DOI:
10.1039/C9RA06594F
(Paper)
RSC Adv., 2019,
9, 30736-30740
5-Bromo-norborn-2-en-7-one derivatives as a carbon monoxide source for palladium catalyzed carbonylation reactions†
Received
22nd August 2019
, Accepted 20th September 2019
First published on 30th September 2019
Abstract
Norbornenone (5b), obtained from the reaction of 2,5-dimethyl-3,4-diphenylcyclopentadienone dimer (3) with bromomaleic anhydride (4b), provides an excellent base-triggered source of carbon monoxide for palladium-catalysed carbonylation reactions. Aminocarbonylation, ketoamide synthesis, and Suzuki–Miyaura reactions of aryl iodides carried out in a two-chamber reaction vessel gave good to excellent yields of carbonylated products.
Introduction
In order to avoid the use of carbon monoxide (CO) gas as a reactant in small scale reactions in situ generation from many sources including carbon dioxide,1 aldehydes,2 formates,3 and formamides4,5 such as N-formylsaccharin developed by Manabe and coworkers6 has been utilized. This circumvents the need for equipping small-scale chemical laboratories with cylinders of this highly toxic, odourless and colourless gas. In early work, the most commonly used CO sources were metal carbonyl compounds such as Mo(CO)6 which proved useful, among others, for palladium catalysed aminocarbonylations and carbonylative Stille reactions of aryl halides.7–9 Recently, we have reported the development of norborneneone derivatives such as oCOm-21 as physiologically relevant carbon monoxide releasing molecules. This water soluble Carbon Monoxide Releasing Molecule (CORM) undergoes an E1cB elimination of HBr to a norbornadienone intermediate that spontaneously undergoes cheletropic release of CO in TRIS-sucrose buffer at pH 7.4 at 37 °C with a half-life of 19 minutes (Scheme 1).10 The rate of release is pH dependent and at higher pH the half-life is much reduced.
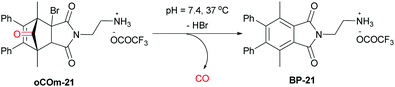 |
| | Scheme 1
oCOm-21, a physiologically relevant CO source.10 | |
In parallel work to the development of these compounds, we viewed that simpler norborneneone constructs could provide readily accessible and bench stable CO sources for various palladium catalyzed carbonylation reactions as originally described by Heck,11 Suzuki and coworkers12 and Yamamoto.13 These would be an alternative to compounds such as silacarboxylic acids 1a–c and 9-methyl-fluorene-9-carbonyl chloride (2) for CO generation for use in two-chamber reaction system such as that developed, and comprehensively studied, by the Skrydstrup group. They have demonstrated the utility of this approach, using SilaCOgen 1b and COgen 2 (along with their 13C and 14C labelled counterparts)14 in a number of palladium catalysed carbonylations including, aminocarbonylation,15,16 double carbonylations,16 Heck,17,18 and Suzuki–Miyaura reactions of both aryl iodides and bromides.19–21 CO release from COgen 2 is triggered in one chamber upon heating with a tri-tert-butylphosphine ligated palladium catalyst while that of silacarboxylic acids such as 1a produces CO at ambient temperature upon treatment with a fluoride source. The synthesis of both COgen (2) and SilaCOgen (1a) requires carboxylation with CO2 to install the carbonyl group (Fig. 1). More recently, this group have developed a tablet form of 2 that contains tri-tert-butylphosphonium tetrafluoroborate and Pd(OAc)2 which allows convenient CO generation without the need for use of a glovebox for carbonylative transformations.22
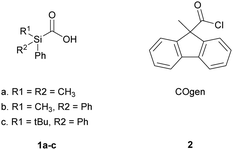 |
| | Fig. 1 CO donor molecules developed by the Skrydstrup group. | |
Herein, we report the development of readily accessible, stable norbornenone derivatives as efficient base-triggered CO donors as alternatives to 1 and 2 for palladium catalysed carbonylation reactions of aryl iodides using a two-chamber reaction system without the need for glovebox equipment.
Results and discussion
Our initial investigations into the viability of norborneneone based CORMs as suitable CO donors for various palladium catalysed carbonylation utilized 5a as an ex situ CO surrogate. 2,5-Dimethyl-3,4-diphenylcyclopentadienone (3), readily available from the condensation of benzil and 3-pentanone,23 exists as a reversible dimer, which reacted smoothly with N-phenyl-3-bromomaleimide (4a) to give cycloadduct 5a in 71% yield as a bench stable solid (Scheme 2). The efficiency of 5a as a CO donor was tested with the aminocarbonylation of p-iodoanisole (6a) and butylamine (7), catalysed by Pd2(dba)3 and using triethylamine as a base. This reaction gave N-butyl p-methoxybenzamide (8a) in 97% yield using a two-chamber reaction system similar to that developed by Skrydstrup et al.24
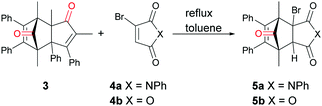 |
| | Scheme 2 Reaction of 3 and 4a or 4b to give corresponding cycloadduct 5a or 5b. | |
Given the success of this reaction, the more readily accessible norbornadienone derivative 5b was synthesized from commercially available bromomaleic anhydride (4b) and cyclopentadienone dimer 3 as an 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of the endo and exo-isomers in 96% yield, again as a bench stable solid. It was viewed that the anhydride moiety of 5b would better facilitate the base induced elimination of HBr due to the electron withdrawing property of the anhydride compared to that of imide 5a.
:1 mixture of the endo and exo-isomers in 96% yield, again as a bench stable solid. It was viewed that the anhydride moiety of 5b would better facilitate the base induced elimination of HBr due to the electron withdrawing property of the anhydride compared to that of imide 5a.
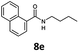
With the synthesis of cycloadduct 5b complete, we investigated the scope of this compound as a CO source for palladium-catalysed carbonylation reactions. Initially, aminocarbonylations using a range of aryl iodides as the limiting reagent with both butylamine and morpholine as coupling partners were investigated (Scheme 3). In general, 1.5 equivalents of CO donor 5b were dissolved in 1,4-dioxane and 2.0 equivalents of base were used to liberate CO in Chamber A. In all cases, complete conversion of 5b into 9 was observed. Furthermore, in Chamber B high conversions of aryl iodide substrates into amides 8a–e and 10a–e were observed with isolated yields after column chromatography ranging from 72–99%, as detailed in Tables 1 and 2.
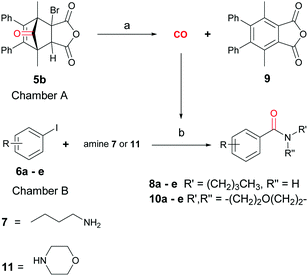 |
| | Scheme 3 General reaction scheme using 5b as the CO source for aminocarbonylation reactions of aryl iodides 6a–e and either n-butylamine (7) and morpholine (11) in 1,4-dioxane. Reagents and conditions: 80 or 100 °C, 20–25 h. (A) Et3N (1.5 eq.), 1,4-dioxane. (B) Pd2(dba)3 (5 mol%), PPh3 (10 mol%), Et3N (2.3 eq.) 1,4-dioxane. | |
Table 1 Palladium-catalysed aminocarbonylation reactions carried out with aryl iodides and n-butylamine. All reactions were carried out at 80 °C for 20 hours in a sealed two-chamber apparatus
| Entry |
Aryl iodide |
Product |
Conversiona (%) |
Yieldb (%) |
|
Measured from 1H NMR spectra of the crude reaction mixture as compared to the limiting reactant. An example is provided in Fig. SI1 (ESI).
Isolated yields after column chromatography.
Yield in brackets have been obtained from reactions using the alternative source of CO in Chamber A as shown in Scheme 4.
6e = 1-iodonaphthalene.
|
| 1 |
6a, R = p-MeO |
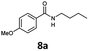
|
100 |
88 (95)c |
| 2 |
6b, R = p-NO2 |
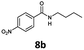
|
100 |
99 (82)c |
| 3 |
6c, R = p-Br |
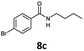
|
100 |
81 |
| 4 |
6d, R = H |
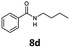
|
100 |
97 |
| 5 |
6e
d
|
|
90 |
88 |
Table 2 Palladium-catalysed aminocarbonylation reactions carried out with aryl iodides 6a–e and morpholine (9). All reactions were carried out at 100 °C for 20 hours in a sealed two-chamber apparatus
| Entry |
Aryl iodide |
Product |
Conversiona (%) |
Yieldb (%) |
|
Measured from 1H NMR spectra of the crude reaction mixture as compared to the limiting reactant. An example is provided in Fig. SI1 (ESI).
Isolated yields after column chromatography.
Reaction carried out at 80 °C for 20 h.
6e = 1-iodonaphthalene.
|
| 1 |
6a, R = p-MeO |
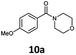
|
100 (80) |
75 (39)c |
| 2 |
6b, R = p-NO2 |
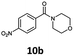
|
100 |
88 |
| 3 |
6c, R = p-Br |
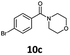
|
100 |
99 |
| 4 |
6d, R = H |
|
100 |
72 |
| 5 |
6e
d
|
|
80 |
75 |
In addition to cycloadduct 5b, a second CO source was investigated which produces a norbornadienone intermediate that spontaneously undergoes chelotropic loss of CO. A Diels–Alder reaction was carried out between cyclopentadienone dimer 3 and diethyl acetylenedicarboxylate in the CO producing chamber (Scheme 4). On heating both chambers in 1,4-dioxane substituted benzene 12 (ref. 25) was produced in Chamber A in 91% yield. The aminocarbonylation reaction in Chamber B (see Table 1; Entries 1 and 2, note c) gave N-butyl-4-iodo- and N-butyl-4-nitro-benzamides (8a and 8b) in 95 and 82% yields, respectively.
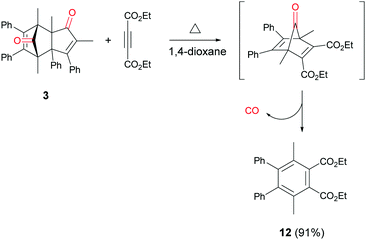 |
| | Scheme 4 Reaction of 3 and diethyl acetylenedicarboxylate to give 12 and CO. | |
In order to show the scope of cycloadduct 5b as a CO donor, other palladium catalysed carbonylation reactions were investigated. Following a 2006 report by Kondo et al. on room temperature double carbonylations,26 the Skrydstrup group have successfully utilized both COgen and SilaCOgen as an ex situ CO sources for double carbonylation reactions.15,27 The reaction of iodoanisole (6a) and butylamine (7) with CO generated from three equivalents of 5b in Chamber A gave doubly carbonylated α-ketoamide product 13 in 64% yield. This result demonstrated that cycloadduct 5b could liberate CO at room temperature (Scheme 5).
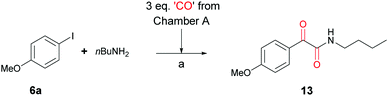 |
| | Scheme 5 The double carbonylation of p-iodoanisole (6a) and butylamine (7) using cycloadduct 5b as a CO source. Chamber A: toluene, rt. (a) DBU (3.0 eq.), Chamber B: Pd2(dba)2 (5 mol%), [(tBu)3PH]BF4 (10 mol%), DBU (2.0 eq.), 64%. | |
Another example of widely utilized palladium catalysed carbonylation reactions is the well-known Suzuki–Miyaura carbonylation. This was first reported in 1998 following development of the cross coupling reaction of the same name.28 A brief investigation into the optimization of reaction conditions for our system showed a ligand-free system could be used and a higher CO/Pd(0) ratio was necessary when compared to the aminocarbonylation reaction. This was achieved by using three equivalents of CO donor in the CO releasing chamber (Scheme 6). With optimized reaction conditions in hand, a series of Suzuki–Miyaura carbonylation reactions were carried out with the goal of synthesizing fenofibrate – a biologically relevant example of an unsymmetrical biaryl ketone. Entry 4, Table 3 shows the synthesis of a model system that involved the synthesis of simpler aryl iodide component 6f reacting with 4-chlorophenylboronic acid (14b) to give the corresponding biaryl ketone 15d in 75% yield. In a similar manner iodide 6g and 14b gave fenofibrate 15e in an 80% yield (Entry 5, Table 3).
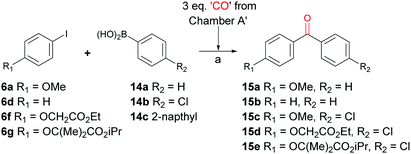 |
| | Scheme 6 Synthesis of aromatic ketones using 5b (3.0 eq.) and Et3N (3.5 eq.) in 1,4-dioxane as the CO source from Chamber A for palladium-catalysed carbonylative Suzuki–Miyaura reactions. Reagents and conditions: 80 °C; Chamber A: 5b (3.0 eq.), NEt3 (3.5 eq.), dioxane. Chamber B: aryl iodides 6 (1 eq.), Pd2(dba)2 (2 mol%), K2CO3 (3.0 eq.), anisole. | |
Table 3 Suzuki–Miyaura carbonylation reactions carried out with selected aryl iodides and boronic acids. All reactions were carried out at 80 °C for 25 hours in a sealed two-chamber apparatus
| Entrya (%) |
Aryl iodide |
Boronic acid |
Product |
Yield |
|
Isolated yields after column chromatography.
|
| 1 |
6a
|
14a
|
15a
|
74 |
| 2 |
6d
|
14a
|
15b
|
78 |
| 3 |
6a
|
14b
|
15c
|
94 |
| 4 |
6f
|
14b
|
15d
|
75 |
| 5 |
6g
|
14b
|
15e
|
80 |
Recent literature has shown only a few examples of cannabimimetic indoles that have been synthesised via carbonylation reactions.29–31 The most relevant to this work involved a tandem C–H bond iodination and carbonylative Suzuki–Miyaura cross-coupling.32 However, their process used gaseous CO and additives such as pyridine and CsF to facilitate the reaction. Trial carbonylation reactions of 3-iodo-1H-indole (16) with phenylboronic acid (14a) and 2-napthylboronic acid (14c) were carried out under the same conditions used for previous Suzuki–Miyaura carbonylations to give 3-acyl indoles (17) and (18) in moderate yields (Fig. 2).
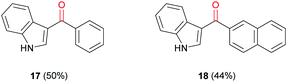 |
| | Fig. 2 3-Acyl indoles synthesised via Suzuki–Miyaura carbonylation reaction. | |
Synthetic acylindole cannabinoids are commonly functionalized at the indole nitrogen with an ethylmorpholine side chain. Early work into the structure activity relationships of synthetic cannabinoids found that this substituent had optimal binding activity at the CB1 receptor.33
N-alkylation of 3-iodo-1H-indole (16) with mesylate 19 gave iodoindole 20 (Scheme 7) which was then subjected to Suzuki–Miyaura carbonylation with 4-methoxyphenylboronic acid (14b) and 2-naphthylboronic acid (14c), respectively under the conditions described in Scheme 6. The corresponding acylindoles 21 and 22 were obtained in moderate yields (Fig. 3).
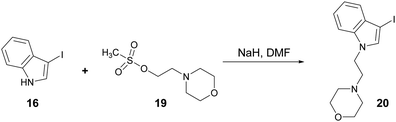 |
| | Scheme 7 Reaction of 3-iodo-1H-indole (16) with 2-morpholinoethyl methanesulfonate (20) to give iodoindole 21. Reagents and conditions: DMF, NaH, 4 h, rt, 90%. | |
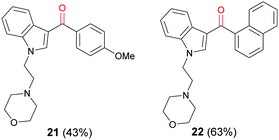 |
| | Fig. 3 Synthetic cannabinoid 3-acyl indoles synthesised via Suzuki–Miyaura carbonylation reaction. | |
Conclusions
In conclusion, we have shown that readily synthesized norbornenones 5a and 5b are excellent alternatives to using CO gas for small scale carbonylation reactions using a two-chamber reaction system. CO release is triggered by organic bases such as triethylamine and DBU. With the latter base release is rapid even at room temperature which is essential for forming α-ketoamides via a double carbonylation reaction. Alternatively, CO gas can be generated by the thermally promoted Diels–Alder reaction of cyclopentadienone 4 and diethyl acetylenedicarboxylate which was also effective for aminocarbonylation reactions.
Experimental
Aminocarbonylation reactions
General procedure A: synthesis of secondary amides from aryl halides and primary amine (n-butylamine) using diethyl acetylenedicarboxylate as CO source.
The two-chamber system, equipped with Teflon® coated magnetic stirrer bars, was loaded with all solid reagents. In Chamber A; diene dimer 3 (0.75 eq.). In Chamber B; Pd2(dba)3 (0.025 eq.) and PPh3 (0.1 eq.). Then, all liquid reagents were added via syringe: to Chamber A: 1,4-dioxane (5 mL) and diethyl acetylenedicarboxylate (1.5 eq.); to Chamber B: 1,4-dioxane (4 mL), aryl halide 6 (1 eq.; aryl halides that were solids at room temperature were added with the catalyst and ligands), n-butylamine (7) (2.3 eq.) and Et3N (2.3 eq.). The system was sealed with screwcaps fitted with silicon seal inserts. The two chamber system was heated in an oil bath at 80 °C with stirring, for 20 h. The reaction mixture was cooled to rt then the solvents in Chamber A and B were individually concentrated in vacuo. The crude material obtained was purified by silica gel column chromatography.
General procedure B: synthesis of secondary and tertiary amides from aryl halides and primary amine (n-butylamine) using cycloadduct 6b as the CO source.
The two-chamber system, equipped with Teflon® coated magnetic stirrer bars, was loaded with all solid reagents. In Chamber A; cycloadduct 5b (1.5 eq.). In Chamber B; Pd2(dba)3 (0.025 eq.) and PPh3 (0.1 eq.), the aryl halide (1 eq.), if solid. Then, all liquid reagents were added to Chamber B via syringe: aryl halide 6 (1 eq.) (if liquid), n-butylamine (2.3 eq.) and 1,4-dioxane (Chamber A: 5 mL, Chamber B: 4 mL). The system was cooled in an ice bath (with sodium chloride) and flushed with argon. Et3N (2.3 eq.) was added to Chamber B via syringe, and the cap (fitted with a silicon seal insert) screwed in place. Et3N (2.3 eq.) was also added to Chamber A via syringe, and the system was sealed. The two chamber system was heated in an oil bath at either 80 or 100 °C with stirring, for 20 h. The reaction mixture was to cooled to rt then the solvent in Chamber B was concentrated in vacuo. The crude material obtained was purified by silica gel column chromatography.
Suzuki–Miyaura carbonylation
General procedure C: synthesis of unsymmetrical biaryl ketones using cycloadduct 6b as the CO source.
The two-chamber system, equipped with Teflon® coated magnetic stirrer bars, was loaded with all solid reagents. In Chamber A; cycloadduct 5b (3 eq.). In Chamber B; Pd2(dba)3 (0.025 eq.), K2CO3 (3 eq.), boronic acid 14 (1.5 eq.) and aryl halide 6 (1 eq.), if solid. Then, all liquid reagents were added to Chamber B via syringe: aryl halide (1 eq.) (if liquid), and anisole (5 mL). The system was cooled in an ice bath (with sodium chloride) and flushed with argon, and the cap (fitted with a silicon seal insert) screwed in place. 1,4-dioxane (5 mL) was added to Chamber A, via syringe followed by Et3N (2.3 eq.) and the system was sealed. The two chamber system was heated in an oil bath at 80 °C with stirring, for 20 h. The reaction mixture was cooled to rt, and the solvent in Chamber B was concentrated in vacuo. The crude material obtained was purified by silica gel column chromatography.
Conflicts of interest
The authors declare that there is no conflict of interest regarding the publication of this paper.
Acknowledgements
CMP thanks the University of Otago for financial support for an MSc stipend and DSL, the Marsden Fund (UOO1728), for financial support.
Notes and references
- P. H. Gehrtz, V. Hirschbeck and I. Fleischer, Chem. Commun., 2015, 51, 12574–12577 RSC.
- T. Shibata, N. Toshida and K. Takagi, Org. Lett., 2002, 4, 1619–1621 CrossRef CAS.
- S. Ko, H. Han and S. Chang, Org. Lett., 2003, 5, 2687–2690 CrossRef CAS.
- Y. Wan, M. Alterman, M. Larhed and A. Hallberg, J. Comb. Chem., 2003, 5, 82–84 CrossRef CAS.
- Y. Wan, M. Alterman, M. Larhed and A. Hallberg, J. Org. Chem., 2002, 67, 6232–6235 CrossRef CAS.
- T. Ueda, H. Konishi and K. Manabe, Org. Lett., 2013, 15, 5370–5373 CrossRef CAS.
- N.-F. K. Kaiser, A. Hallberg and M. Larhed, J. Comb. Chem., 2002, 4, 109–111 CrossRef CAS.
- K. Yamazaki and Y. Kondo, J. Comb. Chem., 2004, 6, 121–125 CrossRef CAS.
- J. Lindh, A. Fardost, M. Almeida and P. Nilsson, Tetrahedron Lett., 2010, 51, 2470–2472 CrossRef CAS.
- J. T. B. Kueh, N. J. Stanley, R. J. Hewitt, L. M. Woods, L. Larsen, J. C. Harrison, D. Rennison, M. A. Brimble, I. A. Sammut and D. S. Larsen, Chem. Sci., 2017, 8, 5454–5459 RSC.
- A. Schoenberg and R. F. Heck, J. Org. Chem., 1974, 39, 3327–3331 CrossRef CAS.
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS.
- F. Ozawa, N. Kawasaki, D. Okamoto, T. Yamamoto and A. Yamamoto, Organometallics, 1987, 6, 1640–1651 CrossRef CAS.
- P. Nordeman, S. D. Friis, T. L. Andersen, H. Audrain, M. Larhed, T. Skrydstrup and G. Antoni, Chem. - Eur. J., 2015, 21, 17601–17604 CrossRef CAS.
- P. Hermange, A. T. Lindhardt, R. H. Taaning, K. Bjerglund, D. Lupp and T. Skrydstrup, J. Am. Chem. Soc., 2011, 133, 6061–6071 CrossRef CAS.
- D. U. Nielsen, K. Neumann, R. H. Taaning, A. T. Lindhardt, A. Modvig and T. Skrydstrup, J. Org. Chem., 2012, 77, 6155–6165 CrossRef CAS PubMed.
- T. M. Gøgsig, D. U. Nielsen, A. T. Lindhardt and T. Skrydstrup, Org. Lett., 2012, 14, 2536–2539 CrossRef.
- P. Hermange, T. M. Gøgsig, A. T. Lindhardt, R. H. Taaning and T. Skrydstrup, Org. Lett., 2011, 13, 2444–2447 CrossRef CAS PubMed.
- A. Ahlburg, A. T. Lindhardt, R. H. Taaning, A. E. Modvig and T. Skrydstrup, J. Org. Chem., 2013, 78, 10310–10318 CrossRef CAS.
- K. M. Bjerglund, T. Skrydstrup and G. A. Molander, Org. Lett., 2014, 16, 1888–1891 CrossRef CAS.
- S. D. Friis, T. L. Andersen and T. Skrydstrup, Org. Lett., 2013, 15, 1378–1381 CrossRef CAS.
- H. Collin, W. Reis, D. U. Nielsen, A. T. Lindhardt, M. Valle, R. Freitas and T. Skrydstrup, Org. Lett., 2019, 21, 5775–5778 CrossRef CAS.
- T. L. Andrew, J. R. Cox and T. M. Swager, Org. Lett., 2010, 12, 5302–5305 CrossRef CAS PubMed.
- S. D. Friis, R. H. Taaning, A. T. Lindhardt and T. Skrydstrup, J. Am. Chem. Soc., 2011, 133, 18114–18117 CrossRef CAS PubMed.
- Y. Kuninobu, M. Nishi, A. Kawata, H. Takata, Y. Hanatani, Y. S. Salprima, A. Iwai and K. Takai, J. Org. Chem., 2010, 75, 334–341 CrossRef CAS.
- M. Iizuka and Y. Kondo, Chem. Commun., 2006, 1739–1741, 10.1039/B600632A.
- S. R. Laursen, M. T. Jensen, A. T. Lindhardt, M. F. Jacobsen and T. Skrydstrup, Eur. J. Org. Chem., 2016, 2016, 1881–1885 CrossRef CAS.
- T. Ishiyama, H. Kizaki, T. Hayashi, A. Suzuki and N. Miyaura, J. Org. Chem., 1998, 63, 4726–4731 CrossRef CAS.
- H.-T. Zhang, L.-J. Gu, X.-Z. Huang, R. Wang, C. Jin and G.-P. Li, Chin. Chem. Lett., 2016, 27, 256–260 CrossRef CAS.
- Z. Wang, Z. Yin and X.-F. Wu, Org. Lett., 2017, 19, 4680–4683 CrossRef CAS.
- F. Zeng and H. Alper, Org. Lett., 2013, 15, 2034–2037 CrossRef CAS PubMed.
- M.-N. Zhao, L. Ran, M. Chen, Z.-H. Ren, Y.-Y. Wang and Z.-H. Guan, ACS Catal., 2015, 5, 1210–1213 CrossRef CAS.
- M. A. Eissenstat, M. R. Bell, T. E. D'Ambra, E. J. Alexander, S. J. Daum, J. H. Ackerman, M. D. Gruett, V. Kumar and K. G. Estep, J. Med. Chem., 1995, 38, 3094–3105 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental protocols, 1H and 13C NMR spectra. See DOI: 10.1039/c9ra06594f |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Kyulee
Cho
and
David S.
Larsen
,
Kyulee
Cho
and
David S.
Larsen



















