
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRetracted Article: Anti-inflammatory potency of Locusta migratoria manilensis cyclopeptides in mast cells and macrophages
Jie
Liu†
ab,
Huailing
Wang†
ab,
Guangjun
Wang
c,
Qiang
Luo
b,
Hui
Cao
b,
Xiaoyu
Liu
b,
Zehuang
Zhang
c,
Pingchang
Yang
b and
Zhigang
Liu
 *ab
*ab
aThe Third Affiliated Hospital of Shenzhen University, Shenzhen University, Shenzhen 518020, China. E-mail: lzgszuniversity@sina.com; Tel: +86 755-86671905
bThe Research Center of Allergy & Immunology, Shenzhen University School of Medicine, Shenzhen 518060, China
cInstitute of Plant Protection, Chinese Academy of Agricultural Sciences, Beijing 100193, China
First published on 2nd October 2019
Abstract
Locusts are esteemed as a traditional Chinese medicine, as well as tonic foods in Asian countries. While searching for natural anti-inflammatory agents in natural products, we isolated four novel locust cyclopeptides (LCPs) and the results show that [cyclo-(Trp-Leu-His-Thr)]∼LCP-3 has potent anti-inflammatory potency in RAW264.7 and HMC-1 cells under LPS (lipopolysaccharide) stimuli. Furthermore, mechanistic studies show that LCP-3 attenuates pro-inflammatory cytokine (TNF-α, IL-6, IL-1β, NO and PGE2) expression. Moreover, LCP-3 attenuates inflammatory damage associated with the direct inhibition of iNOS and COX-2 expression. LCP-3 also regulates the MAPK, PI3K/AKT and NF-κB pathways to attenuate LPS-induced damage. Of note, our study first reports the anti-inflammatory potency of LCPs and elucidates their underlying molecular mechanisms.
1 Introduction
Inflammation is an involved vascular response to various types of harmful stimulation and plays an important role in the progression of a wide array of diseases.1 The inflammatory process involves different immune cells such as Th1, Th2, macrophages, and B cells.2 Many studies have demonstrated that activated macrophages, key inflammatory cells, play a pivotal role in diseases via the secretion of various inflammatory mediators such as nitric oxide (NO) and prostaglandin (PG) E2, as well as pro-inflammatory cytokines, including tumor necrosis factor-α (TNF-α), interleukin-1beta (IL-1β) and interleukin-6 (IL-6).3 Studies in animal models and patients suggest that pro-inflammatory factors, including NO, TNF-α, PGE2 and IL-6 may be produced by macrophages or by the surrounding tissues, and also induce the expression of cyclooxygenase-2 (COX-2) and inducible NO synthase (iNOS), which leads to the up-regulation of NO and PGE2.4 The activated forms of COX, namely COX-1 and COX-2, catalyze the production of leukotrienes, which are arachidonic acid metabolites. COX-1 is expressed in most tissues and is a constitutive enzyme, whereas COX-2 is mainly present in inflammatory cells.5There are various signaling pathways involved in the inflammation progress, and NF-κB is the central hub molecule for inflammation. NF-κB, a nuclear transcription factor, regulates the expression of various genes, including cytokines, iNOS and COX-2, which play critical roles in inflammation.6 NF-κB exists in most cells as homodimeric or heterodimeric complexes of p50 and p65 subunits and remains inactive in the cytoplasm of cells associated with the NF-κB inhibitory protein (IκB).7 The arachidonic acid pathway is the downstream signaling of NF-κB and can be trans-activated by NF-κB upon its activation by pro-inflammatory cytokines, such as TNF-α and IL-6.8 Moreover, it has been demonstrated that upstream mitogen-activated protein kinases (MAPKs), such as extracellular signal regulated kinase (ERK), p38 and c-Jun NH2-terminal kinase (JNK), and PI3K/AKT are also implicated in increasing the transcriptional activity of NF-κB.9
Up to now, reports have shown that strictum can produce cyclic hexapeptide verlamelin 6 and be used as a microbial fungicide with mycoparasitism and antibiosis mechanisms.10 A new linear peptide, simplicilliumtide, and four new cyclic peptide simplicilliumtides from a deep-sea-derived fungus have shown significant anti-fungal activity toward Aspergillus versicolor and Curvularia australiensis and also obvious antiviral activity toward HSV-1.11 Both compounds from Bacillus amyloliquefaciens L-H15 have shown good inhibitory activities against three plant fungal pathogens in a cylinder-plate diffusion assay.12
So far, more than 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 species of locusts have been identified and are widely distributed in tropical and subtropical regions.13 The locust is a highly nutritional insect, as it is an excellent source of protein.14 According to the records of traditional Chinese medicine, locusts are widely used in relieving coughs and asthma, and enhancing immunity.15 Recently, studies have revealed that locusts are a good source of edible protein and are rich in functional proteins, such as antifreeze proteins, storage proteins, and antimicrobial and antioxidant peptides.16,17 However, the anti-inflammatory potency and mechanisms of locust cyclopeptides have been rarely explored. To solve this issue, we isolated the LCPs and explored their anti-inflammatory potency in HMC-1 and RAW264.7 cells.
000 species of locusts have been identified and are widely distributed in tropical and subtropical regions.13 The locust is a highly nutritional insect, as it is an excellent source of protein.14 According to the records of traditional Chinese medicine, locusts are widely used in relieving coughs and asthma, and enhancing immunity.15 Recently, studies have revealed that locusts are a good source of edible protein and are rich in functional proteins, such as antifreeze proteins, storage proteins, and antimicrobial and antioxidant peptides.16,17 However, the anti-inflammatory potency and mechanisms of locust cyclopeptides have been rarely explored. To solve this issue, we isolated the LCPs and explored their anti-inflammatory potency in HMC-1 and RAW264.7 cells.
2 Materials and methods
2.1 Reagents
Locusta migratoria manilensis (Meyen, 1835) was acquired from the Institute of Plant Protection, Chinese Academy of Agricultural Sciences (Beijing). Iscove's modified Dulbecco's medium (IMDM), Dulbecco's modified Eagle's medium (DMEM) and fetal bovine serum (FBS) were purchased from Invitrogen (Thermo Fisher Scientific, MA, USA). All the chemicals used in the current study were of analytical reagent grade and were purchased from Aldrich or Adamas and used without further purification. All the antibodies were purchased from Cell Signaling Technology (CST, USA). RAW 264.7 cells (derived from murine macrophages) and HMC-1 cells (human mast cell line-1) were obtained from Cell Bank of the Chinese Academy of Sciences (Shanghai, China).2.2 Isolation of LCPs
All extraction and separation procedures were carried out at 4 °C, and after we got the total locust protein, the LCPs were isolated using a series of chromatographies. The extraction scheme of the LCPs is summarized in Fig. 1. | ||
| Fig. 1 Extraction scheme of LCPs and the structures of LCP-1–4. All extraction and separation procedures were carried out as in the flow chart, at 4 °C. 250 g of Locusta migratoria manilensis (Meyen, 1835) was minced to a homogenate and mixed with iso-propanol in a ratio of 1:6 (w/v) and stirred for 1 h, replacing the solvent every 15 min. The supernatant was removed, and the sediment was freeze-dried. The defatted precipitate (110 g) was dissolved (10%, w/v) in 0.50 M phosphate-buffered saline (PBS, pH 7.2). After centrifugation (10000g, 20 min), the supernatant was collected and stored as total protein at −80 °C. The total protein was fractionated by ultrafiltration using 1 kDa molecular weight (MW) cut-off membranes (Millipore, Hangzhou, China). Two peptide fractions, named LCP-A (MW < 1 kDa) and LCP-B (MW > 1 kDa) were collected and lyophilized. Hydrophobic chromatography: LCP-A was dissolved in 1.40 M (NH4)2SO4 prepared with 30 mM PBS (pH 7.5) and loaded into a phenyl sepharose CL-4B hydrophobic chromatography column (2.5 cm × 110 cm). Stepwise elution was carried out with decreasing concentrations of (NH4)2SO4 (1.40, 0.70 and 0 M) dissolved in 30 mM PBS (pH 7.5) at a flow rate of 2.5 mL min−1. Each fraction was collected at a volume of 100 mL and monitored at 300 nm. Finally, five fractions were collected, freeze-dried and their anti-inflammatory potency was detected. The fraction with the strongest anti-inflammation activity was collected and used for anion-exchange chromatography. LCP-A-III solution (18 mL, 2.44 g mL−1) was loaded into a DEAE-52 cellulose (Shanghai, China) anion-exchange column (2.0 × 100 cm) pre-equilibrated with deionized water, and was stepwise eluted with distilled water, 0.35, 0.70, and 1.40 M (NH4)2SO4 solutions at a flow rate of 2.0 mL min−1. Eight fractions were collected, freeze-dried and their anti-inflammatory potency was detected. The fraction with the strongest anti-inflammatory potency was collected, and prepared for gel filtration chromatography. The LCP-A-III-IV solution (5 mL, 2.30 mg mL−1) was loaded into a Sephadex G-25 (Sigma-Aldrich, China) column (2.0 × 100 cm) at a flow rate of 2.0 mL min−1. Each eluate (90 mL) was collected and monitored at 300 nm. Six fractions were collected, freeze-dried and their anti-inflammatory potency was detected. The fraction with the strongest anti-inflammatory potency was collected, and prepared for reversed phase high performance liquid chromatography (RP-HPLC). LCP-A-III-IV-II was finally separated using RP-HPLC (Agilent 1100, HPLC) on a Zorbax, SB C-18 column (4.6 × 250 mm, 5 μm). The elution solvent system was composed of water–trifluoroacetic acid (solvent A; 100:0.1, v/v) and acetonitrile–trifluoroacetic acid (solvent B; 100:0.1, v/v). The peptides were separated using a gradient elution from 30% to 80% of solvent B for 60 min at a flow rate of 1.0 mL min−1. The detection wavelength was set at 300 nm. | ||
After we obtained the LCPs, the amino acid sequences were analyzed using high-performance liquid chromatography-electrospray ionization-mass spectrometry (HPLC-ESI-MS). Prior to HPLC-ESI-MS analysis, freeze-dried LCPs were rehydrated with 1.0 mL of Milli-Q water. Before being used, the water was boiled for 5 min and then cooled to 4 °C. The rehydrated solution was stored at −20 °C until analysis.
HPLC-ESI-MS was carried out on a SCIEX X500R Q-TOF mass spectrometer (Framingham, U.S.A.) and the MS conditions were as follows: ESI-MS analysis was performed using a SCIEX X500R Q-TOF mass spectrometer equipped with an ESI source. The mass range was set at m/z 50–1500. The Q-TOF MS data were acquired in positive mode and conditions of MS analysis were as follows: CAD gas flow-rate, 7 L min−1; drying gas temperature, 550 °C; ion spray voltage, 5500 V; declustering potential, 80 V. The data file was generated using SCIEX OS 1.0 software. Furthermore, the purity (>85.6%) was determined by HPLC. The LCPs were dissolved in phosphate-buffered saline (PBS) at a concentration of 2.0 mM as a stock solution and stored at −20 °C.
2.3 Cell culture and cell viability
RAW 264.7 and HMC-1 cells were cultured in DMEM and IMDM supplemented with 10% FBS (GIBCO, Grand Island, NY, USA), 100 U per mL penicillin, and 100 μg per mL streptomycin and were grown in a humidified incubator containing 5% CO2 at 37 °C. Cells (2 × 104 per well in 96-well plates) were treated with the LCPs (50 μM) for 24 h, and then treated with LPS (1 μg mL−1) for 12 h. The cell viability was detected based on the MTT method.182.4 Inflammatory cytokine production assay
RAW264.7 and HMC-1 cells were seeded in 6-well plates (4 × 106 cells per mL) and cultured as the experimental design. The inflammatory cytokines (NO, PGE2, TNF-α, IL-6 and IL-1β) were detected using ELISA kits (Beyotime Biotechnology, Shanghai, China) according to the manufacturer's instructions.2.5 Mitochondrial transmembrane potential (ΔΨm) assay
RAW264.7 cells and HMC-1 cells (3 × 105 per well) were cultured into 6-well plates overnight. After exposure to LPS (1 μg mL−1) for 12 h, they were treated with the LCPs (50 and 200 μM) for 12 h. The cells were collected, washed with cold PBS and incubated with 1 μg mL−1 of JC-1 at 37 °C for 30 min in the dark. The supernatant was removed and then assayed using flow cytometry (BD FACS Calibur, Franklin Lakes, CA, USA).2.6 Western blotting
Proteins were extracted for analysis by western blotting. After the cells were treated as per the experimental design, the cells were harvested and the total protein was collected. The protein samples were separated by via sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). The immunoblots were blocked with 5% nonfat milk and subsequently incubated with the primary antibodies (1:1000) at 4 °C overnight, followed by incubation with peroxidase conjugated second antibodies (1:5000) at room temperature for 2 h.19 The protein bands were measured and β-actin was used as an internal standard of process control. The blot band densitometry was analyzed using the ImageJ software.
2.7 Statistical analysis
The evaluation of statistical significance of observed differences was performed by one-way analysis of variance (one-way ANOVA), using SPSS software 21.0 (SPSS Inc., Chicago, IL, USA), where statistical significance was set at p < 0.05. The graphs were analyzed using the Graph Pad INSTAT software (GraphPad software, San Diego, CA, USA).3 Results
3.1 Anti-inflammatory potency of the LCPs
The extraction scheme of LCP is summarized in Fig. 1. Finally, four novel LCPs and sixteen known LCPs were obtained and structurally identified. The purity was determined by HPLC at >98%. The LCPs were dissolved in PBS at a concentration of 20 mM as a stock solution and stored at −20 °C.The cytotoxic potency of the LCPs was detected based on the MTT method. Interestingly, the MTT results show that all the LCPs exhibit no harmful effects up to 6.0 mM. Then, we detected the anti-inflammatory potency of the LCPs in RAW264.7 and HMC-1 cells. As can be seen in Table 1, most of the LCPs displayed a protective effect. Of interest, LCP-3 showed the best protective effect against LPS induced inflammatory damage, both in RAW264.7 and HMC-1 cells. Compared with the LPS group, the survival rates of RAW264.7 and HMC-1 cells were effectively elevated by 16.7% and 16.6%, respectively. We inferred that LCP-3 attenuates the inflammatory injury induced by LPS. Hence, further experiments were carried out with LCP-3 on the RAW264.7 and HMC-1 cells.
| Compounds | Survival rate (%) | Compounds | Survival rate (%) | ||
|---|---|---|---|---|---|
| RAW264.7 cells | HMC-1 cells | RAW264.7 cells | HMC-1 cells | ||
| LCP-1 | 89.2 ± 7.85 | 91.3 ± 7.51 | LCP-11 | 90.6 ± 7.11 | 89.9 ± 7.28 |
| LCP-2 | 92.7 ± 7.93 | 89.6 ± 8.28 | LCP-12 | 87.5 ± 8.74 | 88.7 ± 8.63 |
| LCP-3 | 93.6 ± 7.63 | 94.9 ± 7.79 | LCP-13 | 83.4 ± 7.07 | 84.7 ± 6.85 |
| LCP-4 | 89.9 ± 8.24 | 89.6 ± 8.02 | LCP-144 | 81.7 ± 7.74 | 82.4 ± 7.83 |
| LCP-5 | 85.2 ± 7.71 | 89.3 ± 8.80 | LCP-15 | 81.7 ± 6.72 | 82.0 ± 7.27 |
| LCP-6 | 87.6 ± 7.54 | 86.6 ± 8.03 | LCP-16 | 87.7 ± 7.97 | 88.8 ± 7.73 |
| LCP-7 | 88.7 ± 6.90 | 87.6 ± 7.27 | LCP-17 | 83.8 ± 7.87 | 87.9 ± 8.03 |
| LCP-8 | 89.9 ± 7.78 | 88.4 ± 8.02 | LCP-18 | 83.2 ± 7.84 | 89.9 ± 7.58 |
| LCP-9 | 91.3 ± 7.15 | 89.5 ± 8.81 | LCP-19 | 81.3 ± 6.60 | 85.8 ± 7.62 |
| LCP-10 | 87.1 ± 7.33 | 88.3 ± 7.46 | LCP-20 | 80.1 ± 8.53 | 83.4 ± 7.57 |
| LPS group | 76.9 ± 7.22 | 78.3 ± 6.80 | Control group | 100 ± 7.09 | 100 ± 8.86 |
3.2 LCP-3 suppresses iNOS and COX-2 expression
It is known that IL-1β, TNF-α and IL-6 are implicated in inflammatory diseases and are associated with the nociceptive activity of certain stimulating factors. In response to LPS stimulation, macrophages produce several cytokines such as TNF-α, IL-6, and IL-1β.20 The inflammatory mediators and NO can disturb the balance between immunity and inflammatory responses, which are associated with abnormal physiological conditions. Further, excessive cytokine and iNOS mediated NO production may be linked to inflammatory diseases.21 We then detected whether LCP-3 attenuated LPS-induced inflammatory cytokine (TNF-α, IL-6, and IL-1β) expression in RAW264.7 and HMC-1 cells, and the results are shown in Fig. 2.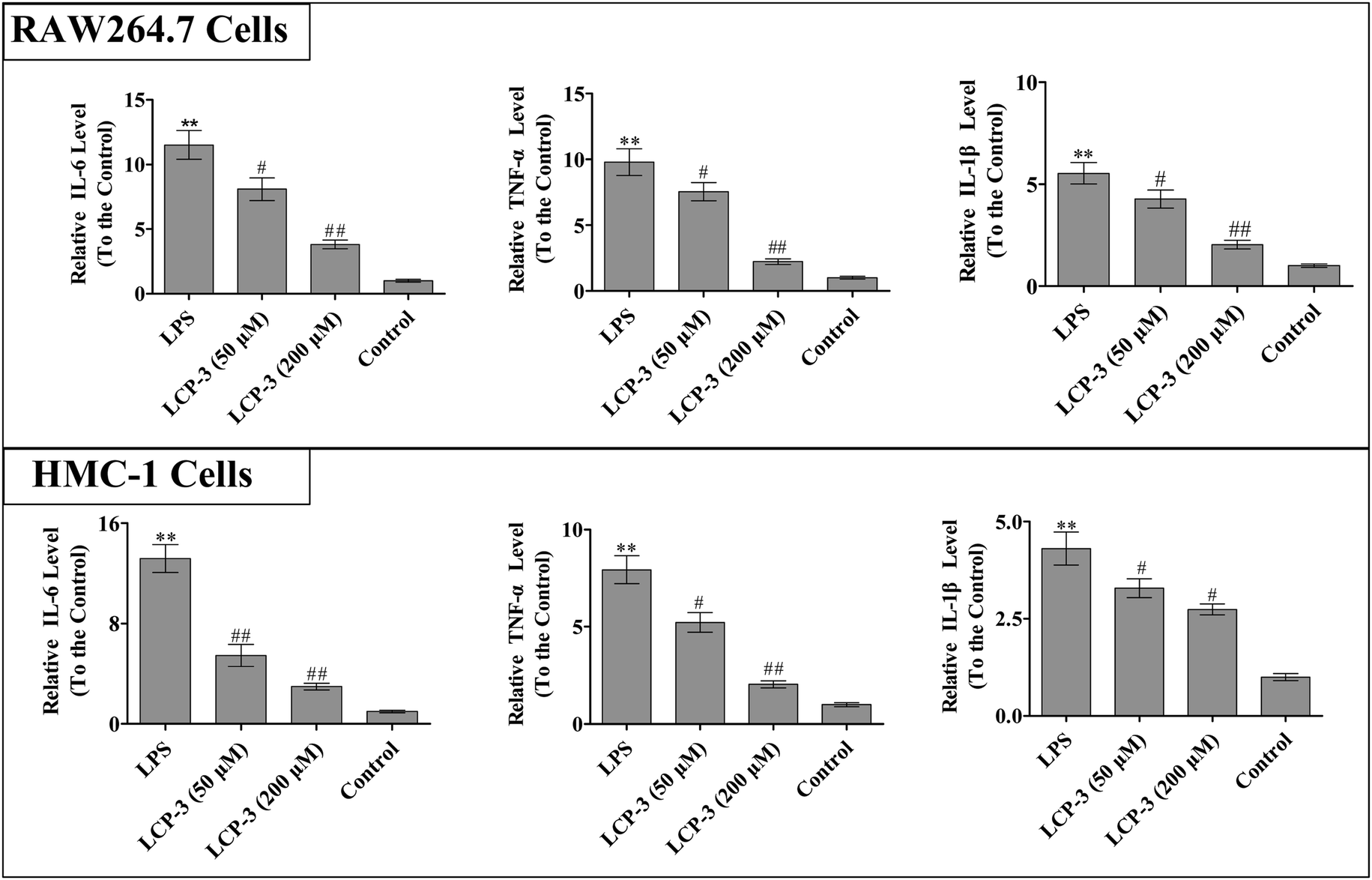 | ||
| Fig. 2 The effect of LCP-3 on IL-6, TNF-α, IL-1β productions in RAW264.7 cells and HMC-1 cells. Both RAW264.7 and HMC-1 cells were treated with LPS (1 μg mL−1) for 12 h, and then treated with LCP-3 (50 and 200 μM) for 12 h. After that, the culture media were collected and the TNF-α, IL-6 and IL-1β concentrations were detected with ELISA kits according to the manufacturer's instructions. The values are expressed as means ± SD of triplicate tests. *p < 0.05 and **p < 0.01 vs. the control, and #p < 0.05 and ##p < 0.01 vs. LPS indicate statistically significant difference. | ||
In RAW264.7 cells, compared with the control group, LPS stimuli markedly increased the TNF-α, IL-6, and IL-1β levels by 8.78-, 10.5- and 4.53-fold, respectively. However, the 200 μM LCP-3 treatments significantly attenuated the levels of TNF-α (77.2%, p < 0.01), IL-6 (66.8%, p < 0.01), and IL-1β (63.1%, p < 0.01), respectively. Similarly, the levels of inflammatory cytokines in HMC-1 cells were also markedly increased upon LPS stimuli. Of interest, after the 200 μM LCP-3 treatments, the levels of TNF-α, IL-6, and IL-1β were down-regulated by 73.8%, (p < 0.01), (58.6%, p < 0.01) and (36.4%, p < 0.05), respectively.
To further explore the anti-inflammatory potency of LCP-3, the levels of prostaglandin E2 (PGE2) and NO were detected. As shown in Fig. 3, compared to the control group, LPS stimuli increased the NO expressions both in RAW264.7 and HMC-1 cells by 4.38- and 5.82-fold, respectively. However, 200 μM LCP-3 treatments remarkably attenuated LPS-induced NO production by 66.3% and 67.7%, respectively. Moreover, 200 μM LCP-3 treatments also down-regulated LPS induced PGE2 production by 66.4% and 71.7% in RAW264.7 and HMC-1 cells, respectively. Of interest, both the PGE2 and NO levels were down-regulated in the two types of experimental cell by the LPC-3 treatments.
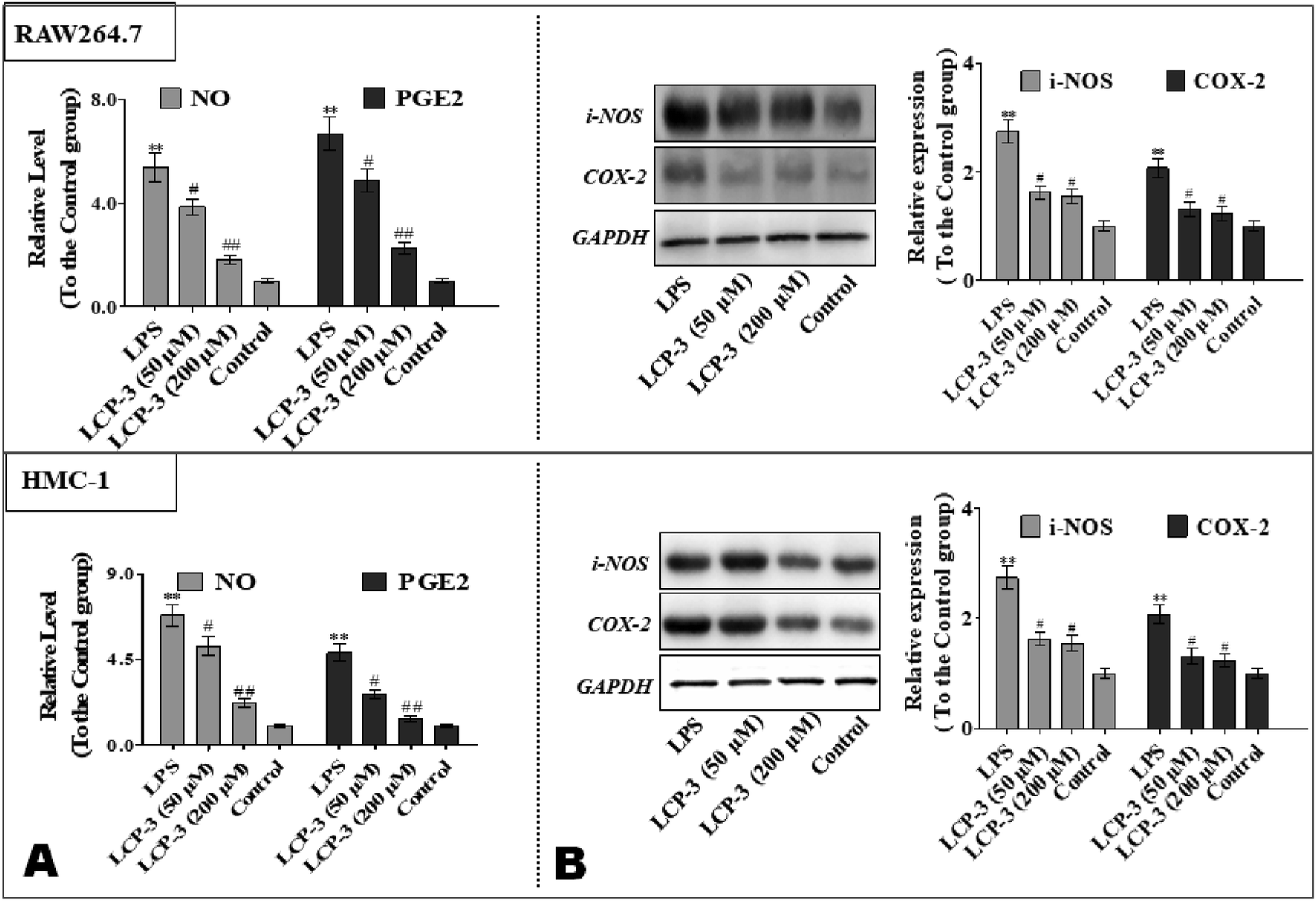 | ||
| Fig. 3 Inhibitory potency of LCP-3 on LPS-induced NO, PGE2 production and iNOS, COX-2 protein expressions in RAW264.7 and HMC-1 cells. RAW264.7 and HMC-1 cells were seeded in 96-well plates (4 × 105 cells per mL). The cells were treated with LPS (1 μg mL−1) for 12 h, and then treated with LCP-3 (50 and 200 μM) for 16 h. After that, 100 μL of culture media were collected for (A) NO and PGE2 assay. (B) The total cell lysate was prepared and the levels of the iNOS or COX-2 proteins were analyzed by western blot analysis. The values are expressed as means ± SD of triplicate tests. *p < 0.05 and **p < 0.01 vs. the control, and #p < 0.05 and ##p < 0.01 vs. LPS, indicate statistically significant difference. GAPDH was used as an internal standard of process control and blot band densitometry was analyzed using the ImageJ software. | ||
iNOS and COX-2 are pivotal enzymes for the production of NO and PGE2, respectively, and therefore the potency of LCP-3 on LPS-induced iNOS and COX-2 expression was investigated. As shown in Fig. 3, after LPS stimuli for 12 h, the expressions of iNOS and COX-2 were markedly increased, whereas both protein levels were significantly attenuated after the LCP-3 treatments. This result is consistent with the reduction in the NO and PGE2 levels detected above. These data suggest that LCP-3 may inhibit iNOS and COX-2 expression through transcriptional mechanism regulation. Of interest, the results suggest that LCP-3 shows anti-inflammatory potency, maybe by inhibiting iNOS mediated NO production, as well as TNF-α, IL-6, and IL-1β production.
3.3 LCP-3 suppresses the activation of NF-κB pathways
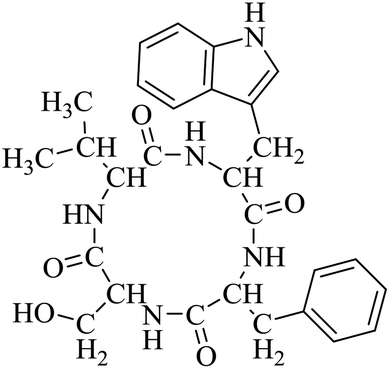
Nuclear factor kappa-B (NF-κB) is a transcriptional factor that regulates the expression of pro-inflammatory cytokines.22 For this reason, NF-κB has become a target of anti-inflammatory treatment. Further, NF-κB activation is significant for regulating gene expression in LPS or other inflammatory cytokine-induced inflammatory responses, including iNOS and COX-2 expression. Hence, we explored the potency of LCP-3 on NF-κB activation upon LPS stimuli (Table 2).| Compounds | Structure | Compounds | Structure | Compounds | Structure | Compounds | Structure |
|---|---|---|---|---|---|---|---|
| LCP-1 |
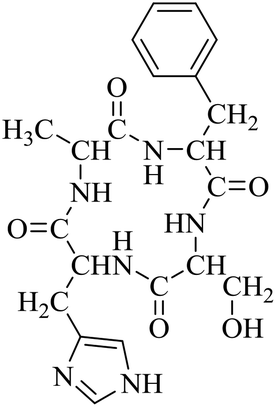
|
LCP-2 |
|
LCP-9 |
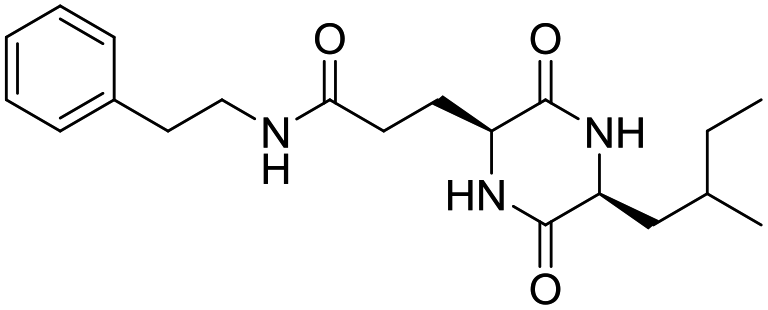
|
LCP-15 |
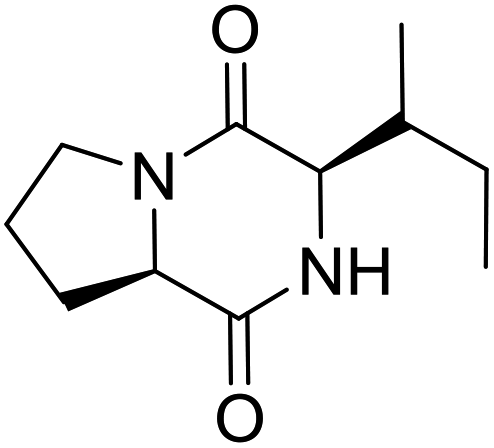
|
| LCP-10 |
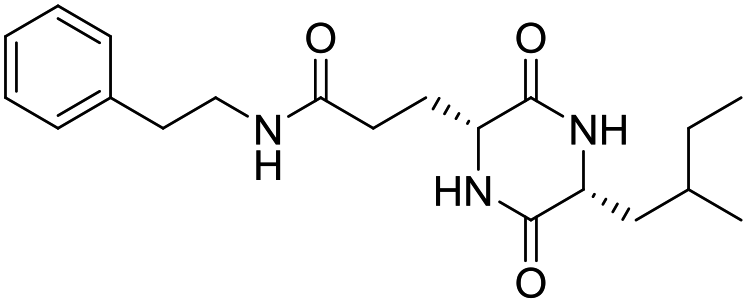
|
LCP-16 |
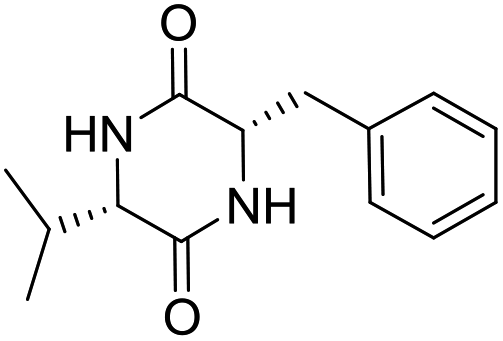
|
||||
| LCP-3 |
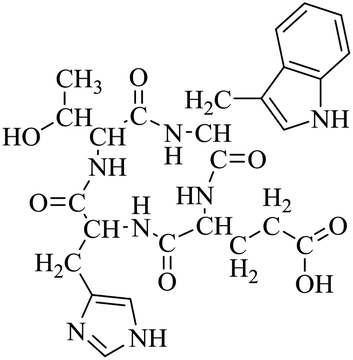
|
LCP-4 |
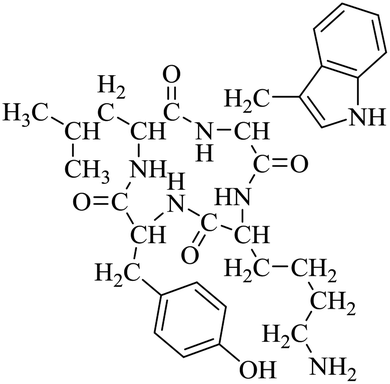
|
LCP-11 |
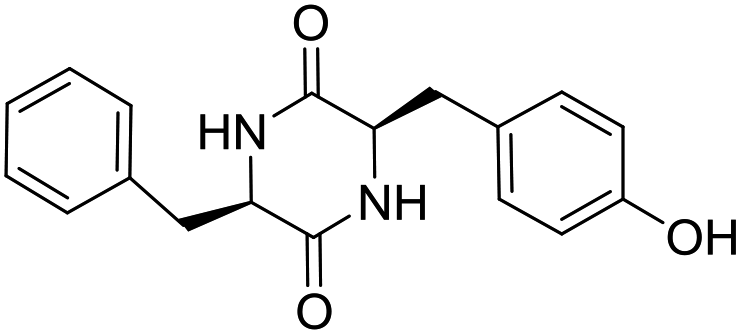
|
LCP-17 |
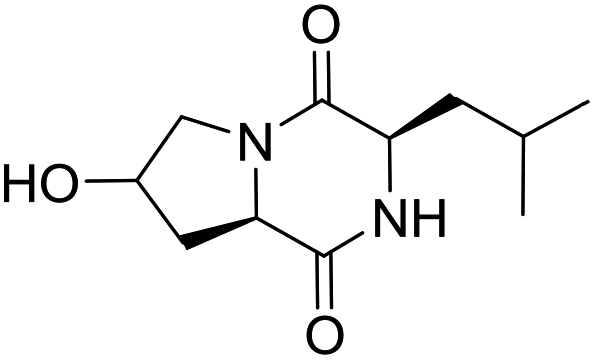
|
| LCP-12 |
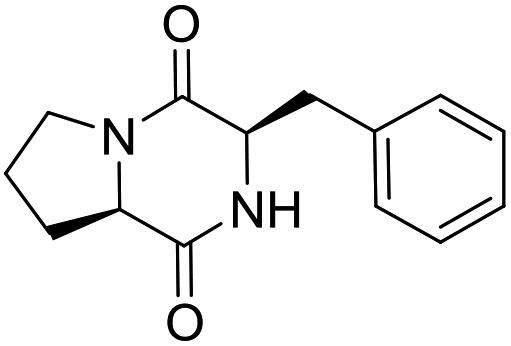
|
LCP-18 |
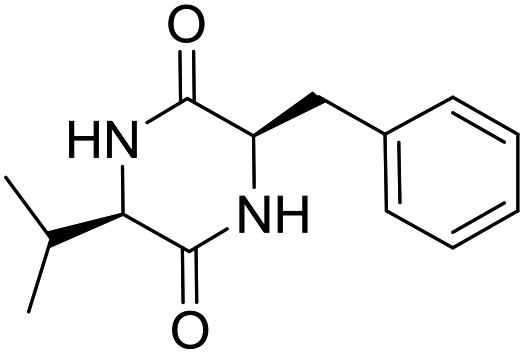
|
||||
| LCP-5 |
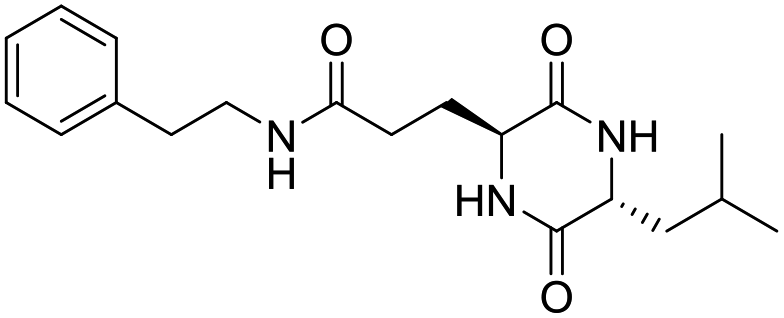
|
LCP-6 |
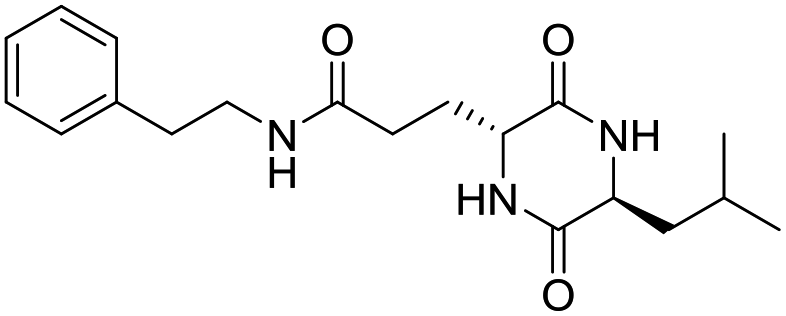
|
LCP-13 |
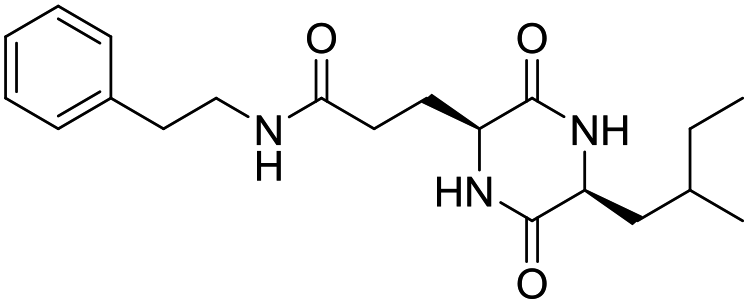
|
LCP-19 |
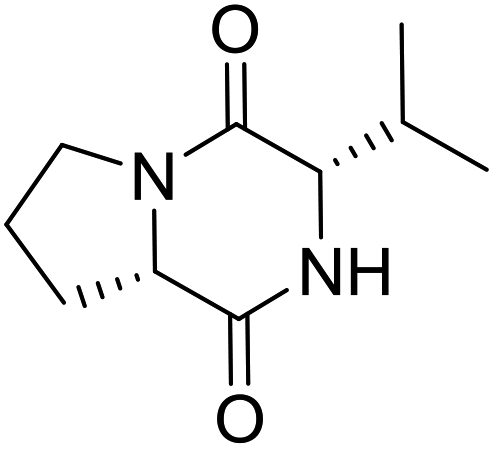
|
| LCP-7 |
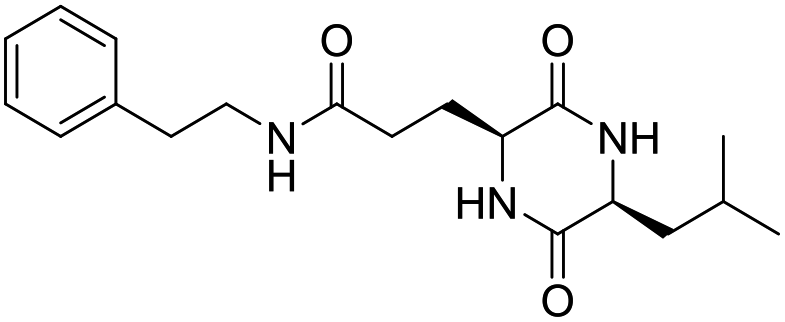
|
LCP-8 |
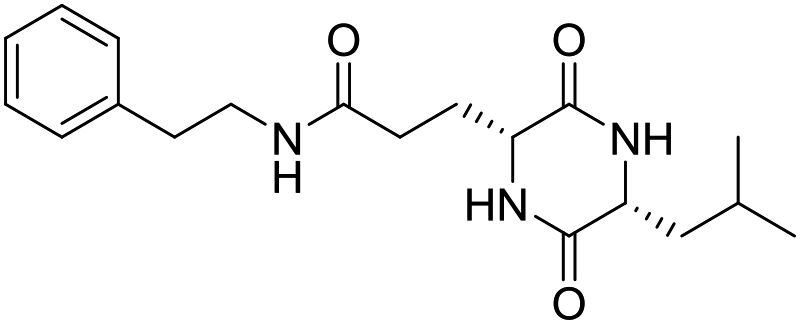
|
LCP-14 |
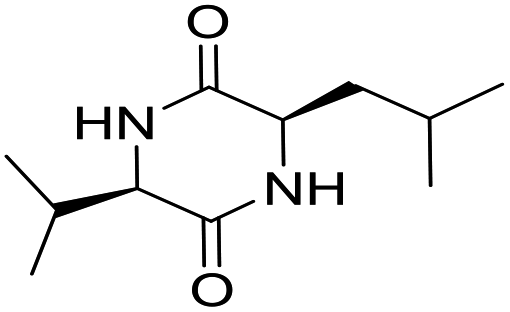
|
LCP-20 |
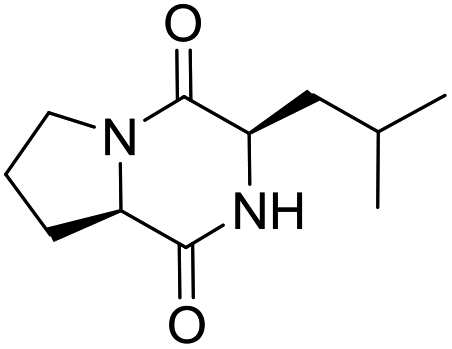
|
As can be seen in Fig. 4, LCP-3 markedly attenuates the expression of p-p65, p-IκBα and p-IKKα/β in LPS induced RAW264.7 and HMC-1 cells. Compared with the LPS groups, 200 μM LCP-3 attenuates the phosphorylation of p65, IκBα and IKKα/β by 42.8% (p < 0.01), 69.5% (p < 0.01), and 16.2% (p < 0.05) in RAW264.7 cells. Further, LCP-3 (200 μM) also significantly down-regulates the expression of the phosphorylation of p65, IκBα and IKKα/β by 38.2% (p < 0.05), 28.3% (p < 0.05), and 25.2% (p < 0.05) in HMC-1 cells induced by LPS.
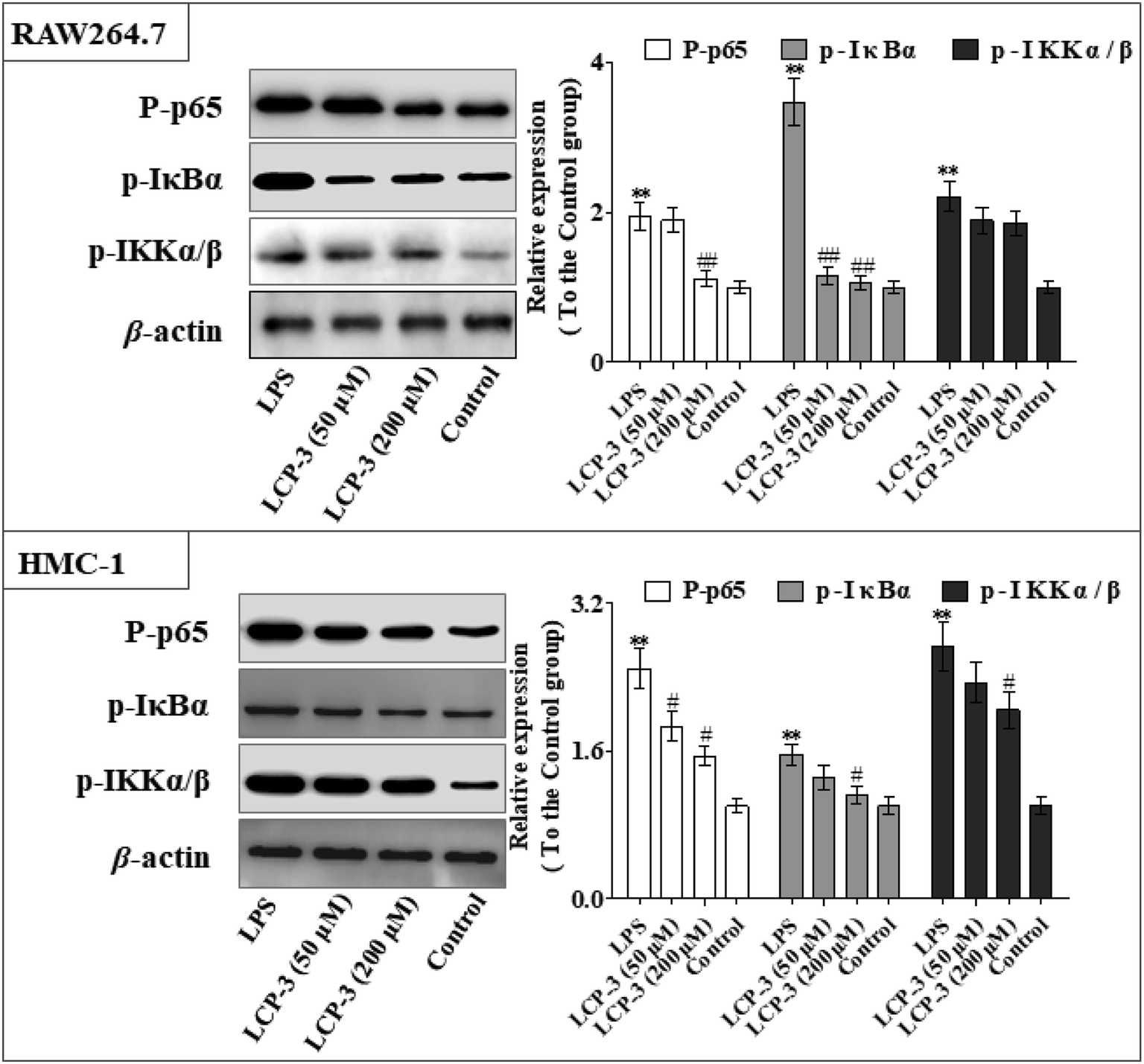 | ||
| Fig. 4 Effect of LCP-3 on the NF-κB signaling pathway in RAW264.7 and HMC-1 cells. RAW264.7 cells were treated with LPS (1 μg mL−1) for 12 h, then treated with LCP-3 (50 and 200 μM) for 16 h. The total protein lysate was prepared, and cell signaling proteins were detected by western blot analysis. The values are expressed as means ± SD of triplicate tests. *p < 0.05 and **p < 0.01 vs. the control, and #p < 0.05 and ##p < 0.01 vs. LPS, indicate statistically significant difference. β-Actin was used as an internal standard of process control and blot band densitometry was analyzed using the ImageJ software. | ||
3.4 LCP-3 activated Nrf2/HO-1 signaling through MAPKs
The MAPKs are highly conserved mediators of eukaryotic signal transducing enzymes that respond to extracellular stimulation and regulate diverse cellular activities in the nucleus.23 MAPKs also play a pivotal role in the regulation of pro-inflammatory cytokines. Phosphoinositide 3-kinase (PI3K), a classical upstream kinase in the mTOR pathway, has been implicated in various immune responses and inflammatory processes. Akt is the main (but not exclusive) intermediate between PI3K and mTOR kinase.24 Signaling pathways mediated by PI3K/Akt, MAPK, and transcription factors such as NF-κB and Nrf2 are the predominant cascades that participate in HO-1 expression. A recent study also indicated that HO-1 expression via the PI3K/Akt and MAPK signaling pathway can induce anti-inflammatory response.25Several studies have indicated that mitochondrial membrane potential (Δψm) decline is involved in the inflammation.26 The Δψm decline is potentially neutralized by antioxidant enzymes such as HO-1, which are regulated by Nrf2. MAPKs are supposed to a major pathway in inflammation and participate in the regulation of Nrf2/HO-1 signaling.27,28 As expected, LCP-3 significantly suppresses the LPS induced phosphorylation of JNK and p38 (Fig. 5). As shown in Fig. 6, LCP-3 markedly attenuates the decline of Δψm in the two sets of experimental cells upon LPS stimuli. Further, we detected the potency of LCP-3 on the Nrf2/HO-1 pathway by western blotting. LCP-3 significantly attenuates the expression of HO-1, suppressing the cytosol translocation of Nrf2 (Fig. 6). Moreover, phosphorylation of PI3K and Akt in the LPS-induced cells was also significantly reduced. Here, the western blotting analysis revealed that LCP-3 might block LPS-induced NF-κB activation by blocking the MAPK and PI3K/Akt pathway, which interrupts the degradation of IκBa and further downstream iNOS and COX-2 expression. These findings demonstrate that p38 MAPK-mediated Nrf2/HO-1 activation contributes to the anti-inflammatory activity of LCP-3 in RAW 264.7 and HMC-1 cells upon LPS stimuli.
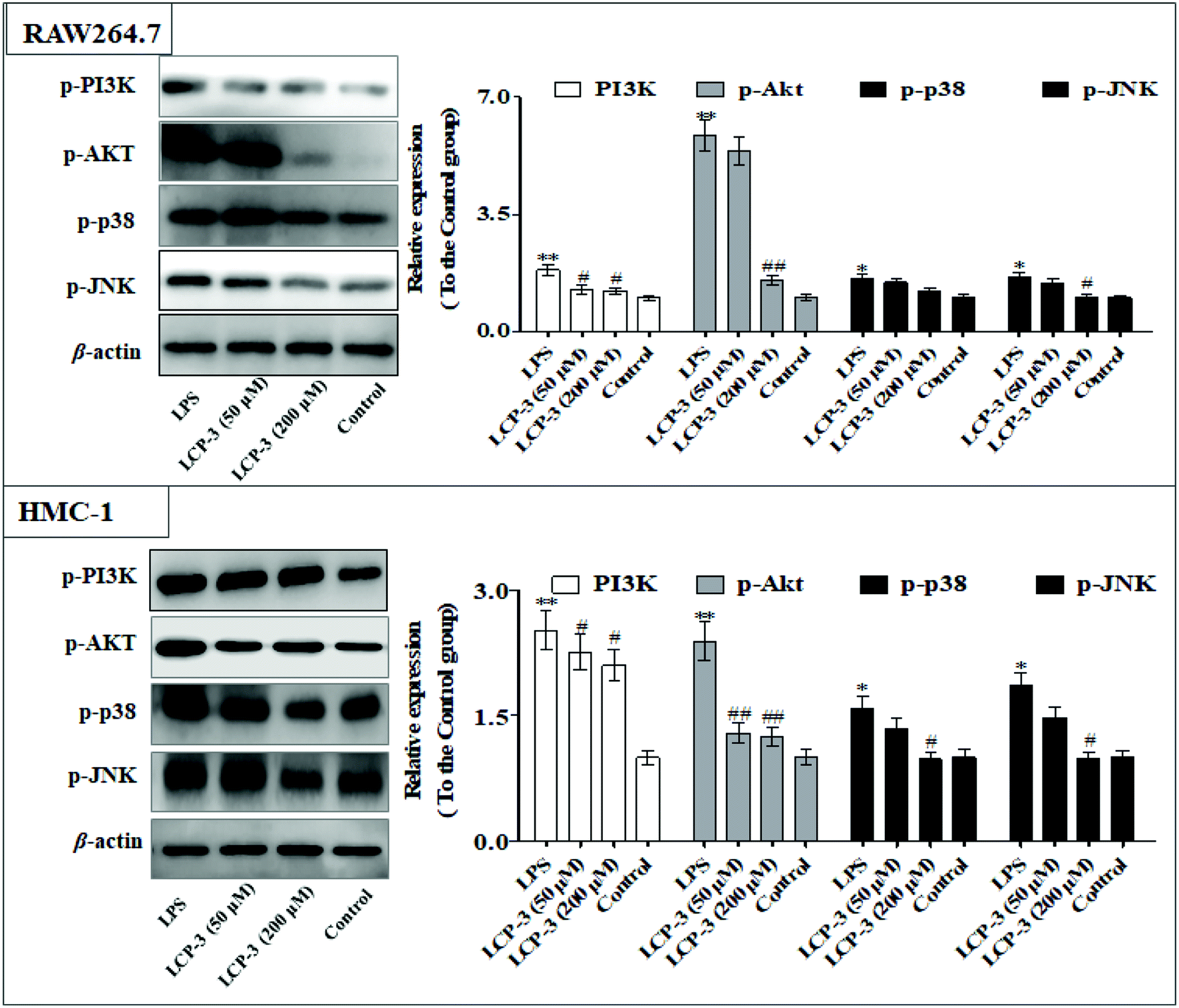 | ||
| Fig. 5 Effect of LCP-3 on the PI3K/Akt and MAPK signaling pathways in RAW264.7 and HMC-1 cells. RAW264.7 and HMC-1 cells were treated with LPS (1 μg mL−1) for 12 h, and then treated with LCP-3 (50 and 200 μM) for 16 h. The total protein lysate was prepared, and cell signaling proteins were detected by western blot analysis. The values are expressed as means ± SD of triplicate tests. *p < 0.05 and **p < 0.01 vs. the control, and #p < 0.05 and ##p < 0.01 vs. LPS, indicate statistically significant difference. β-Actin was used as an internal standard of process control and blot band densitometry was analyzed using the ImageJ software. | ||
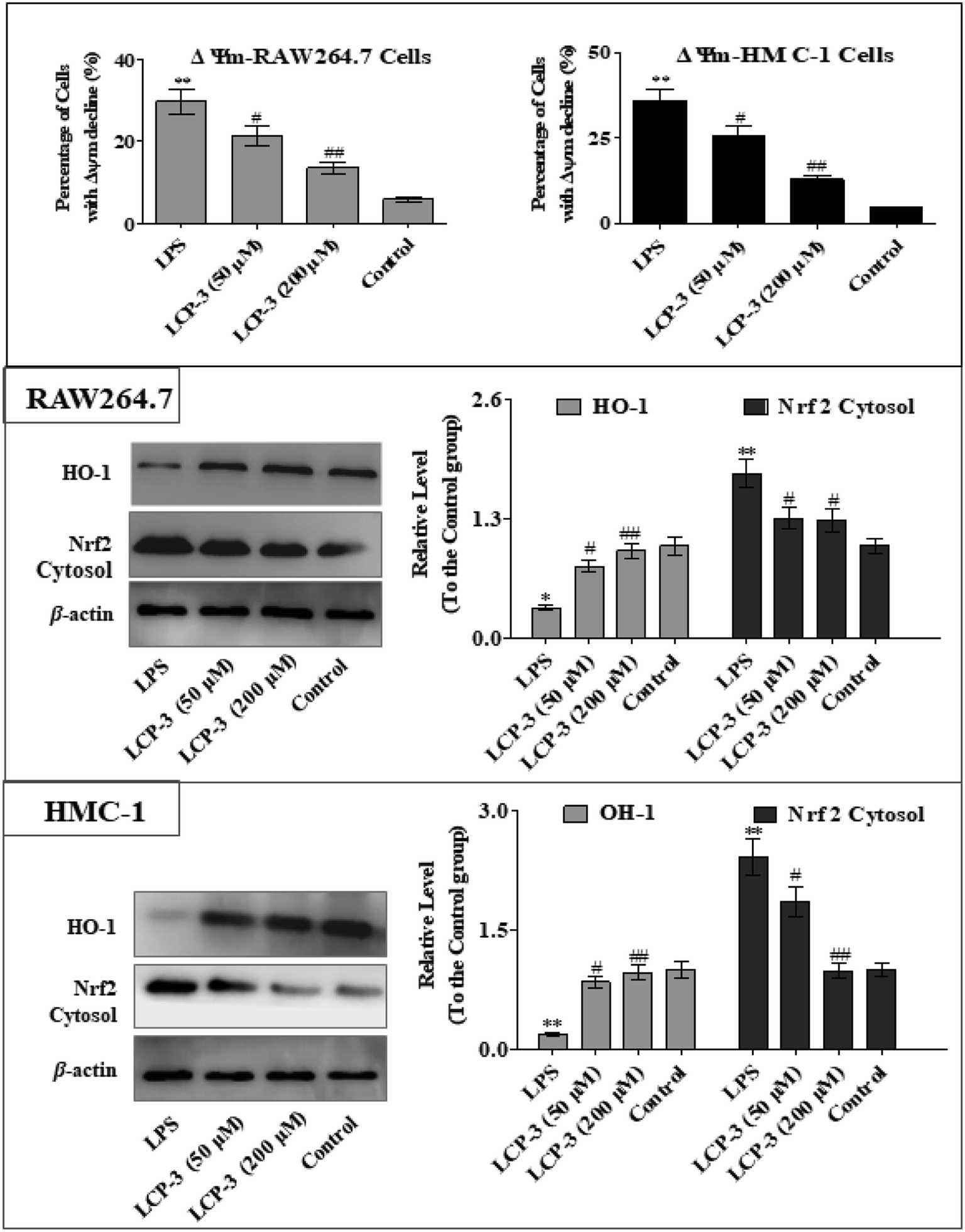 | ||
| Fig. 6 Effect of LCP-3 on Nrf2/HO-1 signaling in RAW264.7 and HMC-1 cells. RAW264.7 and HMC-1 cells were treated with LPS (1 μg mL−1) for 12 h, and then treated with LCP-3 (50 and 200 μM) for 16 h. The total protein lysate was prepared, and cell signaling proteins were detected by western blot analysis. The values are expressed as means ± SD of triplicate tests. *p < 0.05 and **p < 0.01 vs. the control, and #p < 0.05 and ##p < 0.01 vs. LPS, indicate statistically significant difference. β-Actin was used as an internal standard of process control and blot band densitometry was analyzed using the ImageJ software. | ||
4 Discussion
Although documents have reported that locusts are widely used in relieving coughs, asthma and enhancing immunity,29 their anti-inflammatory effects and the underlying molecular mechanisms of their bio-active peptides have been poorly explored. In the current study, we identify the bio-active peptides in locusts and clarify the potential anti-inflammatory mechanisms in RAW 264.7 and HMC-1 cells upon LPS stimuli. A novel cyclopeptide [cyclo-(Trp-Leu-His-Thr)]∼LCP-3 has been isolated from the locust and shows excellent anti-inflammatory potency.Studies have shown that a high concentration of NO can cause oxidative damage or inflammatory diseases.30 Pro-inflammatory cytokines such as TNF-α and IL-1β play crucial roles in the development of inflammatory diseases. Therefore, the levels of NO and pro-inflammatory cytokines are regarded as indicators of inflammation.31 In the current study, the results indicate that LCP-3 noticeably inhibits NO, TNF-α and IL-1β production in RAW 264.7 and HMC-1 cells upon LPS stimuli. Inflammatory cell exposure to endotoxins and cytokines producing excessive NO by iNOS have been linked to the pathogenesis of atherogenesis. The enzyme COX-2 and its product prostaglandin also promote cell proliferation, tumour angiogenesis and growth.32 The present data show that LCP-3 strongly inhibits LPS-induced NO and PGE2 production by decreasing the expression of iNOS and COX-2. These results indicate that the inhibition of iNOS and COX-2 protein expression by LCP-3 might be through regulating the transcriptional levels in RAW 264.7 and HMC-1 cells.
NF-κB plays a pivotal role in regulating inflammatory mediators during inflammatory response. The nuclear translocation of NF-κB, which dissociated from its inhibitory protein IκB, is the most crucial step for NF-κB signaling activation.33 It has been found that LCP-3 suppresses LPS activated NF-κB signaling by inhibiting phosphorylation of IκBα. Moreover, LCP-3 attenuates the phosphorylation of IKKα/β in both types of cells. In addition, LCP-3 reduces phosphorylation of AKT induced by LPS. Studies have shown that AKT can simulate the activation of NF-κB through IKKα/β.34 Taken together, LCP-3 might alleviate the activation of LPS-stimulated NF-κB and prevent the phosphorylation of AKT, which contributes to its inhibitory potency on inflammatory mediators. MAPK signaling pathways can collaborate synergistically to promote the expression and release target genes. To confirm the anti-inflammatory mechanisms of LCP-3, we further evaluated the inhibitory potency of LCP-3 on the activation of the MAPK signaling pathway. MAPKs, including ERK, JNK and p38 also take part in the regulation of the expression of inflammatory-related genes, leading to the overproduction of pro-inflammatory cytokines.35 The results we obtained show that LCP-3 obviously inhibits LPS-induced phosphorylation of p38 and JNK; it exerts its anti-inflammatory potency through the inhibition of both NF-κB and MAPKs in LPS induced cells.
Accumulated evidence indicates that Δψm decline aggravates the expression of inflammatory mediators such as iNOS and COX-2, eliciting an inflammatory process, which may be modulated by HO-1.36 The degradation of Keap1 induced by autophagy can provoke Nrf2 stabilization and the release of Nrf2 from Keap1, which subsequently accumulates in the nucleus, regulating various target genes such as HO-1 and participating in anti-inflammatory response.37 As expected, LCP-3 not only attenuates LPS-induced Δψm decline, but also suppresses cytosol translocation of Nrf2, and the following induction of HO-1 in RAW264.7 and HMC-1 cells. Hence, the results above suggest that Nrf2/HO-1 signaling participates in the anti-inflammatory effect of LCP-3. It has been reported that the activation of MAPKs participates in the induction of Nrf2-mediated HO-1. Moreover, the activation of p38 is well recognized as participating in inflammation through mediating Nrf2/HO-1 signaling.38 Thus, these findings also suggest that the activation of p38 MAPK is likely to participate in activating Nrf2/HO-1 signaling.
Of note, our data demonstrate that LCP-3 is the most effective anti-inflammatory cyclopeptide among the LCPs. LCP-3 significantly suppresses NO, TNF-α and IL-1β production in LPS-induced RAW264.7 and HMC-1 cells, and the possible mechanisms might involve the down-regulation of pro-inflammatory cytokine expression via blocking of the NF-κB and MAPK signaling pathways.
5 Conclusions
Locusts are esteemed in traditional Chinese medicine, and our results show that [cyclo-(Trp-Leu-His-Thr)]∼LCP-3 has potent anti-inflammatory potency in RAW264.7 and HMC-1 cells upon LPS stimuli. In addition, mechanistic studies show that LCP-3 attenuates pro-inflammatory cytokine (TNF-α, IL-6, IL-1β, NO and PGE2) expression. Moreover, LCP-3 attenuates inflammatory damage associated with the direct inhibition of iNOS and COX-2 expression. LCP-3 also regulates the MAPK, PI3K/AKT and NF-κB pathways to attenuate LPS-induced damage. Future investigations will aim to provide a better understanding of the function of LCP-3. Indeed, both animal and human studies must be designed, as extrapolations cannot be made from cell culture studies to humans.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
We thank Professor Chen (Jinan University, Guangzhou, China) for technical assistance as well as critical editing of the manuscript. The present study was supported in part by a grant from the National 863 Plan (No. 2006 AA10Z236).References
- S. I. Grivennikov, F. R. Greten and M. Karin, Cell, 2010, 140, 883–899 CrossRef CAS.
- O. Palomares, M. Akdis, M. Martin-Fontecha and C. A. Akdis, Immunol. Rev., 2017, 278, 219–236 CrossRef CAS.
- C. H. Kang, Y. H. Choi, S. K. Moon, W. J. Kim and G. Y. Kim, Toxicol. In Vitro, 2013, 27, 782–787 CrossRef CAS.
- D. Lee and J. Y. Imm, J. Food Biochem., 2017, 41(2), e12293 CrossRef.
- N. K. Bassal, B. P. Hughes and M. Costabile, Prostaglandins, Leukotrienes Essent. Fatty Acids, 2012, 87, 119–126 CrossRef CAS.
- P. Hye-Jeong and Y. Hyung-Sun, Toxicol. Ind. Health, 2013, 29, 169 CrossRef.
- N. Perkins, J. Am. Soc. Mass Spectrom., 2014, 74, 651–659 Search PubMed.
- S. S. Chung, Y. Wu, Q. Okobi, D. Adekoya, M. Atefi, O. Clarke, P. Dutta and J. V. Vadgama, Mediators Inflammation, 2017, 2017, 1–11 CrossRef.
- T. Oranuch, W. Pornthip, S. Hiroaki and S. Ikuo, Int. J. Oncol., 2011, 39, 279–285 Search PubMed.
- Q. Le Dang, T. S. Shin, M. S. Park, Y. H. Choi, G. J. Choi, K. S. Jang, I. S. Kim and J.-C. Kim, J. Agric. Food Chem., 2014, 62, 3363–3370 CrossRef CAS.
- X. Liang, X.-H. Nong, Z.-H. Huang and S.-H. Qi, J. Agric. Food Chem., 2017, 65, 5114–5121 CrossRef CAS.
- Y. Han, B. Zhang, Q. Shen, C. You, Y. Yu, P. Li and Q. Shang, Appl. Biochem. Biotechnol., 2015, 176, 2202–2212 CrossRef CAS.
- Z. Zhao and L. Geng-si, Agric. Sci. China, 2007, 6, 825–831 CrossRef CAS.
- G. Bosch, S. Zhang, D. G. A. B. Oonincx and W. H. Hendriks, J. Nutr. Sci., 2014, 3, 1–4 CrossRef.
- X. Jin, A. Brandazza, A. Navarrini, L. Ban, S. Zhang, R. A. Steinbrecht, L. Zhang and P. Pelosi, Cell. Mol. Life Sci., 2005, 62, 1156–1166 CrossRef CAS.
- E. Zielińska, B. Baraniak and M. Karaś, Nutrients, 2017, 9, 1–14 CrossRef.
- R. J. Dillon and A. K. Charnley, J. Invertebr. Pathol., 1995, 66, 72–75 CrossRef CAS.
- E. Kim, I. S. Jeon, J. W. Kim, J. Kim, J. H. Sam and S. J. Lee, Oral Diseases, 2010, 13, 495–499 CrossRef.
- G. Gallo-Oller, R. Ordoñez-Ciriza and J. Dotor, J. Immunol. Methods, 2018, 457, 1–5 CrossRef CAS.
- Z. Xichun, B. Rami and L. Dan, Cephalalgia, 2012, 32, 66–72 CrossRef.
- O. Sareila, R. Korhonen, H. Auvinen, M. HMLInen, H. Kankaanranta, E. Nissinen and E. Moilanen, Br. J. Pharmacol., 2010, 155, 884–895 CrossRef.
- M. P. Sang, B. G. Min, Y. J. Ji, K. H. Jegal, C. W. Lee, K. Y. Kim, Y. W. Kim, Y. W. Choi, I. J. Cho and S. K. Ku, BMC Complementary Altern. Med., 2018, 18, 20 CrossRef.
- I. T. Lee and C. M. Yang, Mediators Inflammation, 2013, 2013, 791231 Search PubMed.
- K. Penelope, L. Georgia, T. Eleni-Andriana, P. Niki, K. Maria, T. Irene, S. Stratigoula, F. Paraskevi, I. Dimitrios and P. Petros, BJU Int., 2013, 110, E1237–E1248 Search PubMed.
- F. Molica, S. Morel, B. R. Kwak, F. Rohner-Jeanrenaud and S. Steffens, Thromb. Haemostasis, 2015, 114, 553–566 Search PubMed.
- J. D. Ly, D. R. Grubb and A. Lawen, Apoptosis, 2003, 8, 115 CrossRef CAS.
- L. Cheng-Ling, C. Yu-Ting and H. Miao-Lin, J. Agric. Food Chem., 2011, 59, 11344–11351 CrossRef.
- J. LCP, R. Ding, L. Zou, C. Zhang, K. Wang, F. Liu, P. Li, M. Chen, J. B. Wan and H. Su, Am. J. Chin. Med., 2016, 44, 1043–1061 CrossRef.
- B. Boerjan, K. Vandingenen, A. D. Loof and L. Schoofs, Peptides, 2012, 34, 57–64 CrossRef CAS.
- K. Shosuke, H. Yusuke, P. Somchai and M. Ning, Biol. Chem., 2006, 98, 117–372 Search PubMed.
- Y. Li, R. Shen, G. Wen, R. Ding, A. Du, J. Zhou, Z. Dong, X. Ren, H. Yao and R. Zhao, Front. Pharmacol., 2017, 8, 139 Search PubMed.
- B. Sebastien, G. M. Soliman and M. Dusica, Nanomedicine, 2012, 7, 1149–1165 CrossRef.
- M. Pang, Y. Yuan, D. Wang, T. Li, D. Wang, X. Shi, M. Guo, C. Wang, X. Zhang and G. Zheng, Acta Biochim. Biophys. Sin., 2017, 49, 435–443 CrossRef CAS PubMed.
- R. Geleziunas, S. Ferrell, X. Lin, Y. Mu, E. T. Cunningham, M. Grant, M. A. Connelly, J. E. Hambor, K. B. Marcu and W. C. Greene, Mol. Cell. Biol., 1998, 18, 5157 CrossRef CAS.
- L. Yonghai, C. Niansheng, H. Aihua, G. Bruno, R. Abdolmohamad and Z. Guang-Xian, Exp. Mol. Pathol., 2008, 84, 1–8 CrossRef.
- H. Ohira, Y. Fujioka, C. Katagiri, R. Mamoto, M. Aoyama-Ishikawa, K. Amako, Y. Izumi, S. Nishiumi, M. Yoshida, M. Usami and M. Ikeda, J. Atheroscler. Thromb., 2013, 20, 425–442 CrossRef CAS.
- S. G. Rhee and S. H. Bae, Free Radical Biol. Med., 2015, 88, 205–211 CrossRef CAS.
- C.-K. Chan, L. T.-H. Tan, S. N. Andy, M. N. A. Kamarudin, B.-H. Goh and H. A. Kadir, Front. Pharmacol., 2017, 8 DOI:10.3389/fphar.2017.00397.
Footnote |
| † Contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |