
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMaterials with the CrVO4 structure type as candidate superprotonic conductors†
Pandu
Wisesa
 ,
Chenyang
Li
,
Chuhong
Wang
and
Tim
Mueller
*
,
Chenyang
Li
,
Chuhong
Wang
and
Tim
Mueller
*
Department of Materials Science and Engineering, Johns Hopkins University, Baltimore, MD 21218, USA. E-mail: tmueller@jhu.edu
First published on 8th October 2019
Abstract
Proton conducting oxides have the potential to improve the efficiency of solid oxide fuel cells and electrolyzers, yet many oxide structures remain relatively unexplored for the ability to conduct protons. To accelerate the search for novel proton-conducting oxides, we have performed a computational screen of the proton migration energy in 41 different commonly-occurring oxide structure types. The results of this screen, which are supported by a comprehensive set of density functional theory calculations, indicate that known materials with the CrVO4 structure type have an average migration energy for proton diffusion of less than 0.2 eV, with several known materials having calculated migration energies below 0.1 eV. These results indicate that materials with the CrVO4 structure type, which to our knowledge have not been previously explored as candidate proton conductors, may exhibit very high proton conductivity that surpasses that of leading proton-conducting oxides. We present the results of our screen as well as diffusion dimensionality analysis and thermodynamic stability analysis for materials with the CrVO4 structure.
1 Introduction
Proton-conducting solids are used in a variety of applications, including as hydrogen separation membranes in chemical processing1–11 and in electrodes or electrolytes in solid oxide fuel cells12–21 and electrolyzers.22–26 There is particular interest in using proton-conducting oxides as electrolytes and electrode materials in fuel cells, where an electrolyte must be electronically insulating but an electrode material may be a mixed ionic-electronic conductor. A widely-used fuel cell technology, proton exchange membrane (PEM) fuel cells, uses polymer electrolytes that operate in an aqueous environment, so their operating temperatures are typically below 100 °C.27,28 At such low operating temperatures PEM cells require the use of expensive catalysts to achieve sufficiently high rates.28 An alternative is to use a solid oxide that conducts oxygen ions for the electrolyte (and ideally the electrode as well), but to achieve sufficiently high rates such fuel cells typically need to be operated at temperatures above about 600 °C.18,29 There is increasing interest in developing fuel cells that can operate at intermediate temperature ranges, which could lower the cost of the fuel cells, reduce start-up times, improve efficiency, and improve long-term durability.22,29–32 As protons are smaller than oxygen ions and carry half the electronic charge, they tend to diffuse more readily than oxygen ions; thus a promising route to improving the rate capability (and lowering the operating temperature) of solid oxide fuel cells is to use proton-conducting oxides as electrolytes and electrode materials.12–21 Fuel cells with proton-conducting electrolytes have additional advantages in that the reaction products are produced at the cathode, rather than the anode, so they are kept separate from the fuel, which can result in gains in efficiency.13,14 For fuel cells that use ammonia as a fuel, the use of proton-conducting electrolytes prevents the formation of NO, an unwanted byproduct.17 Despite their advantages, fuel cells that use proton-conducting oxides are limited by a number of factors including poor proton transport through grain boundaries and poor stability in fuel cell operating conditions.20,33–36 To address these issues, there is a need to develop new proton-conducting oxides that are stable in fuel cell operating conditions and have high proton conductivity.The most widely-studied class of proton-conducting oxides contains materials with the perovskite structure or closely related structures.20,34–42 A perovskite-structured material, SrCeO3, was one of the first proton-conducting oxides with high proton conductivity,43 and two of the current leading families of proton conducting oxides, based on BaZrO3 and BaCeO3, have the perovskite structure.20,34,36 Perovskite-structured oxides can accommodate a wide variety of cations in different stoichiometries,44 providing researchers great flexibility in tailoring their properties. Many perovskite-structured oxides can also accommodate high concentrations of oxygen vacancies and allow for relatively facile diffusion of oxygen ions, providing a mechanism to introduce protons into the host material through a reaction with water. Importantly, the arrangement of oxygen ions in the perovskite structure provides a continuous three-dimensional network for protons to migrate via the Grotthuss mechanism,45 in which they rotate around oxygen ions and then hop between them. In the search for new proton-conducting oxides, researchers have increasingly been exploring materials that do not have the perovskite structure. These efforts have revealed proton conductivity in materials such as solid acids,21,46–48 ortho-niobates,33 ortho-tantalates,33,49 orthophosphates,50,51 pyrochlores,52 sesquioxides,53 (oxy)sulfides,54 nitrides,55 tungstates,56 tungsten oxide, where the Grotthuss mechanism was not observed,57 and recently Li13.9Sr0.1Zn(GeO4)4 (LSZG).58
There are a number of criteria that must be met for a proton conductor to work in a fuel cell, electrolysis cell, or related technology. Among these, the material must be stable at operating temperatures, resistant to poisoning, and selective. It must also have sufficiently high proton solubility. It is important to consider the electronic conductivity when assessing whether the material would be suitable for use as an electrolyte, membrane, or electrode material. However in all applications, it is necessary that the bulk mobility of protons through the material is high. There are on the order of 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 known inorganic materials,59 but the bulk proton mobility is unknown for most materials, making it possible that there are entire classes of known materials that are good proton conductors but are currently being overlooked.
000 known inorganic materials,59 but the bulk proton mobility is unknown for most materials, making it possible that there are entire classes of known materials that are good proton conductors but are currently being overlooked.
In this work we perform a high-throughput computational search to identify structure types that are likely to have high proton mobility. We focus on structure types rather than individual compounds as the ionic conductivity of a material is determined to a large extent by whether arrangement of cations and anions in the material is conducive to creating a potential energy surface with a low activation energy for ionic migration.36,60–62 Once a structure type conducive to proton mobility is identified, materials with that structure type and different chemical compositions can be designed and/or synthesized to try to find a material with high stability and proton solubility while maintaining a relatively high probability of having high proton mobility.
2 Methodology
We searched for promising structure types for proton conducting oxides using a multi-level screening approach. We first identified over 37000 unique oxide materials with fully occupied sites from the Inorganic Crystal Structure Database (ICSD).59 The structures were grouped by their structure types using an algorithm developed by our group to compare crystalline structures, resulting in 3663 different oxide structure types. As we are most interested in structure types that can accommodate a variety of different elements, we selected from these the structure types for which the ICSD contained at least 20 unique materials, resulting in a data set of 41 structure types and 1946 structures in total.
We use the classical activation energy for proton migration (within the Born–Oppenheimer approximation) as our primary descriptor for proton mobility. We will refer to this value as the “migration energy”. As the contribution to the material energy from the zero-point vibrational energy of protons can be large (∼100–200 meV),63,64 there is likely a significant quantum contribution to proton mobility that is not captured by classical transition state theory. However even if quantum corrections are taken to account, it can be expected that materials with a lower classical activation energies will generally have faster proton migration.65–67 Thus for the purposes of screening for promising proton conductors it is sufficient to use the classical migration energy.
The 1946 different oxide materials we consider contain 75 different elements. To calculate activation energies for proton migration in all 1946 materials it is necessary to use an energy model that works for most of the periodic table. As the use of first-principles methods such as density functional theory (DFT)68,69 for all materials in our data set would have been prohibitively expensive, we developed a simpler model based on the bond valence method,70–74 which has been parameterized for nearly all elements.75 In the bond valence model, the bond valence between two neighboring atoms is given by
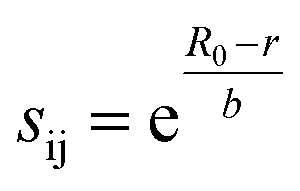 | (1) |
The bond valence method relates the valence of a bond to the equilibrium bond length, but it cannot be used directly to calculate energies.74,76 However there is a natural relationship between the bond valence approach and electrostatic interactions77 that makes it possible to construct a simple pairwise interatomic potential that is largely consistent with the bond valence model. Adams and Rao have developed a method for using the bond-valence method to construct a pairwise interatomic potential by combining a Morse-type term with a repulsive electrostatic term,78 and they have used their approach to study lithium ion diffusion in battery materials.79–81 Here we use a similar approach. The main difference between our approach and that of Adams and Rao is that the repulsive part of our potential is exponential, and attraction is represented by a screened Coulomb potential rather than a Morse potential. All electrostatic interactions, both attractive and repulsive, are thus combined in a single term:
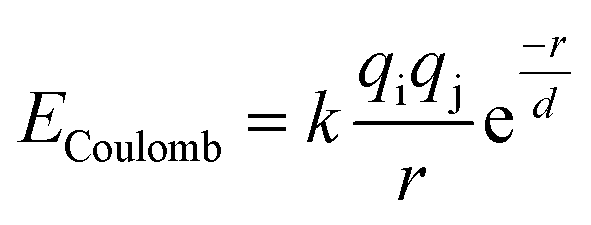 | (2) |
The repulsive part of the potential is given by:
| Eexp = Ae−rC | (3) |
| Epair = Eexp + ECoulomb | (4) |
To determine approximate values for A and C in eqn (3), we use the fact that for a given set of atomic valences, the equilibrium bond length in eqn (4) should match the bond length used to generate those valences in eqn (1). For a given value of R0, we find the values for A and C that minimize the mean squared difference between the equilibrium bond length and the value of r in eqn (1) over seven different binary crystal structures: rhenium trioxide, cristabolite, cuprite, wurtzite (hexagonal), rutile, fluorite, rock salt, and cesium chloride. The values of A and C were then further refined by linearly scaling them using universal scale factors fit to DFT data. To account for atomic relaxations from ideal crystalline sites in the presence of a proton, we connected each atom to its ideal site through a virtual spring, where the spring constant was a linear function of absolute value of the atomic charge. The parameters of this function were also fit to DFT data. Additional details about our approach, including the parameters used for our calculations, are provide in the ESI.†
We used the above energy model to construct the potential energy surface for a single proton in the different oxide materials studied. We used a combination of a grid search, gradient descent, and structural symmetry to find saddle points, local minima, and diffusion pathways through each material. Starting from the proton site with the lowest potential energy, all possible diffusion pathways that fully crossed a unit cell were evaluated, and the pathway with the lowest maximum potential energy was selected as the most likely diffusion path. The migration energy for proton diffusion through the material was then calculated as the difference between the maximum and minimum potential energies along the most likely diffusion path.
2.1 Density functional theory calculations
All DFT calculations were done using the Vienna Ab initio Simulation Package (VASP)82–86 versions 5.3 and 5.4 using “accurate” precision. The electronic minimization was done using a combination of blocked Davidson iteration scheme and RMM-DIIS.87,88 Perdew–Burke–Ernzerhof (PBE)89 projector augmented wave (PAW)90 potentials were used for all calculations. To take into account of possible spin polarization in the material, all calculations are set to be spin polarized. The electronic self-consistency cutoff was set to 10−5 eV and the relaxation cutoff was set to 10−3 eV. The relaxation was done using the RMM-DIIS algorithm. The k-point grid was generated using the k-point grid server91 with a minimum periodic distance of 28.1 Å. For thermodynamic stability calculations, we used the GGA + U approach of Dudarev.92 We used a U-J value of 3.25 eV for vanadium, as determined by the Materials Project by fitting to the experimental formation energies of different vanadium oxides.93 For cerium we used a value of 4.5 eV, which was determined self-consistently for Ce2O3 by Fabris et al.94 and is close to the value of 5 eV determined empirically for BaCeO3 by Shishkin and Ziegler.952.2 Nudged elastic band calculations
We used the climbing image nudged elastic band (CI-NEB) method as implemented in VASP by the Henkelman group.96–100 The spring constant used for the elastic band was 5 eV Å−2 and atomic relaxation was performed using damped molecular dynamics with scaling constant (POTIM) of 0.01. The energy model described above was run using experimental lattice parameters and atomic positions, as these are the values available from the ICSD. As the equilibrium structure in DFT calculations is typically slightly different from the experimentally-determined structure, we relaxed the volume of all structures using DFT prior to running NEB calculations. By relaxing the volume while leaving the fractional atomic coordinates unchanged, we ensured that it was straightforward to use the diffusion paths discovered using our energy model to initialize NEB calculations while avoiding spurious relaxations in DFT due to large stresses on the unit cell.All NEB calculations were run in supercells of the relaxed unit cells that ensured there were at least 8 Å between periodic images. All NEB calculations were run at fixed volume and atomic positions were allowed to relax to create realistic models of proton diffusion in the dilute limit.35,101 For each diffusive hop, the NEB images were initialized by placing hydrogen atoms along the path of the hop with no more than 0.5625 Å between successive hydrogen locations. The end points of the NEB calculation had their lattice vector frozen and atomic positions relaxed.
Oxides are typically doped with an electron acceptor to incorporate protons into the lattice. As this would break symmetry and significantly increase the computational expense of our calculations, we generated training data for our energy model using a perfect (undoped) crystal in which a single electron was removed per diffusing hydrogen atom in the NEB calculations. To minimize discontinuities due to spin flips during the NEB calculations, the magnetic moments on all atoms in each image were fixed to the values of the relaxed, empty (without hydrogen) structure. The conjugate gradient algorithm was used for atomic relaxation in all NEB runs. The relaxation was stopped when forces converged within 0.05 eV Å−1. All other parameters were the same as those listed in the above section on density functional theory calculations.
2.3 Stability of doped phases
We used PyMatGen102 to identify the likely decomposition products of the doped compositions from the Materials Project database, and we calculated the energies of the doped phases and possible decomposition products using DFT as described above. For the DFT calculations we assume the dopants are distributed in a way that maximizes the distance between dopant atoms in the resulting materials, and we use our energy model to calculate the likely position of H in the doped material.3 Results
3.1 Migration energies of the training data
The parameterized potential model was able to predict the migration barriers energies of the 52 structures in our training set (Table 3-S in the ESI†) with a mean absolute error of 0.126 eV (Fig. 1) relative to DFT. Within the set of 52 training structures, there are four structure types that are represented by at least five structures: cubic perovskite, hexagonal perovskite, elpasolite, and spinel. Much of the noise in the fit disappears when we evaluate the average activation energies for each of these structure types (Fig. 1b), and the mean absolute error between the potential model and DFT decreases to 0.032 eV. This reduction in error can be understood by considering a simple model in which the migration energy can be expressed as the sum of two components: the average migration energy for materials with that structure type, and the deviation from this average due to the chemical composition of the material. The total prediction error (Fig. 1a) will be a sum of the errors for each of these components. If the parameter fitting produces a model for which the mean error in the chemical composition component is nearly zero, then even if the variance of this error is large its contribution to the overall error can be greatly reduced by averaging over different chemical compositions.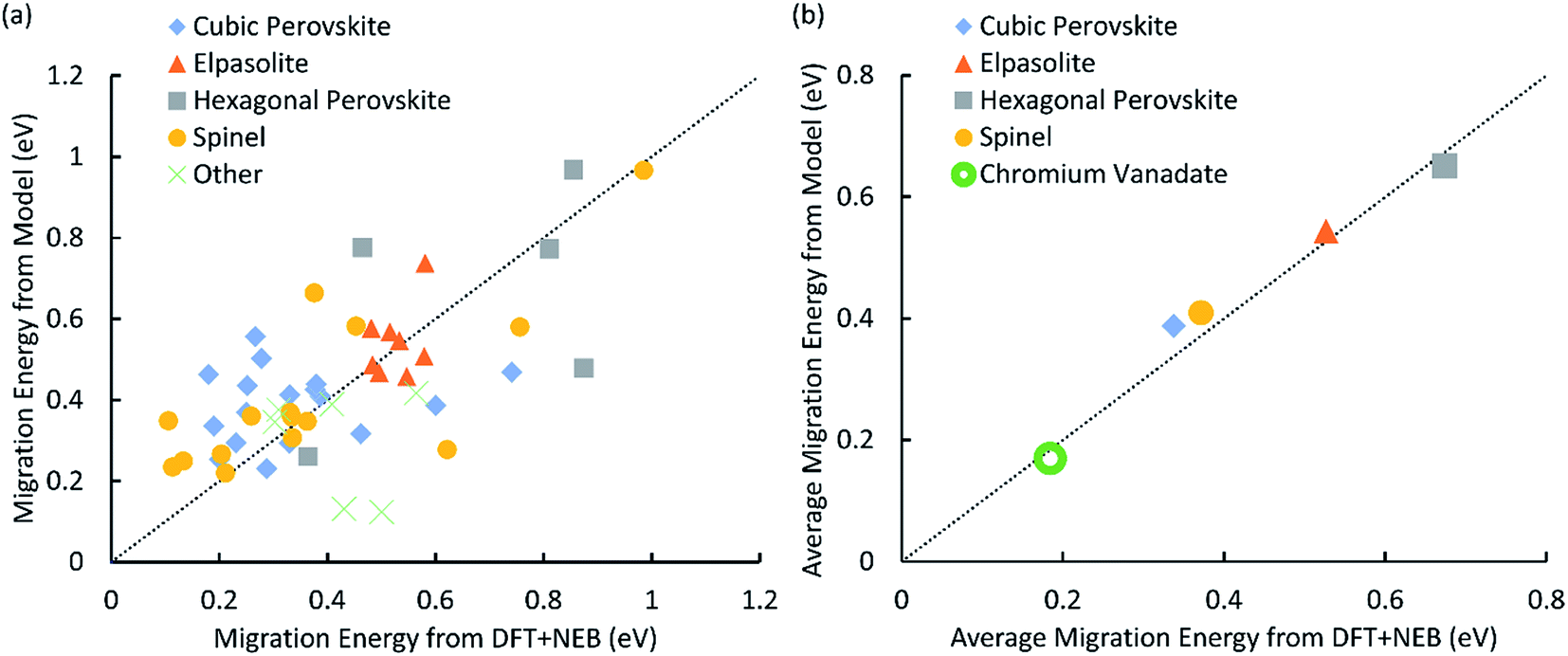 | ||
| Fig. 1 (a) A plot of activation energy for proton migration calculated by DFT + NEB vs. predicted by the energy model. Structure types with less than 5 members in the dataset are put together and represented by the green cross. (b) A plot of average migration energy by structure type calculated by DFT + NEB vs. predicted by the energy model. The filled shapes correspond to the training data in part (a), and the empty green circle represents the CrVO4 structure type discovered by our screen, with the DFT activation energy averaged over all 29 structures in Table 2. In both (a) and (b) the diagonal dotted line represents perfect agreement. | ||
As expected, our calculations show that the average migration energy for cubic perovskites, the most studied class of proton conductors, is relatively low. Our DFT-calculated activation energies are consistent with those reported by Bork et al.103 A typical diffusion pathway in a cubic perovskite, BaZrO3, is shown in Fig. 2. The pathway predicted by the energy model, including the locations of the end points and saddle points, corresponds well to DFT + NEB pathways. Diffusion occurs via a Grotthuss-type mechanism, where the proton rotates around the oxygen atom before hopping to the next oxygen atom. Protons also diffuse through hexagonal perovskites via a Grotthuss-type mechanism (Fig. 2), but the hexagonal perovskites in our training data have nearly twice the average activation energies of cubic perovskites even though they share the same composition of ABX3, providing an indication of the importance of structure type in determining activation energies.
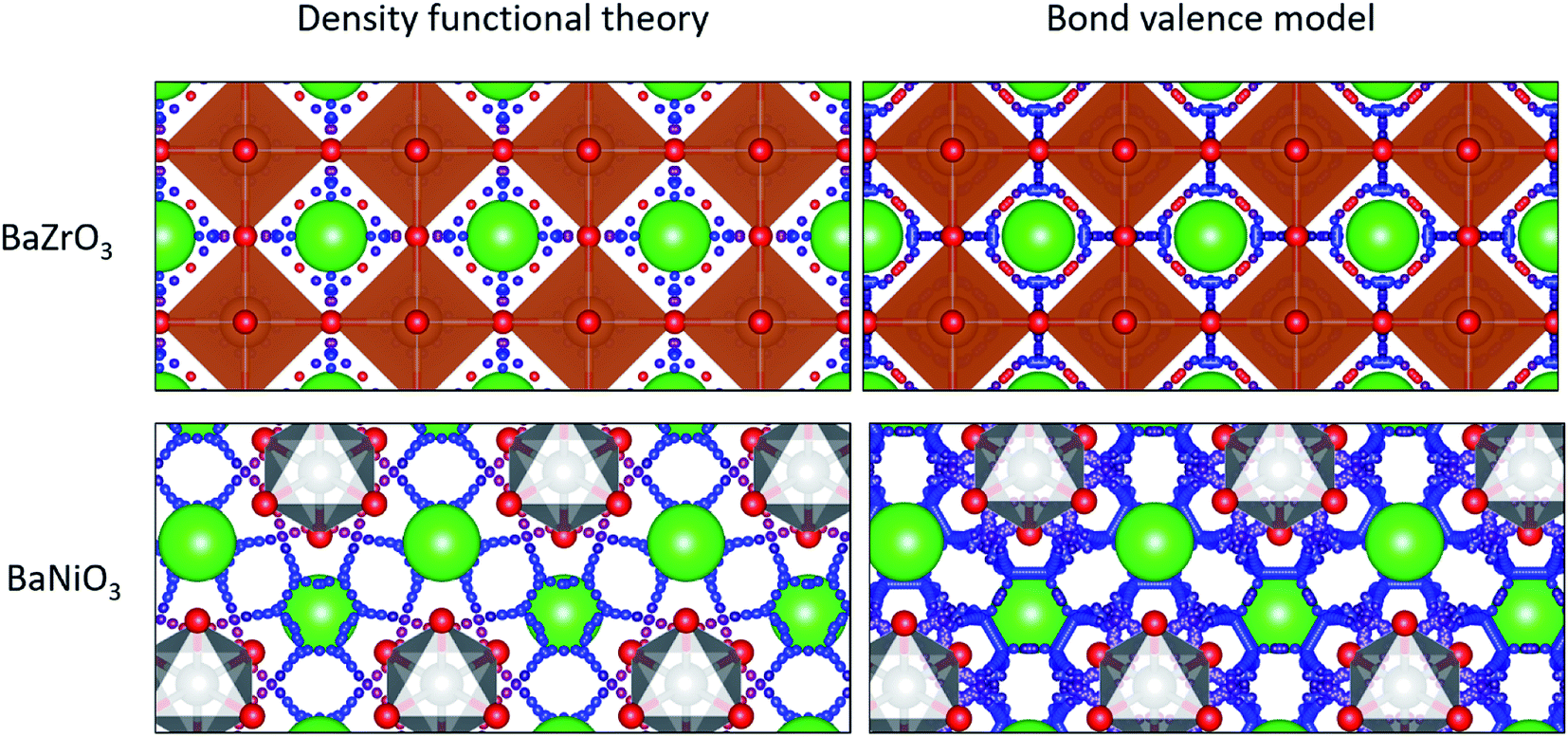 | ||
| Fig. 2 Ab initio (left) and model-predicted (right) diffusion pathways in a cubic perovskite (top) and hexagonal perovskite (bottom). Large green spheres represent barium atoms, orange polyhedra contain zirconium, and grey polyhedra contain nickel. Small spheres represent the possible diffusion pathways with warm (more red) colors representing high energy sites and cool (more blue) colors representing low energy sites. | ||
3.2 Screening structure types
We used our model to predict the activation energies for proton diffusion in all 1946 test structures, representing 41 different structure types with at least 20 oxide materials per type. On average, predicting the average migration energy for a structure type took around 20 minutes on a single core, which is a small fraction of the cost of DFT + NEB calculations. The average activation energies per by structure type are provide in Table 1 and Fig. 3. We note that the average activation energies for cubic perovskite, hexagonal perovskite, spinel, and elpasolite are different than the averages shown in Fig. 1, as these averages were taken over all oxide structures in the ICSD rather than just those in our training set.| Rank | Average migration energy (eV) | Sample standard deviation (eV) | Structure type name | Number of materials | Space group |
|---|---|---|---|---|---|
| 1 | 0.193 | 0.105 | CrVO4 | 29 | Cmcm |
| 2 | 0.312 | 0.099 | Calcite | 20 |
R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) c c |
| 3 | 0.326 | 0.084 | Zircon | 64 | I41/amd |
| 4 | 0.332 | 0.067 | NaMn7O12 | 30 | Im |
| 5 | 0.333 | 0.074 | Fergusonite | 27 | C2/c |
| 6 | 0.359 | 0.154 | Spinel–Al2MgO4 | 64 |
Fdm |
| 7 | 0.364 | 0.105 | Rutile | 41 | P42/mnm |
| 8 | 0.382 | 0.089 | LiYb(WO4)2 | 30 | P2/n |
| 9 | 0.391 | 0.100 | Pyrochlore | 91 |
Fdm |
| 10 | 0.402 | 0.129 | Scheelite | 43 | I41/a |
| 11 | 0.421 | 0.093 | ZrCuSiAs–CuHfSi2 | 57 | P4/nmm |
| 12 | 0.432 | 0.241 | Cubic perovskite | 88 |
Pmm |
| 13 | 0.439 | 0.107 | Barite–BaSO4 | 23 | Pnma |
| 14 | 0.445 | 0.129 | Sc2Si2O7 | 23 | C2/m |
| 15 | 0.458 | 0.121 | Th2TeN2 | 24 | I4/mmm |
| 16 | 0.460 | 0.094 | Olivine | 44 | Pnma |
| 17 | 0.461 | 0.101 | Tilted perovskite | 161 | Pnma |
| 18 | 0.477 | 0.231 | PbClF | 29 | P4/nmm |
| 19 | 0.479 | 0.067 | Quaternary double perovskite | 47 | P21/n |
| 20 | 0.484 | 0.118 | Pyroxene–CaMg(SiO3)2 | 35 | C2/c |
| 21 | 0.491 | 0.093 | Monazite | 20 | P21/n |
| 22 | 0.492 | 0.085 | CaFe2O4 | 30 | Pnma |
| 23 | 0.492 | 0.091 | BaCuY2O5 | 26 | Pnma |
| 24 | 0.493 | 0.068 | Double perovskite | 96 | P21/n |
| 25 | 0.501 | 0.271 | Fluorite–CaF2 | 15 |
Fmm |
| 26 | 0.505 | 0.064 | Bi2ErO4I | 38 | P4/mmm |
| 27 | 0.513 | 0.052 | Sr2NiWO6 | 34 | I4/m |
| 28 | 0.516 | 0.181 | Rocksalt | 28 |
Fmm |
| 29 | 0.517 | 0.079 | Elpasolite | 121 |
Fmm |
| 30 | 0.522 | 0.133 | La2O3 | 40 |
Pm1 |
| 31 | 0.533 | 0.035 | Apatite | 17 | P63/m |
| 32 | 0.536 | 0.231 | K2MgF4 | 39 | I4/mmm |
| 33 | 0.545 | 0.075 | La3NbO7(OS) | 21 | Cmcm |
| 34 | 0.553 | 0.131 | K4CdCl6 | 56 |
Rc |
| 35 | 0.578 | 0.081 | Melilite | 39 |
P![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 21m 21m |
| 36 | 0.580 | 0.108 | K2SO4 | 41 | Pnma |
| 37 | 0.595 | 0.170 | Delafossite | 45 |
Rm |
| 38 | 0.598 | 0.088 | Bixbyite–Mn2O3 | 19 |
Ia |
| 39 | 0.658 | 0.296 | Delafossite–NaCrS2 | 26 |
Rm |
| 40 | 0.667 | 0.169 | CuFeO2 | 14 | P63/mmc |
| 41 | 0.681 | 0.124 | Hexagonal perovskite | 40 | P63/mmc |
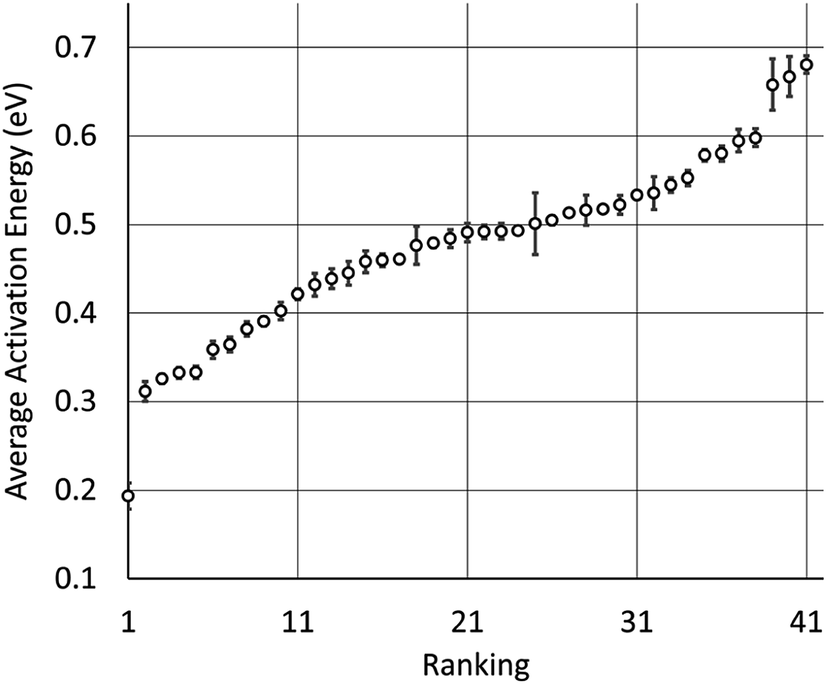 | ||
| Fig. 3 Plot of the ranking shown in Table 1, along with calculated standard errors of the averages. CrVO4 is represented by the dot near 0.2 on the left. | ||
The structure types with the highest predicted average activation energies are elpasolite, hexagonal perovskite, CuFeO2, delafossite–NaCrS2, and Sr2NiWO6. The average activation energies for proton diffusion in these structure types is about 200 meV higher than the average for cubic spinel, which at 600 °C would correspond to a more than 10-fold decrease in proton mobility assuming an Arrhenius-type dependence on migration energy. However within these structure types there can be significant variation in activation energies among individual materials (Table 1), leaving open the possibility that some materials with these structure types may be competitive as proton conductors.
There are 11 oxide structure types for which the predicted average activation energies are below that of cubic perovskite, and nearly 20 more with average activation energies within 100 meV. This suggests that materials with a variety of different structure types could have proton mobilities competitive with cubic perovskites. The fifth-best structure type, a monoclinic fergusonite, has been experimentally investigated for proton conductivity. Several rare-earth niobates and tantalates with this structure type have demonstrated proton conductivity on the order of 10−4 to 10−3 S cm−1 at about 700 °C, making them among the best known proton conductors outside the Ba- and Sr-based cubic perovskites.33 Many materials of this class transform to a high-temperature scheelite phase (#10 on our list) that has similar proton conductivity.33,104 The ninth-best structure type, pyrochlore, has also been investigated for proton conductivity.52,105 Materials of this type have demonstrated proton conductivity on the order of 10−4 to 10−3 S cm−1 at about 800 °C.105
The four structure types with the lowest average activation energies are CrVO4, calcite, zircon, and NaMn7O12. NaMn7O12 is a cubic double-perovskite structure closely related to the cubic perovskites.106 To our knowledge, the remaining three structure types have never been studied for proton conductivity. Of these, the CrVO4 structure type is an outlier in our analysis (Fig. 3), with a predicted average migration energy more than 0.1 eV below that of the next-best structure type, suggesting exceptionally high proton mobility. We have investigated the prediction of our model by using DFT to calculate the activation energies for proton migration in all 29 materials with this structure type in the ICSD (Table 2). We have used both DFT and DFT + U to calculate proton migration barriers (Section 4 in the ESI†) and found that the mean absolute difference is only 34 meV and the mean difference is only 2 meV, suggesting low sensitivity of these results to the U values. The average DFT-calculated migration energy, 184 meV, is in excellent agreement with the model prediction (Fig. 1). These low activation energies for diffusion suggest that some materials with the CrVO4 structure type may be superprotonic conductors.
| ICSD ID | Composition | Energy above hull (eV per atom) | Predicted activation migration energy for 1D diffusion (eV) | Predicted activation migration energy for 2D diffusion (eV) | Predicted activation migration energy for 3D diffusion (eV) | DFT calculated migration energy (eV) |
|---|---|---|---|---|---|---|
| a The CrVO4 structure type has been identified as that of the high-pressure phase.107 b The CrVO4 structure type was computationally determined to be the lowest-energy polymorph,108 but to our knowledge it has never been synthesized. | ||||||
| 16618 | InPO4 | 0.000 | 0.084 | 0.427 | 0.870 | 0.340 |
| 16619 | TlPO4 | 0.000 | 0.159 | 0.473 | 0.841 | 0.270 |
| 16741 | NiSO4 | 0.000 | 0.067 | 0.389 | 0.865 | 0.161 |
| 16759 | MgSO4 | 0.000 | 0.048 | 0.433 | 0.959 | 0.234 |
| 18117 | MgCrO4 | 0.000 | 0.259 | 0.430 | 0.775 | 0.094 |
| 18118 | CdCrO4 | 0.000 | 0.212 | 0.518 | 0.620 | 0.056 |
| 23492 | CoCrO4 | 0.000 | 0.232 | 0.414 | 0.873 | 0.094 |
| 23493 | NiCrO4 | 0.000 | 0.228 | 0.306 | 0.875 | 0.053 |
| 25700 | NiSeO4 | 0.000 | 0.219 | 0.421 | 0.906 | 0.249 |
| 31231 | MnSO4 | 0.000 | 0.386 | 0.386 | 0.669 | 0.217 |
| 60571 | CdSO4 | 0.000 | 0.135 | 0.470 | 0.924 | 0.177 |
| 82286 | VPO4 | 0.000 | 0.070 | 0.367 | 0.810 | 0.196 |
| 155162 | InVO4 | 0.000 | 0.246 | 0.494 | 0.881 | 0.054 |
| 416147 | HgCrO4 | 0.000 | 0.270 | 0.551 | 0.943 | 0.085 |
| 23507 | FeSO4 | 0.001 | 0.075 | 0.437 | 0.725 | 0.159 |
| 33736 | CoSO4 | 0.002 | 0.048 | 0.437 | 0.752 | 0.229 |
| 109070 | MgSeO4 | 0.002 | 0.140 | 0.447 | 0.935 | 0.282 |
| 109071 | MnSeO4 | 0.002 | 0.133 | 0.465 | 0.923 | 0.165 |
| 109072 | CoSeO4 | 0.005 | 0.158 | 0.444 | 0.918 | 0.256 |
| 36244 | CrVO4 | 0.007 | 0.275 | 0.338 | 0.881 | 0.172 |
| 109073 | CuSeO4 | 0.007 | 0.128 | 0.462 | 0.898 | 0.295 |
| 82161 | FeVO4 | 0.015 | 0.214 | 0.459 | 0.897 | 0.262 |
| 62159 | CrPO4 | 0.018 | 0.048 | 0.366 | 0.848 | 0.242 |
| 155065 | FePO4 | 0.021 | 0.037 | 0.428 | 0.862 | 0.195 |
| 183216 | CuCrO4 | 0.031 | 0.152 | 0.383 | 0.853 | 0.154 |
| 82282 | TiPO4 | 0.074 | 0.167 | 0.311 | 0.627 | 0.321 |
| 159272a | AlPO4 | 0.106 | 0.091 | 0.218 | 0.846 | 0.161 |
| 89505 | LiMnO4 | 0.143 | 0.242 | 0.444 | 0.831 | 0.086 |
| 166436b | TiSiO4 | 0.156 | 0.054 | 0.437 | 0.695 | 0.091 |
CrVO4 has an orthorhombic lattice, with one-dimensional columns of CrO6 octahedra linked together by sharing common oxygen atoms with VO4 tetrahedra. It is generally the stable structure for compositions in which the crystal radius of the octahedrally-coordinated ion is between 0.75 and 1.1 Å, and the crystal radius of the tetrahedrally-coordinated ion is between 0.25 and 0.5 Å.109 Proton diffusion is predicted to preferentially occur along a one-dimensional path through the lattice perpendicular to the columns of CrO6 octahedra (Fig. 4). As with most oxides, proton conduction is predicted to occur via a Grotthuss-type mechanism where the proton rotates around on oxygen ion and then jumps to another. Large rotations of the tetrahedra, as observed in CsH2(PO4),110 are not required for proton migration. Because the potential energy surface along the diffusion path is fairly flat, the local minimum varies from material to material, and the jump between the oxygen ions sometimes represents the local minimum along the diffusion path (Fig. 4). As our energy model sometimes identifies the wrong local minimum along this path due the small energy differences between the local minima and the transition states (Fig. 4), we manually evaluated different possible local minima for many of the DFT nudged-elastic-band calculations.
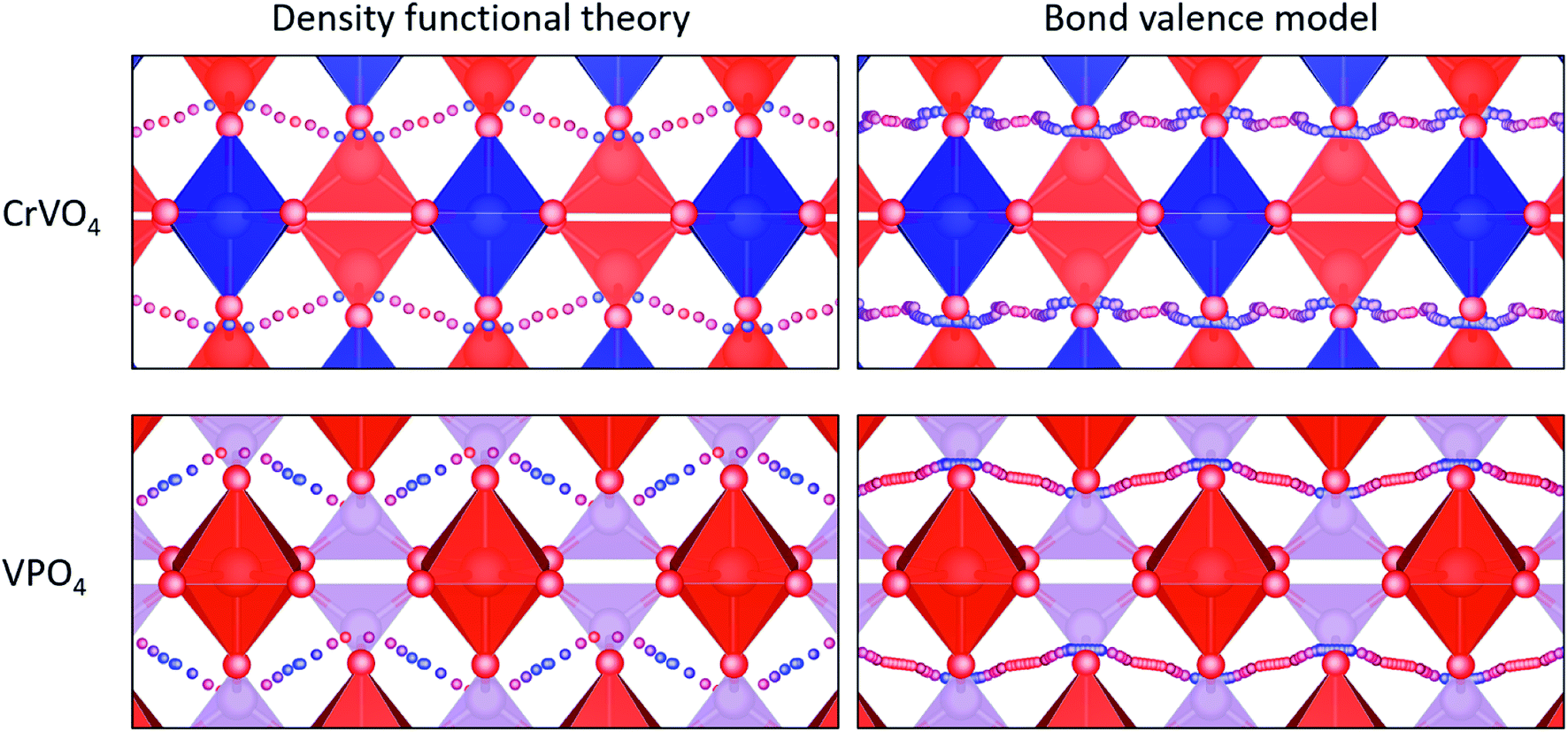 | ||
| Fig. 4 Fastest diffusion paths for VPO4 and CrVO4, which are the representatives of the CrVO4 structure type. Red tetrahedra represent the bonds between vanadium and oxygen blue tetrahedra represent the bonds between chromium and oxygen, and purple tetrahedra represent PO4−3. Small blue and red spheres represent the diffusion pathway, with blue indicating low energy sites and red indicating high energy sites. | ||
Ionic conductivity in one dimension presents practical challenges, as defects that block the diffusion channel could significantly inhibit diffusivity.111 For this reason it is important to assess the ability of protons to migrate around defects by diffusing in a second dimension. We have used our model to calculate the minimum migrations energies required for one-, two-, and three-dimensional diffusion in each of the 29 materials with the CrVO4 structure type (Table 2). As we ran some of these calculations with denser grids to ensure convergence in all three dimensions, the average migration energy in one dimension is slightly below that reported in Table 1. The directions through the crystal along which one-, two-, and three-dimensional diffusion are predicted to occur most readily are shown in Fig. 5. The average migration energy required for diffusion in two dimensions is 419 meV, which is approximately the calculated average migration energy for diffusion in cubic perovskites. This suggests that in many of these materials there will likely be an acceptably fast path around any defects that block the fastest diffusion channels. Diffusion in three dimensions has an average predicted migration energy of 838 meV, indicating that diffusion at competitive rates will likely be limited to two dimensions in these materials. In practice, the degree to which the single dimensionality of the highly conductive channel limits ionic conductivity will vary by material based on both the two-dimensional migration energy in that material and the defect density.
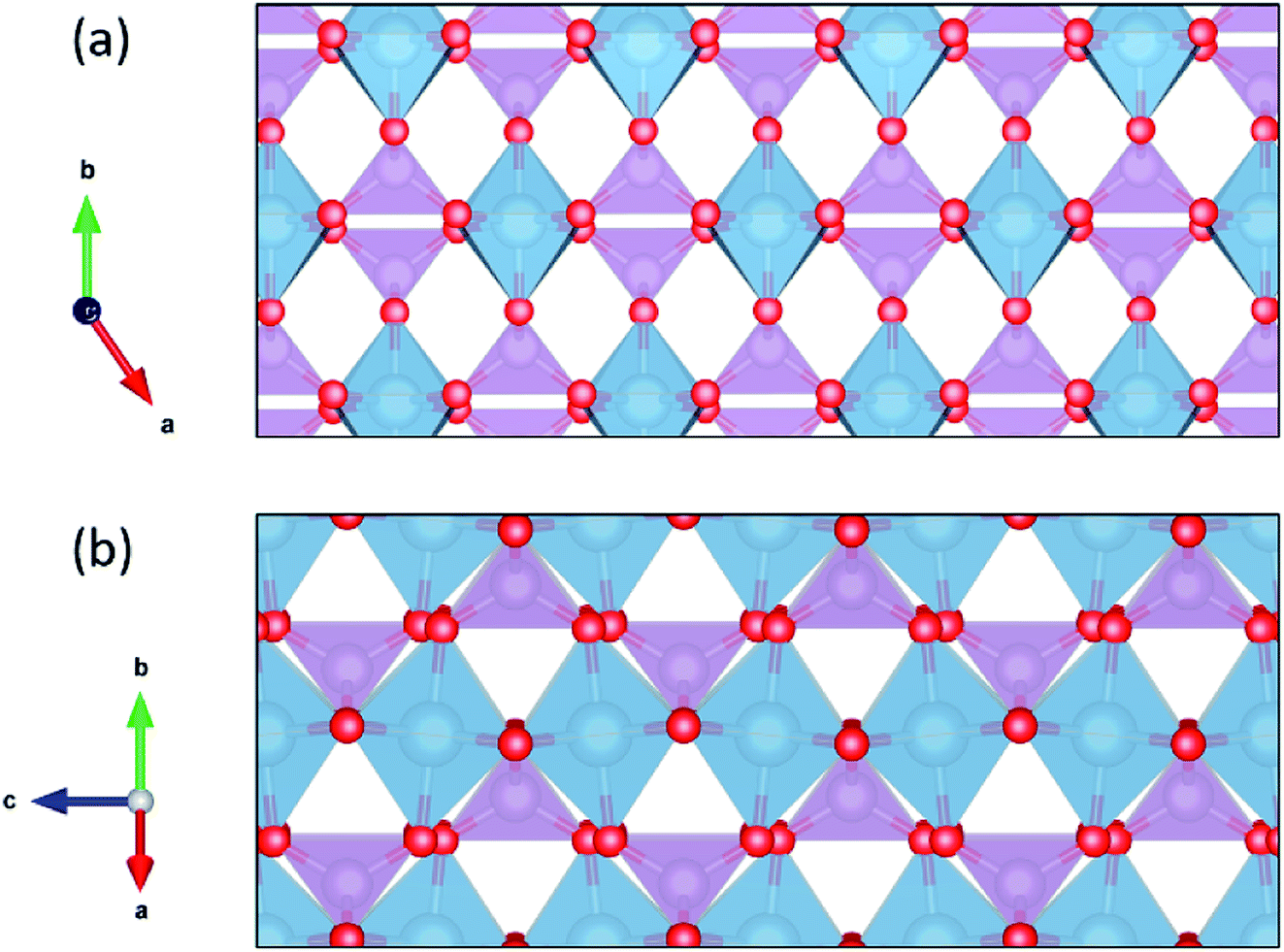 | ||
| Fig. 5 The structure of CrVO4 family represented by AlPO4. Blue spheres in the tetrahedra represent aluminum, and purple tetrahedra represents PO4−3 ions. The fastest diffusion path is left-to-right in (a), and the second-fasted diffusion path is left-to-right (along the c axis) in (b). | ||
Proton conduction depends on both proton mobility and proton concentration in the material. As none of the known CrVO4-structured materials intrinsically contain hydrogen atoms, it would be necessary to introduce protons into these materials. In some materials this might be accomplished through redox reactions with transition metals or by hydrating intrinsic oxygen vacancies. However in many cases it will likely be necessary to introduce protons by doping the materials with electron acceptors, as is commonly done in proton-conducting oxides.12 The conductivity of the doped materials will depend on the concentration of protons (and dopants) that can be introduced without sacrificing stability.
To assess the stability of the doped and undoped materials, we use a DFT-calculated convex hull of stable phases, which provides the energy of the thermodynamically stable phase or combination of phases as a function of composition.112–114 The 0 K DFT-calculated energy relative to the convex hull has been shown to be a useful descriptor for stability and synthesizability.115 This value is an estimate of the thermodynamic driving force for decomposition, with a value of 0 indicating a stable material. Most known oxides have energies within about 15 meV per atom of the convex hull, and 90% of known oxides have energies within about 62 meV per atom of the convex hull.115 The energies above the convex hull for the 29 known CrVO4-structured materials, calculated using data from the Materials Project, are provided in Table 2. Fourteen of the twenty-nine materials are on the DFT-calculated convex hull, and another eleven are within 35 meV per atom of the hull. Of the remaining four, one (AlPO4) is a high-pressure phase, and another (TiSiO4) has never been synthesized to our knowledge. The CrVO4 structure type is predicted to be the lowest-energy polymorph of TiSiO4,108 but as it is calculated to be 156 meV per atom above the convex hull we believe TiSiO4 is unlikely to exist in a form stable enough for practical use.
We assess the thermodynamic stability of the doped phases in a similar way. The widely-studied proton conducting oxides BaZrO3 and BaCeO3 are typically doped with about 10% (mole fraction) Y,12 resulting in nominal compositions of H0.1Y0.1BaZr0.9O3 and H0.1Y0.1BaCe0.9O3. We predict these materials to be 19 meV per atom and 36 meV per atom above the convex hull, respectively. For comparison, we have performed similar calculations on two promising CrVO4-structured materials, VPO4 and InVO4, doped with 10% Mg. The doped materials, with nominal compositions of H0.1Mg0.1V0.9PO4 and H0.1Mg0.1In0.9VO4, have DFT-calculated energies that are 5 meV per atom and 18 meV per atom above the hull, respectively. These results indicate that doping is likely to be a viable strategy for incorporating practically high concentrations of protons into materials with the CrVO4 structure type.
Materials with the CrVO4 structure type are generally composed of common, non-precious elements (Table 2). Our calculations indicate that most of them are electronically insulating, with a band gap of greater than 1 eV as calculated using GGA/GGA + U (Section 4 in the ESI†). They have been studied for their magnetic properties116,117 and as possible battery electrodes.118–120 However although all of the known CrVO4-structured materials are predicted to have low migration barriers for protons, with DFT-calculated values ranging from 53 to 340 meV, to our knowledge no materials in this class have been investigated as possible proton conductors. Of particular note is InVO4, a compound with high chemical stability and a melting point of 1134 °C,109 that has been extensively studied as a photocatalyst for H2 production.121–123 It has recently been shown that thin, single crystals of InVO4 also serve as efficient catalysts for CO2 photoreduction in the presence of water vapor.121 The DFT-calculated proton migration barrier in InVO4 is only 54 meV, and its ability to rapidly transport protons may play a role in its catalytic properties for reactions involving protons.
4 Conclusions
A high-throughput computational screen has identified materials with the CrVO4 structure type as likely to have very low migration energies for the one-dimensional diffusion of protons, and this prediction is supported by density functional theory calculations on known CrVO4-structured materials. In practice proton conduction may be limited by the one-dimensional nature of the path for rapid migration; the average migration energy for two-dimensional diffusion is predicted to be comparable to that of leading structure types for proton conduction. A thermodynamic assessment of acceptor-doped CrVO4-structured materials indicates that this structure type is capable of hosting competitively high concentrations of protons without creating an unacceptably large thermodynamic driving force for decomposition. Known materials with this structure type are typically composed of common, non-precious elements, providing flexibility to chemically tailor their properties materials and investigate different strategies for proton incorporation. These results indicate that CrVO4-structured materials are a promising new area for exploration in the search for new proton-conducting oxides.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was funded by ONR Grant N00014-15-1-2494 and supported by XSEDE Grant DMR-140068. All 3D models were generated using VESTA.124References
- R. Bredesen, K. Jordal and O. Bolland, Chem. Eng. Process., 2004, 43, 1129–1158 CrossRef CAS.
- R. W. Baker, Ind. Eng. Chem. Res., 2002, 41, 1393–1411 CrossRef CAS.
- J. W. Phair and S. P. S. Badwal, Sci. Technol. Adv. Mater., 2006, 7, 792–805 CrossRef CAS.
- J. W. Phair and S. P. S. Badwal, Ionics, 2006, 12, 103–115 CrossRef CAS.
- P. o. S. T. f. I. Reuse, Recycling, C. o. I. T. Assessments, C. o. Engineering, T. Systems and N. R. Council, Separation Technologies for the Industries of the Future, The National Academies Press, 1998 Search PubMed.
- M. T. a. R. Inc., Case Study: Hydrogen Separations in Syngas Processes, http://www.mtrinc.com/case_study/h2_co_ratio_adjustment_in_syngas_production.html, 2014 Search PubMed.
- S. Tosti, A. Basile, G. Chiappetta, C. Rizzello and V. Violante, Chem. Eng. J., 2003, 93, 23–30 CrossRef CAS.
- G. Barbieri, A. Brunetti, A. Caravella and E. Drioli, RSC Adv., 2011, 1, 651–661 RSC.
- S. Uemiya, N. Sato, H. Ando and E. Kikuchi, Ind. Eng. Chem. Res., 1991, 30, 585–589 CrossRef CAS.
- H. Malerød-Fjeld, D. Clark, I. Yuste-Tirados, R. Zanón, D. Catalán-Martinez, D. Beeaff, S. H. Morejudo, P. K. Vestre, T. Norby, R. Haugsrud, J. M. Serra and C. Kjølseth, Nat. Energy, 2017, 2, 923–931 CrossRef.
- S. H. Morejudo, R. Zanón, S. Escolástico, I. Yuste-Tirados, H. Malerød-Fjeld, P. K. Vestre, W. G. Coors, A. Martínez, T. Norby, J. M. Serra and C. Kjølseth, Science, 2016, 353, 563–566 CrossRef CAS.
- E. Fabbri, D. Pergolesi and E. Traversa, Chem. Soc. Rev., 2010, 39, 4355–4369 RSC.
- A. Demin and P. Tsiakaras, Int. J. Hydrogen Energy, 2001, 26, 1103–1108 CrossRef CAS.
- W. G. Coors, J. Power Sources, 2003, 118, 150–156 CrossRef CAS.
- W. Jamsak, S. Assabumrungrat, P. L. Douglas, N. Laosiripojana, R. Suwanwarangkul, S. Charojrochkul and E. Croiset, Chem. Eng. J., 2007, 133, 187–194 CrossRef CAS.
- M. Ni, D. Y. C. Leung and M. K. H. Leung, J. Power Sources, 2008, 183, 133–142 CrossRef CAS.
- Q. Ma, R. Peng, Y. Lin, J. Gao and G. Meng, J. Power Sources, 2006, 161, 95–98 CrossRef CAS.
- C. Duan, J. Tong, M. Shang, S. Nikodemski, M. Sanders, S. Ricote, A. Almansoori and R. O'Hayre, Science, 2015, 349, 1321–1326 CrossRef CAS.
- J. R. G. Taillades, J. Dailly, N. Fukatsu, and R. H. A. Magrasó, and P. R. Slater, in Proton-Conducting Electrolytes for Solid Oxide Fuel Cell Applications, ed. M. Marrony, 2016, ch. 2, pp. 73–146, DOI:10.1007/978-3-319-46146-5_3.
- N. Kochetova, I. Animitsa, D. Medvedev, A. Demin and P. Tsiakaras, RSC Adv., 2016, 6, 73222–73268 RSC.
- S. M. Haile, D. A. Boysen, C. R. I. Chisholm and R. B. Merle, Nature, 2001, 410, 910–913 CrossRef CAS.
- L. Bi, S. Boulfrad and E. Traversa, Chem. Soc. Rev., 2014, 43, 8255–8270 RSC.
- M. A. Laguna-Bercero, J. Power Sources, 2012, 203, 4–16 CrossRef CAS.
- S. Giddey, S. P. S. Badwal and A. Kulkarni, Int. J. Hydrogen Energy, 2013, 38, 14576–14594 CrossRef CAS.
- I. Garagounis, V. Kyriakou, A. Skodra, E. Vasileiou and M. Stoukides, Frontiers in Energy Research, 2014, 2, 1–10 CrossRef.
- E. Vøllestad, R. Strandbakke, M. Tarach, D. Catalán-Martínez, M.-L. Fontaine, D. Beeaff, D. R. Clark, J. M. Serra and T. Norby, Nat. Mater., 2019, 18, 752–759 CrossRef.
- D. Carter and J. Wing, The Fuel Cell Industry Review 2013, 2013 Search PubMed.
- A. Hauch, S. D. Ebbesen, S. H. Jensen and M. Mogensen, J. Mater. Chem., 2008, 18, 2331–2340 RSC.
- Z. Gao, L. V. Mogni, E. C. Miller, J. G. Railsback and S. A. Barnett, Energy Environ. Sci., 2016, 9, 1602–1644 RSC.
- D. J. L. Brett, A. Atkinson, N. P. Brandon and S. J. Skinner, Chem. Soc. Rev., 2008, 37, 1568–1578 RSC.
- E. D. Wachsman and K. T. Lee, Science, 2011, 334, 935–939 CrossRef CAS.
- J. A. Kilner and M. Burriel, Annu. Rev. Mater. Res., 2014, 44, 365–393 CrossRef CAS.
- R. Haugsrud and T. Norby, Nat. Mater., 2006, 5, 193–196 CrossRef CAS.
- M. Marrony and P. Pan Stanford, Proton-conducting ceramics from fundamentals to applied research, Pan Stanford Publishing, Singapore, 2016 Search PubMed.
- W. Munch, K. D. Kreuer, G. Seifert and J. Maier, Solid State Ionics, 2000, 136, 183–189 CrossRef.
- K. D. Kreuer, Annu. Rev. Mater. Res., 2003, 33, 333–359 CrossRef CAS.
- Q. Zhang, G. Wahnstrom, M. E. Bjorketun, S. Gao and E. Wang, Phys. Rev. Lett., 2008, 101, 215902 CrossRef.
- M. A. Gomez and F. J. Liu, Solid State Ionics, 2013, 252, 40–47 CrossRef CAS.
- J. A. Dawson and I. Tanaka, J. Mater. Chem. A, 2015, 3, 10045–10051 RSC.
- K. D. Kreuer, Solid State Ionics, 1997, 97, 1–15 CrossRef CAS.
- F. Shimojo, Solid State Ionics, 1998, 113–115, 319–323 CrossRef CAS.
- E. Matsushita and T. Sasaki, Solid State Ionics, 1999, 125, 31–37 CrossRef CAS.
- H. Iwahara, T. Esaka, H. Uchida and N. Maeda, Solid State Ionics, 1981, 3–4, 359–363 CrossRef CAS.
- M. A. Peña and J. L. G. Fierro, Chem. Rev., 2001, 101, 1981–2018 CrossRef.
- C. J. T. de Grotthuss, Mémoire sur la décomposition de l'eau: et des corps qu' elle tient en dissolution à l'aide de l'électricité galvanique, 1805 Search PubMed.
- C. R. I. Chisholm, Y. H. Jang, S. M. Haile and W. A. Goddard, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72 Search PubMed.
- B. C. Wood and N. Marzari, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 134301 CrossRef.
- O. Barron, H. N. Su, V. Linkov, B. G. Pollet and S. Pasupathi, J. Appl. Electrochem., 2014, 44, 1037–1045 CrossRef CAS.
- R. Haugsrud and T. Norby, J. Am. Ceram. Soc., 2007, 90, 1116–1121 CrossRef CAS.
- K. Amezawa, Y. Tomii and N. Yamamoto, Solid State Ionics, 2003, 162, 175–180 CrossRef.
- N. Hatada, K. Toyoura, T. Onishi, Y. Adachi and T. Uda, J. Phys. Chem. C, 2014, 118, 29629–29635 CrossRef CAS.
- K. Toyoura, A. Nakamura and K. Matsunaga, J. Phys. Chem. C, 2015, 119, 8480–8487 CrossRef CAS.
- T. Norby, O. Dyrlie and P. Kofstad, J. Am. Ceram. Soc., 1992, 75, 1176–1181 CrossRef CAS.
- J. M. Polfus, T. Norby and R. Bredesen, J. Phys. Chem. C, 2015, 119, 23875–23882 CrossRef CAS.
- J. M. Polfus and R. Haugsrud, Solid State Commun., 2012, 152, 1921–1923 CrossRef CAS.
- R. Haugsrud, Solid State Ionics, 2007, 178, 555–560 CrossRef CAS.
- H. Lin, F. Zhou, C. P. Liu and V. Ozolins, J. Mater. Chem. A, 2014, 2, 12280–12288 RSC.
- T. Wei, L. A. Zhang, Y. Chen, P. Yang and M. L. Liu, Chem. Mater., 2017, 29, 1490–1495 CrossRef CAS.
- Inorganic Crystal Structure Database, Fiz Karlsruhe, http://www.fiz-karlsruhe.de/icsd.html Search PubMed.
- Y. Wang, W. D. Richards, S. P. Ong, L. J. Miara, J. C. Kim, Y. Mo and G. Ceder, Nat. Mater., 2015, 14, 1026–1031 CrossRef CAS.
- K.-D. Kreuer, Chem. Mater., 1996, 8, 610–641 CrossRef CAS.
- A. Van der Ven, J. Bhattacharya and A. A. Belak, Acc. Chem. Res., 2013, 46, 1216–1225 CrossRef CAS.
- C. Wolverton, V. Ozoliņš and M. Asta, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 144109 CrossRef.
- D. E. Jiang and E. A. Carter, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 064102 CrossRef.
- G. Henkelman, A. Arnaldsson and H. Jonsson, J. Chem. Phys., 2006, 124, 044706–044709 CrossRef.
- B. C. Garrett and D. G. Truhlar, J. Phys. Chem., 1979, 83, 200–203 CrossRef CAS.
- E. Wigner, Trans. Faraday Soc., 1938, 34, 29–41 RSC.
- P. Hohenberg and W. Kohn, Phys. Rev. B, 1964, 136, B864–B871 CrossRef.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133–A1138 CrossRef.
- L. Pauling, J. Am. Chem. Soc., 1929, 51, 1010–1026 CrossRef CAS.
- I. D. Brown and R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1973, 29, 266–282 CrossRef CAS.
- D. Altermatt and I. D. Brown, Acta Crystallogr., Sect. B: Struct. Sci., 1985, 41, 240–244 CrossRef.
- W. L. Bragg, 1930, 74, 237.
- I. D. Brown, Chem. Rev., 2009, 109, 6858–6919 CrossRef CAS.
- M. O'Keefe and N. E. Brese, J. Am. Chem. Soc., 1991, 113, 3226–3229 CrossRef.
- S. Adams and J. Swenson, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 63, 054201 CrossRef.
- C. Preiser, J. Losel, I. D. Brown, M. Kunz and A. Skowron, Acta Crystallogr., Sect. B: Struct. Sci., 1999, 55, 698–711 CrossRef.
- S. Adams and R. P. Rao, Phys. Chem. Chem. Phys., 2009, 11, 3210–3216 RSC.
- S. Adams and R. P. Rao, J. Mater. Chem., 2012, 22, 1426–1434 RSC.
- S. Adams and R. P. Rao, Solid State Ionics, 2011, 184, 57–61 CrossRef CAS.
- S. Adams and R. P. Rao, Phys. Status Solidi A, 2011, 208, 1746–1753 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251–14269 CrossRef CAS PubMed.
- G. Kresse and J. Furthmuller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- G. Kresse and J. Furthmuller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- D. M. Wood and A. Zunger, J. Phys. A: Math. Gen., 1985, 18, 1343–1359 CrossRef CAS.
- P. Pulay, Chem. Phys. Lett., 1980, 73, 393–398 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- P. E. Blochl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef.
- P. Wisesa, K. A. McGill and T. Mueller, Phys. Rev. B, 2016, 93, 155109 CrossRef.
- S. L. Dudarev, G. A. Botton, S. Y. Savrasov, C. J. Humphreys and A. P. Sutton, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 1505–1509 CrossRef CAS.
- A. Jain, S. P. Ong, G. Hautier, W. Chen, W. D. Richards, S. Dacek, S. Cholia, D. Gunter, D. Skinner, G. Ceder and K. A. Persson, APL Mater., 2013, 1, 011002 CrossRef.
- S. Fabris, G. Vicario, G. Balducci, S. de Gironcoli and S. Baroni, J. Phys. Chem. B, 2005, 109, 22860–22867 CrossRef CAS PubMed.
- M. Shishkin and T. Ziegler, Surf. Sci., 2012, 606, 1078–1087 CrossRef CAS.
- G. Henkelman, B. P. Uberuaga and H. Jonsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
- G. Henkelman and H. Jonsson, J. Chem. Phys., 2000, 113, 9978–9985 CrossRef CAS.
- D. Sheppard, R. Terrell and G. Henkelman, J. Chem. Phys., 2008, 128, 134106 CrossRef.
- D. Sheppard and G. Henkelman, J. Comput. Chem., 2011, 32, 1769–1771 CrossRef CAS PubMed , author reply 1772-1763.
- D. Sheppard, P. Xiao, W. Chemelewski, D. D. Johnson and G. Henkelman, J. Chem. Phys., 2012, 136, 074103 CrossRef.
- P. G. Sundell, M. E. Björketun and G. Wahnström, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 094301 CrossRef.
- S. P. Ong, W. D. Richards, A. Jain, G. Hautier, M. Kocher, S. Cholia, D. Gunter, V. L. Chevrier, K. A. Persson and G. Ceder, Comput. Mater. Sci., 2013, 68, 314–319 CrossRef CAS.
- N. Bork, N. Bonanos, J. Rossmeisl and T. Vegge, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 014103 CrossRef.
- R. Haugsrud and T. Norby, Solid State Ionics, 2006, 177, 1129–1135 CrossRef CAS.
- T. Omata and S. Otsuka-Yao-Matsuo, J. Electrochem. Soc., 2001, 148, E252–E261 CrossRef CAS.
- M. Marezio, P. D. Dernier, J. Chenavas and J. C. Joubert, J. Solid State Chem., 1973, 6, 16–20 CrossRef CAS.
- J. Pellicer-Porres, A. M. Saitta, A. Polian, J. P. Itié and M. Hanfland, Nat. Mater., 2007, 6, 698 CrossRef CAS.
- L. Gracia, A. Beltrán and D. Errandonea, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 094105 CrossRef.
- E. J. Baran, J. Mater. Sci., 1998, 33, 2479–2497 CrossRef CAS.
- G. Kim, J. M. Griffin, F. Blanc, S. M. Haile and C. P. Grey, J. Am. Chem. Soc., 2015, 137, 3867–3876 CrossRef CAS.
- R. Malik, D. Burch, M. Bazant and G. Ceder, Nano Lett., 2010, 10, 4123–4127 CrossRef CAS.
- A. A. Emery and C. Wolverton, Sci. Data, 2017, 4, 170153 CrossRef CAS.
- S. Curtarolo, D. Morgan and G. Ceder, Calphad, 2005, 29, 163–211 CrossRef CAS.
- D. Hildebrandt and D. Glasser, Chem. Eng. J. Biochem. Eng. J., 1994, 54, 187–197 CrossRef CAS.
- W. Sun, S. T. Dacek, S. P. Ong, G. Hautier, A. Jain, W. D. Richards, A. C. Gamst, K. A. Persson and G. Ceder, Sci. Adv., 2016, 2, e1600225 CrossRef.
- B. C. Frazer and P. J. Brown, Phys. Rev., 1962, 125, 1283–1291 CrossRef CAS.
- M. J. Isasi, R. Saezpuche, M. L. Veiga, C. Pico and A. Jerez, Mater. Res. Bull., 1988, 23, 595–601 CrossRef CAS.
- M. E. Arroyo-de Dompablo, J. M. Gallardo-Amores, M. T. Azcondo, F. García-Alvarado and U. Amador, J. Phys. Chem. Solids, 2006, 67, 1243–1247 CrossRef CAS.
- X. Liang, X. Ou, H. Dai, F. Zheng, Q. Pan, P. Liu, X. Xiong, M. Liu and C. Yang, Chem. Commun., 2017, 53, 12696–12699 RSC.
- S. Denis, E. Baudrin, M. Touboul and J. M. Tarascon, J. Electrochem. Soc., 1997, 144, 4099–4109 CrossRef CAS.
- Q. Han, X. Bai, Z. Man, H. He, L. Li, J. Hu, A. Alsaedi, T. Hayat, Z. Yu, W. Zhang, J. Wang, Y. Zhou and Z. Zou, J. Am. Chem. Soc., 2019, 141, 4209–4213 CrossRef CAS.
- J. Ye, Z. Zou, M. Oshikiri, A. Matsushita, M. Shimoda, M. Imai and T. Shishido, Chem. Phys. Lett., 2002, 356, 221–226 CrossRef CAS.
- R. V. d. Krol, J. Ségalini and C. S. Enache, J. Photonics Energy, 2011, 1, 1–11 Search PubMed.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra06291b |
| This journal is © The Royal Society of Chemistry 2019 |