
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTheoretical study on the excited state decay properties of iron(II) polypyridine complexes substituted by bromine and chlorine†
Yuan
Li
,
Xue-Wen
Fan
,
Jie
Chen
,
Fu-Quan
Bai
 * and
Hong-Xing
Zhang
*
* and
Hong-Xing
Zhang
*
International Joint Research Laboratory of Nano-Micro Architecture Chemistry, Laboratory of Theoretical and Computational Chemistry, Institute of Theoretical Chemistry, Jilin University, Changchun 130023, People's Republic of China. E-mail: baifq@jlu.edu.cn; zhanghx@jlu.edu.cn
First published on 4th October 2019
Abstract
Transition metal iron(II) polypyridyl complexes with quintet ground states were deeply investigated by density functional theory (DFT) and time-dependent density functional theory (TDDFT). Compared with the parent complex [Fe(tpy)2]2+ (tpy = 2,2′:6′,2′′-terpyridine), the ground states of the complexes substituted by halogen atoms changed from singlet states to quintet states with rare high spin excited state lifetimes. The substituted complex [Fe(dbtpy)2]2+ (1) results in a high spin metal–ligand charge transfer lifetime of 17.4 ps, which is 1.4 ps longer than that of [Fe(dctpy)2]2+ (2) with the substitution of chlorine atoms. The reason for this is explored by a combination of electronic structures, absorption spectra, extended transition state coupled with natural orbitals for chemical valence (ETS-NOCV) studies and potential energy curves (PECs). The distortion of 1 in the angles and dihedrals of the ligands is slightly larger than that in 2, although the average metal–ligand bond lengths of the latter are larger. The twisted octahedron decreases the interactions between the d orbitals of iron(II) and the n/π orbitals of the ligands. Compared with 2, the enlarged energy gaps among the different PECs of 1 and the increased energy crossing points caused by the larger distortion result in the increase of its excited state lifetime. The different pairwise orbital interaction contributions between the metal center and the ligands in their singlet states are qualitatively estimated by ETS-NOCV. The results show that the substitution of bromine atoms will decrease the electrostatic attraction between the metal and ligands but not significantly impact the orbital interactions.
1 Introduction
Transition metal complexes have demonstrated their capabilities in organic light emitting devices (OLEDs) on account of their phosphorescence mechanism.1,2 Their luminescence quantum efficiencies are as high as 100%, and they have seen large-scale commercial production since the first phosphorescent OLED was reported in 1996.3 Most studies have been performed on precious transition metals, such as ruthenium, iridium and platinum.4–7 However, their high cost and environment pollution limit their applications. Recently, the use of inexpensive metals such as copper, zinc and iron has seen an increasing amount of studies to promote light-harvesting applications.8–16 Although iron is the second most Earth-abundant metal on Earth and is inexpensive and environmentally benign, its subpicosecond nonradiative relaxation from the metal-to-ligand charge transfer (MLCT) state into a low-lying metal-centered (MC) state results in more difficult utilization.17,18 Investigations of the excited state decay path of the prototype [Fe(bpy)3]2+ (bpy = 2,2-bipyridine) complex have proved that ultrafast intersystem crossing (ISC) occurs from the 1,3MLCT state to the 3MC state and then to the 5MC state. Increasing the ligand field strength is expected to change the energetic accessibility of the MC states, which results in a decrease in the nonradiative decay.19 Haacke and Gros et al. used N-heterocyclic carbine ligands to dramatically increase the Fe(II) MLCT excited state lifetime to 26 ps by strengthening the metal–ligand bonds.20 More recently, using a combination of cyano ligands and bpy ligands has been demonstrated as a way of separately controlling the 3,5MC and 1,3MLCT state energies to achieve a 20 ps lifetime.9 However, an opposite strategy of decreasing the ligand-field strength is still used to expand the charge-transfer excited state lifetimes, which increases the excited state lifetimes effectively. Damrauer et al. introduced this strategy for extending MLCT lifetimes by using different halogen atoms connected to the pyridyls which interact with the opposing ligand.6 The insertion of halogen atoms destabilizes the singlet ground state and results in a quintet ground state and an optically accessible 5,7MLCT. In this work, the conclusion is based on two ideas. Firstly, the 5,7MLCT happens to be less coupled to intermediates such as 3MC, which is necessary for nonradiative decay processes. Secondly, the sizes of the different halogen substituents diminish the conformational freedom of the system within the excited state manifold, thus preventing the dynamics necessary for intersystem crossing within the relaxation process.It is an enormous challenge to explore the luminescence of Fe(II) complexes in detail because of their mixed multiple spin states, such as singlet, triplet, quintet and septet states. Moreover, it is difficult to detect the corresponding photophysical and decay mechanisms by experimental methods. With improving computational power and theoretical approaches, quantum chemical calculations can provide significant insights not only into the fundamental structures, but also into the electronic and photophysical properties of metal complexes. Here, we study the complexes [Fe(dbtpy)2]2+ (dbtpy is the abbreviation of 6,6′′-dibromo-2,2’;6′2′′-terpyridine), simplified to 1, and [Fe(dctpy)2]2+ (dctpy is the abbreviation of 6,6′′-dichloro-2,2’:6′2′′-terpyridine), named 2, to explore how halogen atoms increase the high spin MLCT lifetimes with larger sizes (Fig. 1). In Fig. 1, Nc and Nt are the central and terminal nitrogen atoms of tpy, respectively. X represents the halogen atoms.
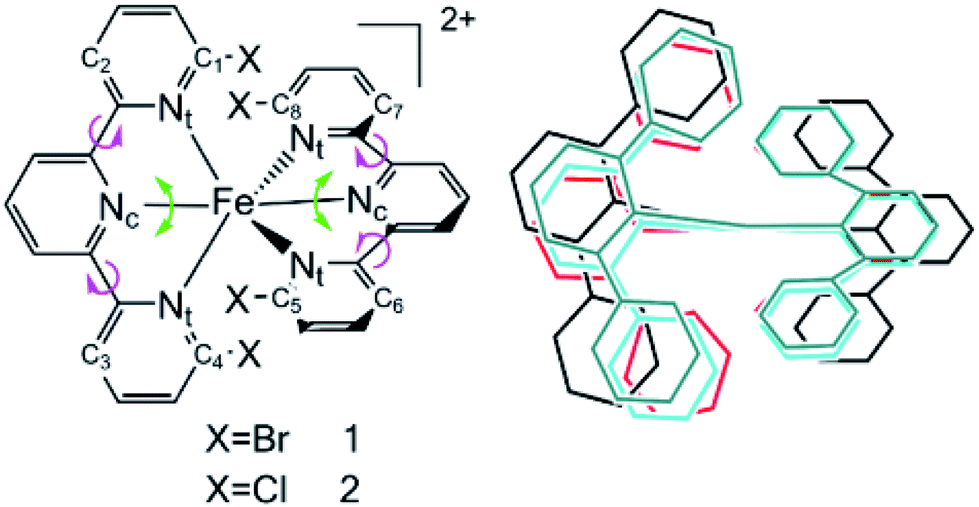 | ||
| Fig. 1 (Left): Two constructions containing halogen substituents. (Right): The skeletons of the fully optimized structures of 1 and 2 with halogens omitted in the ground state 5MC (black), singlet state (red), triplet state 3MC (cyan) and septet state 7MLCT (green). | ||
When the parent complex [Fe(tpy)2]2+ is substituted by halogen atoms, quintet ground states are accessible with rare high spin MLCT state lifetimes as long as 14–17.4 ps. The singlet ground state is favoured by enthalpy and the quintet ground state is populated due to entropic contributions to the free energy (3: 97 at room temperature); thus, the fluorine-substituted complex is complicated to discuss. The special characteristic and ligand field of the F-substituted complex [Fe(dftpy)2] may result from the small bulk of F with weaker steric hindrance of the interactions compared to Cl or Br. Weakly sterically hindered interactions result in higher conformational freedom, which gives rise to an enthalpically favoured singlet ground state. Before analyzing the effects of halogen substitution in the coexistence of mixed spin states, we may obtain insights into the mechanism of the particularity of the spin crossover equilibrium after solving the decay mechanisms of 1 and 2 with pure quintet ground states well in advance in this work.
The distortion in the angles and dihedrals of the ligands of 1, increased by bromine atoms with greatly sterically hindered interaction, is slightly larger than in 2, although the average metal–ligand bond lengths of the latter are larger. The increase of the CT proportion from the chlorine to bromine substituents causes hypsochromic shifts of the lowest absorption peaks. The hole and electron analysis simplifies the manifold orbitals as one main transition mode. The potential energy curves (PECs) consist of the average values of the Fe–Nc bond lengths as the x-axis and the single-point energies as the y-axis. This shows that the sum of the energy gaps of intersystem crossing according to the Franck–Condon rules and Kasha's rule on the PECs of the different spin states of 1 is larger than that of 2, which results in the 1.4 ps longer lifetime. The higher energy crossing points of 1 improve the energy barriers and hinder the thermodynamic hopping from the optimized structures to the energy crossing points, which results in faster non-radiative decay. ETS-NOCV studies and the pictures of the deformation density show that the substitution of bromine atoms will decrease the electrostatic attraction between the metal and ligands but only slightly impact the orbital interactions.
2 Computational details
The geometrical optimizations of the ground quintet states, singlet states, triplet states and septet states of 1 and 2 were separately simulated by DFT and UDFT with the hybrid functional PBE0.21 The quasi-relativistic effective core potential (ECP) of Fe proposed by Hay and Wadt with 18 valence electrons with the double-ζ quality basis set LANL2DZ22 and the 6-31G(d)23 basis set for H, C, N, and Cl and LANL08 (ref. 24) for Br were adopted. Vibrational frequencies were calculated at the same level to confirm that all the optimized structures were at the minimal points of their potential energy surfaces without imaginary frequencies. Furthermore, the solvent effect of acetonitrile (CH3CN) was considered by the polarized continuum model (PCM)25 for all the calculations. The absorption spectra and potential energy surfaces were simulated by LANL2TZ(f)26 and 6-311g(d, p)27 for higher accuracy. The above calculations were all performed using Gaussian 16 Rev. A.03.28 The hole and electron analysis function of Multiwfn 3.6 (ref. 29) was used to analyse the complicated transition orbitals of the absorption processes.The bond properties among the metal center, tpy and halogen atoms at different spin states were determined by the ETS-NOCV30–33 algorithm of the Amsterdam Density Functional (ADF) 2016.104 program.34,35 The BP86 (ref. 36–38) functional with a Slater type all-electron triple-ζ basis set with polarizations (TZP)39 was used with one-component zeroth-order regular approximation (ZORA)40–42 on the scalar relativistic singlet geometries. Moreover, the conductor-like screening model (COSMO)43 of solvent with CH3CN was adopted in the ETS-NOCV calculations.
3 Results and discussion
3.1 Geometrical structures
The main optimized geometrical parameters of the different spin excited states of [Fe(tpy)2]2+ ,44 [Fe(dbtpy)2]2+ (1) and [Fe(dbtpy)2]2+ (2) are listed in Table 1 with the corresponding experimental X-ray characterization results. The calculated data of Fe–Nc, Fe–Nt and Nt–Nc–Nt are in good agreement with the experimental values, with deviations of 0.9% to 2.4%. Also, the quintet metal-center states of 1 and 2 have the lowest energies compared with the singlet, triplet and septet states.| Fe–Nc(Å)b | Fe–Nt(Å)b | Fe–X(Å) | Nt–Nc–Nt(deg) | C1–C2–C3–C4, C5–C6–C7–C8/Nt–Nc–Nc–Nt(deg)a | θ(deg)b | X–pyridyl(Å) | |
|---|---|---|---|---|---|---|---|
| a Nc is the central nitrogen atom of tpy. Nt represents the terminal nitrogen atoms of tpy. X is the halogen atoms. The line of dihedrals includes the average dihedrals of single dxtpy (positive values) and the dihedrals between two dxtpy ligands. θ is the bond angle between the iron(II) metal center and the central nitrogen atoms of the ligands. All the bond lengths are average values, except for Fe–Nc of the septet states. b See ref. 12 and 42. | |||||||
| [Fe(tpy) 2 ] 2+ b | |||||||
| Quintet | 2.16 | 2.20 | — | — | — | 10.8 | — |
| Singlet | 1.91 | 2.01 | — | — | — | 0 | — |
| Exp. | 1.89 to 1.99 | 1.89 to 1.99 | — | — | — | — | — |
| Triplet | 1.94 | 2.14 | — | — | — | 0.2 | — |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||||||
| 1 | |||||||
| Quintet | 2.11 | 2.34 | 3.81 | 113.7 | 29.8/−90.7 | 2.9 | 1.93 |
| Exp. | 2.07 | 2.30 | — | 112.7 | — | 11.7 | 3.18 |
| Singlet | 1.90 | 2.12 | 3.83 | 107.4 | 37.4/−93.8 | 0 | 1.93 |
| Triplet | 1.93 | 2.25 | 3.88 | 112.1 | 32.1/−92.8 | 0.7 | 1.93 |
| Septet | 1.95/2.06 | 2.22/2.25 | 3.78/3.82 | 111.2/111.8 | 31.2, 34.8/88.4 | 1.7 | 1.92/1.93 |
|
|||||||
| 2 | |||||||
| Quintet | 2.13 | 2.32 | 3.62 | 112.3 | 15.8/−90.2 | 0.4 | 1.73 |
| Exp. | 2.08 | 2.27 | — | 111.4 | — | 4.1 | 3.07 |
| Singlet | 1.90 | 2.11 | 3.66 | 106.7 | 25.0/−92.1 | 0 | 1.72 |
| Triplet | 1.93 | 2.23 | 3.71 | 111.2 | 16.3/−90.8 | 0 | 1.73 |
| Septet | 1.96/2.08 | 2.21/2.24 | 3.61/3.64 | 110.4/111.2 | 16.4, 20.4/−89.8 | 0.9 | 1.72/1.73 |
In the ground quintet states, the bond lengths between the nitrogen atoms and iron(II) center are longer than those of the other spin excited states. The bond lengths of Fe–Nc of the quintet states, triplet states and singlet states of 1 and 2 are separately 2.11/2.13, 1.93 and 1.90 Å. Meanwhile, the bond lengths of Fe–Nt have the same trend as those of Fe–Nc. When they have the same spin states, there are only 0 to 0.02 Å deviations between Fe–N of 1 and 2. Also, the shorter bond lengths enhance the interaction between the ligands and iron(II) metal center. In terms of the bond angles, the angles of terpyridine of the different spin excited state structures still gradually decrease, with small deviations (0.7 to 1.4 degrees), in the order of quintet, triplet and singlet. The changing rules of the bond lengths and bond angles may result from the Jahn–Teller effect. The repulsion between the d orbitals of iron(II) and the n/π orbitals of terpyridine result in the extension of the Fe–N bonds of 1 and 2.
Compared with [Fe(tpy)2]2+, Fe–Nt and Nt–Nc–Nt of 1 and 2 show obvious extension due to the halogen substituents. [Fe(tpy)2]2+ has only 0.01 Å deviations of Fe–Nc in the singlet and triplet states from [Fe(dxtpy)2]2+. However, the bond lengths of Fe–Nt of [Fe(tpy)2]2+ are 0.09 to 0.14 Å longer than those of [Fe(dxtpy)2]2+ due to the substitution of Br and Cl. The singlet ground state of [Fe(tpy)2]2+ may result from the increased ligand field split, which improves the energy gap of the eg and t2g orbitals. In comparison with [Fe(tpy)2]2+, the octahedral structures of 1 and 2 are more twisted, which weakens the interaction between iron(II) and terpyridine because there is less orbital overlap among the d orbitals and n/π orbitals. In addition, the dihedral angles of the single terpyridine and two terpyridines separately increase from quintet to triplet to singlet, which may decrease the effective radiative process ands increase the non-radiative decay. Furthermore, for the dihedral angles of the single terpyridine, the large deviations of 12.4 to 15.8 degrees between 1 and 2 in different spin excited states indicate that the replacement by bromine can result in larger distortion of the structures and change the population of the potential energy surfaces. Furthermore, θ, the bond angle between the iron(II) center and central nitrogen atoms, shows that the larger distortion of the structures of 1 than of 2 may be caused by the replacement of bromine. Concerning the bond lengths of Fe–X and X–pyridyl, the larger bromine atom has greater steric repulsion interactions and weaker influence on the iron(II) center. We found that the geometries of the quintet, triplet and singlet states have the same average values of their bond lengths and bond angles. However, the geometrical characteristics of the septet states of the two complexes are obviously special because one β electron is flipped onto the π* of the closer ligand as α spin. 400 nm excitation can easily cause the complexes to reach the energy levels of the septet states. The deactivating pathway beginning from 7MLCT will be discussed in the next part.
3.2 Absorption properties and hole–electron analysis
[Fe(tpy)2]2+ with the singlet ground state has been theoretically proved by the Jakubikova et al.44 The differences in the absorption properties between the singlet ground state and quintet ground states are another interesting topic that may be investigated in the future. Herein, only the absorption properties based on the optimized quintet ground states of 1 and 2 are discussed. The electron transition configurations, excitation energies, oscillator strengths (f), and assignments of the main absorption bands are listed in Table S1.† The corresponding absorption spectra of 1 and 2 and the alpha and beta spin molecular orbitals are displayed in Fig. 2 and 3.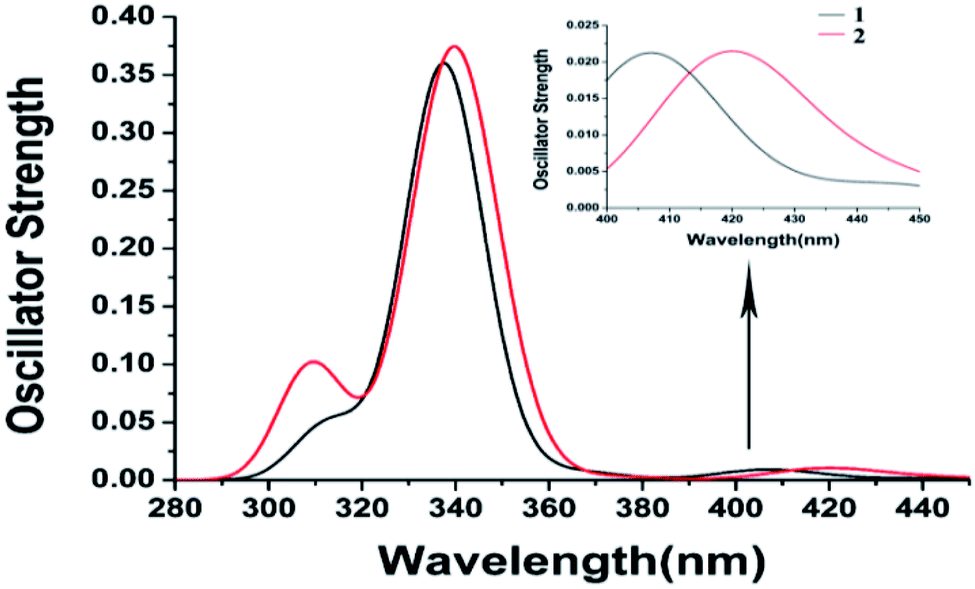 | ||
| Fig. 2 Calculated absorption spectra and enlargement of the lowest absorption peaks of 1 and 2. | ||
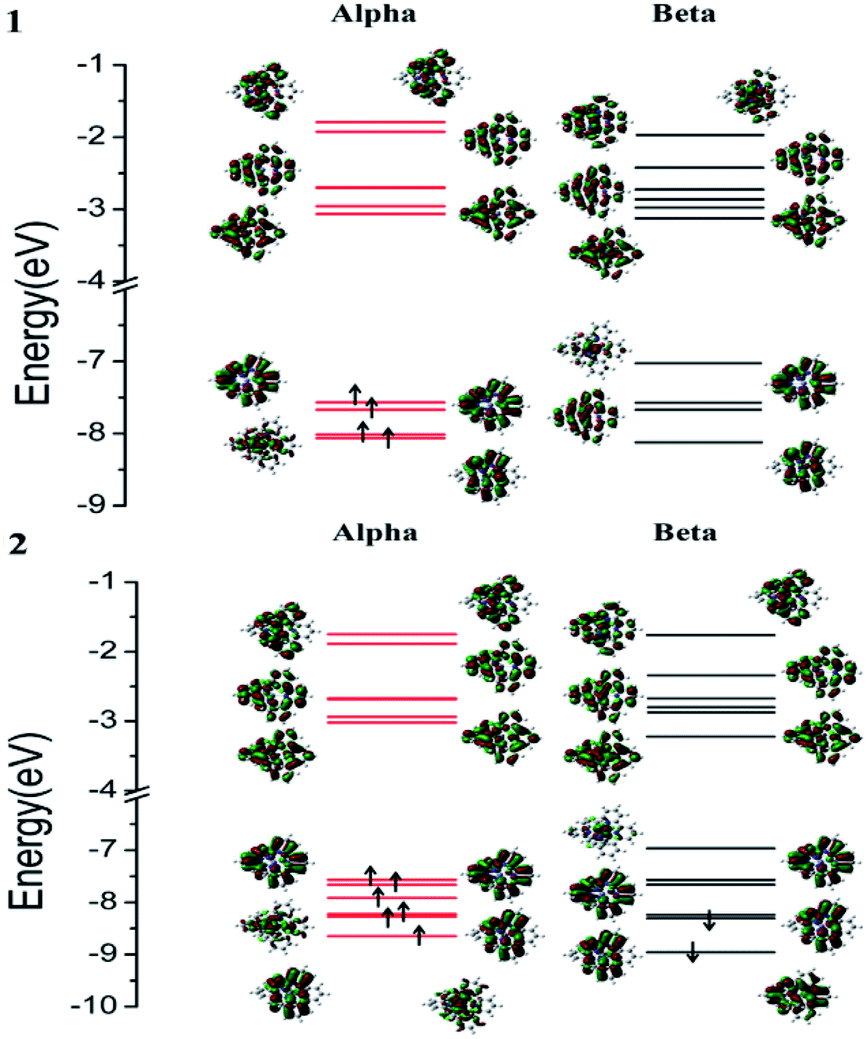 | ||
| Fig. 3 The main alpha and beta spin molecular orbitals of 1 and 2. | ||
The calculated absorption spectra are in good agreement with the experimental values.6 It is obvious that the bromine substituents result in a slight blue-shift of the lowest peak compared to the chlorine substituents. The lowest absorption peak of 1 is mainly contributed by Q10, with the transitions of H(B) → L(B) and H(B) → L+5(B). The assignment of H(B) → L(B) is MLCT/LLCT (ligand-to-ligand charge transfer) and that of H(B) → L+5(B) is MLCT/LLCT/MC. However, the lowest absorption peak of 2 is mainly contributed by Q7 with the transitions of H(B) → L(B), whose assignment is MLCT/MC. The hypsochromic shift may result from the increase of LLCT. Meanwhile, the strongest absorption peaks of 1 and 2 consist of similar high excited states to almost degenerate energy levels. Therefore, they have similar strongest absorption peaks to the main transition configuration of ILCT (intraligand charge transfer).
Nevertheless, there are too many transition configurations with trivial proportions because of the special quintet ground state excitations, although we have a rough conclusion about the absorption properties. The hole and electron analysis performed with Multiwfn can conclude and transfer the manifold orbital transition characters into one main transition contribution, such as a natural transition orbital (NTO). The corresponding data and visual pictures are displayed in Table 2 and Fig. 4, where D is the total magnitude of the charge transfer length between the centroids of the hole and electron and t is the overall measure of the separation degree of the hole and electron in the charge transfer (CT) direction, which is from electrons to holes. The negative values of t and small values of D show that the separation degrees of the hole and electron in the CT direction of 1 and 2 are very small. The D of the quintet excited state Q10 of 1 is 0.04 Å longer than that of the quintet excited state Q7 of 2. The absolute value of t of Q10 of 1 is about 0.11 Å longer than that of Q7 of 2. This indicates that the CT process of 1 is more obvious, which may result in the hypsochromic shift of the lowest absorption peak of 1. Sr is the geometric average of the overlap function between the hole and electron distribution. The range of Sr values is [0, 1]. This indicates complete overlap of the hole and electron when Sr is 1. So, the overlap extent of Q10 of 1 is slightly smaller than that of Q7 of 2. Δσ is the overall root-mean-square deviation of the extent of spatial distribution of the hole and electron. The smaller Δσ and smaller Sr of 1 illustrate that the MLCT from the dyz of iron(II) ion to the π* of tpy of 1 is more distinct. Therefore, the hypochromatic shift from 2 to 1 depends on a stronger CT process. The charge transfer process based on a single molecule occurs before exciton recombination among polymolecules. We will simulate exciton recombination in a future article. In this work, the charge transfer state and excited state relaxation pathways are discussed as the main points.
| Excited state | S r | D (Å) | t (Å) | Δσ (Å) | Assignment |
|---|---|---|---|---|---|
| a S r is the geometric average of the overlap function between the hole and electron distributions. D is the total magnitude of the charge transfer length between the centroids of the hole and electron. t is the overall measure of the separation degree of the hole and electron in the charge transfer direction. Δσ is the overall root-mean-square deviation of the extent of spatial distribution of the hole and electron. ILCT/LLCT represent intraligand/ligand-to-ligand charge transfer. LMCT/MLCT are ligand-to-metal/metal-to-ligand charge transfer. MC represents metal-center charge transfer. | |||||
| 1 | |||||
| Q0 → Q6 | 0.647 | 0.006 | −2.079 | 0.292 | MLCT/LLCT (82.3%) |
| Q0 → Q10 | 0.453 | 0.048 | −1.674 | 1.697 | MLCT/LLCT (88.7%) |
| Q0 → Q13 | 0.533 | 0.058 | −2.013 | −0.566 | LLCT/MLCT (84.7%) |
| Q0 → Q14 | 0.647 | 0.092 | −2.062 | −0.072 | LLCT (93.7%) |
|
|||||
| 2 | |||||
| Q0 → Q5 | 0.672 | 0.009 | −2.077 | 0.376 | ILCT (85.4%) |
| Q0 → Q7 | 0.497 | 0.008 | −1.568 | 1.754 | MLCT/LLCT (92.9%) |
| Q0 → Q13 | 0.611 | 0.013 | −2.198 | −0.241 | MLCT/LLCT (86.4%) |
| Q0 → Q14 | 0.515 | 0.021 | −1.902 | 1.340 | MLCT/LLCT (94.4%) |
| Q0 → Q15 | 0.621 | 0.021 | −2.155 | −0.358 | MLCT/LLCT (88.2%) |
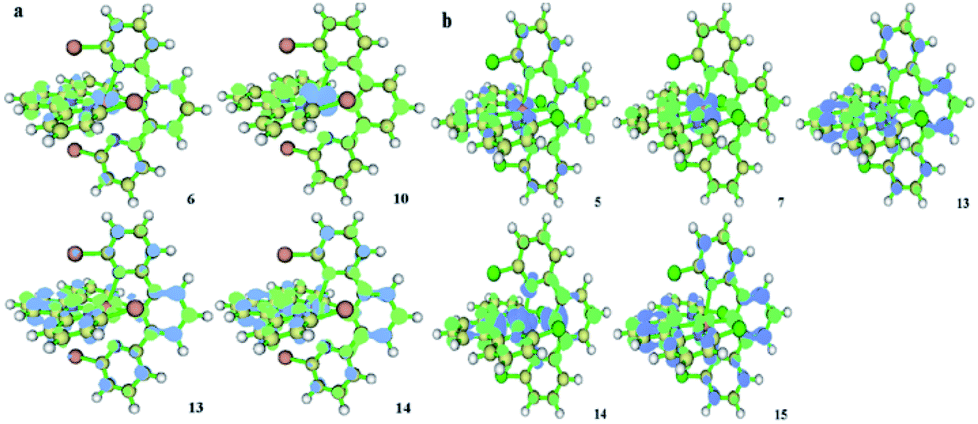 | ||
| Fig. 4 The hole and electron populations of 1 (a) and 2 (b). The numbers at the bottom of each structure show the different quintet excited states. Blue indicates the population of holes. Green represents the population of electrons. The charge direction is from hole to electron. | ||
3.3 The potential energy curves (PECs) of 1 and 2
Although the reason for the differences in the lowest absorption peaks has been explained, the deviation of the excited state lifetimes of 1 and 2 is still obscure. Thus, the PECs with the same values of Fe–Nc in Fig. 5 were used to qualitatively explore the corresponding decay mechanism. The detailed data of the PECs are listed in Table S2.† It is obvious that the energy gaps among the PECs of 1MLCT, 3MC and 5MC of 1 are slightly larger than those of 2. Meanwhile, the 1.4 ps lifetime deviation between 1 and 2 may result from the intersystem crossing among the PECs of the different spin states. If we follow Kasha's rule and the Frank–Condon rule, the order of intersystem crossing will be from septet to singlet to triplet to quintet, and all the intersystem crossing will occur from the minimum points of the previous PECs to the points with the same structures of the next PECs. If the decay path is 7MLCT–5MLCT–3MLCT–1MLCT through crossing of the potential energy surfaces, it may be difficult due to probable high minimum energy crossing points and large structural distortion in the ultrafast process. In contrast, the transition by vertical decay in the Franck–Condon regions may be feasible because of the more distinct d orbital degeneration caused by the tridentate ligand tpy than by the bidentate ligand.45 Therefore, we think a vertical transition from the high spin excited states in the Franck–Condon regions may be easier than the deactivated process. The smaller energy gap of 2 results in a faster vertical decay process than in 1 (in Table S2†). Furthermore, the energy crossing point between the singlet and quintet states of 1 is much higher than that of 2. The excitons decay from 7MLCT by a vertical process and rapidly relax to the minimum points. However, the energy crossing points among 1MLCT, 3MC and 5MC of 2 are very close, and the deactivation process may directly go through the crossing point between 1MLCT and 5MC as non-radiative decay. Compared with 2, a higher energy crossing point between 1MLCT and 5MC of 1 can effectively decrease this process and promote the radiative decay, which will increase the excited state lifetime.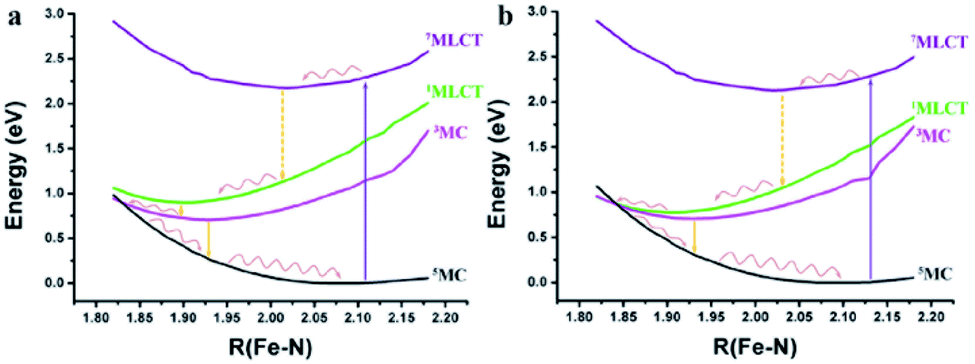 | ||
| Fig. 5 The PECs of (a) 1 and (b) 2 consisting of the average values of two Fe–Nc. | ||
3.4 The analysis of the transition state coupled with natural orbitals for chemical valence (ETS-NOCV)
To explore the interactions between the metal centers and the terpyridines quantitatively, ETS-NOCV analysis was performed. All the detailed ETS-NOCV data of the singlet states of 1 and 2 are listed in Table 3. The graphical information of the deformation densities of different pairwise orbital interactions of the corresponding modes of 1 and 2 are displayed in Fig. 6. ETS-NOCV is an effective and convenient method to analyze the decomposition of the interaction energy (ΔEint) between the fragments of molecules, where ΔEint can be expressed as follows:| ΔEint = ΔEpauli + ΔEelstat + ΔEorb | (1) |
| 1 | 2 | |
|---|---|---|
| ΔEint | −499.2 | −507.9 |
| ΔEpauli | 330.6 | 333.7 |
| ΔEelstat | −345.1 (41.6%) | −355.8 (42.3%) |
| ΔEorb | −484.7 (58.4%) | −485.8 (57.7%) |
| ΔE1 | −107.4 (22.2%) | −110.8 (22.8%) |
| ΔE2 | −115.7 (23.9%) | −114.3 (23.5%) |
| ΔE9 | −19.4 (4.0%) | −19.8 (4.1%) |
| ΔErest | −261.6 (49.9%) | −260.7 (49.6%) |
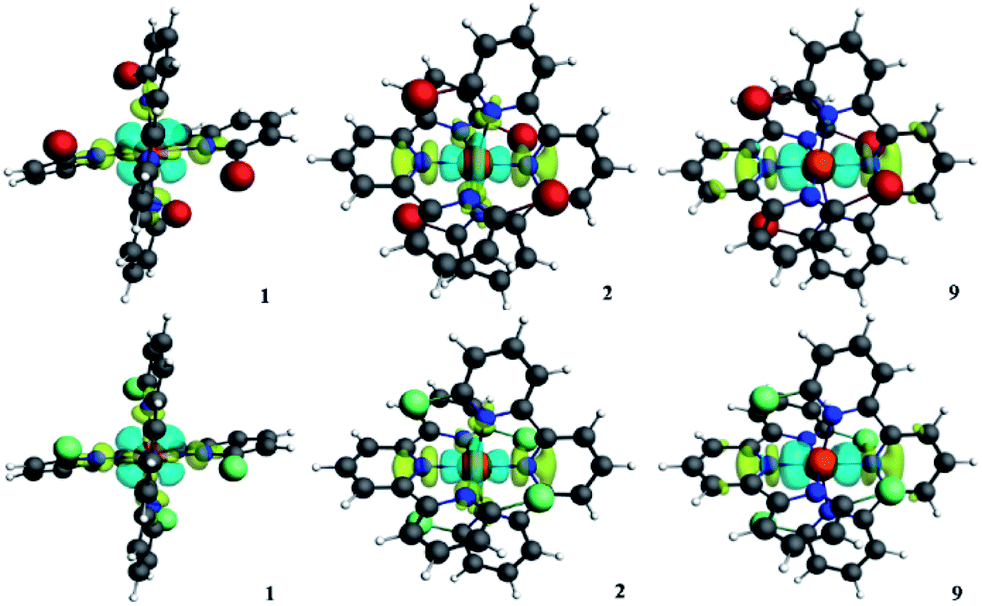 | ||
| Fig. 6 Plots of the deformation density Δρ of the pairwise orbital interactions. 1 is at the top and 2 is at the bottom. The numbers at the bottom of each construction represent the different modes of deformation density. The direction of the charge flow is from yellow to cyan. | ||
ΔEpauli is the exchange (Pauli) repulsion energy. ΔEelstat is the quasiclassical Coulomb interaction. ΔEorb originates from the orbital mixing and is identified as the covalent contribution to the chemical bond. In energy and visualization, ΔEorb developed by Ziegler and Rauk can be analyzed to determine which part of the orbital energy is involved in the metal–ligand interactions. The deformation density Δρ in the NOCV representation is a sum of pairs of complimentary eigenfunctions (ψ−k, ψk) corresponding to the eigenvalues −λk and λk as follows:
| Δρ = ΣΔρk(r) = Σλk(−ψ−k2(r) + ψk2) | (2) |
| ΔEorb = ΣΔEorbk = Σλk(−FTS−k + FTSk) | (3) |
In terms of ΔEint, we found that the metal–ligand interactions of 1 are 8.7 kcal mol−1 weaker than those of 2. The occupied 3d orbitals of the iron(II) center encounter not only the stabilizing interaction of the n orbital on the 6 nitrogen atoms of tpy, but also the repulsive interaction with the occupied 2s and 2p orbitals on the other centers. As a result, the additional dctpys of 2 result in stronger electrostatic attraction and weaker Pauli repulsion. However, this does not play a significant role in enhancing the orbital interactions. The deviation of ΔEorb between 1 and 2 is only 1.1 kcal mol−1. Due to the stronger non-metallic character of Cl and the longer bond lengths of Fe–N in 2, the interactions between Fe and the ligand may lie in a strong electrostatic attraction state and weak repulsion state on the interaction potential curves.
Also, there are similar pairwise orbital interactions. In Fig. 6, we can find that the main orbital contributions of the singlet states of 1 and 2 originate from dx2−y2 of iron(II) ion and the n orbitals of the nitrogen atoms to dz2 or dxy of iron(II) ion as σ* or non-bonding with similar contributions. Thus, we found that the substitution of bromine and chlorine impacts the metal–ligand interactions by electrostatic attraction to a greater extent.
4 Conclusions
1 and 2 were studied to explore how different halogen atoms impact the MLCT lifetimes by a combination of electronic structures, absorption spectra, ETS-NOCV and PECs. The larger distortion of 1 than 2 is caused by the sterically hindered interactions of bromine atoms. Taking the advantage of the combination of the Kohn–Sham molecular orbitals of the absorption properties and the hole and electron analysis, the increase in the CT proportion from the chlorine to bromine substituents causes a hypsochromic shift of the lowest absorption peaks. The PECs show that the total energy gaps of intersystem crossing following the Franck–Condon rule and Kasha's rule on the PECs of the different spin states of 1 are larger than those of 2, which results in the 1.4 ps longer lifetime. The higher energy crossing points of 1 improve the energy barriers and hinder the thermodynamic hopping from the optimized structures to the energy crossing points, which results in faster non-radiative decay. ETS-NOCV and the pictures of the deformation density show that the substitution of bromine atoms will decrease the electrostatic attractions between the metal and ligands but will slightly impact the orbital interactions. Unfortunately, the analysis of the MLCT progress and lifetimes is only qualitative. As different metal center analogues, [Ru(tpy)2]2+ with 602 nm emission at 77 K only has a singlet ground state, a triplet metal–ligand charge transfer state (3MLCT) and a triplet metal center state (3MC).46 However, [Fe(dxtpy)2]2+ without luminescence in this article has ground quintet states, singlet metal–ligand charge transfer states (1MLCT), and 3MC and septet metal–ligand charge transfer states (7MLCT). Due to the abundant excited states, exploration of the complicated excited states of the deactivated pathways in detail is difficult. The photophysical properties of the complex containing fluorine still remain to be discussed. We will make more effort to resolve these problems and explore more properties and mechanisms of iron complexes in the future.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is funded by the Natural Science Foundation of China (Grant Nos. 21873038 and 21573088), and the “13th Five-Year” science and technology project of the education department of Jilin province (Grant No. JJKH20180123KJ) and the Young Scholar Training Program of Jilin University.References
- J.-Y. Hung, C.-H. Lin, Y. Chi, M.-W. Chung, Y.-J. Chen, G.-H. Lee, P.-T. Chou, C.-C. Chen and C.-C. Wu, J. Mater. Chem., 2010, 20, 7682 RSC.
- G. Cheng, S. C. F. Kui, W.-H. Ang, M.-Y. Ko, P.-K. Chow, C.-L. Kwong, C.-C. Kwok, C. Ma, X. Guan, K.-H. Low, S.-J. Su and C.-M. Che, Chem. Sci., 2014, 5, 4819–4830 RSC.
- K. M. Maness, R. H. Terrill, T. J. Meyer, R. W. Murray and R. M. Wightman, J. Am. Chem. Soc., 1996, 118, 10609–10616 CrossRef CAS.
- Y. Wang, J. Wang, H.-X. Zhang, I. M. Szilágyi and F.-Q. Bai, Organometallics, 2018, 37, 2491–2499 CrossRef CAS.
- Z.-X. Zhang, W. Wei, F.-Q. Bai, S. Bibi and H.-X. Zhang, Org. Chem. Front., 2017, 4, 2191–2201 RSC.
- S. M. Fatur, S. G. Shepard, R. F. Higgins, M. P. Shores and N. H. Damrauer, J. Am. Chem. Soc., 2017, 139, 4493–4505 CrossRef CAS.
- M. Abrahamsson, H. C. Becker and L. Hammarstrom, Dalton Trans., 2017, 46, 13314–13321 RSC.
- T. He, A. Ren, S. Chen, Z. Yang, P. Yu, Y. Chen, X. Ding, L. Shen, H. Zhang and L. Zou, Org. Electron., 2017, 45, 9–19 CrossRef CAS.
- W. Zhang, K. S. Kjaer, R. Alonso-Mori, U. Bergmann, M. Chollet, L. A. Fredin, R. G. Hadt, R. W. Hartsock, T. Harlang, T. Kroll, K. Kubicek, H. T. Lemke, H. W. Liang, Y. Liu, M. M. Nielsen, P. Persson, J. S. Robinson, E. I. Solomon, Z. Sun, D. Sokaras, T. B. van Driel, T. C. Weng, D. Zhu, K. Warnmark, V. Sundstrom and K. J. Gaffney, Chem. Sci., 2017, 8, 515–523 RSC.
- Y. Liu, T. Harlang, S. E. Canton, P. Chabera, K. Suarez-Alcantara, A. Fleckhaus, D. A. Vithanage, E. Goransson, A. Corani, R. Lomoth, V. Sundstrom and K. Warnmark, Chem. Commun., 2013, 49, 6412–6414 RSC.
- X. C. Ma, Y. Dai, L. Yu and B. B. Huang, Light Sci Appl, 2016, 5, e16017 CrossRef CAS.
- X. Wang, F.-Q. Bai, Y.-T. Liu, J. Wang and H.-X. Zhang, Synth. Met., 2016, 213, 18–24 CrossRef CAS.
- A. Baker and H. Goodwin, Aust. J. Chem., 1985, 38, 207–214 CrossRef CAS.
- L. A. Buldt and O. S. Wenger, Dalton Trans., 2017, 46, 15175–15177 RSC.
- I. M. Dixon, F. Alary, M. Boggio-Pasqua and J. L. Heully, Dalton Trans., 2015, 44, 13498–13503 RSC.
- W. Zhang, Y. Li, V. P. Nguyen, Z. Huang, Z. Liu, X. Wang and Y. M. Paulus, Light Sci Appl, 2018, 7, 103 CrossRef.
- G. Auböck and M. Chergui, Nat. Chem., 2015, 7, 629 CrossRef PubMed.
- A. Cannizzo, C. J. Milne, C. Consani, W. Gawelda, C. Bressler, F. van Mourik and M. Chergui, Coordin. Chem. Rev., 2010, 254, 2677–2686 CrossRef CAS.
- W. Zhang, R. Alonso-Mori, U. Bergmann, C. Bressler, M. Chollet, A. Galler, W. Gawelda, R. G. Hadt, R. W. Hartsock, T. Kroll, K. S. Kjær, K. Kubiček, H. T. Lemke, H. W. Liang, D. A. Meyer, M. M. Nielsen, C. Purser, J. S. Robinson, E. I. Solomon, Z. Sun, D. Sokaras, T. B. van Driel, G. Vankó, T.-C. Weng, D. Zhu and K. J. Gaffney, Nature, 2014, 509, 345 CrossRef CAS.
- L. Liu, T. Duchanois, T. Etienne, A. Monari, M. Beley, X. Assfeld, S. Haacke and P. C. Gros, Phys. Chem. Chem. Phys., 2016, 18, 12550–12556 RSC.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS.
- G. A. Petersson, A. Bennett, T. G. Tensfeldt, M. A. Al-Laham, W. A. Shirley and J. Mantzaris, J. Chem. Phys., 1988, 89, 2193–2218 CrossRef CAS.
- L. E. Roy, P. J. Hay and R. L. Martin, J. Chem. Theory Comput., 2008, 4, 1029–1031 CrossRef CAS.
- G. Scalmani and M. J. Frisch, J. Chem. Phys., 2010, 132, 114110 CrossRef.
- A. W. Ehlers, M. Böhme, S. Dapprich, A. Gobbi, A. Höllwarth, V. Jonas, K. F. Köhler, R. Stegmann, A. Veldkamp and G. Frenking, Chem. Phys. Lett., 1993, 208, 111–114 CrossRef CAS.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, Williams, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Rev. A.03, Wallingford, CT, 2016 Search PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS.
- T. Ziegler and A. Rauk, Theore. Chim. Acta, 1977, 46, 1–10 CrossRef CAS.
- T. Ziegler and A. Rauk, Inorg. Chem., 1979, 18, 1755–1759 CrossRef CAS.
- T. Ziegler and A. Rauk, Inorg. Chem., 1979, 18, 1558–1565 CrossRef CAS.
- M. P. Mitoraj, A. Michalak and T. Ziegler, J. Chem. Theory Comput., 2009, 5, 962–975 CrossRef CAS PubMed.
- C. Fonseca Guerra, J. G. Snijders, G. te Velde and E. J. Baerends, Theore. Chem. Acc., 1998, 99, 391–403 Search PubMed.
- G. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. Fonseca Guerra, S. J. A. van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931–967 CrossRef CAS.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS.
- J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822–8824 CrossRef.
- J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 34, 7406 CrossRef.
- E. Van Lenthe and E. J. Baerends, J. Comput. Chem., 2003, 24, 1142–1156 CrossRef CAS.
- E. v. Lenthe, E. J. Baerends and J. G. Snijders, J. Chem. Phys., 1993, 99, 4597–4610 CrossRef.
- E. v. Lenthe, E. J. Baerends and J. G. Snijders, J. Chem. Phys., 1994, 101, 9783–9792 CrossRef.
- E. v. Lenthe, A. Ehlers and E.-J. Baerends, J. Chem. Phys., 1999, 110, 8943–8953 CrossRef.
- C. C. Pye and T. Ziegler, Theore. Chem. Acc., 1999, 101, 396–408 CrossRef CAS.
- J. Nance, D. N. Bowman, S. Mukherjee, C. T. Kelley and E. Jakubikova, Inorg. Chem., 2015, 54, 11259–11268 CrossRef CAS PubMed.
- D. G. Brown, N. Sanguantrakun, B. Schulze, U. S. Schubert and C. P. Berlinguette, J. Am. Chem. Soc., 2012, 134, 12354–12357 CrossRef CAS.
- A. Soupart, I. M. Dixon, F. Alary and J.-L. Heully, Theor. Chem. Acc., 2018, 137 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra06366h |
| This journal is © The Royal Society of Chemistry 2019 |