleophilicity of the diaminophosphinoboranes increases in the order 3 < 2 < 1, where the values of fN for the P atoms are 0.175, 0.198 and 0.277, respectively. Interestingly, boron is neither a nucleophilic nor an electrophilic center; the values of both the fN and fE Fukui functions are close to zero. Taking into account the results of all the structural and NBO analyses, 1–3 can be described as nucleophilic borylphosphine-like species.
Next, we studied the reactivity of diaminophosphinoboranes 1–3 towards CO2. The reactions of 1, 2 or 3 with CO2 (1 atm) at room temperature in toluene resulted in the formation of phosphinoboration products 1a, 2a and 3a, respectively (Scheme 3).
 | ||
| Scheme 3 Phosphinoboration of CO2. | ||
The analytically pure 1a, 2a, and 3a were isolated in high yields (87–94%) by evaporation of the solvent as colorless oils solidified below −20 °C. The 31P{1H} spectra of 1a, 2a and 3a exhibit sharp singlets at 52.4 ppm, 23.5 and −1.0 ppm, respectively. The signals are strongly downfield shifted in comparison to the corresponding resonance of the parent diaminophosphinoboranes, indicating a change in the coordination environment of the P atom. 1a, 2a, and 3a have very similar 11B NMR spectra, which show one resonance at a shift of approximately 27 ppm. Furthermore, the 13C{1H} spectra of the reaction products show a downfield doublet in the range of 180.2–177.9 ppm with 1JPC (30.9–7.3 Hz). Notably, the 31P{1H} and 13C{1H} NMR data of 3a are very similar to those reported for Ph2P–C(O)–O–Bpin.41 Moreover, the IR data for 1a and 2a showed absorption bands at 1644 cm−1 and 1664 cm−1, respectively, which are very characteristic for carbonyl groups.
X-ray diffraction studies of 1a and 2a unambiguously confirmed that the CO2 molecule is incorporated between the B and P atoms (Fig. 2). The X-ray structures of 1a and 2a are very similar and will be discussed collectively. The CO2 moiety is bound to the phosphanyl group via a carbon atom, whereas one oxygen atom links this moiety with the boryl group. In comparison to parent compounds 1 and 2, the planar geometries of the B and N atoms and the pyramidal geometry of the P atom are retained. The geometry of C1 is also planar. The P1–C1 (1.863(1) Å (1a), 1.851(5) Å (2a)) and B1–O1 (1.459(2) Å (1a), 1.452(6) Å (2a)) bonds values are consistent with single covalent bonds.45 The P1–C1 distances in 1a and 2a are very similar to corresponding bond distances in Ph2PCO2Bpin and Ph2PCO2BMes2, however B1–O1 bond lengths in 1a and 2a are about 0.05 Å longer than in mentioned compounds.42 The C1–O1 (1.345(2) Å (1a), 1.351(5) Å (2a)) and C2O2 (1.215(2) (1a), 1.218(6) Å (2a)) bond distances are very similar to those observed for carboxylic esters (∼1.33–1.41 Å and ∼1.19–1.20 Å, respectively).47 The B–N bond lengths in 1a and 2a are in the range of 1.408(1)–1.438(2) Å and are slightly shorter than the corresponding distances in parent species 1 and 2. The structures of 1a and 2a differ significantly from known structures of CO2 activation products; typically, inter- and intramolecular P,B-based frustrated Lewis pairs form zwitterionic adducts containing tetra-coordinated P and B centers.27 Compounds 1a, 2a, and 3a are stable under argon or vacuum, and even heating these products up to 60 °C did not regenerate the parent diaminophosphinoboranes.
 | ||
| Fig. 2 Molecular structures of 1a and 2a. | ||
To investigate the mechanism of CO2 capture by the diaminophosphinoboranes and elucidate the differences in their reactivities, we carried out DFT calculations. According to the Gibbs energy profile, the insertion of carbon dioxide into the P–B bond of 1 proceeds via a simple two-step mechanism (Fig. 3).
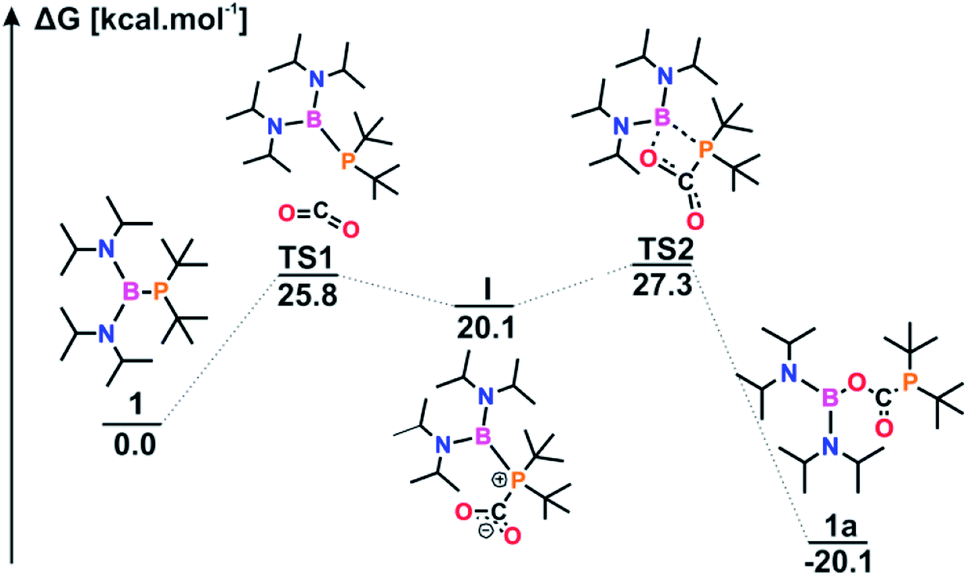 | ||
| Fig. 3 The Gibbs energy profile of the formation of 1a. | ||
The reaction starts with a nucleophilic attack of P on the electrophilic CO2 atom. Upon attachment of C to the PtBu2 atom, the electron density shifts towards the PCO2 moiety; the value of the B(iPr2N)2 electrophilic Fukui function, fE, increases from 0.001 for 1 to 0.152 for I (Table S10†). Consequently, the B(iPr2N)2 atom becomes the electrophilic center of the molecule, facilitating the formation of the B–O bond, and the simultaneous cleavage of the P–B bond leads to the generation of 1a. Although the values of the free energies (ΔG298) confirmed that in the reaction with CO2, all three systems form stable products via exergonic processes, the kinetics of these reactions are notably different (Table 1).
| Reaction | ΔG‡ [kcal mol−1] | ΔG [kcal mol−1] |
|---|---|---|
| a ωB97XD/6-31G+(d,p). | ||
| 1a | 27.3 | −20.1 |
| 2a | 26.4 | −17.8 |
| 3a | 30.2 | −10.8 |
In general, as the nucleophilicity of the phosphorus atom in 1–3 decreases, the energy barrier ΔG‡ increases. A small aberration is observed for 2a; PtBuPh is less nucleophilic than PtBu2 but is also less sterically hindered, and therefore, the value of ΔG‡ is slightly lower for 2a (Fig. S41†). While 1 and 2 react with CO2 in an analogous manner involving the generation of adduct I, a PES scan of 3a revealed that the corresponding intermediate is not formed along the reaction path (Fig. S42†). In this case, the activation of CO2 proceeds via a single four-membered ring transition state. This path requires the simultaneous interaction of both reactive centers with CO2, which in conjunction with the PPh2 atom being the least nucleophilic, justifies the highest energy barrier and the extremely long reaction time for 3.
We found it very interesting to compare the reactivities of trivalent species containing single and double boron-phosphorus bonds (Scheme 4).
 | ||
| Scheme 4 Comparison of the reactivities of species possessing P–B or PB bonds towards H2 and CO2. | ||
B(C6F5)2 (R = tBu, Cy) can activate dihydrogen34,37 (Scheme 4, bottom left); however, we did not find any reports on the reactivity of such species towards CO2.
For this reason, we reacted a representative species, tBu2PB(C6F5)234,37 (A), with CO2 under the same conditions as described for 1. The monitoring of the reaction mixture by 31P{1H} and 11B spectroscopy showed that compound A does not activate CO2. Furthermore, in the reaction of 1 with H2 conducted under the same conditions, the 31P{1H} and 11B NMR spectra revealed only signals of unreacted 1. The differences in the reactivity of 1 and tBu2PB(C6F5)2 towards H2 and CO2 can be explained by their electronic structures. According to the mechanism of H2 activation proposed by Stephan, the reaction starts with the addition of H2 (H–H bond acts as a Lewis base) to the Lewis acidic B-center of tBu2PB(C6F5)2.34,37 In the case of 1, the Lewis acidity of boron is quenched due to the strong donation from both N atoms, which explains its lack of reactivity towards H2. In regard to CO2, our mechanistic study reveals that the presence of a strong nucleophilic P center with an accessible lone pair is crucial for the activation of electrophilic CO2. While compound 1 meets these conditions, in the case of tBu2PB(C6F5)2, the lone pair on the P atom is involved in a strong donor–acceptor interaction with the B atom, resulting in π-bonding.
Conclusions
We have synthesized a series of novel monomeric diaminophosphinoboranes which are not only effective species for functionalization of CO2 but are also promising systems for activation of other small molecules. Our studies revealed which factors are crucial for the activation of small molecules by trivalent phosphorus and boron species with direct P–B bond and how to design effective systems of this kind. Studies on the reactivity of diaminophosphinoboranes towards a wide range of small, electrophilic molecules are currently in progress.Conflicts of interest
There are no conflicts to declare.Acknowledgements
R. G., N. S., and A. O. thank the National Science Centre NCN, Poland (Grant 2016/21/B/ST5/03088) for their financial support. The authors thank TASK Computational Center for access to computational resources.Notes and references
- F. G. Fontaine, M. A. Courtemanche, M. A. Légaré and É. Rochette, Coord. Chem. Rev., 2017, 334, 124–135 CrossRef CAS.
- D. J. Scott, M. J. Fuchter and A. E. Ashley, Chem. Soc. Rev., 2017, 46, 5689–5700 RSC.
- L. Greb, P. Oña-Burgos, B. Schirmer, S. Grimme, D. W. Stephan and J. Paradies, Angew. Chem., Int. Ed., 2012, 51, 10164–10168 CrossRef CAS.
- D. W. Stephan, J. Am. Chem. Soc., 2015, 137, 10018–10032 CrossRef CAS.
- S. Mummadi, D. K. Unruh, J. Zhao, S. Li and C. Krempner, J. Am. Chem. Soc., 2016, 138, 3286–3289 CrossRef CAS PubMed.
- D. W. Stephan, Acc. Chem. Res., 2015, 48, 306–316 CrossRef CAS PubMed.
- E. Theuergarten, T. Bannenberg, M. D. Walter, D. Holschumacher, M. Freytag, C. G. Daniliuc, P. G. Jones and M. Tamm, Dalton Trans., 2014, 43, 1651–1662 RSC.
- M. A. Dureen and D. W. Stephan, J. Am. Chem. Soc., 2010, 132, 13559–13568 CrossRef CAS.
- G. Ménard and D. W. Stephan, J. Am. Chem. Soc., 2010, 132, 1796–1797 CrossRef PubMed.
- W. B. Tolman, Activation of Small Molecules, Wiley, 2006 Search PubMed.
- F. Buß, P. Mehlmann, C. Mück-Lichtenfeld, K. Bergander and F. Dielmann, J. Am. Chem. Soc., 2016, 138, 1840–1843 Search PubMed.
- S. Bontemps, Coord. Chem. Rev., 2016, 308, 117–130 CrossRef CAS.
- H. Sabet-Sarvestani, M. Izadyar and H. Eshghi, J. CO2 Util., 2017, 21, 459–466 CrossRef CAS.
- D. W. Stephan and G. Erker, Chem. Sci., 2014, 5, 2625–2641 RSC.
- M. Sajid, A. Klose, B. Birkmann, L. Liang, B. Schirmer, T. Wiegand, H. Eckert, A. J. Lough, R. Fröhlich, C. G. Daniliuc, S. Grimme, D. W. Stephan, G. Kehr and G. Erker, Chem. Sci., 2013, 213–219 RSC.
- P. A. Chase and D. W. Stephan, Angew. Chem., Int. Ed., 2008, 47, 7433–7437 CrossRef CAS PubMed.
- D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2010, 49, 46–76 CrossRef CAS PubMed.
- G. C. Welch, R. R. S. Juan, J. D. Masuda and D. W. Stephan, Science, 2006, 314, 1124–1126 CrossRef CAS PubMed.
- T. A. Rokob, I. Bakó, A. Stirling, A. Hamza and I. Pápai, J. Am. Chem. Soc., 2013, 135, 4425–4437 CrossRef CAS.
- M. A. Courtemanche, M. A. Légaré, L. Maron and F. G. Fontaine, J. Am. Chem. Soc., 2014, 136, 10708–10717 CrossRef CAS PubMed.
- T. Mahdi and D. W. Stephan, J. Am. Chem. Soc., 2014, 136, 15809–15812 CrossRef CAS PubMed.
- R. Declercq, G. Bouhadir, D. Bourissou, M. A. Légaré, M. A. Courtemanche, K. S. Nahi, N. Bouchard, F. G. Fontaine and L. Maron, ACS Catal., 2015, 5, 2513–2520 CrossRef CAS.
- T. Wang and D. W. Stephan, Chem. Commun., 2014, 50, 7007–7010 RSC.
- T. Privalov, Chem.–Eur. J., 2009, 15, 1825–1829 CrossRef CAS PubMed.
- W.-H. Wang, X. Feng and M. Bao, in SpringerBriefs in Molecular Science, 2018, pp. 7–42 Search PubMed.
- D. Chen, Y. Wang and J. Klankermayer, Angew. Chem., Int. Ed., 2010, 49, 9475–9478 CAS.
- C. M. Mömming, E. Otten, G. Kehr, R. Fröhlich, S. Grimme, D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2009, 48, 6643–6646 Search PubMed.
- F. Bertini, V. Lyaskovskyy, B. J. J. Timmer, F. J. J. de Kanter, M. Lutz, A. W. Ehlers, J. C. Slootweg and K. Lammertsma, J. Am. Chem. Soc., 2012, 134, 201–204 CAS.
- Y. Wang, Z. H. Li and H. Wang, RSC Adv., 2018, 8, 26271–26276 RSC.
- R. T. Paine and H. Nöth, Chem. Rev., 1995, 95, 343–379 Search PubMed.
- J. A. Bailey and P. G. Pringle, Coord. Chem. Rev., 2015, 297–298, 77–90 CrossRef CAS.
- J. A. Bailey, M. Ploeger and P. G. Pringle, Inorg. Chem., 2014, 53, 7763–7769 CAS.
- M. Kaaz, C. Bäucker, M. Deimling, S. König, S. H. Schlindwein, J. Bender, M. Nieger and D. Gudat, Eur. J. Inorg. Chem., 2017, 2017, 4525–4532 CrossRef CAS.
- S. J. Geier, T. M. Gilbert and D. W. Stephan, Inorg. Chem., 2011, 50, 336–344 CrossRef CAS PubMed.
- P. P. Power, Angew. Chem., Int. Ed. Engl., 1990, 29, 449–460 CrossRef.
- D. C. Pestana and P. P. Power, J. Am. Chem. Soc., 1991, 113, 8426–8437 CrossRef CAS.
- S. J. Geier, T. M. Gilbert and D. W. Stephan, J. Am. Chem. Soc., 2008, 130, 12632–12633 CrossRef CAS PubMed.
- E. N. Daley, C. M. Vogels, S. J. Geier, A. Decken, S. Doherty and S. A. Westcott, Angew. Chem., Int. Ed., 2015, 54, 2121–2125 CrossRef CAS PubMed.
- M. B. Kindervater, J. F. Binder, S. R. Baird, C. M. Vogels, S. J. Geier, C. L. B. Macdonald and S. A. Westcott, J. Organomet. Chem., 2019, 880, 378–385 CAS.
- S. J. Geier, C. M. Vogels, N. R. Mellonie, E. N. Daley, A. Decken, S. Doherty and S. A. Westcott, Chem.–Eur. J., 2017, 23, 14485–14499 CAS.
- S. J. Geier, J. H. W. Lafortune, D. Zhu, S. C. Kosnik, C. L. B. Macdonald, D. W. Stephan and S. A. Westcott, Dalton Trans., 2017, 46, 10876–10885 CAS.
- J. Lafortune, Z.-W. Qu, K. Bamford, A. Trofimova, S. Westcott and D. W. Stephan, Chem.–A Eur. J., 2019 DOI:10.1002/chem.201903407.
- N. Szynkiewicz, Ł. Ponikiewski and R. Grubba, Dalton Trans., 2018, 47, 16885–16894 CAS.
- N. Szynkiewicz, Ł. Ponikiewski and R. Grubba, Chem. Commun., 2019, 55, 2928–2931 RSC.
- P. Pyykkö and M. Atsumi, Chem.–Eur. J., 2009, 15, 186–197 Search PubMed.
- P. Pyykkö and M. Atsumi, Chem.–Eur. J., 2009, 15, 12770–12779 Search PubMed.
- F. H. Allen, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, in International Tables for Crystallography, ed. T. Hahn, International Union of Crystallography, Chester, England, 2006, vol. A, pp. 790–811 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental, crystallographic, spectroscopic and computational details. CCDC 1906496 and 1906498–1906500. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra06638a |
| This journal is © The Royal Society of Chemistry 2019 |