
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanism and kinetics of the atmospheric reaction of 1,3,5-trimethylbenzene bicyclic peroxy radical with OH†
Xiaoxiao
Lin
a,
Zhenli
Yang
ab,
Hui
Yu
ab,
Yanbo
Gai
 *a and
Weijun
Zhang
*a
*a and
Weijun
Zhang
*a
aLaboratory of Atmospheric Physico-Chemistry, Anhui Institute of Optics and Fine Mechanics, Chinese Academy of Sciences, Hefei, 230031, Anhui, China. E-mail: gaiyanbo@aiofm.ac.cn; wjzhang@aiofm.ac.cn
bUniversity of Science and Technology of China, Hefei, 230026, Anhui, China
First published on 11th October 2019
Abstract
The bicyclic peroxy radical (BPR) is the key intermediate during atmospheric oxidation of aromatics. In this paper, the reaction mechanisms and kinetics of the atmospheric reaction of the 1,3,5-trimethylbenzene (1,3,5-TMB) BPR with the OH radical were studied by density functional theory (DFT) and conventional transition-state theory (CTST) calculations. The product channels of formation of the 1,3,5-TMB trioxide (ROOOH), OH-adducts and Criegee intermediate (CI) have been identified, and the geometries and energies of all the stationary points were calculated at the M08-HX/6-311 + g(2df,2p) level of theory. In addition, the rate constants for the individual reaction pathway at 298 K were calculated. The results showed that OH addition reactions including the formation of ROOOH and OH-adducts are the main pathways, whereas Criegee intermediate formation is of minor importance.
1. Introduction
Aromatics constitute an important fraction (20–40%) of the total non-methane hydrocarbons (NMHCs) in the urban atmosphere which are mostly emitted by fuel combustion and industrial solvent evaporation.1–3 1,3,5-Trimethylbenzene is a significant component of the suite of aromatic hydrocarbons that leads to the production of tropospheric ozone and secondary organic aerosols (SOA).4 Atmospheric oxidation of 1,3,5-trimethylbenzene is mainly initiated by the OH radical, and the bicyclic peroxy radical (BPR) is the key intermediate during the oxidation.5 The detailed gas-phase chemical processes including the degradation mechanism of 1,3,5-trimethylbenzene can be described by MCMv3.1 which is the Master Chemical Mechanism version 3.1. As shown in Scheme 1, in the OH-initiated oxidation of 1,3,5-trimethylbenzene (TMB), the yields of the initial products formed after the OH addition to the aromatic ring in MCMv3.1 are 0.79 for the 1,3,5-TMB bicyclic peroxy radical (BPR) which have been detected experimentally.4,6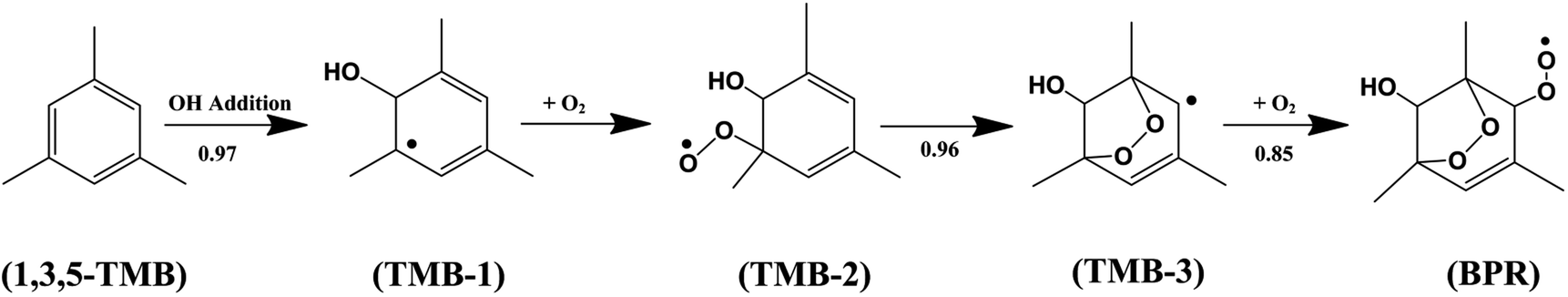 | ||
| Scheme 1 | ||
The main chemical fate of the bicyclic peroxy radical (BPR) depends on levels of NOx (NOx = NO2 + NO). The major pathway is the reaction with NO in polluted urban areas, which is responsible for tropospheric ozone production.7 While in low NOx conditions, the reaction with HO2 or with other peroxy radicals plays a larger role.8,9 Recently, the reaction with OH radicals in clean environments has been considered another important loss path. Indeed, the rate constants for the reactions of the C1–C4 aliphatic peroxy radicals and OH radicals have been measured experimentally.10–14 The very high rate constants show that the reaction with OH can be competitive to other reaction paths for peroxy radicals.15,16 Both theoretical and experimental studies of alkylperoxy radicals and OH reactions have shown that the main reaction channel is formation of ROOOH while the H-abstraction channel giving Criegee intermediate is to be of minor importance.14,17,18 However, the mechanism and kinetics for the reactions of aromatic peroxy radicals with OH radical are still unknown. Due to the effect of O2-bridged bicyclic group substituent, the reactivity of aromatic peroxy radicals toward OH radical may be different. In this article, we apply density functional theory (DFT) and conventional transition-state theory (CTST) to investigate the reaction mechanisms and kinetics of the 1,3,5-TMB bicyclic peroxy radical (BPR) with OH radical.
2. Computational details
Modern quantum chemical calculations can obtain quantitative kinetics of atmospheric reactions,19–21 where quantitative accurate barrier heights are very key for obtaining quantitative rate constants. However, for the large and complex diradical reactions, it's particularly difficult to obtain quantitative barrier heights based on couple cluster theory. For example, the highly accurate CCSD(T) and CBS level can not obtain the quantitative barrier heights in the small-sized reaction of the simplest Criegee intermediate (CH2O2) with H2O.19 Although post-CCSD(T) level of theory is able to deal with such radical–radical system, the computational costs are prohibitive. In contrast, the density functional theory (DFT) is the commonly used method in theoretical study on atmospheric oxidation of aromatic compounds.22–24 M08-HX hybrid functional shows excellent performance for thermochemistry, kinetics, noncovalent interactions.25 Xu et al. carried out extensive systematic study on 48 transition state geometries of small reactions with a variety of methods.26 It was found that M08-HX is the most highly recommended functional having a lower cost when compared to doubly hybrid functionals. Moreover, this method has been confirmed reliable by investigation of the small diradical reaction of CH3O2 and OH. The kinetic results show good agreement with the experimental values (see ESI† for details).Thus, all stationary points including the reactants, reactant complexes, transition states and products on the potential energy surface of 1,3,5-TMB BPR + OH reaction have been fully optimized by the M08-HX functional with the 6-311 + g(2df,2p) basis set. Harmonic vibrational frequency computations were carried out at the same level in order to characterize the nature of the stationary points as either minima (all positive frequencies) or transition states (one and only one imaginary frequency), to provide the zero-point energies (ZPEs), and the thermodynamic contributions to the enthalpy and free energy. Besides, the intrinsic reaction coordinate (IRC)27,28 calculations were performed to ensure that the transition states connect the relevant reactants and products. The electronic structure and energy computations were carried out using the Gaussian 16 package.29 The rate constants were calculated by conventional transition-state theory (CTST)30–32 with the Eckart tunneling correction33 using the TheRate program.34,35
3. Results and discussion
The 1,3,5-TMB bicyclic peroxy radical can undergo both OH-addition and H-abstraction reactions. As shown in Scheme 2, the addition of OH radical to the terminal oxygen atom of the bicyclic peroxy radical forms a trioxide (ROOOH). Assaf et al.36 suggested that for larger peroxy radicals with more than two or three C-atoms, the trioxide (ROOOH) intermediates undergo mostly colliosional stabilization. Thus the ROOOH produced by OH addition on 1,3,5-TMB bicyclic peroxy radical is considered stabilized by collisional energy loss instead of prompt unimolecular reactions at atmospheric conditions. Different from the alkylperoxy radicals, there is a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond on the ring structure of the 1,3,5-TMB bicyclic peroxy radical. Thus the addition of the OH radical to the double bond to form OH-adduct may also occur in the title reaction. In addition, the title reaction can be initiated through H-abstraction by OH radical to give Criegee intermediates which are mainly formed from the ozonolysis reaction of unsaturated substances37,38 due to the presence of CH group adjacent to the –OO˙ radical as shown in Scheme 3.
C double bond on the ring structure of the 1,3,5-TMB bicyclic peroxy radical. Thus the addition of the OH radical to the double bond to form OH-adduct may also occur in the title reaction. In addition, the title reaction can be initiated through H-abstraction by OH radical to give Criegee intermediates which are mainly formed from the ozonolysis reaction of unsaturated substances37,38 due to the presence of CH group adjacent to the –OO˙ radical as shown in Scheme 3.
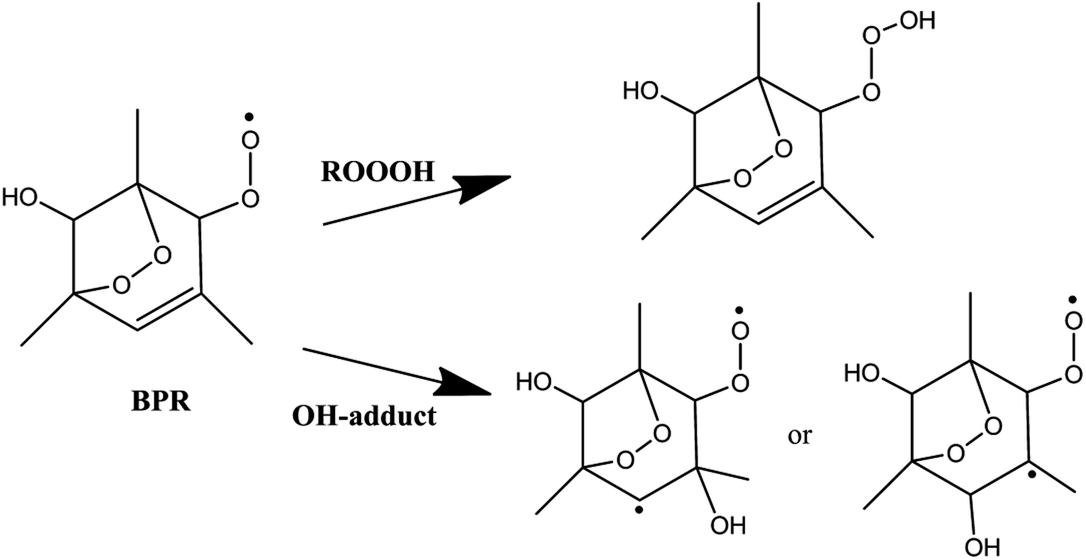 | ||
| Scheme 2 | ||
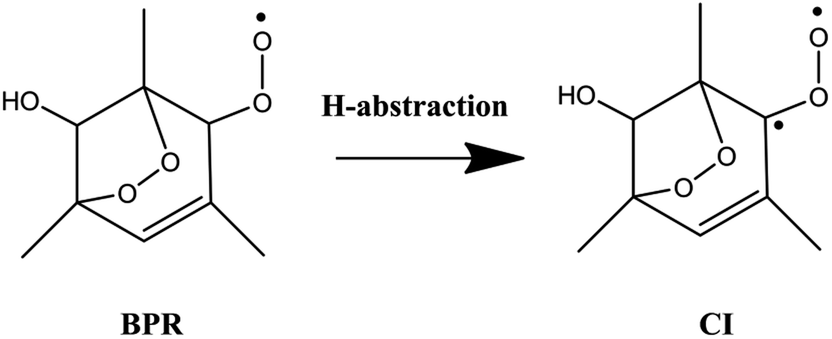 | ||
| Scheme 3 | ||
The schematic energy diagrams obtained at M08-HX/6-311 + g(2df,2p) level of theory are displayed in Fig. 1. The most relevant geometrical parameters of all stationary points described in Fig. 1 have been drawn in Fig. 2. Table 1 presents the thermodynamic parameters including the relative energies (Δ(E + ZPE) and ΔE298 K), enthalpies (ΔH298 K), and Gibbs free energies (ΔG298 K). Then CTST rate constants of each channel at 298 K are listed in Table 2.
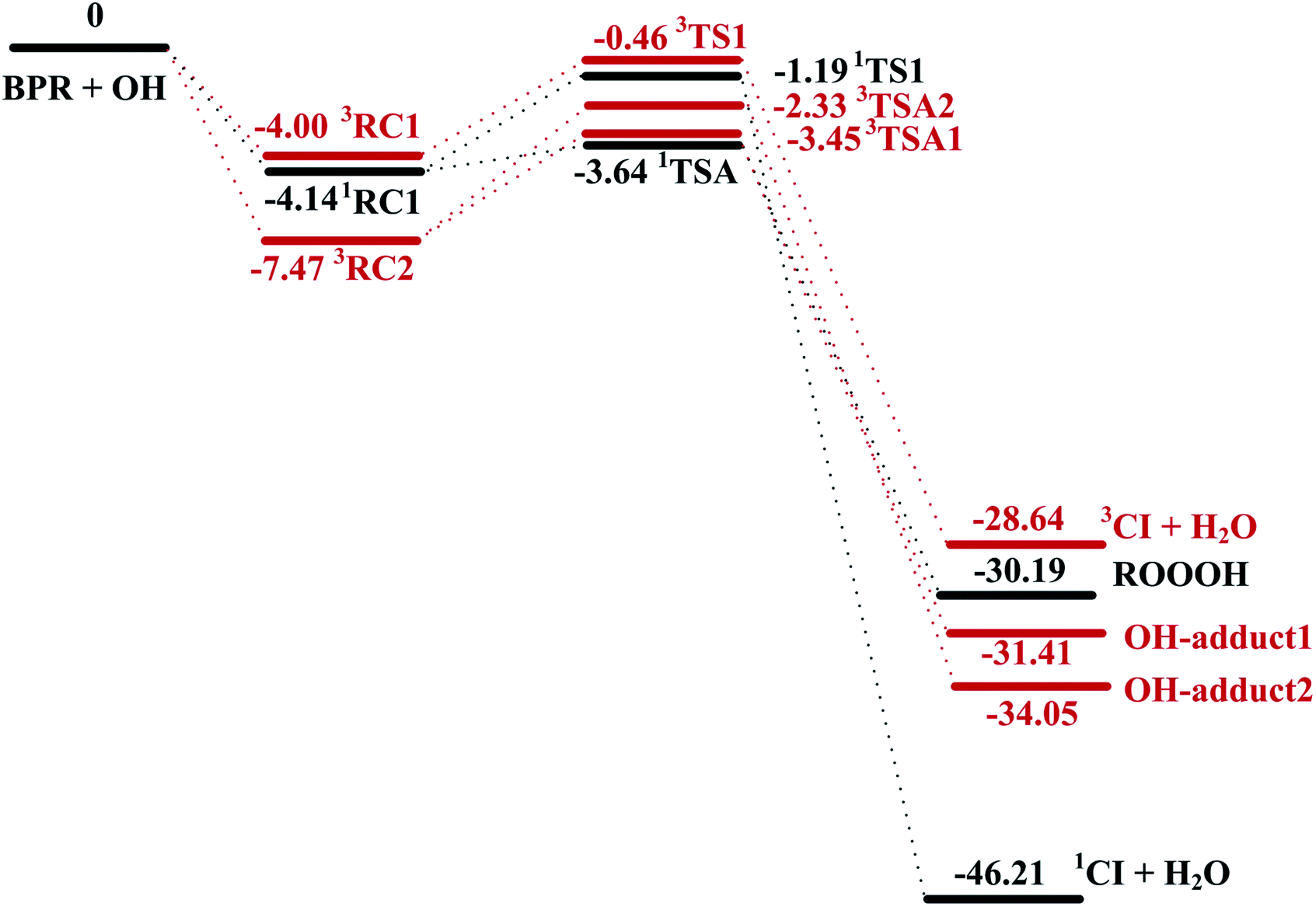 | ||
| Fig. 1 Potential energy surface for BPR + OH biradical reactions. The energies (kcal mol−1) relative to separated reactants BPR and OH at M08-HX/6-311 + g(2df,2p) level of theory. RC, reactant complex; TS, transition state; CI, Criegee intermediate. The singlet reaction pathways are depicted in black, and the triplet reaction pathways are depicted in red for clarity. | ||
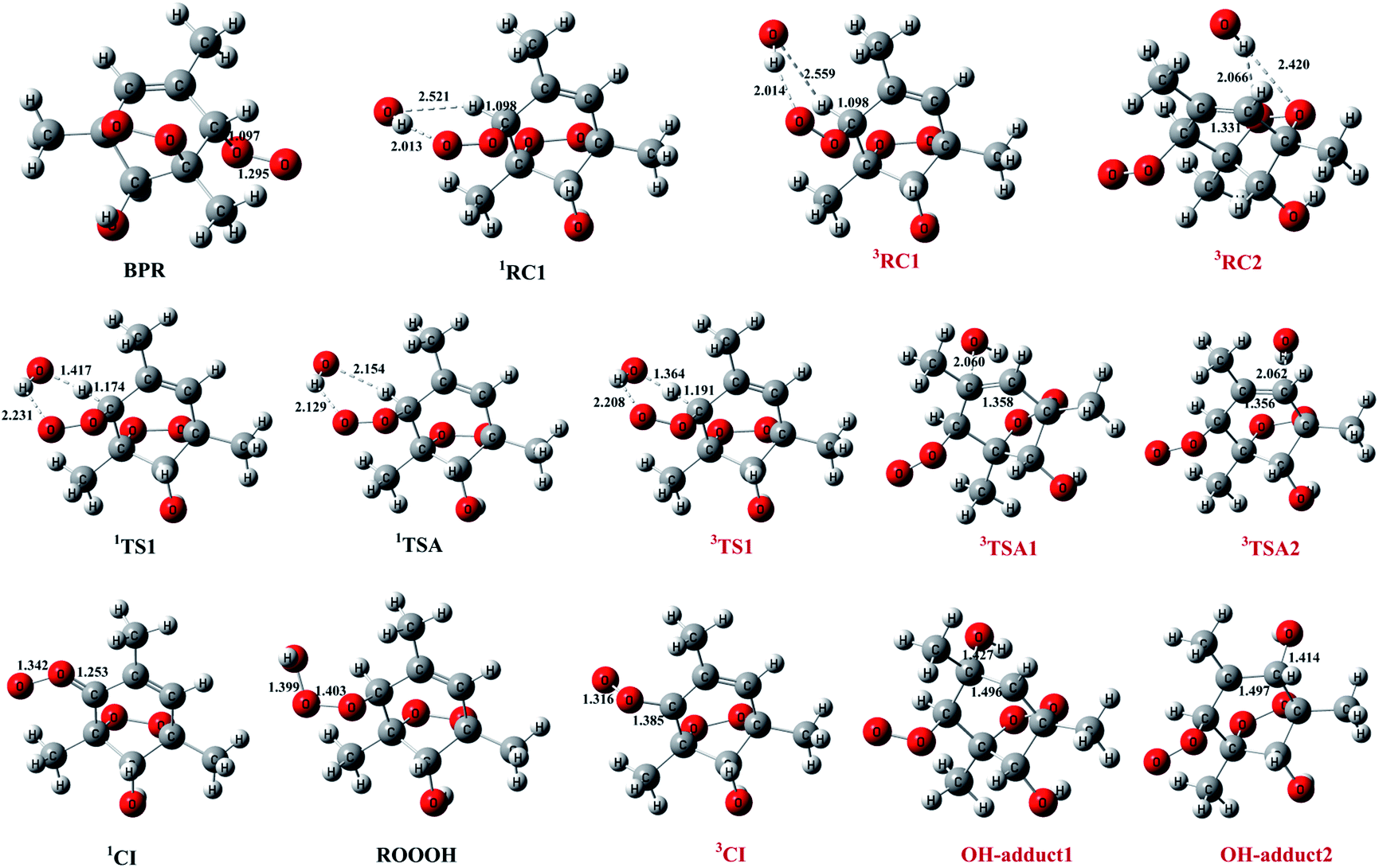 | ||
| Fig. 2 Structures of key species in the reaction of bicyclic peroxy radical (BPR) with OH optimized at M08-HX/6-311 + g(2df,2p) level of theory. The singlet species are named in black, while the triplet ones are named in red for clarity. Bond distances are in angstrom. | ||
| Compound | Δ(E + ZPE) | ΔE298 K | ΔH298 K | ΔG298 K |
|---|---|---|---|---|
| BPR + OH | 0 | 0 | 0 | 0 |
| 1RC1 | −4.14 | −3.92 | −4.51 | 4.53 |
| 1TS1 | −1.19 | −1.44 | −2.04 | 8.30 |
| 1CI + H2O | −46.21 | −46.19 | −46.19 | −45.86 |
| 1TSA | −3.64 | −3.77 | −4.36 | 5.65 |
| ROOOH | −30.19 | −30.74 | −31.33 | −20.17 |
| 3RC1 | −4.00 | −3.85 | −4.44 | 4.10 |
| 3TS1 | −0.46 | −0.65 | −1.24 | 8.19 |
| 3CI + H2O | −28.64 | −28.09 | −28.09 | −30.01 |
| 3RC2 | −7.47 | −7.15 | −7.74 | 0.60 |
| 3TSA1 | −3.45 | −3.82 | −4.41 | 5.77 |
| OH-adduct 1 | −31.41 | −31.93 | −32.52 | −21.84 |
| 3TSA2 | −2.33 | −2.58 | −3.17 | 6.48 |
| OH-adduct 2 | −34.05 | −34.32 | −34.91 | −25.62 |
| Reaction pathways | K eq | κ | k 2 | k TS | k Total |
|---|---|---|---|---|---|
| 1TS1 | 1.95 × 10−23 | 2.10 | 2.23 × 1010 | 4.35 × 10−13 | 5.01 × 10−11 |
| 1TSA | 1.95 × 10−23 | 1.02 | 9.62 × 1011 | 1.88 × 10−11 | |
| 3TS1 | 4.02 × 10−23 | 2.52 | 1.57 × 1010 | 6.31 × 10−13 | |
| 3TSA1 | 1.49 × 10−20 | 1.56 | 1.55 × 109 | 2.31 × 10−11 | |
| 3TSA2 | 1.49 × 10−20 | 1.60 | 4.81 × 108 | 7.17 × 10−12 |
3.1. Trioxide formation between 1,3,5-TMB BPR and OH
The important reaction between 1,3,5-TMB BPR and OH radical is addition of OH to the terminal oxygen atom of the peroxy end to produce ROOOH shown in Scheme 3. The large aromatic ROOOH is stabilized as Assaf et al.36 predicted that the trioxide, ROOOH containing more than three carbons are nearly exclusively collisionally stabilized in atmospheric conditions. This reaction starts with a H-bonded reactant complex 1RC1 which is 4.14 kcal mol−1 stabilized with respect to the separated reactants before the transition state 1TSA and the corresponding product ROOOH, which is clearly displayed in Fig. 1. The structures of key species of the title reaction on the potential energy surface have been drawn in Fig. 2. In 1RC1 (see Fig. 2), the O⋯HO bonding occurs between the terminal O atom of BPR and the H atom of OH radical with the distance of 2.013 Å. In 1TSA, the vibration mode of the imaginary frequency shows reorientation of OH fragment and then addition to the terminal oxygen atom of the peroxy end forming a new O–O bond to give a trioxide, ROOOH. The formation of this trioxide is exothermic by 31.33 kcal mol−1 in terms of ΔH298 K (see Table 1). Seen from Fig. 1, the energy barrier of 1TSA is computed to be −3.64 kcal mol−1 with respect to the 1RC1 and the reactants, respectively, which is much lower than the energy barrier of 1TS1 (−0.46 kcal mol−1) or 3TS1 (−1.19 kcal mol−1).3.2. OH-adduct formation between 1,3,5-TMB BPR and OH
As the 1,3,5-TMB BPR contains a double bond in the ring, the reaction of OH addition to the double bond producing OH-adduct should also be considered. Due to the substitution of the O2-bridged group in 1,3,5-TMB BPR, the OH addition on the CC bond proceeds through a reactant complex (3RC2) by strong hydrogen bond interactions between the H from the OH radical and the O from the O2-bridged group (see Fig. 1 and 2). The distances of the hydrogen bonds are 2.066 Å and 2.420 Å, and the binding energy of 3RC2 is −7.47 kcal mol−1. Then the reaction continues by addition of the OH radical to either of the carbon atoms of the double bond through the triplet transition states 3TSA1 and 3TSA2. Due to this addition, the OH-adduct 1 is formed by a shortening of C⋯OH bond from 2.060 Å in 3TSA1 to 1.427 Å, simultaneously, the CC bond length is elongated from 1.358 Å in 3TSA1 to 1.496 Å in the product OH-adduct 1. Similarly, the OH-adduct 2 is formed by a shortening of C⋯OH bond from 2.062 Å in 3TSA2 to 1.414 Å, at the same time, the CC bond length is elongated from 1.356 Å in 3TSA2 to 1.497 Å in the product OH-adduct 2. The formation of OH-adduct 1 and OH-adduct 2 are exothermic by 32.52 and 34.91 kcal mol−1 respectively in terms of ΔH298 K (see Table 1). The energy barriers of 3TSA1 and 3TSA2 are −3.45 kcal mol−1 and -2.33 kcal mol−1 respectively, which are also lower than that of 1TS1 or 3TS1, indicating that OH addition reactions including the formation of ROOOH and OH-adducts are the dominant pathways. This conclusion can be supported by the study of kinetics in the following section.
3.3. Criegee intermediate formation between 1,3,5-TMB BPR and OH
The hydrogen abstraction reaction of 1,3,5-TMB bicyclic peroxy radical with OH to form Criegee intermediate can occur via both singlet and triplet potential energy surfaces (PESs). On the singlet PES, as shown in Fig. 1, the H-abstraction pathway starts with the reactant complex 1RC1. Thereafter, 1RC1 is transformed into 1CI and water through a transition state 1TS1 with the energy barrier of −1.19 kcal mol−1 (with respect to the reactants). In 1TS1, the C–H bond is breaking with the distance of 1.174 Å. Simultaneously, the H atom is abstracted by the O atom from OH with the bond length of 1.417 Å. In addition, this reaction producing singlet CI (1CI) is significantly exothermic (ΔH298 K = −46.19 kcal mol−1, see Table 1). The H-abstraction mechanism is also found on the triplet PES. As shown in Fig. 1, the initial step is formation of a reactant complex 3RC1 stabilized by 4.00 kcal mol−1 with respect to the reactants. As seen from Fig. 2, the 3RC1 is characterized by H-bond interaction with the bond length of 2.014 Å between the hydrogen atom of OH and the terminal oxygen atom of BPR. Then the H atom of BPR is abstracted by OH radical, leading to the formation of 3CI + H2O via3TS1. This transition state 3TS1 is lower located than the reactants by 0.46 kcal mol−1 which involves a shortening of H⋯OH bond (from 2.559 to 1.364 Å) and an elongation of the C–H bond (from 1.098 to 1.191 Å) which will eventually break. Additionally, this reaction producing triplet CI (3CI) is moderately exothermic (ΔH298 K = −28.09 kcal mol−1, see Table 1).As seen from Fig. 2, the singlet 1RC1 and triplet 3RC1, as well as the corresponding 1TS1 and triplet 3TS1 all show six-membered ring forms though there are some structural discrepancies in the reactant complexes and transition states. The energy barrier of 1TS1 and 3TS1 differs by 0.73 kcal mol−1, indicating that, for the hydrogen abstraction mechanism, the reaction rate of singlet Criegee intermediate 1CI formation is close to that of triplet 3CI formation.
3.4. Kinetics
It has been shown that either OH addition or H-abstraction pathway consists of a reversible first step involving the barrierless formation of a reactant complex (RC), followed by the irreversible formation of products. Therefore, the reaction pathway is a two-step process as described by Scheme 4, where the reactant complex is in equilibrium with the reactants.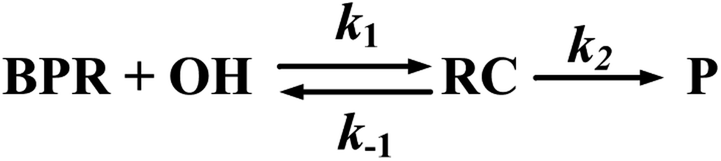 | ||
| Scheme 4 | ||
If k1 and k−1 are the rate constants for the first step and k2 is the rate constant for the second, a steady-state analysis leads to a rate constant for the reaction pathway under consideration, which can be approximated as
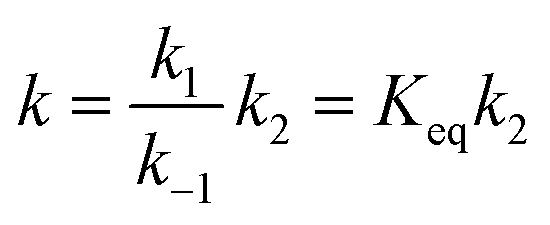 | (1) |
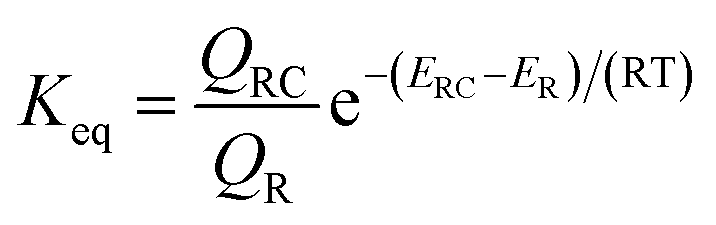 | (2) |
According to the conventional transition state theory, the unimolecular rate constant k2 with the Eckart tunneling correction is given by
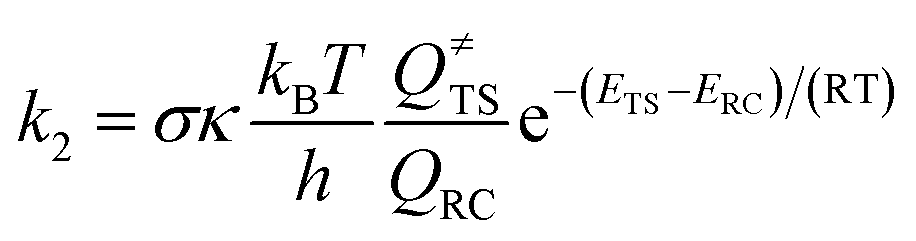 | (3) |
The rate coefficients were calculated using the M08-HX/6-311 + g(2df,2p) optimized geometrical parameters, frequencies and energies listed in Table 2. From Table 2, the rate constants of OH addition reactions by 1TSA, 3TSA1 and 3TSA2 are computed to be 1.88 × 10−11 cm3 mol−1 s−1, 2.31 × 10−11 cm3 mol−1 s−1, and 7.17 × 10−12 cm3 mol−1 s−1 respectively. While the rate constants of H-abstractions via singlet 1TS1 and triplet 3TS1 at 298 K are 4.35 × 10−13 cm3 mol−1 s−1 and 6.31 × 10−13 cm3 mol−1 s−1 respectively. The computed overall rate constant is 5.01 × 10−11 cm3 mol−1 s−1. The atmospheric lifetime of the studied 1,3,5-trimethylbenzene bicyclic peroxy radical due to reaction with OH can be estimated to be 5.54 hour using the following expression: τ = 1/(k[X]), where X is the average concentration of OH in the atmosphere ([OH] = 1.0 × 106 molecules per cm−3).39 The branching ratios for H-abstraction pathways via1TS1 and 3TS1 are only 8.68 × 10−3 and 1.26 × 10−2 respectively. Hence the OH addition reactions including the formation of ROOOH and OH-adducts are the major paths in contrast with H-abstraction channels producing Criegee intermediate which are negligible. The degradation of the ROOOH species in the atmosphere is significant which might help explain the difference in simulated and observed OH concentrations under low NOx environment.40 The OH-adducts may contribute to the formation and growth of SOA through subsequent reactions. Therefore, research is urgently needed into the atmospheric fate and possible influence of the 1,3,5-TMB ROOOH and OH-adducts on atmospheric chemistry.
4. Conclusions
In the present study, the mechanism and kinetics of the atmospheric reaction of 1,3,5-TMB bicyclic peroxy radical with OH have been studied by M08-HX/6-311 + g(2df,2p) method and CTST including Eckart tunneling correction. The conclusions are drawn in the following:(1) Two types of mechanisms including OH addition producing ROOOH and OH-adducts, and H-abstraction for Criegee intermediate formation are considered. Both the OH addition to the peroxy end and to the double bond of 1,3,5-TMB bicyclic peroxy radical are easier than H-abstraction reaction due to the lower energy barrier.
(2) The rate constants of OH addition to BPR are computed to be 1.88 × 10−11 cm3 mol−1 s−1, 2.31 × 10−11 cm3 mol−1 s−1, and 7.17 × 10−12 cm3 mol−1 s−1 by 1TSA, 3TSA1 and 3TSA2 respectively, while the abstraction rate constants via1TS1 and 3TS1 are calculated to be 4.35 × 10−13 cm3 mol−1 s−1 and 6.31 × 10−13 cm3 mol−1 s−1 respectively at 298 K. Kinetically, the abstraction reactions are much less favorable than OH-addition reactions, thus can be negligible.
(3) The 1,3,5-TMB ROOOH and OH-adducts are the most dominant products. The atmospheric fate and possible influence of the ROOOH and OH-adducts on atmospheric chemistry have not been explored yet.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank financial support from the National Natural Science Foundation of China (41605102, 91544228, 41575125, 21876177, 41775125), the National Key Research and Development Program of China (2016YFC0202205, 2017YFC0209401), the Youth Innovation Promotion Association CAS (2019439), and the Science and Technology Foundation of Guizhou Province, China ([2019]5648 and [2018]1080).References
- R. G. Derwent, T. J. Davies, M. Delaney, G. J. Dollard, R. A. Field, P. Dumitrean, P. D. Nason, B. M. R. Jones and S. A. Pepler, Atmos. Environ., 2000, 34, 297–312 CrossRef CAS.
- L. T. Molina, C. E. Kolb, B. de Foy, B. K. Lamb, W. H. Brune, J. L. Jimenez, R. Ramos-Villegas, J. Sarmiento, V. H. Paramo-Figueroa, B. Cardenas, V. Gutierrez-Avedoy and M. J. Molina, Atmos. Chem. Phys., 2007, 7, 2447–2473 CrossRef CAS.
- E. Velasco, B. Lamb, H. Westberg, E. Allwine, G. Sosa, J. L. Arriaga-Colina, B. T. Jobson, M. L. Alexander, P. Prazeller, W. B. Knighton, T. M. Rogers, M. Grutter, S. C. Herndon, C. E. Kolb, M. Zavala, B. de Foy, R. Volkamer, L. T. Molina and M. J. Molina, Atmos. Chem. Phys., 2008, 7, 329–353 CrossRef.
- A. Metzger, J. Dommen, K. Gaeggeler, J. Duplissy, A. S. H. Prevot, J. Kleffmann, Y. Elshorbany, A. Wisthaler and U. Baltensperger, Atmos. Chem. Phys., 2008, 8, 6453–6468 CrossRef CAS.
- M. J. Elrod, J. Phys. Chem. A, 2011, 115, 8125–8130 CrossRef CAS.
- K. P. Wyche, P. S. Monks, A. M. Ellis, R. L. Cordell, A. E. Parker, C. Whyte, A. Metzger, J. Dommen, J. Duplissy, A. S. H. Prevot, U. Baltensperger, A. R. Rickard and F. Wulfert, Atmos. Chem. Phys., 2009, 9, 635–665 CrossRef CAS.
- M. E. Jenkin and K. C. Clemitshaw, Atmos. Environ., 2000, 34, 2499–2527 CrossRef CAS.
- J. J. Orlando and G. S. Tyndall, Chem. Soc. Rev., 2012, 41, 6294–6317 RSC.
- J. A. Thornton, P. J. Wooldridge, R. C. Cohen, M. Martinez, H. Harder, W. H. Brune, E. J. Williams, J. M. Roberts, F. C. Fehsenfeld, S. R. Hall, R. E. Shetter, B. P. Wert and A. Fried, J. Geophys. Res., 2002, 107, ACH 7-1–ACH 7-17 Search PubMed.
- E. Assaf, B. Song, A. Tomas, C. Schoemaecker and C. Fittschen, J. Phys. Chem. A, 2016, 120, 8923–8932 CrossRef CAS.
- E. Assaf, S. Tanaka, Y. Kajii, C. Schoemaecker and C. Fittschen, Chem. Phys. Lett., 2017, 684, 245–249 CrossRef CAS.
- A. Bossolasco, E. P. Faragó, C. Schoemaecker and C. Fittschen, Chem. Phys. Lett., 2014, 593, 7–13 CrossRef CAS.
- E. P. Faragó, C. Schoemaecker, B. Viskolcz and C. Fittschen, Chem. Phys. Lett., 2015, 619, 196–200 CrossRef.
- C. Yan, S. Kocevska and L. N. Krasnoperov, J. Phys. Chem. A, 2016, 120, 6111–6121 CrossRef CAS.
- A. T. Archibald, A. S. Petit, C. J. Percival, J. N. Harvey and D. E. Shallcross, Atmos. Sci. Lett., 2009, 10, 102–108 CrossRef.
- C. Fittschen, L. K. Whalley and D. E. Heard, Environ. Sci. Technol., 2014, 48, 7700–7701 CrossRef CAS.
- E. Assaf, L. Sheps, L. Whalley, D. Heard, A. Tomas, C. Schoemaecker and C. Fittschen, Environ. Sci. Technol., 2017, 51, 2170–2177 CrossRef CAS.
- J. F. Müller, Z. Liu, V. S. Nguyen, T. Stavrakou, J. N. Harvey and J. Peeters, Nat. Commun., 2016, 7, 13213 CrossRef.
- B. Long, J. L. Bao and D. G. Truhlar, J. Am. Chem. Soc., 2016, 138, 14409–14422 CrossRef CAS PubMed.
- B. Long, J. L. Bao and D. G. Truhlar, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 6135–6140 CrossRef CAS.
- B. Long, J. L. Bao and D. G. Truhlar, J. Am. Chem. Soc., 2019, 141, 611–617 CrossRef CAS.
- M. Altarawneh, E. M. Kennedy, B. Z. Dlugogorski and J. C. Mackie, J. Phys. Chem. A, 2008, 112, 6960–6967 CrossRef CAS.
- T. J. Frankcombe, J. Phys. Chem. A, 2008, 112, 1572–1575 CrossRef CAS.
- J. M. Andino, J. N. Smith, R. C. Flagan, W. A. Goddard and J. H. Seinfeld, J. Phys. Chem., 1996, 100, 10967–10980 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, J. Chem. Theory Comput., 2008, 4, 1849–1868 CrossRef CAS.
- X. F. Xu, I. M. Alecu and D. G. Truhlar, J. Chem. Theory Comput., 2011, 7, 1667–1676 CrossRef CAS PubMed.
- C. Gonzalez and H. B. Schlegel, J. Phys. Chem., 1989, 90, 2154–2161 CrossRef CAS.
- C. Gonzalez and H. B. Schlegel, J. Phys. Chem., 1990, 94, 5523–5527 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, and D. J. Fox, Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- M. G. Evans and M. Polanyi, Trans. Faraday Soc., 1935, 31, 875–894 RSC.
- H. Eyring, J. Chem. Phys., 1935, 3, 107–115 CrossRef CAS.
- D. G. Truhlar, B. C. Garrett and S. J. Klippenstein, J. Phys. Chem., 1996, 100, 12771–12800 CrossRef CAS.
- C. Eckart, Phys. Rev., 1930, 35, 1303–1309 CrossRef CAS.
- W. T. Duncan, R. L. Bell and T. N. Truong, J. Comput. Chem., 1998, 19, 1039–1052 CrossRef CAS.
- S. Zhang and T. N. Truong, VKLab, version 1.0. University of Utah: Salt Lake City, UT, 2001 Search PubMed.
- E. Assaf, C. Schoemaecker, L. Vereecken and C. Fittschen, Int. J. Chem. Kinet., 2018, 50, 670–680 CrossRef CAS.
- M. A. H. Khan, C. J. Percival, R. L. Caravan, C. A. Taatjesc and D. E. Shallcross, Environ. Sci.: Processes Impacts, 2018, 20, 437–453 RSC.
- D. Osborn and C. A. Taatjes, Int. Rev. Phys. Chem., 2015, 34, 309–360 Search PubMed.
- W. J. Bloss, M. J. Evans, J. D. Lee, R. Sommariva, D. E. Heard and M. J. Pilling, Faraday Discuss., 2005, 130, 425–436 RSC.
- C. Fittschen, M. A. Ajami, S. Batut, V. Ferracci, S. Archer-Nicholls, A. T. Archibald and C. Schoemaecker, Atmos. Chem. Phys., 2019, 19, 349–362 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra06562h |
| This journal is © The Royal Society of Chemistry 2019 |