DOI:
10.1039/C9RA06212B
(Paper)
RSC Adv., 2019,
9, 30277-30291
Oxone promoted dehydrogenative Povarov cyclization of N-aryl glycine derivatives: an approach towards quinoline fused lactones and lactams†
Received
9th August 2019
, Accepted 17th September 2019
First published on 25th September 2019
Abstract
Oxone promoted intramolecular dehydrogenative imino Diels–Alder reaction (Povarov cyclization) of alkyne tethered N-aryl glycine esters and amides has been explored, thus affording biologically significant quinoline fused lactones and lactams. The reaction is simple, scalable, and high yielding (up to 88%). The method was further extended to prepare biologically important luotonin-A analogues and the quinoline core of uncialamycin.
Introduction
Substituted quinolines are a ubiquitous heterocyclic motif present in a plethora of natural products and medicinal agents,1 among which quinoline fused lactones and lactams are of great importance due to their presence in complex natural products and pharmaceutically relevant molecules. In addition, they serve as a valuable precursor in the synthesis of biologically active natural products and their analogues such as luotonin-A (cytotoxic alkaloid),2 uncialamycin (antibiotic),3 aza podophyllotoxin4 analogues (antitumor agents) and quinoline carboxamides (radio ligands for molecular imaging).5 (Fig. 1). Consequently, there is a great deal of attention on the synthesis of these privileged structures. In general, the synthesis of these frameworks is associated with multistep processes as well as usage of toxic reagents.2e,g,6 Therefore, the development of a general, sustainable and efficient synthetic approach to achieve these functionalized quinoline-fused lactones/lactams is of high value, and it would provide an appropriate platform for the detail biological investigation of these valuable molecules.
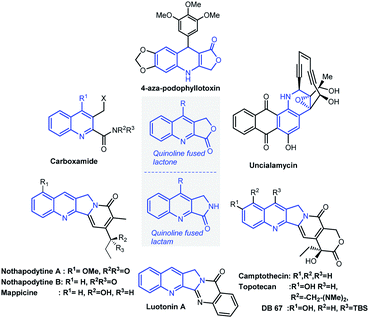 |
| | Fig. 1 Examples of pharmaceuticals and natural products containing quinoline-fused lactone/lactam moiety. | |
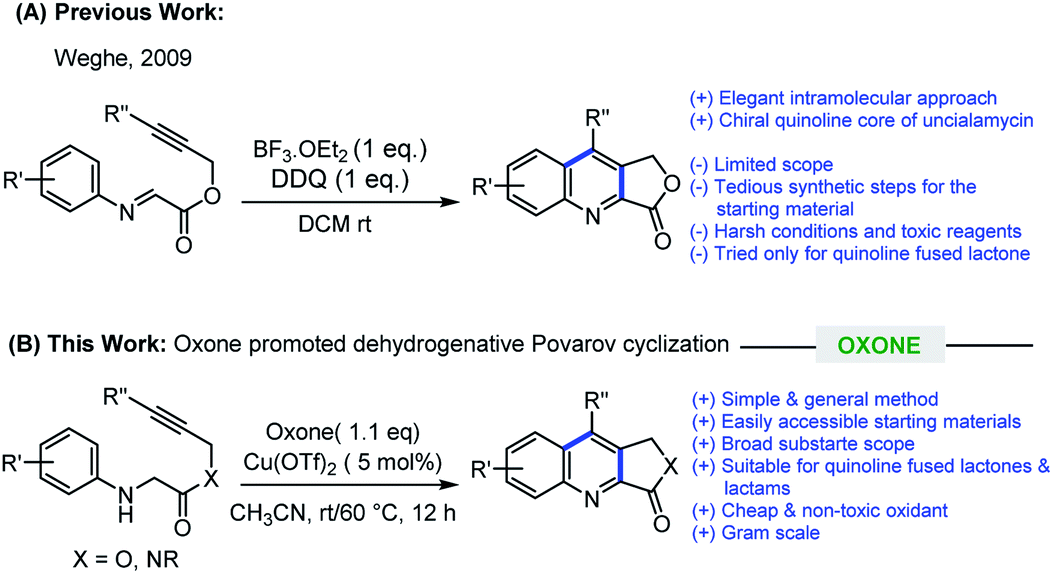
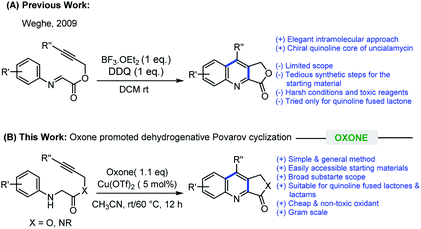
In recent years, imino Diels–Alder reaction (Povarov reaction) has received a renewed interest to construct quinoline scaffolds, in which electron-rich alkenes (or alkynes) were added to the electron-deficient aromatic imines followed by oxidation.7 However, an intramolecular variant of this transformation is less explored compared to intermolecular.8 In 2009, Weghe et al. elegantly utilized intramolecular imino Diels–Alder reaction promoted by BF3·OEt2/DDQ for the construction of quinoline fused lactones employing an alkene or alkyne as a dienophile.9 Despite the merits, there are certain drawbacks associated with this method such as, non-ready availability of the requisite starting materials, requiring a multistep reaction sequences for their synthesis, hazardous and expensive reagents, limited substrate scope, and the method have not been explored for the synthesis of quinoline fused lactams. Later, Jia and Zhang group also explored the concept of cross dehydrogenative coupling (CDC) for the construction quinoline fused lactone/lactam in the presence of TBPA radical cation salt and visible-light photoredox conditions, respectively.10 In the context of our ongoing research program dealing with drug discovery, we were encountered with a need for an efficient methodology for the synthesis of quinoline fused lactones and lactams for our internal screening program. While studying the suitability of Weghe's method for our library synthesis, we encountered a difficulty in the preparation of starting imine compounds as the procedures are lengthy along with issues pertinent to their stability. With regard to practicality, we envisioned that alkyne tethered N-aryl glycine derivatives would be an ideal substrate for our library synthesis as (i) these substrates are stable and can be easily prepared (ii) in suitable condition, these substrates can undergo oxidative dehydrogenation11 followed by Povarov cyclization could lead to quinoline fused lactones/lactams.
In this manuscript, we wish to disclose the successful realization of this new strategy, which involves Oxone promoted oxidative dehydrogenation followed by intramolecular Povarov cyclization of alkyne tethered N-aryl glycine derivatives for the efficient synthesis of quinoline fused lactones/lactams (Scheme 1). Furthermore, to the best of our knowledge, Oxone promoted intramolecular Povarov cyclization of hitherto unknown alkyne tethered N-aryl glycine derivatives has not been reported.
 |
| | Scheme 1 Intramolecular Povarov cyclization for the synthesis of quinoline fused lactones and lactams. | |
Results and discussion
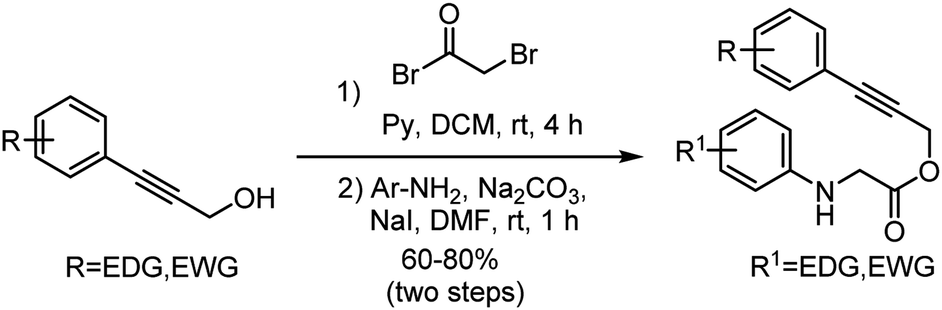
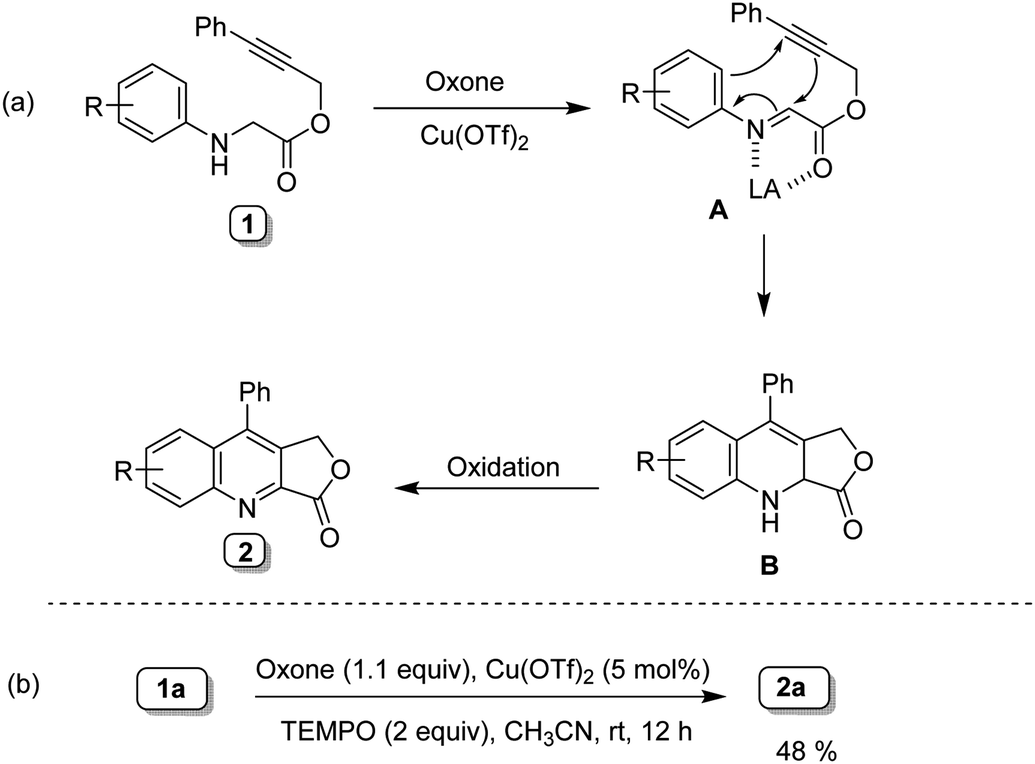
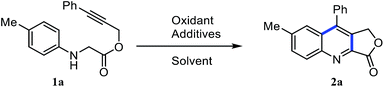
Accordingly, the required starting material alkyne tethered N-aryl glycine derivatives were conveniently prepared in two steps from substituted propargyl alcohols (Scheme 2). Initially, we examined the dehydrogenative Povarov cyclization of N-aryl glycine ester 1a as a model substrate by employing 5 mol% BF3·OEt2 as a Lewis acid in the presence of IBX as an oxidant at room temperature (Table 1, entry 1).12 To our delight, as expected, the reaction proceeded smoothly to give the desired product 3a in 58% yield. Inspired by this initial result, we screened other Lewis acids and found that Cu(OTf)2 is the best among other Lewis acids tried (Table 1, entries 1–4). Next, we studied the effect of various oxidants and observed that peroxide-based oxidants also suitable for this transformation (Table 1, entries 5–8). Interestingly, the reaction proceeded well in the presence of Oxone and 5 mol% of Cu(OTf)2 at room temperature afforded the required product 3a in 88% yield (Table 1, entry 9). Notably, Oxone would be a favourable oxidant as it is cheap, non-toxic, and easy to handle.13 Screening of other solvents for this transformation reveals that CH3CN is the best solvent of choice (Table 1, entry 9 vs. 10–12). Further, not significant improvement in the yield has been observed while using more equiv. of Oxone (Table 1, entry 13). Notably, when we use only Oxone, the desired product formed in a 60% yield albeit in 24 hours (entry 14), which indicates that the Oxone alone can trigger this transformation presumably due to the slightly acidic nature of Oxone (2KHSO5-KHSO4-K2SO4). Finally, in the absence of Oxone there is only a trace amount of product formation has been observed, which reveals the crucial role of Oxone in this transformation (Table 1, entry 15).
 |
| | Scheme 2 Synthesis of alkyne tethered N-aryl glycine derivatives. | |
Table 1 Optimization studiesa,b
|
|
| Entry |
Oxidant |
Additives |
Solvent |
Yield (%) 3a |
|
Reaction conditions: (1) 0.18 mmol 1a, 0.20 mmol oxidant, additive (5 mol%) solvent (3.0 mL), 12 h.
Isolated yields; rt.
1.3 equiv. of Oxone was employed.
24 h; IBX: 2-iodoxybenzoic acid.
|
| 1 |
IBX |
BF3·OEt2 |
CH3CN |
58 |
| 2 |
IBX |
Sc(OTf)3 |
CH3CN |
53 |
| 3 |
IBX |
Cu(OTf)2 |
CH3CN |
68 |
| 4 |
IBX |
Cu(OAc)2 |
CH3CN |
51 |
| 5 |
PhI(OAc)2 |
Cu(OTf)2 |
CH3CN |
23 |
| 6 |
PhI(OCOCF3)2 |
Cu(OTf)2 |
CH3CN |
21 |
| 7 |
Na2S2O8 |
Cu(OTf)2 |
CH3CN |
41 |
| 8 |
BPO |
Cu(OTf)2 |
CH3CN |
49 |
| 9 |
Oxone |
Cu(OTf)2 |
CH3CN |
88 |
| 10 |
Oxone |
Cu(OTf)2 |
THF |
42 |
| 11 |
Oxone |
Cu(OTf)2 |
Toluene |
Traces |
| 12 |
Oxone |
Cu(OTf)2 |
CHCl3 |
<5 |
| 13 |
Oxone |
Cu(OTf)2 |
CH3CN |
89c |
| 14 |
Oxone |
— |
CH3CN |
60d |
| 15 |
— |
Cu(OTf)2 |
CH3CN |
Traces |
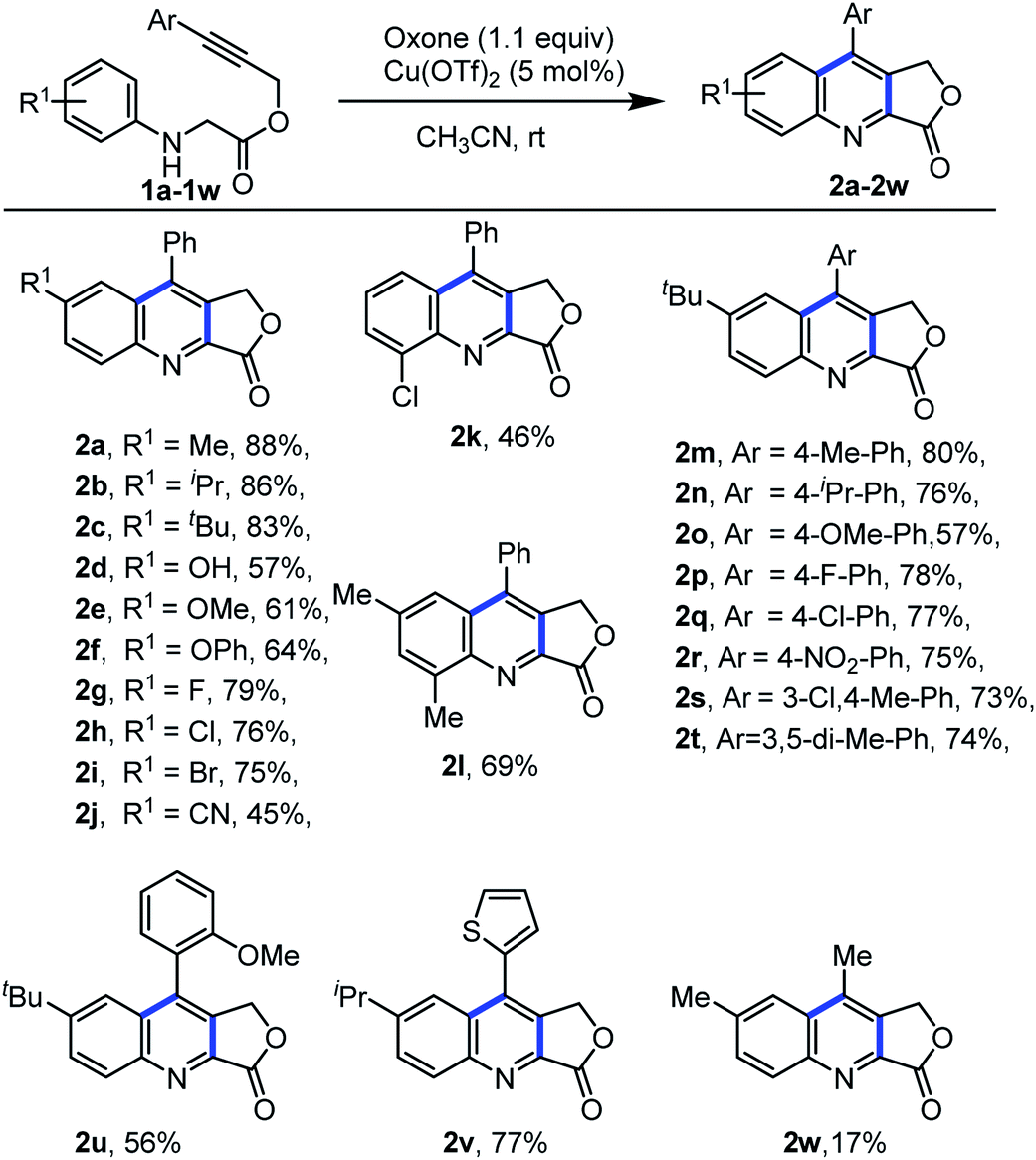
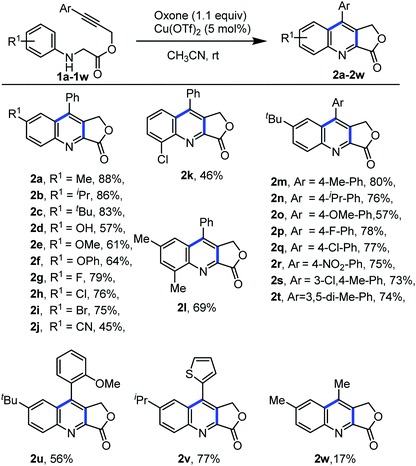
After the optimal reaction condition was established for the construction of quinoline-fused lactones, the scope and generality of this protocol were investigated (Scheme 3). Different electron-donating and electron-withdrawing functional groups on the aniline ring as well as the aryl alkyne part were well tolerated. For example, substrates bearing electron-donating groups such as 4-methyl, 4-isopropyl, and 4-tbutyl on aniline ring were compatible with the reaction conditions and provided the desired products 2a, 2b and 2c in 88%, 86% and 83% yields, respectively. However, strong electron-donating such as hydroxy, methoxy, and phenoxy substrates resulted the desired products in moderate yield (2d–2f, 57–64%). Additionally, substrates bearing halogen atoms such as F, Cl, and Br successfully reacted under the optimized condition to give the desired product (2g–2i) in good yield (75–79%). However, the electron-withdrawing group at the aniline ring also gave the desired product in moderate yield (2j, 45%). Furthermore, substitution on the ortho position of the aniline ring provided the desired product in moderate yield (2k, 46%). Moreover, the di-substituted substrate also underwent smoothly, to achieve the desired product 2l in 69% yield. Next, we examined the scope of our protocol by altering the substitution on aryl alkyne part of N-aryl glycine derivatives. Electron donating and withdrawing substituents on the aryl alkyne part of N-aryl glycine derivatives also underwent the reaction smoothly and afforded the product in good yield (2m–2r). Further, the present protocol is suitable for ortho substitution, disubstitution as well as heteroaryl substitution on aryl alkyne part of the glycine derivatives (2s–2v). Alkyl substitution on the alkyne moiety under optimal condition provided the required product 2w, in less yield.
 |
| | Scheme 3 Intramolecular Povarov cyclization of N-aryl glycine estersa,b. aReaction conditions: 0.18 mmol 1a, 0.20 mmol oxidant, 5 mol% of Cu(OTf)2, CH3CN (3.0 mL), rt, 12 h; bisolated yields. | |
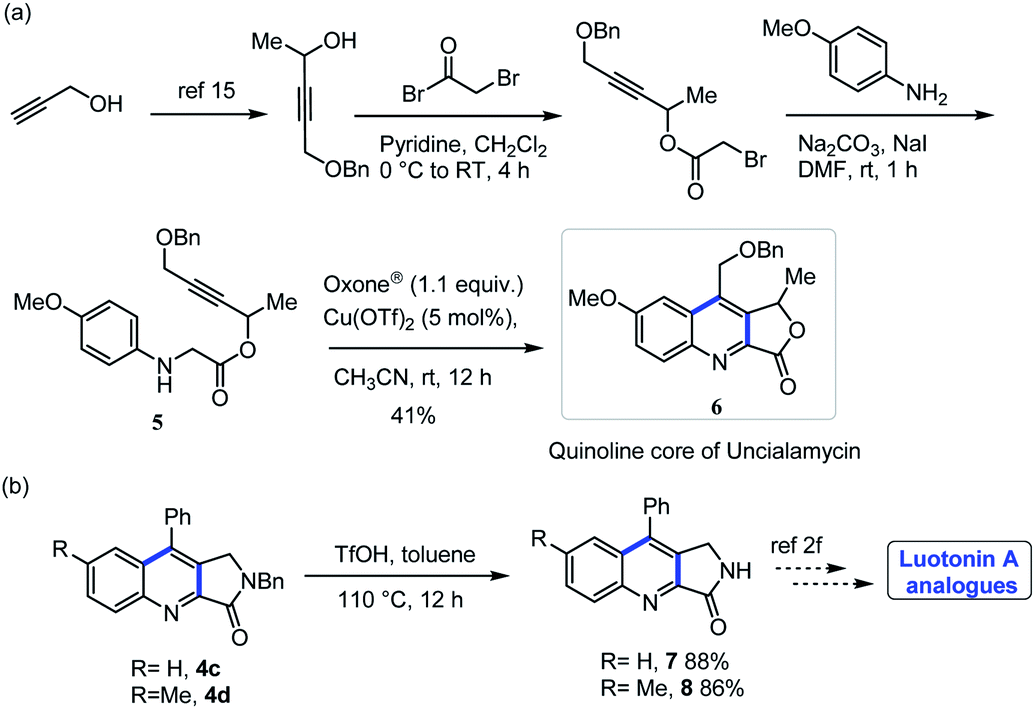
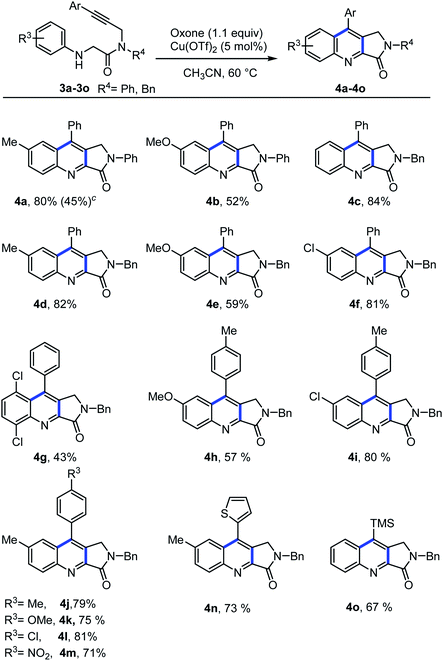
After successful synthesis of quinoline-fused lactones, we further explored the suitability of the present protocol for the synthesis of quinoline-fused lactams. Accordingly, we examined the intramolecular dehydrogenative Povarov cyclization of N-aryl glycine amide substrates under our optimized reaction condition and delighted to find that the present protocol is suitable for the construction of quinoline fused lactam as well. However, the reaction requires higher temperature (60 °C) for completion. Various electron donating as well as electron withdrawing substituents on the aniline ring as well as aryl alkyne part of N-protected glycine amide (Ph- and Bn-) such as methyl, methoxy, chloro, di chloro, nitro and thienyl underwent cyclization to furnish the corresponding quinoline-fused lactams in moderate to good yields (Scheme 4). It is noteworthy to mention here that, the substrate bearing a TMS group is also well tolerated (Scheme 4, 4o) and the resulted product can conveniently be converted into the natural product luotonin A in two steps.2 To demonstrate the synthetic practicality of the present protocol, we conducted a gram-scale experiment by employing 3c (2.82 mmol, 1.0 g) under the optimal reaction conditions in which the desired product 4c was obtained in 80% yield (0.79 g) showing that the present method could be easily adapted for the large-scale synthesis with high efficiency.
 |
| | Scheme 4 Intramolecular Povarov cyclisation of N-aryl glycine amides.a,b. aReaction conditions: 0.14 mmol 3a, 0.15 mmol Oxidant, 5 mol% of Cu(OTf)2, CH3CN (3.0 mL), 60 °C, 12 h; bisolated yields; creaction carried out at room temperature for 12 h. | |
Although the mechanism of this transformation is not fully understood, however, based on the above experiments (Table 1. Entry 14 and 15) and previous literature reports,14 a tentative reaction mechanism is proposed in Scheme 5a. The first step involves the in situ generation of highly reactive imine intermediate A, followed by intramolecular cycloaddition to form an intermediate B.7q–t Further, intermediate B undergoes oxidation to form the corresponding fused quinoline 2. A radical trapping experiment was conducted by employing TEMPO as a radical scavenger and obtained 2a in 48% yield (Scheme 5b), which indicates that the reaction proceeds via non-radical pathway.
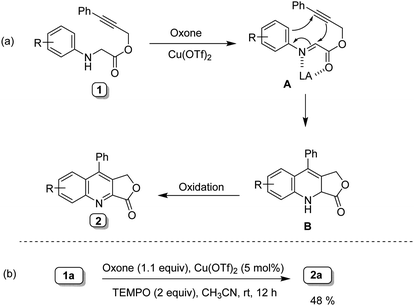 |
| | Scheme 5 A plausible mechanism and control experiment. | |
The quinoline fused lactone/lactam produced from the method described here may find utility in complex molecule synthesis. Some representative examples are shown in Scheme 6. We utilized this method for the preparation of quinoline core precursor of antitumor antibiotic uncialamycin 6.9 Also, compounds 4c, 4d can easily be converted into cytotoxic alkaloid luotonin-A analogues.2f
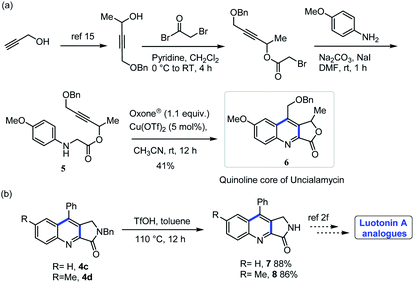 |
| | Scheme 6 Utility of the reaction. | |
Conclusions
In conclusion, we have demonstrated Oxone promoted intramolecular dehydrogenative Povarov cyclization of various alkyne tethered N-aryl glycine derivatives to furnish biologically relevant quinoline-fused lactones and lactams. This operationally simple, scalable protocol utilizes non-toxic, inexpensive Oxone as an oxidant to furnish the required products in high yield. The method was further utilized for the preparation of cytotoxic alkaloid luotonin-A analogues and quinoline core of uncialamycin. Efforts are underway in our laboratory to extend the application of this method as well as the detail mechanistic investigation.
Experimental section
General information
Where stated, all reagents were purchased from commercial sources and used without further purification. All the substituted propargyl alcohols were prepared using a known literature procedure.15,161H NMR, 13C NMR, and 19F NMR spectra were recorded on Bruker AV, 200/400/500, JEOL 400 MHz spectrometers in appropriate solvents using TMS as an internal standard or the solvent signals as secondary standards and the chemical shifts are shown in δ scales. Chemical shifts (δ) are quoted in parts per million (ppm). The residual solvent peak, 1H NMR δH 7.26 and 13C {1H} NMR δC 77.0 for CDCl3 were used as a reference. Coupling constants (J) are reported in Hertz (Hz) to the nearest 0.1 Hz. Multiplicities of 1H NMR signals are designated as s (singlet), d (doublet), dd (doublet of doublet), t (triplet), quin (quintet), sept (septet) bs (broad singlet), m (multiplet), etc. Melting points were determined using digital Buchi Melting Point Apparatus B-540 and are uncorrected. Thin layer chromatography was carried out on Merck silica gel 60F254 pre-coated aluminum foil sheets and was visualized using UV light (254 nm) and stained with Ninhydrin. Flash column chromatography was carried out through silica gel (100–200 mesh) using ethyl acetate/hexane as eluent. Structures of the products were identified by 1H NMR, 13C {1H} NMR, 19F NMR, HRMS.
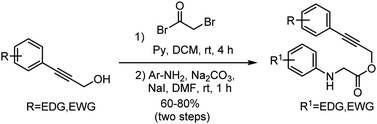
General procedure for the synthesis of substituted phenyl propargyl bromo acetate
To a solution of substituted propargyl alcohol (7.56 mmol) and pyridine (9.0 mmol) in anhydrous DCM (15 mL) was added 2-bromoacetyl bromide (8.31 mmol) in DCM (5 mL) at 0 °C under N2 atmosphere over 30 min. After the addition was complete, the reaction mixture was stirred at room temperature for 4 h. After completion of the reaction (monitored by TLC), the crude reaction mixture was then poured into water (30 mL) and extracted with DCM (3 × 20 mL). The organic extracts were dried over Na2SO4 and concentrated in vacuo, affording the substituted phenyl propargyl bromoacetate which was used directly without further purification.
General procedure for the synthesis of substituted N-aryl glycine ester (1a–1w)
A 5 mL Screw Top V vial® was charged with substituted bromoacetate (0.39 mmol), Na2CO3 (0.47 mmol), NaI (0.08 mmol), and substituted aniline (0.38 mmol), DMF (2 mL). The solution was stirred at room temperature. After 1 h, the reaction mixture was poured into ice water (10 mL) and extracted with EtOAc (2 × 20 mL). The organic extracts were combined, washed with brine (15 mL), dried over Na2SO4, concentrated in vacuo and purified by flash column chromatography by eluting 5–15% of ethyl acetate/petroleum ether (silica gel, 100–200 mesh), to afford substituted N-aryl glycine esters (1a–1w).
General procedure for the synthesis of substituted N-aryl glycine amide (3a–3o)
A 5 mL glass vial® was charged with substituted bromo acetamide (0.3 mmol), Na2CO3 (0.36 mmol), NaI (0.061 mmol), substituted aniline (0.29 mmol), DMF (3 mL). The mixture was stirred for 12 h at 60 °C. After completion of the reaction (monitored by TLC), the reaction mixture was cooled to room temperature, then poured into ice water, extracted with ethyl acetate (20 mL × 2). The organic extracts were combined, washed with brine (15 mL), dried over Na2SO4, concentrated in vacuo and purified by flash column chromatography by eluting 10–20% of ethyl acetate/petroleum ether (silica gel, 100–200 mesh), to afford the substituted N-aryl glycine amides (3a–3o).
General procedure for the synthesis of quinoline fused lactones (2a–2w)
To a 5 mL Screw Top V vial® containing a stirring mixture of substituted N-aryl glycine ester (0.18 mmol), Oxone® (122 mg, 0.20 mmol) in CH3CN (3 mL) was added Cu(OTf)2 (3.25 mg, 0.009 mmol) and the vial cap was wrapped tightly with a Teflon. The solution was then stirred at room temperature. After 12 h (colorless to dark brown color was observed), the solvent was removed under reduced pressure. The crude reaction mixture was purified by flash column chromatography by eluting 20–30% of ethyl acetate/petroleum ether (silica gel, 100–200 mesh), to afford the pure substituted quinoline fused lactones (2a–2w).
General procedure for the synthesis of quinoline fused lactams (4a–4o)
To a 5 mL Screw Top V vial® containing a stirring mixture of substituted N-aryl glycine amide (0.14 mmol), oxone® (92 mg, 0.15 mmol) in CH3CN (3 mL) was added Cu(OTf)2 (2.64 mg, 0.0073 mmol) and the vial cap was wrapped tightly with a Teflon. The solution was stirred at 60 °C. After 12 h (colorless to dark brown color was observed), the solvent was removed under reduced pressure. The crude reaction mixture was purified by flash column chromatography by eluting 25–40% of ethyl acetate/petroleum ether (silica gel, 100–200 mesh), to afford the pure substituted quinoline fused lactams (4a–4o).
3-Phenylprop-2-yn-1-yl p-tolylglycinate (1a).
Compound 1a was isolated in 84% yield (93 mg, yellow solid); mp = 88–90 °C; Rf = 0.55 (VPE/VEA = 90/10); 1H NMR (500 MHz, CDCl3) δ = 7.48 (d, J = 7.3 Hz, 2H), 7.38–7.33 (m, 3H), 7.03 (d, J = 7.8 Hz, 2H), 6.58 (d, J = 8.1 Hz, 2H), 5.02 (s, 2H), 4.18 (bs, 1H), 3.99 (s, 2H), 2.26 (s, 3H); 13C {1H} NMR (125 MHz, CDCl3) δ = 170.7, 144.6, 131.9, 129.8, 128.8, 128.3, 127.6, 121.9, 113.2, 86.9, 82.3, 53.4, 46.1, 20.3; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C18H18NO2 280.1332; found 280.1325.
3-Phenylprop-2-yn-1-yl(4-iso-propylphenyl)glycinate (1b).
Compound 1b was isolated in 81% yield (98 mg, viscous liquid); Rf = 0.55 (VPE/VEA = 90/10); 1H NMR (500 MHz, CDCl3) δ = 7.48 (d, J = 6.3 Hz, 2H), 7.34 (m, 3H), 7.09 (d, J = 8.0 Hz, 2H), 6.61 (d, J = 8.0 Hz, 2H), 5.03 (s, 2H), 4.07 (bs, 1H), 4.00 (s, 2H), 2.83 (sept, J = 6.8 Hz, 1H), 1.23 (d, J = 6.8 Hz, 6H); 13C {1H} NMR (125 MHz, CDCl3) δ = 170.7, 144.7, 138.9, 131.8, 128.8, 128.3, 127.1, 121.9, 113.2, 86.9, 82.3, 53.4, 46.1, 33.1, 24.1; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C20H22NO2 308.1645; found 308.1646.
3-Phenylprop-2-yn-1-yl(4-(tert-butyl)phenyl)glycinate (1c).
Compound 1c was isolated in 80% yield (102 mg, yellow solid); mp = 89–91 °C; Rf = 0.55 (VPE/VEA = 90/10); 1H NMR (500 MHz, CDCl3) δ = 7.43 (dd, J = 7.7, 1.7 Hz, 2H), 7.29 (m, 3H), 7.20 (d, J = 8.7 Hz, 2H) 6.55 (d, J = 8.7 Hz, 2H), 4.96 (s, 2H), 4.20 (bs, 1H), 3.93 (s, 2H), 1.26 (s, 9H); 13C {1H} NMR (125 MHz, CDCl3) δ = 170.7, 144.4, 140.9, 131.8, 128.8, 128.2, 125.9, 121.8, 112.7, 86.9, 82.4, 53.3, 45.9, 33.7, 31.39; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C21H24NO2 322.1802; found 322.1801.
3-Phenylprop-2-yn-1-yl(4-hydroxyphenyl)glycinate (1d).
Compound 1d was isolated in 70% yield (78 mg, viscous liquid); Rf = 0.40 (VPE/VEA = 60/40); 1H NMR (500 MHz, CDCl3) δ = 7.46–7.42 (m, 2H), 7.36–7.30 (m, 3H), 6.68 (d, J = 8.7 Hz, 2H), 6.52 (d, J = 8.7 Hz, 2H), 4.99 (s, 2H), 3.92 (s, 2H); 13C {1H} NMR (125 MHz, CDCl3) δ = 171.2, 148.7, 140.6, 131.8, 128.8, 128.3, 121.8, 116.2, 114.9, 86.9, 82.3, 53.5, 46.8; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C17H16NO3 282.1125; found 282.1126.
3-Phenylprop-2-yn-1-yl(4-methoxyphenyl)glycinate (1e).
Compound 1e was isolated in 60% yield (note: decomposition was observed after keeping prolong time in column; brown liquid); Rf = 0.40 (VPE/VEA = 80/20); 1H NMR (500 MHz, CDCl3) δ = 7.47–7.44 (m, 2H), 7.37–7.31 (m, 3H), 6.80 (d, J = 8.8 Hz, 2H), 6.61 (d, J = 8.8 Hz, 2H), 5.00 (s, 2H), 3.96 (s, 2H), 3.74 (s, 3H); 13C {1H} NMR (125 MHz, CDCl3) δ = 170.8, 152.7, 141.0, 131.8, 128.8, 128.3, 121.9, 114.9, 114.4, 86.9, 82.4, 55.6, 53.4, 46.7; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C18H18NO3 296.1281; found 296.1280.
3-Phenylprop-2-yn-1-yl(4-phenoxyphenyl)glycinate (1f).
Compound 1f was isolated in 75% yield (106 mg, off white solid); mp = 109–111 °C; Rf = 0.40 (VPE/VEA = 80/20); 1H NMR (400 MHz, CDCl3) δ = 7.48 (dd, J = 7.6, 1.7 Hz, 2H), 7.39–7.27 (m, 5H), 7.04 (t, J = 7.4 Hz, 1H), 6.95 (dt, J = 7.7, 3.4 Hz, 4H), 6.66–6.61 (m, 2H), 5.04 (s, 2H), 4.26 (bs, 1H), 4.00 (s, 2H); 13C{1H} NMR (100 MHz, CDCl3) δ = 170.6, 158.7, 148.4, 143.3, 131.8, 129.4, 128.9, 128.3, 122.0, 121.8, 121.1, 117.2, 114.1, 87.0, 82.3, 53.5, 46.2; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C23H20NO3 358.1438; found 358.1429.
3-Phenylprop-2-yn-1-yl(4-fluorophenyl)glycinate (1g).
Compound 1g was isolated in 78% yield (87 mg, off white solid); mp = 86–88 °C; Rf = 0.40 (VPE/VEA = 90/10); 1H NMR (400 MHz, CDCl3) δ = 7.48 (d, J = 7.3 Hz, 2H), 7.38–7.34 (m, 3H), 6.92 (t, J = 8.6 Hz, 2H), 6.56 (dd, J = 8.7 Hz, 4.2 Hz, 2H), 5.02 (s, 2H), 4.15 (bs, 1H), 3.95 (s, 2H); 13C {1H} NMR (100 MHz, CDCl3) δ = 170.5, 156.2 (d, J = 235.9 Hz) 143.2, 131.8, 128.8, 128.3, 121.8, 115.7 (d, J = 22.5 Hz), 113.9 (d, J = 7.5 Hz), 86.9, 82.3, 53.4, 46.2; 19F NMR (376 MHz, CDCl3) δ = −126.8; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C17H15FNO2 284.1081; found 284.1075.
3-Phenylprop-2-yn-1-yl(4-chlorophenyl)glycinate (1h).
Compound 1h was isolated in 79% yield (93 mg, off white solid); mp = 83–85 °C; Rf = 0.40 (VPE/VEA = 90/10); 1H NMR (400 MHz, CDCl3) δ = 7.45 (d, J = 7.3 Hz, 2H), 7.37–7.31 (m, 3H), 7.14 (d, J = 8.6 Hz, 2H), 6.55 (d, J = 8.6 Hz, 2H), 5.02 (s, 2H), 4.31 (bs, 1H), 3.97 (s, 2H); 13C {1H} NMR (100 MHz, CDCl3) δ = 170.3, 145.4, 131.9, 129.2, 128.9, 128.3, 123.1, 121.8, 114.1, 87.1, 82.2, 53.6, 45.8; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C17H15ClNO2 300.0786; found 300.0780.
3-Phenylprop-2-yn-1-yl(4-bromophenyl)glycinate (1i).
Compound 1i was isolated in 78% yield (107 mg, brown solid); mp = 86–88 °C; Rf = 0.40 (VPE/VEA = 90/10); 1H NMR (500 MHz, CDCl3) δ = 7.44 (d, J = 6.8 Hz, 2H), 7.37–7.29 (m, 3H), 7.27 (d, J = 8.5 Hz, 2H), 6.48 (d, J = 8.5 Hz, 2H), 5.00 (s, 2H), 4.31 (bs, 1H), 3.94 (s, 2H); 13C {1H} NMR (125 MHz, CDCl3) δ = 170.2, 145.8, 132.0, 131.9, 128.9, 128.3, 121.8, 114.6, 110.1, 87.1, 82.2, 53.6, 45.6; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C17H15BrNO2 344.0281; found 344.0284.
3-Phenylprop-2-yn-1-yl(4-cyanophenyl)glycinate (1j).
Compound 1j crude (90 mg, viscous liquid); Rf = 0.40 (VPE/VEA = 80/20) Rf = 0.40 (VPE/VEA = 80/20); 1H NMR (500 MHz, CDCl3) δ = 7.44 (d, J = 7.2 Hz, 4H), 7.34 (t, J = 8.1 Hz, 3H), 6.58 (d, J = 8.2 Hz, 2H), 5.03 (s, 2H), 4.87 (s, 1H), 4.01 (d, J = 4.9 Hz, 2H); 13C {1H} NMR (125 MHz, CDCl3) δ = 169.5, 149.9, 133.7, 131.8, 129.0, 128.3, 121.6, 120.0, 112.5, 100.0, 87.2, 81.9, 53.9, 44.7; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C18H15N2O2 291.1178; found 291.1140.
3-Phenylprop-2-yn-1-yl(2-chlorophenyl)glycinate (1k).
Compound 1k was isolated in 68% yield (80 mg, off white solid); mp = 69–71 °C; Rf = 0.40 (VPE/VEA = 90/10); 1H NMR (400 MHz, CDCl3) δ = 7.54–7.44 (m, 2H), 7.43–7.27 (m, 4H), 7.15 (t, J = 7.7 Hz, 1H), 6.71 (t, J = 7.6 Hz, 1H), 6.56 (d, J = 8.1 Hz, 1H), 5.04 (s, 2H), 4.98 (bs, 1H), 4.05 (s, 2H); 13C {1H} NMR (100 MHz, CDCl3) δ = 169.9, 142.7, 131.8, 129.3, 128.9, 128.3, 127.8, 121.8, 119.5, 118.2, 111.2, 87.0, 82.2, 53.6, 45.3; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C17H15ClNO2 300.0778; found 300.0796.
3-Phenylprop-2-yn-1-yl(2,4-dimethylphenyl)glycinate (1l).
Compound 1l was isolated in 72% yield (83 mg, viscous liquid); Rf = 0.50 (VPE/VEA = 90/10); 1H NMR (500 MHz, CDCl3) δ = 7.47 (d, J = 7.7 Hz, 2H), 7.38–7.31 (m, 3H), 6.94 (s, 1H), 6.92 (s, 1H), 6.43 (d, J = 7.9 Hz, 1H), 5.02 (s, 2H), 4.03 (s, 2H) 2.24 (s, 3H), 2.21 (s, 3H); 13C {1H} NMR (125 MHz, CDCl3) δ = 170.9, 142.6, 131.9, 131.2, 128.9, 128.3, 127.3, 127.2, 122.8, 121.9, 110.2, 86.9, 82.3, 53.5, 46.1, 20.3, 17.3; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C19H20NO2 294.1489, found 294.1476.
3-(p-Tolyl)prop-2-yn-1-yl(4-(tert-butyl)phenyl)glycinate (1m).
Compound 1m was isolated in 77% yield (97 mg, off white solid); mp = 85–87 °C; Rf = 0.55 (VPE/VEA = 90/10); 1H NMR (500 MHz, CDCl3) δ = 7.34 (d, J = 7.8 Hz, 2H), 7.21 (d, J = 8.3 Hz, 2H), 7.11 (d, J = 7.7 Hz, 2H), 6.57 (d, J = 8.3 Hz, 2H), 4.99 (s, 2H), 3.96 (s, 2H), 2.34 (s, 3H), 1.27 (s, 9H); 13C {1H} NMR (125 MHz, CDCl3) δ = 170.8, 144.4, 141.1, 139.1, 131.8, 129.0, 126.1, 118.8, 112.8, 87.2, 81.7, 53.6, 46.0, 33.8, 31.5, 21.4; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C22H26NO2 336.1958; found 336.1969.
3-(4-iso-Propylphenyl)prop-2-yn-1-yl(4-(tert-butyl)phenyl)glycinate (1n).
Compound 1n was isolated in 76% yield (93 mg, off white solid); mp = 86.5–88.5 °C; Rf = 0.55 (VPE/VEA = 90/10); 1H NMR (500 MHz, CDCl3) δ = 7.38 (d, J = 8.2 Hz, 2H), 7.21 (d, J = 8.6 Hz, 2H), 7.17 (d, J = 8.1 Hz, 2H), 6.58 (d, J = 8.6 Hz, 2H), 4.99 (s, 2H), 3.96 (s, 2H), 3.86 (bs, 1H), 2.89 (sept, J = 6.9 Hz, 1H), 1.26 (s, 9H), 1.23 (d, J = 6.9 Hz, 6H); 13C {1H} NMR (125 MHz, CDCl3) δ = 170.8, 149.9, 144.4, 141.1, 131.9, 126.4, 126.1, 119.2, 112.8, 87.2, 81.6, 53.6, 46.0, 34.0, 33.8, 31.4, 23.7; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C24H30NO2 364.2271; found 364.2272.
3-(4-Methoxyphenyl)prop-2-yn-1-yl(4-(tert-butyl)phenyl)glycinate (1o).
Compound 1o was isolated in 75% yield (93 mg, viscous liquid); Rf = 0.45 (VPE/VEA = 90/10); 1H NMR (400 MHz, CDCl3) δ = 7.49 (d, J = 7.7 Hz, 2H), 7.29 (d, J = 7.5 Hz, 2H), 6.93 (d, J = 7.9 Hz, 2H), 6.67 (d, J = 8.1 Hz, 2H), 5.08 (s, 2H), 4.31 (bs, 1H), 4.05 (s, 2H), 3.86 (s, 3H), 1.38 (s, 9H); 13C {1H} NMR (100 MHz, CDCl3) δ = 170.7, 159.9, 144.4, 140.9, 133.3, 125.9, 114.7, 113.8, 112.7, 86.9, 81.0, 55.1, 53.5, 45.8, 33.7, 31.4; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C22H26NO3 352.1907; found 352.1906.
3-(4-Fluorophenyl)prop-2-yn-1-yl(4-(tert-butyl)phenyl)glycinate (1p).
Compound 1p was isolated in 72% yield (90 mg, off white solid); mp = 78.5–80.5 °C; Rf = 0.50 (VPE/VEA = 90/10); 1H NMR (500 MHz, CDCl3) δ = 7.42 (dd, J = 8.7, 5.4 Hz, 2H), 7.21 (d, J = 8.6 Hz, 2H), 7.00 (t, J = 8.7 Hz, 2H), 6.58 (d, J = 8.6 Hz, 2H), 4.98 (s, 2H), 3.97 (s, 2H), 1.27 (s, 9H); 13C {1H} NMR (125 MHz, CDCl3) δ = 170.8, 162.8 (d, J = 250.2 Hz), 144.4, 141.2, 133.9 (d, J = 8.5 Hz), 126.1, 117.9 (d, J = 3.4 Hz), 115.6 (d, J = 22.2 Hz), 112.8, 85.9, 82.1, 53.3, 45.9, 33.8, 31.4; 19F NMR (376 MHz, CDCl3) δ = −109.8; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C21H23FNO2 340.1707; found 340.1709.
3-(4-Chlorophenyl)prop-2-yn-1-yl(4-(tert-butyl)phenyl)glycinate (1q).
Compound 1q was isolated in 72% yield (89 mg, off white solid); mp = 76–78 °C; Rf = 0.52 (VPE/VEA = 90/10); 1H NMR (400 MHz, CDCl3) δ = 7.39 (d, J = 8.6 Hz, 2H), 7.30 (d, J = 8.5 Hz, 2H), 7.23 (d, J = 8.7 Hz, 2H), 6.59 (d, J = 8.7 Hz, 2H), 5.00 (s, 2H), 4.00 (s, 2H), 1.28 (s, 9H); 13C {1H} NMR (100 MHz, CDCl3) δ = 170.8, 144.4, 141.2, 135.0, 133.1, 128.7, 126.1, 120.4, 112.8, 85.8, 83.3, 53.3, 46.0, 33.9, 31.5; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C21H23ClNO2 356.1412; found 356.1405.
3-(4-Nitrophenyl)prop-2-yn-1-yl(4-(tert-butyl)phenyl)glycinate (1r).
Compound 1r was isolated in 70% yield (86 mg, yellow solid); mp = 76.5–78.5 °C; Rf = 0.45 (VPE/VEA = 90/10); 1H NMR (400 MHz, CDCl3) δ = 8.17 (d, J = 8.6 Hz, 2H), 7.57 (d, J = 8.6 Hz, 2H), 7.22 (d, J = 8.4 Hz, 2H), 6.58 (d, J = 8.4 Hz, 2H), 5.02 (s, 2H), 4.00 (s, 2H), 1.27 (s, 9H); 13C {1H} NMR (100 MHz, CDCl3) δ = 170.7, 147.4, 144.3, 141.2, 132.5, 128.7, 126.1, 123.5, 112.8, 87.6, 84.8, 52.9, 45.9, 33.8, 31.4; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C21H23N2O4 367.1652; found 367.1658.
3-(3-Chloro-4-methylphenyl)prop-2-yn-1-yl(4-(tert-butyl)phenyl)glycinate (1s).
Compound 1s was isolated in 68% yield (83 mg, off white solid); mp = 66.5–68.5 °C; Rf = 0.55 (VPE/VEA = 90/10); 1H NMR (500 MHz, CDCl3) δ = 7.47 (d, J = 1.0 Hz, 1H), 7.29–7.24 (m, 3H), 7.20 (d, J = 7.8 Hz, 1H), 6.62 (d, J = 8.6 Hz, 2H), 5.02 (s, 2H), 4.01 (s, 2H), 3.46 (bs, 1H), 2.40 (s, 3H), 1.30 (s, 9H); 13C {1H} NMR (125 MHz, CDCl3) δ = 170.8, 144.4, 141.2, 137.3, 134.2, 132.2, 130.8, 130.0, 126.1, 120.9, 112.8, 85.7, 82.8, 53.4, 46.0, 33.9, 31.5, 20.1; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C22H25ClNO2 370.1568; found 370.1570.
3-(3,5-Dimethylphenyl)prop-2-yn-1-yl(4-(tert-butyl)phenyl)glycinate (1t).
Compound 1t was isolated in 69% yield (86 mg, off white solid); mp = 89–91 °C; Rf = 0.55 (VPE/VEA = 90/10); 1H NMR (400 MHz, CDCl3) δ = 7.21 (d, J = 8.2 Hz, 2H), 7.08 (s, 2H), 6.96 (s, 1H), 6.57 (d, J = 8.1 Hz, 2H), 4.97 (s, 2H), 3.95 (s, 2H), 2.27 (s, 6H), 1.26 (s, 9H); 13C {1H} NMR (100 MHz, CDCl3) δ = 170.7, 144.4, 141.0, 137.8, 130.7, 129.5, 126.0, 121.5, 112.8, 87.3, 81.6, 53.5, 45.9, 33.8, 31.4, 20.9; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C23H28NO2 350.2115; found 350.2109.
3-(2-Methoxyphenyl)prop-2-yn-1-yl(4-(tert-butyl)phenyl)glycinate (1u).
Compound 1u was isolated in 69% yield (86 mg, yellow solid); mp = 96–98 °C; Rf = 0.45 (VPE/VEA = 90/10); 1H NMR (400 MHz, CDCl3) δ = 7.42 (d, J = 7.5 Hz, 1H), 7.30 (t, J = 7.9 Hz, 1H), 7.21 (d, J = 8.4 Hz, 2H), 6.88 (dd, J = 7.5 Hz, 8.5 Hz, 2H), 6.57 (d, J = 8.3 Hz, 2H), 5.05 (s, 2H), 4.18 (bs, 1H), 3.96 (s, 2H), 3.86 (s, 3H), 1.27 (s, 9H); 13C {1H} NMR (100 MHz, CDCl3) δ = 170.7, 160.2, 144.4, 140.9, 133.9, 130.4, 126.0, 120.4, 112.7, 110.9, 110.6, 86.2, 83.4, 55.7, 53.7, 45.9, 33.8, 31.4; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C22H26NO3 352.1907; found 352.1899.
3-(Thiophen-2-yl)prop-2-yn-1-yl(4-iso-propylphenyl)glycinate (1v).
Compound 1v was isolated in 69% yield (84 mg, viscous liquid); Rf = 0.50 (VPE/VEA = 95/5); 1H NMR (500 MHz, CDCl3) δ = 7.28 (d, J = 5.1 Hz, 1H), 7.25 (d, J = 3.8 Hz, 1H), 7.06 (d, J = 8.4 Hz, 2H), 6.97 (dd, J = 4.9, 3.9 Hz, 1H), 6.57 (d, J = 8.4 Hz, 2H), 5.01 (s, 2H), 3.97 (s, 2H), 3.54 (bs, 1H), 2.80 (sept, J = 6.9 Hz, 1H), 1.20 (d, J = 6.9 Hz, 6H); 13C {1H} NMR (125 MHz, CDCl3) δ = 170.7, 144.8, 138.9, 133.1, 128.0, 127.2, 126.9, 121.8, 113.2, 86.4, 80.4, 53.5, 46.1, 33.1, 24.1; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C18H20NO2S 314.1209; found 314.1210.
But-2-yn-1-yl p-tolylglycinate (1w).
Compound 1w was isolated in 60% yield (94 mg, viscous liquid); Rf = 0.5 (VPE/VEA = 90/10); 1H NMR (500 MHz, CDCl3) δ = 6.99 (d, J = 8.1 Hz, 2H), 6.52 (d, J = 8.3 Hz, 2H), 4.72 (s, 2H), 4.09 (bs, 1H), 3.91 (s, 2H), 2.23 (s, 3H), 1.85 (s, 3H); 13C {1H} NMR (125 MHz, CDCl3) δ = 170.7, 144.5, 129.7, 127.4, 113.1, 83.7, 72.6, 53.4, 45.9, 20.3, 3.5; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C13H16NO2 218.1178.; found 218.1176.
N-Phenyl-N-(3-phenylprop-2-yn-1-yl)-2-(p-tolylamino)acetamide (3a).
Compound 3a was isolated in 73% yield (79 mg, off white solid); mp = 94.2–96.2 °C; Rf = 0.40 (VPE/VEA = 70/30); 1H NMR (500 MHz, CDCl3) δ = 7.55–7.47 (m, 3H), 7.38 (d, J = 7.0 Hz, 2H), 7.35 (d, J = 7.3 Hz, 2H), 7.32–7.26 (m, 3H), 6.93 (d, J = 8.0 Hz, 2H), 6.39 (d, J = 8.0 Hz, 2H), 4.77 (s, 2H), 4.59 (bs, 1H), 3.58 (s, 2H), 2.21 (s, 3H); 13C {1H} NMR (125 MHz, CDCl3) δ = 169.3, 145.1, 140.0, 131.6, 129.9, 129.6, 129.1, 128.4, 128.3, 128.2, 126.9, 122.6, 113.1, 84.5, 83.9, 46.6, 39.4, 20.3; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C24H23N2O 355.1805; found 355.1807.
2-((4-Methoxyphenyl)amino)-N-phenyl-N-(3-phenylprop-2-yn-1-yl)acetamide (3b).
Compound 3b was isolated in 41% yield (46 mg, brown liquid); Rf = 0.35 (VPE/VEA = 70/30); 1H NMR (400 MHz, CDCl3, starting material was not stable) δ = 7.49 (d, J = 7.3 Hz, 2H), 7.39–7.33 (m, 5H), 7.32–7.25 (m, 3H), 6.71 (d, J = 8.8 Hz, 2H), 6.43 (d, J = 8.8 Hz, 2H), 4.75 (s, 2H), 3.69 (s, 3H), 3.55 (s, 2H); 13C {1H} NMR (100 MHz, CDCl3) δ = 169.4, 152.3, 141.6, 139.9, 131.7, 131.6, 129.9, 129.1, 128.8, 128.4, 128.3, 128.2, 114.7, 114.3, 84.4, 83.9, 55.7, 47.1, 39.3; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C24H23N2O2 371.1754; found 371.1755.
N-Benzyl-2-(phenylamino)-N-(3-phenylprop-2-yn-1-yl)acetamide (3c)17,18.
Compound 3c was isolated in 76% yield (79 mg, off white solid); mp = 95.5–97.5 °C; Rf = 0.40 (VPE/VEA = 70/30); 1H NMR (500 MHz, CDCl3, rotameric mixture found in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.98 ratio) δ = 7.44–7.39 (rotameric m, 6H, aromatic) 7.34 (rotameric m, 14H, aromatic), 7.21 (rotameric quin, 4H, aromatic), 6.79–6.73 (rotameric m, 2H, aromatic), 6.71 (d, J = 8.0 Hz, 2H, aromatic) and 6.61 (d, J = 7.9 Hz, 2H, aromatic) (rotameric), 4.93 (rotameric bs, 2H, NH), 4.84 (s, 2H, CH2) and 4.76 (s, 2H, CH2) (rotameric), 4.56 (s, 2H, CH2) and 4.00 (s, 2H, CH2) (rotameric), 4.21 (s, 2H, CH2) and 4.16 (s, 2H, CH2) (rotameric); 13C {1H} NMR (125 MHz, CDCl3) δ = 169.5 and 162.4 (rotameric C
:0.98 ratio) δ = 7.44–7.39 (rotameric m, 6H, aromatic) 7.34 (rotameric m, 14H, aromatic), 7.21 (rotameric quin, 4H, aromatic), 6.79–6.73 (rotameric m, 2H, aromatic), 6.71 (d, J = 8.0 Hz, 2H, aromatic) and 6.61 (d, J = 7.9 Hz, 2H, aromatic) (rotameric), 4.93 (rotameric bs, 2H, NH), 4.84 (s, 2H, CH2) and 4.76 (s, 2H, CH2) (rotameric), 4.56 (s, 2H, CH2) and 4.00 (s, 2H, CH2) (rotameric), 4.21 (s, 2H, CH2) and 4.16 (s, 2H, CH2) (rotameric); 13C {1H} NMR (125 MHz, CDCl3) δ = 169.5 and 162.4 (rotameric C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e001.gif) O), 147.4 and 147.3 (rotameric), 136.5 and 135.6 (rotameric), 131.9 (rotameric), 129.4 and 129.4 (rotameric), 129.2 and 128.9 (rotameric), 128.8 and 128.6 (rotameric), 128.5 and 128.4 (rotameric), 128.2 and 127.9 (rotameric), 126.8 (rotameric), 122.6 and 122.0 (rotameric), 117.8 (rotameric), 113.2 and 113.1 (rotameric), 85.2 and 84.5 (rotameric Calkyne), 83.7 and 82.7 (rotameric Calkyne), 49.4 and 49.3 (rotameric CH2), 45.6 and 45.5 (rotameric CH2), 36.4 and 35.7 (rotameric CH2); HRMS (ESI-TOF) m/z: [M + H]+ calcd for C24H23N2O; 355.1805 found 355.1798.
O), 147.4 and 147.3 (rotameric), 136.5 and 135.6 (rotameric), 131.9 (rotameric), 129.4 and 129.4 (rotameric), 129.2 and 128.9 (rotameric), 128.8 and 128.6 (rotameric), 128.5 and 128.4 (rotameric), 128.2 and 127.9 (rotameric), 126.8 (rotameric), 122.6 and 122.0 (rotameric), 117.8 (rotameric), 113.2 and 113.1 (rotameric), 85.2 and 84.5 (rotameric Calkyne), 83.7 and 82.7 (rotameric Calkyne), 49.4 and 49.3 (rotameric CH2), 45.6 and 45.5 (rotameric CH2), 36.4 and 35.7 (rotameric CH2); HRMS (ESI-TOF) m/z: [M + H]+ calcd for C24H23N2O; 355.1805 found 355.1798.
N-Benzyl-N-(3-phenylprop-2-yn-1-yl)-2-(p-tolylamino)acetamide (3d).
Compound 3d was isolated in 74% yield (80 mg, off white solid); mp = 93–95 °C; Rf = 0.40 (VPE/VEA = 70/30); 1H NMR (500 MHz, CDCl3, rotameric mixture found in 1:0.98 ratio) δ = 7.43 (rotameric m, 6H, aromatic), 7.35 (rotameric m, 14H, aromatic), 7.06 (d, J = 8.0 Hz, 2H, aromatic) and 7.02 (d, J = 8.0 Hz, 2H, aromatic) (rotameric), 6.66 (d, J = 8.1 Hz, 2H, aromatic) and 6.56 (d, J = 8.1 Hz, 2H, aromatic) (rotameric), 4.85 (s, 2H, CH2) and 4.76 (s, 2H, CH2) (rotameric), 4.57 (s, 2H, CH2) and 4.00 (s, 2H, CH2) (rotameric) 4.21 (s, 2H, CH2) and 4.15 (s, 2H, CH2) (rotameric), 2.29 (s, 3H, 4-CH3 aniline) and 2.27 (s, 3H, 4-CH3 aniline) (rotameric); 13C {1H} NMR (125 MHz, CDCl3 rotamers) δ = 169.5 and 169.4 (rotameric CO), 145.1 and 145.0 (rotameric) 136.4 and 135.5 (rotameric), 131.7, 129.7 and 129.6 (rotameric), 129.0 and 128.7 (rotameric), 128.6 and 128.4 (rotameric), 128.3 and 128.2 (rotameric), 127.9 and 127.7 (rotameric), 126.8 and 128.6 (rotameric), 122.4 and 121.9 (rotameric), 113.1, 84.9 and 84.2 (rotameric Calkyne), 83.6 and 82.6 (rotameric Calkyne), 49.2 and 49.1 (rotameric CH2), 45.8 and 45.7 (rotameric CH2), 36.2 and 35.5 (rotameric CH2), 20.3; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C25H25N2O 369.1961; found 369.1958.
N-Benzyl-2-((4-methoxyphenyl)amino)-N-(3-phenylprop-2-yn-1-yl)acetamide (3e).
Compound 3e crude (note: crude was recrystallize in n-hexane) mp = 79–81 °C; Rf = 0.35 (VPE/VEA = 70/30); 1H NMR (400 MHz, CDCl3, rotameric mixture found in 1:0.97 ratio) δ = 7.45–7.26 (m, 20H, aromatic) (rotameric), 6.85–6.72 (m, 4H, aromatic) (rotameric), 6.66 (d, J = 8.6 Hz, 2H, aromatic) and 6.56 (d, J = 8.5 Hz 2H, aromatic) (rotameric), 4.80 (s, 2H, CH2) and 4.74 (s, 2H, CH2) (rotameric), 4.53 (s, 2H, CH2) and 3.95 (s, 2H, CH2), (rotameric), 4.19 (s, 2H, CH2) and 4.11 (s, 2H, CH2) (rotameric), 3.75 (s, 3H, OMe) and 3.73 (s, 3H, OMe) (rotameric); 13C {1H} NMR (100 MHz, CDCl3, rotamers) δ = 169.7 and 169.6 (rotameric CO), 152.3 and 152.2 (rotameric), 141.7 and 141.6 (rotameric), 136.4 and 135.5 (rotameric), 131.7 (rotameric), 129.1 and 128.3 (rotameric), 128.8 and 128.7 (rotameric), 128.5 and 128.4 (rotameric), 128.0 and 127.8 (rotameric), 126.7 (rotameric), 122.5 and 121.9 (rotameric), 114.9 and 114.9 (rotameric), 114.4 (rotameric), 84.9 and 84.3 (rotameric Calkyne), 83.6 and 82.6 (rotameric Calkyne), 55.8 (OMe) 49.2 and 49.1 (rotameric CH2), 46.5 and 46.4 (rotameric CH2), 36.3 and 35.5 (rotameric CH2); HRMS (ESI-TOF) m/z: [M + H]+ calcd for C25H25N2O2 385.1911; found 385.1911.
N-Benzyl-2-((4-chlorophenyl)amino)-N-(3-phenylprop-2-yn-1-yl)acetamide (3f).
Compound 3f was isolated in 74% yield (42 mg, brown solid); mp = 91–93 °C; Rf = 0.40 (VPE/VEA = 70/30); 1H NMR (400 MHz, CDCl3, rotameric mixture found in 1:0.98 ratio) δ = 7.40–7.26 (m, 20H, aromatic) (rotameric), 7.11 (dd, J = 11.5, 8.6 Hz, 4H, aromatic) (rotameric), 6.59 (d, J = 8.3 Hz, 2H) and 6.48 (d, J = 8.4 Hz, 2H, aromatic) (rotameric), 4.80 (s, 2H, CH2) and 4.73 (s, 2H, CH2) (rotameric), 4.53 (s, 2H, CH2) and 3.93 (s, 2H, CH2) (rotameric), 4.17 (s, 2H, CH2) and 4.09 (s, 2H, CH2) (rotameric); 13C {1H} NMR (100 MHz, CDCl3, rotamers) δ = 169.0 and 168.8 (rotameric CO), 145.8 and 145.7 (rotameric), 136.2 and 135.3 (rotameric), 131.7 (rotameric), 129.1 (rotameric) 129.1 and 129.0 (rotameric), 128.8 and 128.7 (rotameric), 128.5 and 128.3 (rotameric), 128.4 and 128.3 (rotameric), 128.1 and 127.8 (rotameric), 126.6 (rotameric), 122.4 and 122.2 (rotameric), 114.0 and 114.0 (rotameric), 85.1 and 84.5 (rotameric Calkyne), 83.4 and 82.4 (rotameric Calkyne), 49.2 (rotameric CH2), 45.4 (rotameric CH2), 36.3 and 35.6 (rotameric CH2); HRMS (ESI-TOF) m/z: [M + H]+ calcd for C24H22ClN2O 389.1415; found 389.1422.
N-Benzyl-2-((2,5-dichlorophenyl)amino)-N-(3-phenylprop-2-yn-1-yl)acetamide (3g).
Compound 3g was isolated in 61% yield (76 mg, viscous liquid); Rf = 0.40 (VPE/VEA = 70/30); 1H NMR (400 MHz, CDCl3, rotameric mixture found in 1:0.97 ratio) δ = 7.49–7.28 (m, 20H, aromatic) (rotameric), 7.18 (t, J = 7.9 Hz, 2H, aromatic), (rotameric), 6.62 (t, J = 8.1 Hz, 2H, aromatic) (rotameric), 6.58 (s, 1H, aromatic) and 6.41 (s, 1H, aromatic) (rotameric), 5.72 (bs, 2H, NH) (rotameric), 4.83 (s, 2H, CH2) and 4.76 (s, 2H, CH2) (rotameric), 4.56 (s, 2H, CH2) and 4.20 (s, 2H, CH2) (rotameric), 4.13 (s, 2H, CH2) and 3.98 (s, 2H, CH2) (rotameric); 13C {1H} NMR (100 MHz, CDCl3, rotamers) δ = 168.4 and 168.3 (rotameric CO), 144.0, 136.3 and 135.3 (rotameric), 133.6 and 133.5 (rotameric), 131.9, 130.1 and 129.4 (rotameric), 129.0, 128.9 and 128.7 (rotameric), 128.6 and 128.4 (rotameric), 128.3 and 128.1 (rotameric), 126.8, 122.5 and 121.9 (rotameric), 118.0, 117.3, 111.3 and 111.2 (rotameric), 85.5 and 84.6 (rotameric Calkyne), 83.4 and 82.4 (rotameric Calkyne), 49.5 and 49.4 (rotameric CH2), 45.0, 36.5 and 35.9 (rotameric CH2); HRMS (ESI-TOF) m/z: [M + H]+ calcd for C24H21Cl2 N2O (M + H)+ 423.1025; found 423.1016.
N-Benzyl-2-((4-methoxyphenyl)amino)-N-(3-(p-tolyl)prop-2-yn-1-yl)acetamide (3h).
Compound 3i was isolated in 68% yield (76 mg, off white solid); mp = 115.5–117.5 °C; Rf = 0.35 (VPE/VEA = 70/30); 1H NMR (500 MHz, CDCl3, rotameric mixture found in 1:0.98 ratio) δ = 7.41–7.31 (m, 14H aromatic) (rotameric), 7.15 (d, J = 7.9 Hz, 4H, aromatic) (rotameric), 6.81 (dd, J = 9.0, 8.1 Hz, 4H, aromatic) (rotameric), 6.69 (d, J = 7.8 Hz, 2H, aromatic) and 6.58 (d, J = 7.8 Hz, 2H, aromatic) (rotameric), 4.83 (s, 2H, CH2) and 4.75 (s, 2H, CH2) (rotameric), 4.63 (bs, 2H, NH) (rotameric), 4.55 (s, 2H, CH2) and 3.96 (s, 2H, CH2) (rotameric), 4.19 (s, 2H, CH2) and 4.12 (s, 2H, CH2) (rotameric), 3.76 (s, 3H, OCH3) and 3.75 (s, 3H, OCH3) (rotameric), 2.37 (s, 3H, 4-MePh) and 2.36, (s, 3H, 4-MePh), (rotameric); 13C {1H} NMR (125 MHz, CDCl3 rotamers) δ = 169.6 and 169.5 (rotameric CO), 152.2, 141.7 and 141.6 (rotameric), 138.9 and 138.5 (rotameric), 136.4 and 135.5 (rotameric), 131.5, 129.0 and 128.9 (rotameric), 128.6 and 128.3 (rotameric), 127.8 and 127.6 (rotameric), 126.6, 119.3 and 118.8 (rotameric), 114.8 and 114.8 (rotameric), 114.2, 85.0 and 84.3 (rotameric Calkyne), 82.8 and 81.9 (rotameric Calkyne), 55.6, 49.1 and 49.0 (rotameric CH2), 46.4 and 46.3 (rotameric CH2), 36.3 and 35.5 (rotameric CH2), 21.3; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C26H27N2O2 399.2067; found 399.2065.
N-Benzyl-2-((4-chlorophenyl)amino)-N-(3-(p-tolyl)prop-2-yn-1-yl)acetamide (3i).
Compound 3j was isolated in 71% yield (80 mg, off white solid); mp = 96–98 °C; Rf = 0.35 (VPE/VEA = 70/30); 1H NMR (400 MHz, CDCl3, rotameric mixture found in 1:0.9 ratio) δ = 1H NMR (400 MHz, DMSO) δ 7.44–7.28 (m, 14H, aromatic) (rotameric), 7.13 (t, J = 8.5 Hz, 8H, aromatic) (rotameric), 6.61 (d, J = 8.5 Hz, 2H, aromatic) and 6.50 (d, J = 8.5 Hz, 2H, aromatic) (rotameric), 4.96 (bs, 2H, NH) (rotameric), 4.82 (s, 2H, CH2) and 4.74 (s, 2H, CH2) (rotameric), 4.54 (s, 2H, CH2) and 3.93 (s, 2H, CH2) (rotameric), 4.18 (s, 2H, CH2) and 4.10 (s, 2H, CH2) (rotameric), 2.36 (s, 6H, NHCH2 4-MePh) (rotameric); 13C {1H} NMR (100 MHz, CDCl3, rotamers) δ = 169.0 and 168.9 (rotameric CO), 145.8 and 145.7 (rotameric), 139.0 and 138.6 (rotameric), 136.3 and 135.4 (rotameric), 131.6, 129.1 and 129.0 (rotameric), 128.7 and 128.4 (rotameric), 128.0 and 127.8 (rotameric), 126.6, 122.2, 119.3 and 118.7 (rotameric), 114.0, 85.3 and 84.5 (rotameric Calkyne), 82.6 and 81.7 (rotameric Calkyne), 49.2, 45.4 and 45.3 (rotameric CH2), 36.3 and 35.7 (rotameric CH2), 21.4; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C25H24ClN2O 403.1572; found 403.1570.
N-Benzyl-N-(3-(p-tolyl)prop-2-yn-1-yl)-2-(p-tolylamino)acetamide (3j).
Compound 3h was isolated in 77% yield (83 mg, off white solid); mp = 106–108 °C; Rf = 0.35 (VPE/VEA = 70/30); 1H NMR (400 MHz, CDCl3, rotameric mixture found in 1:0.97 ratio) δ = 7.42–7.29 (m, 14H, aromatic) (rotameric), 7.14 (t, J = 7.1 Hz, 4H, aromatic) (rotameric), 7.03 (dd, J = 12.4, 8.1 Hz, 4H, aromatic) (rotameric), 6.65 (d, J = 7.9 Hz, 2H, aromatic) and 6.54 (d, J = 7.9 Hz, 2H, aromatic) (rotameric), 4.83 (s, 2H, CH2) and 4.76 (s, 2H, CH2) (rotameric), 4.55 (s, 2H, CH2) and 3.98 (s, 2H, CH2) (rotameric), 4.20 (s, 2H, CH2) and 4.14 (s, 2H, CH2) (rotameric), 2.38 (s, 3H, NHCH2 4-MePh) and 2.37 (s, 3H, NHCH2 4-MePh) (rotameric), 2.28 (s, 3H, 4-Me aniline) and 2.26 (s, 3H, 4-Me aniline) (rotameric); 13C {1H} NMR (100 MHz, CDCl3 rotamers) δ = 169.5 and 169.4 (rotameric C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 145.1 and 145.0 (rotameric), 138.9 and 138.5 (rotameric), 136.4 and 135.5 (rotameric), 131.6, 129.7 and 129.7 (rotameric), 129.1 and 129.0 (rotameric), 128.9, 128.6 and 128.4 (rotameric), 127.9 and 128.4 (rotameric), 126.8 and 126.7 (rotameric), 119.3 and 118.8 (rotameric), 113.2, 85.1 and 84.4 (rotameric Calkyne), 82.8 and 81.9 (rotameric Calkyne), 49.1 and 49.0 (rotameric CH2), 45.8 and 45.8 (rotameric CH2), 36.3 and 35.5 (rotameric CH2), 21.4 and 20.3 (rotameric, N–CH2, 4-PhCH3); HRMS (ESI-TOF) m/z: [M + H]+ calcd for C26H27N2O 383.2118; found 383.2115.
O), 145.1 and 145.0 (rotameric), 138.9 and 138.5 (rotameric), 136.4 and 135.5 (rotameric), 131.6, 129.7 and 129.7 (rotameric), 129.1 and 129.0 (rotameric), 128.9, 128.6 and 128.4 (rotameric), 127.9 and 128.4 (rotameric), 126.8 and 126.7 (rotameric), 119.3 and 118.8 (rotameric), 113.2, 85.1 and 84.4 (rotameric Calkyne), 82.8 and 81.9 (rotameric Calkyne), 49.1 and 49.0 (rotameric CH2), 45.8 and 45.8 (rotameric CH2), 36.3 and 35.5 (rotameric CH2), 21.4 and 20.3 (rotameric, N–CH2, 4-PhCH3); HRMS (ESI-TOF) m/z: [M + H]+ calcd for C26H27N2O 383.2118; found 383.2115.
N-Benzyl-N-(3-(4-methoxyphenyl)prop-2-yn-1-yl)-2-(p-tolylamino)acetamide (3k).
Compound 3k was isolated in 71% yield (76 mg, viscous liquid); Rf = 0.35 (VPE/VEA = 60/40); 1H NMR (400 MHz, CDCl3, rotameric mixture found in 1:0.96 ratio) δ = 7.39–7.24 (m, 14H, aromatic) (rotameric), 6.98 (dd, J = 13.3, 8.1 Hz, 4H, aromatic) (rotameric), 6.80 (dd, J = 7.9, 6.0 Hz, 4H, aromatic) (rotameric), 6.60 (d, J = 8.0 Hz, 2H, aromatic) and 6.49 (d, J = 8.0 Hz, 2H, aromatic) (rotameric), 4.78 (s, 2H, CH2) and 4.70 (s, 2H, CH2) (rotameric), 4.50 (s, 2H, CH2) and 3.93 (s, 2H, CH2) (rotameric), 4.13 (s, 2H, CH2) and 4.09 (s, 2H, CH2) (rotameric), 3.76 (s, 6H, OCH3) (rotameric), 2.23 (s, 3H, PhCH3) and 2.21 (s, 3H, PhCH3) (rotameric); 13C {1H} NMR (100 MHz, CDCl3, rotamers) δ = 169.5 and 169.3 (rotameric), 159.8 and 159.6 (rotameric), 145.0 and 144.9 (rotameric), 136.4 and 135.5 (rotameric), 133.1, 129.7 and 129.6 (rotameric), 128.9 and 128.7 (rotameric), 128.6 and 128.3 (rotameric), 127.8 and 127.6 (rotameric), 126.7 and 126.6 (rotameric), 114.4 and 114.3 (rotameric), 113.9 and 113.8 (rotameric), 113.2 and 113.1 (rotameric), 84.8 and 84.1 (rotameric Calkyne), 82.0 and 81.2 (rotameric Calkyne), 55.14 (rotameric OCH3), 49.1 and 49.0 (rotameric CH2), 45.8 and 45.7 (rotameric CH2), 36.3 and 35.5 (rotameric CH2), 20.3; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C26H27N2O2 (M + H)+ 399.2067; found 399.2075.
N-Benzyl-N-(3-(4-chlorophenyl)prop-2-yn-1-yl)-2-(p-tolylamino)acetamide (3l).
Compound 3l was isolated in 73% yield (81 mg, viscous liquid); Rf = 0.40 (VPE/VEA = 70/30); 1H NMR (400 MHz, CDCl3, rotameric mixture found in 1:0.88 ratio) δ = 7.42–7.21 (m, 18H, aromatic) (rotameric), 6.99 (dd, J = 12.0, 8.2 Hz, 4H, aromatic) (rotameric), 6.60 (d, J = 8.1 Hz, 2H, aromatic) and 6.51 (d, J = 8.1 Hz, 2H, aromatic) (rotameric), 4.78 (s, 2H, CH2) and 4.71 (s, 2H, CH2) (rotameric), 4.50 (s, 2H, CH2) and 3.96 (s, 2H, CH2) (rotameric), 4.16 (s, 2H, CH2) and 4.09 (s, 2H, CH2) (rotameric), 2.24 (s, 3H, NHPhCH3) and 2.22 (s, 3H, NHPhCH3) (rotameric); 13C {1H} NMR (100 MHz, CDCl3, rotamers) δ = 169.5, 145.0 and 144.9 (rotameric), 136.3 and 135.4 (rotameric), 134.8 and 134.4 (rotameric), 132.9, 129.7 and 129.6 (rotameric), 129.0, 128.7, 128.6 and 128.4 (rotameric), 128.0 and 127.7 (rotameric), 126.9 and 126.6 (rotameric), 120.9 and 120.3 (rotameric), 113.16, 84.7 and 83.0 (rotameric Calkyne), 83.8 and 83.7 (rotameric Calkyne), 49.3 and 49.2 (rotameric CH2), 45.81, 36.2 and 35.5 (rotameric CH2), 20.3; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C25H24ClN2O (M + H)+ 403.1572; found 403.1584.
N-Benzyl-N-(3-(4-nitrophenyl)prop-2-yn-1-yl)-2-(p-tolylamino)acetamide (3m).
Compound 3m was isolated in 65% yield (69 mg, viscous liquid); Rf = 0.40 (VPE/VEA = 70/30); 1H NMR (500 MHz, CDCl3, rotameric mixture found in 1:0.86 ratio) δ = 8.20 (m, 4H, aromatic) (rotameric), 7.52 (d, J = 7.5 Hz, 4H, aromatic) (rotameric), 7.46–7.29 (m, 10H, aromatic) (rotameric), 7.08–6.99 (m, 4H, aromatic) (rotameric), 6.65 (d, J = 6.8 Hz, 2H, aromatic) and 6.56 (d, J = 7.4 Hz, 2H, aromatic) (rotameric), 4.84 (s, 2H, CH2) and 4.77 (s, 2H, CH2) (rotameric), 4.58 (s, 2H, CH2) and 4.28 (s, 2H, CH2), (rotameric), 4.15 (s, 2H, CH2) and 4.03 (s, 2H, CH2) (rotameric), 2.27 (s, 6H, NHPhCH3) (rotameric). 13C {1H} NMR (100 MHz, CDCl3, rotamers) δ = 169.7 and 169.5 (rotameric CO), 147.4 and 147.2 (rotameric), 145.0 and 144.9 (rotameric), 136.1 and 135.2 (rotameric), 132.5 and 132.4 (rotameric), 129.8 and 129.7 (rotameric), 129.3 and 129.1 (rotameric), 128.8 and 128.4 (rotameric), 128.2 and 127.9 (rotameric), 127.2 and 127.1 (rotameric), 126.8 and 126.7 (rotameric), 123.6 and 123.5 (rotameric), 113.2 (rotameric), 89.3 and 88.1 (rotameric Calkyne), 83.1 and 82.3 (rotameric Calkyne), 49.7 and 49.1 (rotameric CH2), 45.9 and 45.8 (rotameric CH2), 36.3 and 35.7 (rotameric CH2), 20.3; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C25H24N3O3 (M + H)+ 414.1812; found 414.1817.
N-Benzyl-N-(3-(thiophen-2-yl)prop-2-yn-1-yl)-2-(p-tolylamino)acetamide (3n).
Compound 3n was isolated in 74% yield (79 mg, viscous liquid); Rf = 0.40 (VPE/VEA = 70/30); 1H NMR (500 MHz, CDCl3, rotameric mixture found in 1:0.98 ratio) δ = 7.36–7.26 (m, 8H) (rotameric), 7.20 (dd, J = 13.2, 6.2 Hz, 3H, aromatic) (rotameric), 7.15 (dd, J = 9.0, 4.6 Hz, 3H, aromatic) (rotameric), 6.98 (d, J = 7.9 Hz, 2H, aromatic) and 6.94 (d, J = 7.9 Hz, 2H, aromatic) (rotameric), 6.92–6.88 (m, 2H, aromatic) (rotameric), 6.58 (d, J = 8.0 Hz, 2H, aromatic) and 6.47 (d, J = 8.0 Hz, 2H, aromatic) (rotameric), 4.74 (s, 2H, CH2) and 4.61 (s, 2H, CH2) (rotameric), 4.47 (s, 2H, CH2) and 3.89 (s, 2H, CH2) (rotameric), 4.11 (s, 2H, CH2) and 4.03 (s, 2H, CH2) (rotameric), 2.22 (s, 3H, NHPhCH3) and 2.20 (s, 3H, NHPhCH3) (rotameric); 13C {1H} NMR (125 MHz, CDCl3, rotamers) δ = 169.7 and 169.6 (rotameric CO), 145.3 and 145.2 (rotameric), 136.5 and 135.6 (rotameric), 132.9 and 132.6 (rotameric), 129.9 and 129.9 (rotameric), 129.2 and 129.0 (rotameric), 128.9 and 128.6 (rotameric), 128.2 and 127.5 (rotameric), 127.92 and 127.84 (rotameric), 127.2 and 127.1 (rotameric), 126.9 and 126.9 (rotameric), 122.6 and 121.9 (rotameric), 113.4 and 113.3 (rotameric), 88.0 and 86.9 (rotameric Calkyne), 78.5 and 77.7 (rotameric Calkyne), 49.5 and 49.4 (rotameric CH2), 45.9, 36.6 and 35.8 (rotameric CH2), 20.6; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C23H23N2OS (M + H)+ 375.1526; found 375.1533.
N-Benzyl-2-(phenylamino)-N-(3-(trimethylsilyl)prop-2-yn-1-yl)acetamide (3o).
Compound 3k was isolated in 65% yield (64 mg, viscous liquid); Rf = 0.40 (VPE/VEA = 80/20); 1H NMR (400 MHz, CDCl3, rotameric mixture found in 1:0.96 ratio) δ = 1H NMR (400 MHz, CDCl3) δ 7.21–6.96 (m, 14H) (rotameric), 6.59–6.54 (m, 2H) (rotameric), 6.50 (d, J = 7.8 Hz, 2H) and 6.39 (d, J = 7.6 Hz, 2H) (rotameric), 4.71 (bs, 2H, NH) (rotameric), 4.57 (s, 2H, CH2) and 4.50 (s, 2H, CH2) (rotameric), 4.17 (s, 2H, CH2) and 3.88 (s, 2H, CH2) (rotameric), 3.79 (s, 2H, CH2) and 3.75 (s, 2H, CH2) (rotameric), 0.00 (s, 9H, TMS) and 0.00 (s, 9H, TMS) (rotameric); 13C {1H} NMR (100 MHz, CDCl3, rotamers) δ = 169.3 and 169.1 (rotameric CO), 147.3 and 147.2 (rotameric), 136.3 and 135.4 (rotameric), 129.2 and 129.1 (rotameric), 128.9 and 126.6 (rotameric), 128.6 and 128.4 (rotameric), 127.9 and 127.7 (rotameric), 117.6 (rotameric), 112.9 (rotameric), 99.7 and 98.8 (rotameric), 90.5 and 89.6 (rotameric), 49.1 and 49.0 (rotameric), 45.4 and 45.3 (rotameric), 36.4 and 35.7 (rotameric), −0.2 and 0.3 (rotameric TMS); HRMS (ESI-TOF) m/z: [M + H]+ calcd for C21H27N2OSi 351.1878; found 351.1895.
5-(Benzyloxy)pent-3-yn-2-yl(4-methoxyphenyl)glycinate (5).
Compound 5 was isolated in 62% yield (70 mg, viscous liquid); Rf = 0.35 (VPE/VEA = 70/30); 1H NMR (400 MHz, CDCl3) δ = 7.33–7.27 (m, 5H), 6.76 (d, J = 8.9 Hz, 2H), 6.62 (d, J = 8.9 Hz, 2H), 5.56 (q, J = 6.7 Hz, 1H), 4.73 (bs, 1H), 4.54 (s, 2H), 4.16 (d, J = 1.6 Hz, 2H), 3.87 (s, 2H), 3.68 (s, 3H), 1.50 (d, J = 6.7 Hz, 3H); 13C {1H} NMR (100 MHz, CDCl3) δ = 169.9, 152.9, 139.9, 137.0, 128.2, 127.9, 127.7, 114.9, 114.6, 84.2, 81.1, 71.4, 61.1, 56.9, 55.4, 46.9, 21.0; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C21H24NO4 354.1700; found 354.1692.
7-Methyl-9-phenylfuro[3,4-b]quinolin-3(1H)-one (2a).
Compound 2a was isolated in 88% yield (43 mg, off white solid); mp = 198–200 °C; Rf = 0.40 (VPE/VEA = 60/40); 1H NMR (400 MHz, CDCl3) δ = 8.30 (d, J = 8.7 Hz, 1H), 7.67 (dd, J = 8.7, 1.7 Hz, 1H), 7.64–7.57 (m, 4H), 7.46–7.42 (m, 2H), 5.35 (s, 2H), 2.50 (s, 3H); 13C {1H} NMR (100 MHz, CDCl3) δ = 168.9, 149.3, 143.2, 142.8, 139.9, 133.7, 133.1, 132.5, 130.9, 129.4, 129.3, 128.8, 127.9, 124.2, 67.8, 22.1; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C18H14NO2 276.1019; found 276.1016.
7-iso-Propyl-9-phenylfuro[3,4-b]quinolin-3(1H)-one (2b).
Compound 2b was isolated in 86% yield (43 mg, off white solid); mp = 197–199 °C; Rf = 0.40 (VPE/VEA = 60/40); 1H NMR (400 MHz, CDCl3) δ = 8.37 (d, J = 8.8 Hz, 1H), 7.77 (d, J = 8.8 Hz, 1H), 7.67–7.58 (m, 4H), 7.45 (d, J = 6.7 Hz, 2H), 5.37 (s, 2H), 3.05 (Sept, J = 6.9 Hz, 1H), 1.28 (d, J = 6.9 Hz, 6H); 13C {1H} NMR (100 MHz, CDCl3) δ = 168.9, 150.5, 149.7, 143.4, 143.1, 133.8, 132.4, 131.3, 130.5, 129.4, 129.3, 128.8, 127.9, 121.7, 67.8, 34.5, 23.6; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C20H18NO2 304.1332; found 304.1328.
7-(tert-Butyl)-9-phenylfuro[3,4-b]quinolin-3(1H)-one (2c).
Compound 2c was isolated in 83% yield (41 mg, off white solid); mp = 236–238 °C; Rf = 0.40 (VPE/VEA = 60/40); 1H NMR (500 MHz, CDCl3) δ = 8.33 (d, J = 8.9 Hz, 1H), 7.93 (dd, J = 8.9, 1.9 Hz, 1H), 7.82 (d, J = 1.9 Hz, 1H), 7.64–7.57 (m, 3H), 7.46 (dd, J = 5.0, 2.8 Hz, 2H), 5.37 (s, 2H), 1.32 (s, 9H); 13C {1H} NMR (125 MHz, CDCl3) δ = 168.9, 152.6, 149.2, 143.4, 133.6, 132.4, 130.7, 129.8, 129.4, 129.2, 128.8, 127.5, 120.3, 67.8, 35.3, 30.8; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C21H20NO2 318.1489; found 318.1483.
7-Hydroxy-9-phenylfuro[3,4-b]quinolin-3(1H)-one (2d).
Compound 2d was isolated in 57% yield (28 mg, off white solid); mp = 259–261 °C; Rf = 0.30 (VPE/VEA = 50/50); 1H NMR (400 MHz, CDCl3) δ = 9.73 (s, 1H), 8.16 (d, J = 9.2 Hz, 1H), 7.64 (m, 2H), 7.61–7.57 (m, 3H), 7.54 (dd, J = 9.2, 2.7 Hz, 1H), 7.17 (d, J = 2.7 Hz, 1H), 5.43 (s, 2H); 13C {1H} NMR (100 MHz, CDCl3) δ = 169.1, 158.9, 146.6, 142.4, 141.4, 134.8, 134.3, 133.3, 130.3, 129.8, 129.7, 129.6, 124.0, 106.8, 68.0; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C17H12NO3 278.0812; found 278.0809.
7-Methoxy-9-phenylfuro[3,4-b]quinolin-3(1H)-one (2e).
Compound 2e was isolated in 61% yield (30 mg, off white solid); mp = 242.5–244.5 °C; Rf = 0.35 (VPE/VEA = 60/40); 1H NMR (500 MHz, CDCl3) δ = 8.31 (d, J = 9.3 Hz, 1H), 7.61 (m, 3H), 7.49 (dd, J = 9.3, 2.6 Hz, 1H), 7.45 (d, J = 7.1 Hz, 2H), 7.10 (d, J = 2.6 Hz, 1H), 5.34 (s, 2H), 3.80 (s, 3H); 13C {1H} NMR (125 MHz, CDCl3) δ = 168.9, 160.1, 147.0, 141.8, 141.7, 133.9, 133.1, 132.8, 129.5, 129.4 (two 13C), 128.6, 123.9, 102.9, 67.7, 55.6; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C18H14NO3 292.0968; found 292.0965.
7-Phenoxy-9-phenylfuro[3,4-b]quinolin-3(1H)-one (2f).
Compound 2f was isolated in 64% yield (32 mg, off white solid); mp = 232–234 °C; Rf = 0.35 (VPE/VEA = 50/50); 1H NMR (400 MHz, CDCl3) δ = 8.39 (d, J = 9.3 Hz, 1H), 7.55 (m, 4H), 7.43–7.33 (m, 5H), 7.16 (t, J = 7.4 Hz, 1H), 7.04 (d, J = 7.9 Hz, 2H), 5.38 (s, 2H); 13C {1H} NMR (100 MHz, CDCl3) δ = 168.7, 158.0, 155.7, 147.5, 143.0, 142.4, 133.3, 133.3, 132.9, 130.0, 129.5, 129.3 (two 13C), 128.7, 124.5, 123.9, 119.6, 110.9, 67.7; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C23H16NO3 354.1125; found 354.1121.
7-Fluoro-9-phenylfuro[3,4-b]quinolin-3(1H)-one (2g).
Compound 2g was isolated in 79% yield (39 mg, off white solid); mp = 218–220 °C; Rf = 0.40 (VPE/VEA = 60/40); 1H NMR (400 MHz, CDCl3) δ = 8.40 (dd, J = 9.4, 5.6 Hz, 1H), 7.65–7.58 (m, 4H), 7.49 (dd, J = 9.8, 2.8 Hz, 1H), 7.46–7.42 (m, 2H), 5.39 (s, 2H); 13C {1H} NMR (100 MHz, CDCl3) δ = 168.4, 162.2 (d, J = 253.1 Hz), 147.7, 143.8 (d, J = 2.8 Hz), 143.3 (d, J = 6.4 Hz), 133.9 (d, J = 9.6 Hz), 132.9 (d, J = 5.9 Hz), 129.8, 129.5, 129.0 (d, J = 9.9 Hz), 128.6, 121.4 (d, J = 26.5 Hz), 109.1 (d, J = 23.6 Hz), 67.7; 19F NMR (376 MHz, CDCl3) δ = −107.1; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C17H11FNO2 280.0768; found 280.0765.
7-Chloro-9-phenylfuro[3,4-b]quinolin-3(1H)-one (2h).
Compound 2h was isolated in 76% yield (38 mg, off white solid); mp = 216–218 °C; Rf = 0.40 (VPE/VEA = 60/40); 1H NMR (400 MHz, CDCl3) δ = 8.23 (d, J = 9.1 Hz, 1H), 7.80 (s, 1H), 7.72–7.65 (m, 1H), 7.65–7.55 (m, 3H), 7.47–7.42 (m, 2H), 5.36 (s, 2H); 13C {1H} NMR (100 MHz, CDCl3) δ = 168.2, 148.7, 144.3, 143.0, 135.6, 133.1, 132.7, 132.5, 131.6, 129.7, 129.4, 128.7, 128.3, 124.4, 67.7; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C17H11ClNO2 296.0473; found 296.0469.
7-Bromo-9-phenylfuro[3,4-b]quinolin-3(1H)-one (2i).
Compound 2i was isolated in 75% yield (37 mg, yellow solid); mp = 219–221 °C; Rf = 0.40 (VPE/VEA = 60/40); 1H NMR (500 MHz, CDCl3) δ = 8.26 (d, J = 9.1 Hz, 1H), 8.03 (d, J = 1.2 Hz, 1H), 7.90 (d, J = 9.0 Hz, 1H), 7.65–7.59 (m, 3H), 7.44 (d, J = 7.4 Hz, 2H), 5.39 (s, 2H); 13C {1H} NMR (125 MHz, CDCl3) δ = 168.2, 149.2, 144.6, 143.1, 134.3, 133.1, 132.8, 129.9, 129.5, 128.9, 128.7, 127.9, 124.2, 67.7; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C17H11BrNO2 339.9968; found 339.9963.
3-Oxo-9-phenyl-1,3-dihydrofuro[3,4-b]quinoline-7-carbonitrile (2j).
Compound 2j was isolated in 45% yield (23 mg, viscous liquid); Rf = 0.40 (VPE/VEA = 50/50); 1H NMR (400 MHz, CDCl3) δ = 8.51 (d, J = 8.8 Hz, 1H), 8.30 (d, J = 1.5 Hz, 1H), 7.97 (dd, J = 8.7, 1.7 Hz, 1H), 7.71–7.62 (m, 3H), 7.47–7.42 (m, 2H), 5.45 (s, 2H); 13C {1H} NMR (100 MHz, CDCl3) δ = 167.7, 151.2, 146.9, 145.1, 133.5, 132.8, 132.4, 131.9, 130.9, 130.4, 129.7, 128.7, 127.2, 117.9, 112.9, 67.7; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C18H11N2O2 287.0778.; found 287.0824.
5-Chloro-9-phenylfuro[3,4-b]quinolin-3(1H)-one (2k).
Compound 2k was isolated in 51% yield (25 mg, off white solid); mp = 246–248 °C; Rf = 0.40 (VPE/VEA = 50/50); 1H NMR (400 MHz, CDCl3) δ = 7.96 (dd, J = 7.4, 0.8 Hz, 1H), 7.86–7.80 (m, 1H), 7.64–7.55 (m, 4H), 7.45 (dd, J = 7.7, 1.6 Hz, 2H), 5.39 (s, 2H); 13C {1H} NMR (100 MHz, CDCl3) δ = 167.9, 147.1, 144.7, 135.8, 133.2, 130.8, 129.8, 129.4, 129.3, 128.9, 128.8 (two 13C), 124.9, 67.5; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C17H11ClNO2 296.0478.; found 296.0480.
5,7-Dimethyl-9-phenylfuro[3,4-b]quinolin-3(1H)-one (2l).
Compound 2l was isolated in 69% yield (34 mg, off white solid); mp = 177–179 °C; Rf = 0.40 (VPE/VEA = 60/40); 1H NMR (400 MHz, CDCl3) δ = 7.63–7.54 (m, 3H), 7.52 (s, 1H), 7.45 (s, 1H), 7.43–7.41 (m, 2H), 5.33 (s, 2H), 2.90 (s, 3H), 2.45 (s, 3H); 13C {1H} NMR (100 MHz, CDCl3) δ = 169.3, 148.6, 142.7, 142.0, 139.5, 139.1, 134.1, 133.1, 132.4, 129.2, 129.1, 128.8, 128.0, 122.2, 67.7, 22.1, 18.5; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C19H16NO2 290.1176; found 290.1173.
7-(tert-Butyl)-9-(p-tolyl)furo[3,4-b]quinolin-3(1H)-one (2m).
Compound 2m was isolated in 80% yield (39 mg, pale yellow solid); mp = 205–207 °C; Rf = 0.40(VPE/VEA = 60/40); 1H NMR (400 MHz, CDCl3) δ = 8.33 (d, J = 8.8 Hz, 1H), 7.93 (dd, J = 9.0, 2.1 Hz, 1H), 7.86 (d, J = 2.0 Hz, 1H), 7.41 (d, J = 8.0 Hz, 2H), 7.35 (d, J = 8.0 Hz, 2H), 5.37 (s, 2H), 2.49 (s, 3H), 1.33 (s, 9H); 13C {1H} NMR (100 MHz, CDCl3) δ = 169.0, 152.4, 149.3, 143.6, 143.4, 139.5, 132.4, 130.7, 130.6, 129.9, 129.7, 128.7, 127.6, 120.4, 67.9, 35.4, 30.9, 21.4; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C22H22NO2 332.1645; found 332.1642.
7-(tert-Butyl)-9-(4-isopropylphenyl)furo[3,4-b]quinolin-3(1H)-one (2n).
Compound 2n was isolated in 76% yield (38 mg, off white solid); mp = 239–241 °C; Rf = 0.40 (VPE/VEA = 60/40); 1H NMR (500 MHz, CDCl3) δ = 8.35 (d, J = 9.0 Hz, 1H), 7.94 (d, J = 9.1 Hz, 1H), 7.89 (s, 1H), 7.46 (d, J = 7.8 Hz, 2H), 7.38 (d, J = 7.9 Hz, 2H), 5.40 (s, 2H), 3.05 (sept, J = 6.8 Hz, 1H), 1.36 (d, J = 7.1 Hz, 6H), 1.35 (s, 9H); 13C {1H} NMR (125 MHz, CDCl3) δ = 169.1, 152.4, 150.3, 149.4, 143.6, 143.5, 132.5, 131.0, 130.8, 129.7, 128.9, 127.7, 127.3, 120.5, 68.0, 35.4, 34.0, 30.9, 23.8; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C24H26NO2 360.1958; found 360.1953.
7-(tert-Butyl)-9-(4-methoxyphenyl)furo[3,4-b]quinolin-3(1H)-one (2o).
Compound 2o was isolated in 57% yield (28 mg, off white solid); mp = 262–264 °C; Rf = 0.35 (VPE/VEA = 60/40); 1H NMR (400 MHz, CDCl3) δ = 8.34 (d, J = 9.0 Hz, 1H), 7.93 (dd, J = 9.0, 1.7 Hz, 1H), 7.88 (s, 1H), 7.40 (d, J = 8.5 Hz, 2H), 7.13 (d, J = 8.5 Hz, 2H), 5.39 (s, 2H), 3.94 (s, 3H), 1.35 (s, 9H); 13C {1H} NMR (100 MHz, CDCl3) δ = 169.1, 160.4, 152.4, 149.3, 143.5, 143.3, 132.5, 130.8, 130.3, 129.7, 127.8, 125.7, 120.4, 114.7, 67.9, 55.4, 35.4, 30.9; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C22H22NO3 348.1594; found 348.1589.
7-(tert-Butyl)-9-(4-fluorophenyl)furo[3,4-b]quinolin-3(1H)-one (2p).
Compound 2p was isolated in 78% yield (39 mg, yellow solid); mp = 299–301 °C; Rf = 0.40 (VPE/VEA = 60/40); 1H NMR (400 MHz, CDCl3) δ = 8.31 (d, J = 9.0 Hz, 1H), 7.93 (dd, J = 9.0, 2.1 Hz, 1H), 7.76 (d, J = 2.0 Hz, 1H), 7.50–7.44 (m, 2H), 7.33 (m, 2H), 5.34 (s, 2H), 1.33 (s, 9H); 13C {1H} NMR (100 MHz, CDCl3) δ = 168.7, 163.2 (d, J = 250.2 Hz), 152.8, 149.2, 143.4, 142.3, 132.5, 130.8, 130.7, 129.9, 129.6 (d, J = 3.6 Hz), 127.6, 120.0, 116.5 (d, J = 21.8 Hz), 67.7, 35.4, 30.8; 19F NMR (376 MHz, CDCl3) δ = −110.9; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C21H18FNO2Na 358.1214; found 358.1208.
7-(tert-Butyl)-9-(4-chlorophenyl)furo[3,4-b]quinolin-3(1H)-one (2q).
Compound 2q was isolated in 77% yield (38 mg, pale yellow solid); mp = 265–267 °C; Rf = 0.40 (VPE/VEA = 60/40); 1H NMR (400 MHz, CDCl3) δ = 8.33 (d, J = 9.0 Hz, 1H), 7.94 (dd, J = 8.9, 1.9 Hz, 1H), 7.76 (d, J = 2.1 Hz, 1H), 7.61 (d, J = 8.4 Hz, 2H), 7.42 (d, J = 8.4 Hz, 2H), 5.35 (s, 2H), 1.34 (s, 9H); 13C {1H} NMR (100 MHz, CDCl3) δ = 168.7, 152.9, 149.2, 143.5, 142.1, 135.7, 132.4, 132.1, 130.8, 130.2, 130.0, 129.6, 127.3, 119.9, 67.6, 35.4, 30.8; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C21H19ClNO2 352.1099; found 352.1096.
7-(tert-Butyl)-9-(4-nitrophenyl)furo[3,4-b]quinolin-3(1H)-one (2r).
Compound 2r was isolated in 75% yield (37 mg, yellow solid); mp = 310–312 °C; Rf = 0.35 (VPE/VEA = 60/40); 1H NMR (500 MHz, CDCl3) δ = 8.50 (d, J = 8.6 Hz, 2H), 8.37 (d, J = 9.0 Hz, 1H), 7.99 (dd, J = 9.0, 2.0 Hz, 1H), 7.70 (d, J = 8.6 Hz, 2H), 7.65 (d, J = 1.9 Hz, 1H), 5.35 (s, 2H), 1.34 (s, 9H); 13C {1H} NMR (125 MHz, CDCl3) δ = 168.2, 153.7, 149.3, 148.5, 143.6, 140.7, 140.5, 132.2, 131.1, 130.4, 130.1, 126.9, 124.5, 119.5, 67.3, 35.5, 30.8; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C21H19N2O4 363.1339; found 363.1333.
7-(tert-Butyl)-9-(3-chloro-4-methylphenyl)furo[3,4-b]quinolin-3(1H)-one (2s).
Compound 2s was isolated in 73% yield (36 mg, off white solid); mp = 241–243 °C; Rf = 0.35 (VPE/VEA = 60/40); 1H NMR (400 MHz, CDCl3) δ = 8.36 (d, J = 9.0 Hz, 1H), 7.96 (dd, J = 9.0, 2.1 Hz, 1H), 7.81 (d, J = 2.0 Hz, 1H), 7.48 (d, J = 7.8 Hz, 1H), 7.46 (d, J = 1.7 Hz, 1H), 7.27 (dd, J = 7.8, 1.8 Hz, 1H), 5.38 (s, 2H), 2.52 (s, 3H), 1.35 (s, 9H); 13C {1H} NMR (100 MHz, CDCl3) δ = 168.7, 152.9, 149.3, 143.5, 141.9, 137.7, 135.4, 132.7, 132.4, 131.8, 130.9, 129.9, 129.2, 127.4, 127.1, 120.0, 67.7, 35.4, 30.9, 20.1; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C22H21ClNO2 366.1255; found 366.1251.
7-(tert-Butyl)-9-(3,5-dimethylphenyl)furo[3,4-b]quinolin-3(1H)-one (2t).
Compound 2t was isolated in 74% yield (37 mg, yellow solid); mp = 214.5–216.5 °C; Rf = 0.45 (VPE/VEA = 60/40); 1H NMR (500 MHz, CDCl3) δ = 8.34 (d, J = 9.0 Hz, 1H), 7.93 (dd, J = 9.0, 1.0 Hz, 1H), 7.86 (s, 1H), 7.19 (s, 1H), 7.05 (s, 2H), 5.38 (s, 2H), 2.43 (s, 6H), 1.34 (s, 9H); 13C {1H} NMR (125 MHz, CDCl3) δ = 169.0, 152.3, 149.3, 143.8, 143.5, 138.9, 133.6, 132.3, 130.9, 130.7, 129.7, 127.7, 126.5, 120.6, 67.9, 35.3, 30.9, 21.3; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C23H24NO2 346.1802; found 346.1797.
7-(tert-Butyl)-9-(2-methoxyphenyl)furo[3,4-b]quinolin-3(1H)-one (2u).
Compound 2u was isolated in 56% yield (28 mg, off white solid); mp = 257–259 °C; Rf = 0.30 (VPE/VEA = 60/40); 1H NMR (400 MHz, CDCl3) δ = 8.35 (d, J = 9.0 Hz, 1H), 7.92 (dd, J = 9.0, 1.9 Hz, 1H), 7.68 (d, J = 1.6 Hz, 1H), 7.57–7.53 (m, 1H), 7.30–7.27 (m, 1H), 7.20–7.13 (m, 2H), 5.35 (d, J = 15.0 Hz, 1H), 5.25 (d, J = 15.0 Hz, 1H), 3.78 (s, 3H), 1.32 (s, 9H); 13C {1H} NMR (100 MHz, CDCl3) δ = 169.2, 156.4, 152.1, 149.2, 143.3, 140.5, 133.7, 131.2, 130.9, 130.7, 129.6, 128.2, 121.9, 120.9, 120.6, 111.6, 68.3, 55.4, 35.3, 30.9; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C22H22NO3 348.1594; found 348.1590.
7-iso-Propyl-9-(thiophen-2-yl)furo[3,4-b]quinolin-3(1H)-one (2v).
Compound 2v was isolated in 77% yield (38 mg, brown solid); mp = 157–159 °C; Rf = 0.40 (VPE/VEA = 60/40); 1H NMR (400 MHz, CDCl3) δ = 8.33 (d, J = 8.8 Hz, 1H), 8.07 (d, J = 1.8 Hz, 1H), 7.77 (dd, J = 8.8, 1.9 Hz, 1H), 7.67 (dd, J = 5.1, 1.1 Hz, 1H), 7.38 (dd, J = 3.5, 1.2 Hz, 1H), 7.33 (dd, J = 5.1, 3.5 Hz, 1H), 5.50 (s, 2H), 3.11 (sept, J = 6.9 Hz, 1H), 1.32 (d, J = 6.9 Hz, 6H); 13C {1H} NMR (100 MHz, CDCl3) δ = 168.7, 150.9, 149.7, 143.3, 136.1, 133.6, 132.6, 131.3, 130.7, 129.9, 128.7, 128.3, 127.8, 121.7, 68.2, 34.5, 23.6; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C18H16NO2S 310.0896; found 310.0891.
7,9-Dimethylfuro[3,4-b]quinolin-3(1H)-one (2w).
Compound 2w was isolated in 17% yield (11 mg, brown solid); mp = 213–215 °C; Rf = 0.40 (VPE/VEA = 50/50); 1H NMR (400 MHz, CDCl3) δ = 8.25 (d, J = 8.8 Hz, 1H), 7.85 (s, 1H), 7.67 (dd, J = 8.7, 1.6 Hz, 1H), 5.49 (s, 2H), 2.69 (s, 3H), 2.63 (s, 3H); 13C {1H} NMR (100 MHz, CDCl3) δ = 169.2, 148.5, 143.0, 139.7, 138.7, 133.0, 131.5, 129.1, 122.4, 67.7, 22.3, 14.6; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C13H12NO2 214.0878; found 214.0869.
7-Methyl-2,9-diphenyl-1,2-dihydro-3H-pyrrolo[3,4-b]quinolin-3-one (4a).
Compound 4a was isolated in 80% yield (39 mg, brown solid); mp = 247–249 °C; Rf = 0.40 (VPE/VEA = 40/60); 1H NMR (500 MHz, CDCl3) δ = 8.32 (d, J = 8.7 Hz, 1H), 7.88 (d, J = 7.9 Hz, 2H), 7.66–7.58 (m, 4H), 7.54 (s, 1H), 7.51–7.47 (m, 2H), 7.40 (t, J = 8.0 Hz, 2H), 7.18 (t, J = 7.4 Hz, 1H), 4.79 (s, 2H), 2.48 (s, 3H); 13C {1H} NMR (125 MHz, CDCl3) δ = 165.6, 150.0, 148.7, 142.8, 139.4, 138.7, 134.6, 132.5, 130.9, 129.3 (two 13C), 129.2, 129.1, 127.9, 127.7, 125.3, 124.5, 119.7, 48.3, 22.1; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C24H19N2O 351.1492; found 351.1494.
7-Methoxy-2,9-diphenyl-1,2-dihydro-3H-pyrrolo[3,4-b]quinolin-3-one (4b).
Compound 4b was isolated in 52% yield (26 mg, yellow solid); mp = 236–238 °C; Rf = 0.30 (VPE/VEA = 30/70); 1H NMR (400 MHz, CDCl3) δ = 8.33 (d, J = 9.3 Hz, 1H), 7.88 (d, J = 8.0 Hz, 2H), 7.61 (m, 3H), 7.50 (d, J = 6.8 Hz, 2H), 7.45–7.38 (m, 3H), 7.17 (t, J = 7.3 Hz, 1H), 7.04 (d, J = 2.5 Hz, 1H), 4.78 (s, 2H), 3.78 (s, 3H); 13C {1H} NMR (100 MHz, CDCl3) δ = 165.5, 159.2, 148.4, 146.1, 141.7, 139.2, 134.6, 132.5, 129.3, 129.2, 129.1, 129.1, 128.8, 128.1, 125.1, 122.7, 119.5, 103.3, 55.5, 48.1; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C24H19 N2O2 367.1441 found 367.1435.
2-Benzyl-9-phenyl-1,2-dihydro-3H-pyrrolo[3,4-b]quinolin-3-one (4c).
Compound 4c was isolated in 84% yield (41 mg, yellow solid); mp = 192–194 °C; Rf = 0.40 (VPE/VEA = 30/70); 1H NMR (500 MHz, CDCl3) δ = 8.45 (d, J = 8.4 Hz, 1H), 7.78 (t, J = 8.5 Hz, 2H), 7.58–7.50 (m, 4H), 7.38 (m, 2H), 7.34–7.27 (m, 5H), 4.89 (s, 2H), 4.24 (s, 2H); 13C {1H} NMR (125 MHz, CDCl3) δ = 166.4, 150.7, 149.6, 143.5, 136.2, 134.3, 131.1, 129.8, 128.9, 128.8, 128.3 (two 13C), 128.0, 127.9, 127.5, 125.6, 47.2, 46.8; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C24H19N2O 351.1492; found 351.1487.
2-Benzyl-7-methyl-9-phenyl-1,2-dihydro-3H-pyrrolo[3,4-b]quinolin-3-one (4d).
Compound 4d was isolated in 82% yield (40 mg, yellow solid); mp = 204–206 °C; Rf = 0.40 (VPE/VEA = 30/70); 1H NMR (400 MHz, CDCl3) δ = 8.29 (d, J = 8.7 Hz, 1H), 7.58 (dd, J = 8.7, 1.5 Hz, 1H), 7.55–7.46 (m, 4H), 7.36–7.33 (m, 2H), 7.29–7.23 (m, 5H), 4.85 (s, 2H), 4.19 (s, 2H), 2.43 (s, 3H); 13C {1H} NMR (100 MHz, CDCl3) δ = 166.5, 149.7, 148.1, 142.6, 138.2, 136.3, 134.4, 132.1, 130.6, 128.9 (two 13C), 128.8, 128.8, 128.4, 128.2, 127.8, 127.4, 124.3, 47.0, 46.7, 21.9; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C25H21N2O 365.1648; found 365.1653.
2-Benzyl-7-methoxy-9-phenyl-1,2-dihydro-3H-pyrrolo[3,4-b]quinolin-3-one (4e).
Compound 4e was isolated in 59% yield (29 mg, brown solid); mp = 179–181 °C; Rf = 0.35 (VPE/VEA = 30/70); 1H NMR (400 MHz, CDCl3) δ = 8.31 (d, J = 9.2 Hz, 1H), 7.57–7.51 (m, 3H), 7.43–7.39 (m, 3H), 7.31–7.27 (m, 5H), 6.98 (d, J = 2.6 Hz, 1H), 4.87 (s, 2H), 4.21 (s, 2H), 3.75 (s, 3H); 13C {1H} NMR (100 MHz, CDCl3) δ = 166.6, 158.9, 148.3, 145.6, 141.7, 136.3, 134.5, 132.4, 129.0, 128.9, 128.8, 128.7 (two 13C), 128.2, 127.8, 122.4, 103.3, 55.4, 46.9, 46.7; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C25H21N2O2; 381.1598 found 381.1597.
2-Benzyl-7-chloro-9-phenyl-1,2-dihydro-3H-pyrrolo[3,4-b]quinolin-3-one (4f).
Compound 4f was isolated in 81% yield (40 mg, brown solid); mp = 224–226 °C; Rf = 0.40 (VPE/VEA = 30/70); 1H NMR (400 MHz, CDCl3) δ = 8.33 (d, J = 8.6 Hz, 1H), 7.70 (s, 1H), 7.68 (d, J = 2.3 Hz, 1H), 7.59–7.51 (m, 3H), 7.37 (m, 2H), 7.33–7.27 (m, 5H), 4.88 (s, 2H), 4.24 (s, 2H); 13C {1H} NMR (100 MHz, CDCl3) δ = 165.9, 150.9, 147.8, 142.7, 136.0, 134.1, 133.5, 132.5, 130.8, 129.3, 129.2, 129.1, 128.9 (two 13C), 128.3, 128.1, 127.9, 124.4, 47.1, 46.7; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C24H18ClN2O 385.1102; found 385.1100.
2-Benzyl-5,8-dichloro-9-phenyl-1,2-dihydro-3H-pyrrolo[3,4-b]quinolin-3-one (4g).
Compound 4g was isolated in 43% yield (21 mg, yellow solid); mp = 231–233 °C; Rf = 0.45 (VPE/VEA = 30/70); 1H NMR (500 MHz, CDCl3) δ = 7.80 (d, J = 8.1 Hz, 1H), 7.53 (d, J = 8.1 Hz, 1H), 7.47–7.44 (m, 3H), 7.34–7.26 (m, 7H), 4.85 (s, 2H), 4.10 (s, 2H); 13C {1H} NMR (125 MHz, CDCl3) δ = 165.3, 151.0, 147.1, 144.2, 136.3, 136.0, 135.1, 132.3, 130.4, 129.7, 129.5, 128.8, 128.6, 128.4, 128.2, 128.1, 127.9, 125.8, 47.2, 46.9; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C24H17Cl2N2O (M + H)+ 419.0712; found 419.0703.
2-Benzyl-9-(4-methoxyphenyl)-7-methyl-1,2-dihydro-3H-pyrrolo [3,4-b]quinolin-3-one (4h).
Compound 4h was isolated in 57% yield (28 mg, yellow solid); mp = 197–199 °C; Rf = 0.35 (VPE/VEA = 30/70); 1H NMR (500 MHz, CDCl3) δ = 8.31 (d, J = 9.3 Hz, 1H), 7.41 (dd, J = 9.2, 2.0 Hz, 1H), 7.34 (d, J = 7.6 Hz, 2H), 7.31–7.27 (m, 7H), 7.03 (d, J = 1.7 Hz, 1H), 4.87 (s, 2H), 4.20 (s, 2H), 3.76 (s, 3H), 2.46 (s, 3H); 13C {1H} NMR (125 MHz, CDCl3) δ = 166.7, 158.9, 148.4, 145.7, 141.9, 138.8, 136.4, 132.4, 131.5, 129.7, 128.9, 128.9, 128.8, 128.7, 128.2, 127.7, 122.4, 103.4, 55.4, 47.0, 46.8, 21.3; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C26H23N2O2 395.1754; found 395.1750.
2-Benzyl-7-chloro-9-(p-tolyl)-1,2-dihydro-3H-pyrrolo[3,4-b]quinoline-3-one (4i).
Compound 4i was isolated in 80% yield (39 mg, yellow solid); mp = 187–189 °C; Rf = 0.45 (VPE/VEA = 30/70); 1H NMR (400 MHz, CDCl3) δ = 8.28 (d, J = 9.0 Hz, 1H), 7.71 (s, 1H), 7.65 (d, J = 9.0 Hz, 1H), 7.37–7.27 (m, 9H), 4.87 (s, 2H), 4.24 (s, 2H), 2.46 (s, 3H); 13C {1H} NMR (100 MHz, CDCl3) δ = 165.9, 150.8, 147.8, 142.8, 139.3, 136.0, 133.9, 132.3, 130.7, 130.5, 129.8, 129.2, 128.8, 128.2, 128.1, 127.8, 124.5, 113.9, 47.1, 46.8, 21.3; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C25H20ClN2O 399.1259; found 399.1261.
2-Benzyl-7-methyl-9-(p-tolyl)-1,2-dihydro-3H-pyrrolo[3,4-b]quinolin-3-one (4j).
Compound 4j was isolated in 79% yield (39 mg, yellow solid); mp = 203–205 °C; Rf = 0.40 (VPE/VEA = 30/70); 1H NMR (500 MHz, CDCl3) δ = 8.33 (d, J = 8.7 Hz, 1H), 7.61 (d, J = 7.8 Hz, 1H), 7.54 (s, 1H), 7.35 (d, J = 7.7 Hz, 2H), 7.33–7.25 (m, 7H), 4.89 (s, 2H), 4.23 (s, 2H), 2.47 (s, 6H); 13C {1H} NMR (125 MHz, CDCl3) δ = 166.6, 149.8, 148.2, 142.8, 138.8, 138.1, 136.4, 132.1, 131.5, 130.7, 129.6, 128.9, 128.8, 128.5, 128.3, 127.8, 127.6, 124.4, 47.1, 46.8, 21.9, 21.3; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C26H23N2O 379.1805; found 379.1801.
2-Benzyl-9-(4-methoxyphenyl)-7-methyl-1,2-dihydro-3H-pyrrolo [3,4-b]quinolin-3-one (4k).
Compound 4k was isolated in 75% yield (37 mg, yellow solid); mp = 190–192 °C; Rf = 0.35 (VPE/VEA = 30/70); 1H NMR (400 MHz, CDCl3) δ = 8.29 (d, J = 8.6 Hz, 1H), 7.58 (dd, J = 8.7, 1.8 Hz, 1H), 7.54 (s, 1H), 7.34–7.27 (m, 7H), 7.07 (d, J = 8.7 Hz, 2H), 4.86 (s, 2H), 4.22 (s, 2H), 3.90 (s, 3H), 2.46 (s, 3H); 13C {1H} NMR (100 MHz, CDCl3) δ = 166.54, 159.91, 149.66, 148.11, 142.47, 138.03, 136.29, 131.98, 130.57, 130.30, 128.75, 128.53, 128.23, 127.74, 127.67, 126.41, 124.34, 114.36, 55.31, 47.00, 46.82, 21.90; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C26H23N2O2 395.1754; found 395.1762.
2-Benzyl-9-(4-chlorophenyl)-7-methyl-1,2-dihydro-3H-pyrrolo[3,4-b]quinolin-3-one (4l).
Compound 4l was isolated in 81% yield (40 mg, yellow solid); mp = 193–195 °C; Rf = 0.40 (VPE/VEA = 30/70); 1H NMR (400 MHz, CDCl3) δ = 8.27 (d, J = 8.6 Hz, 1H), 7.59 (dd, J = 8.5, 1.3 Hz, 1H), 7.53 (d, J = 8.3 Hz, 2H), 7.43 (s, 1H), 7.36–7.26 (m, 7H), 4.85 (s, 2H), 4.19 (s, 2H), 2.46 (s, 3H); 13C {1H} NMR (100 MHz, CDCl3) δ = 166.3, 149.6, 147.9, 141.3, 138.5, 136.1, 135.1, 132.8, 132.2, 130.7, 130.4, 129.3, 128.8, 128.4, 128.3, 127.9, 127.2, 123.9, 47.0, 46.6, 21.9; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C25H20ClN2O 399.1259; found 399.1271.
2-Benzyl-7-methyl-9-(4-nitrophenyl)-1,2-dihydro-3H-pyrrolo[3,4-b]quinolin-3-one (4m).
Compound 4m was isolated in 71% yield (35 mg, yellow solid); mp = 200–202 °C; Rf = 0.30 (VPE/VEA = 30/70); 1H NMR (400 MHz, CDCl3) δ = 8.43 (d, J = 8.8 Hz, 2H), 8.34 (d, J = 8.7 Hz, 1H), 7.65 (dd, J = 8.7, 1.8 Hz, 1H), 7.60 (d, J = 8.8 Hz, 2H), 7.36–7.29 (m, 6H), 4.86 (s, 2H), 4.18 (s, 2H), 2.48 (s, 3H). 13C {1H} NMR (100 MHz, CDCl3) δ = 166.10, 149.92, 148.32, 148.19, 141.38, 140.16, 139.33, 136.10, 132.75, 131.06, 130.30, 129.03, 128.49, 128.31, 128.12, 126.74, 124.40, 123.63, 47.26, 46.55, 22.11; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C25H20N3O3 410.1499; found 410.1505.
2-Benzyl-7-methyl-9-(thiophen-2-yl)-1,2-dihydro-3H-pyrrolo[3,4-b]quinolin-3-one (4n).
Compound 4n was isolated in 73% yield (36 mg, yellow solid); mp = 221–223 °C; Rf = 0.40 (VPE/VEA = 30/70); 1H NMR (400 MHz, CDCl3) δ = 8.31 (d, J = 8.7 Hz, 1H), 7.84 (s, 1H), 7.61 (dd, J = 8.7, 1.9 Hz, 1H), 7.59–7.56 (m, 1H), 7.36–7.31 (m, 4H), 7.31–7.23 (m, 4H), 4.90 (s, 2H), 4.36 (s, 2H), 2.51 (s, 3H); 13C {1H} NMR (100 MHz, CDCl3) δ = 166.42, 149.87, 148.32, 138.82, 136.35, 135.73, 134.26, 132.42, 130.88, 129.55, 129.27, 128.96, 128.41, 128.01, 127.97, 127.95, 127.72, 124.34, 47.41, 47.20, 22.16; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C23H19N2OS 371.1213; found 371.1221.
2-Benzyl-9-(trimethylsilyl)-1,2-dihydro-3H-pyrrolo[3,4-b]quinolin-3-one (4o).
Compound 4o was isolated in 67% yield (33 mg, off white solid); mp = 202–204 °C; Rf = 0.40 (VPE/VEA = 30/70); 1H NMR (400 MHz, CDCl3) δ = 8.42 (d, J = 8.2 Hz, 1H), 8.16 (d, J = 8.3 Hz, 1H), 7.75 (ddd, J = 8.3, 7.0, 1.3 Hz, 1H), 7.62 (ddd, J = 8.3, 7.0, 1.3 Hz, 1H), 7.41–7.27 (m, 5H), 4.94 (s, 2H), 4.46 (s, 2H), 0.51 (s, 9H); 13C {1H} NMR (100 MHz, CDCl3) δ = 166.0, 149.3, 148.1, 142.9, 136.2, 135.7, 132.3, 132.1, 129.1, 128.9, 128.3, 127.9, 127.8, 127.4, 49.3, 47.1, 1.5; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C21H23N2OSi (M + H)+ 347.1578; found 347.1581.
9-((Benzyloxy)methyl)-7-methoxy-1-methylfuro[3,4-b]quinolin-3(1H)-one (6).
Compound 7 was isolated in 41% yield (21 mg, yellow solid); mp = 174–176 °C; Rf = 0.35 (VPE/VEA = 30/70); 1H NMR (400 MHz, CDCl3) δ = 8.26 (d, J = 9.3 Hz, 1H), 7.47 (dd, J = 9.3, 2.7 Hz, 1H), 7.41–7.36 (m, 5H), 7.18 (d, J = 2.7 Hz, 1H), 5.88 (q, J = 6.5 Hz, 1H), 5.06 (ABq, J = 13.20 Hz, 2H), 4.71 (ABq, J = 11.74 Hz, 2H), 3.92 (s, 3H), 1.68 (d, J = 6.5 Hz, 3H); 13C {1H} NMR (100 MHz, CDCl3) δ = 168.2, 160.1, 146.3, 142.3, 137.8, 136.7, 136.3, 133.2, 129.0, 128.7, 128.4, 128.1, 123.7, 101.2, 76.8, 73.7, 65.9, 55.7, 21.1; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C21H20NO4 350.1387; found 350.1380.
9-Phenyl-1,2-dihydro-3H-pyrrolo[3,4-b]quinolin-3-one (7).
Compound 7 was isolated in 88% yield (65 mg, grey solid); mp = 279–281 °C (decomposed); Rf = 0.40 (VDCM/VMeOH = 90/10); 1H NMR (400 MHz DMSO d6) δ = 9.26 (bs, 1H), 8.26 (d, J = 8.4 Hz, 1H), 7.87 (t, J = 7.4 Hz, 1H), 7.79 (d, J = 8.3 Hz, 1H), 7.67 (t, J = 7.4 Hz, 1H), 7.58–7.62 (m, 5H), 4.38 (s, 2H); 13C {1H} NMR (100 MHz, DMSO d6) δ = 167.6, 151.4, 148.7, 142.9, 133.9, 131.0, 130.3, 129.7, 129.2, 128.9 (two 13C), 128.1, 126.6, 125.4, 42.0; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C17H13N2O 261.1022; found 261.1019.
7-Methyl-9-phenyl-1,2-dihydro-3H-pyrrolo[3,4-b]quinolin-3-one (8).
Compound 8 was isolated in 86% yield (65 mg, grey solid); mp = 317–319 °C (decomposed); Rf = 0.40 (VDCM/VMeOH = 90/10); 1H NMR (400 MHz, DMSO d6) δ = 9.19 (bs, 1H), 8.15 (d, J = 8.5 Hz, 1H), 7.70 (d, J = 8.4 Hz, 1H), 7.62–7.52 (m, 6H), 4.34 (s, 2H), 2.45 (s, 3H); 13C {1H} NMR (100 MHz, DMSO d6) δ = 168.2, 151.0, 147.9, 142.7, 138.3, 134.5, 132.4, 131.7, 130.6, 129.7, 129.4, 129.3, 127.0, 124.3, 42.5, 22.0; HRMS (ESI-TOF) m/z: [M + H]+ calcd for C18H15N2O 275.1179; found 275.1176.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Generous financial support from CSIR-New Delhi (HCP0008) is gratefully acknowledged. DAM and ACS thank UGC and CSIR for the award of a senior research fellowship.
Notes and references
-
(a) R. Sharma, P. Kour and A. Kumar, J. Chem. Sci., 2018, 130, 73 CrossRef;
(b) V. R. Solomon and H. Lee, Curr. Med. Chem., 2011, 18, 1488–1508 CrossRef CAS;
(c) R. Musiol, M. Serda, S. Hensel-Bielowka and J. Polanski, Curr. Med. Chem., 2010, 17, 1960 CrossRef CAS;
(d) R. Musiol, J. Jampilek, K. Kralova, D. R. Richardson, D. Kalinowski, B. Podeszwa, J. Finster, H. Niedbala, A. Palka and J. Polanski, Bioorg. Med. Chem., 2007, 15, 1280–1288 CrossRef CAS;
(e) J. P. Michael, Nat. Prod. Rep., 2007, 24, 223–246 RSC;
(f) V. V. Kouznetsov, L. Y. V. Mendez and C. M. M. Gomez, Curr. Org. Chem., 2005, 9, 141–161 CrossRef CAS.
-
(a) T. Boisse, L. Gavara, P. Gautret, B. Baldeyrou, A. Lansiaux, J.-F. Goossens, J.-P. Hénichart and B. Rigo, Tetrahedron Lett., 2011, 52, 1592–1596 CrossRef CAS;
(b) K. C. Jahng, S. I. Kim, D. H. Kim, C. S. Seo, J.-K. Son, S. H. Lee, E. S. Lee and Y. Jahng, Chem. Pharm. Bull., 2008, 56, 607–609 CrossRef CAS;
(c) A. Cagir, B. M. Eisenhauer, R. Gao, S. J. Thomas and S. M. Hecht, Bioorg. Med. Chem., 2004, 12, 6287–6299 CrossRef CAS;
(d) E. S. Lee, J.-G. Park and Y. Jahng, Tetrahedron Lett., 2003, 44, 1883–1886 CrossRef CAS;
(e) S. Dallavalle and L. Merlini, Tetrahedron Lett., 2002, 43, 1835–1837 CrossRef CAS;
(f) J. S. Yadav and B. V. S. Reddy, Tetrahedron Lett., 2002, 43, 1905–1907 CrossRef CAS;
(g) H. Wang and A. Ganesan, Tetrahedron Lett., 1998, 39, 9097–9098 CrossRef CAS;
(h) M.-C. Tseng, Y.-W. Chu, H.-P. Tsai, C.-M. Lin, J. Hwang and Y.-H. Chu, Org. Lett., 2011, 13, 920–923 CrossRef CAS;
(i) S. H. Kwon, H.-A. Seo and C.-H. Cheon, Org. Lett., 2016, 18, 5280–5283 CrossRef CAS.
-
(a) K. C. Nicolaou, Y. Wang, M. Lu, D. Mandal, M. R. Pattanayak, R. Yu, A. A. Shah, J. S. Chen, H. Zhang, J. J. Crawford, L. Pasunoori, Y. B. Poudel, N. S. Chowdari, C. Pan, A. Nazeer, S. Gangwar, G. Vite and E. N. Pitsinos, J. Am. Chem. Soc., 2016, 138, 8235–8246 CrossRef CAS;
(b) K. C. Nicolaou, J. S. Chen, H. Zhang and A. Montero, Angew. Chem., Int. Ed., 2008, 47, 185–189 CrossRef CAS PubMed;
(c) K. C. Nicolaou, H. Zhang, J. S. Chen, J. J. Crawford and L. Pasunoori, Angew. Chem., Int. Ed., 2007, 46, 4704–4707 CrossRef CAS.
-
(a) Q. Chen, S. Zhang, T. Zhang, K. He, Y. Yuan and X. Jia, Asian J. Org. Chem., 2019, 8, 115–118 CrossRef CAS;
(b) Y. Q. Liu, L. Yang and X. Tian, Curr. Bioact. Compd., 2007, 3, 37 CrossRef CAS;
(c) M. Gordaliza, P. A. Garcia, J. M. Miguel del Corral, M. A. Castro and M. A. Gomez-Zurita, Toxicon, 2004, 44, 441 CrossRef CAS;
(d) A. S. Feliciano, J. M. Miguel del Corral, M. Gordaliza and M. A. Castro, Phytochemistry, 1989, 28, 659–660 CrossRef.
-
(a) A. Blair, F. Zmuda, G. Malviya, A. A. S. Tavares, G. D. Tamagnan, A. J. Chalmers, D. Dewar, S. L. Pimlott and A. Sutherland, Chem. Sci., 2015, 6, 4772–4777 RSC;
(b) A. Blair, L. Stevenson, D. Dewar, S. L. Pimlott and A. Sutherland, MedChemComm, 2013, 4, 1461–1466 RSC;
(c) L. Stevenson, A. A. S. Tavares, A. Brunet, F. I. McGonagle, D. Dewar, S. L. Pimlott and A. Sutherland, Bioorg. Med. Chem. Lett., 2010, 20, 954–957 CrossRef CAS;
(d) M. Anzini, A. Cappelli, S. Vomero, M. Seeber, M. C. Menziani, T. Langer, B. Hagen, C. Manzoni and J.-J. Bourguignon, J. Med. Chem., 2001, 44, 1134–1150 CrossRef CAS.
-
(a) L. Stevenson, A. A. S. Tavares, A. Brunet, F. I. McGonagle, D. Dewar, S. L. Pimlott and A. Sutherland, Bioorg. Med. Chem. Lett., 2010, 20, 954–957 CrossRef CAS PubMed;
(b) D. Osborne and P. J. Stevenson, Tetrahedron Lett., 2002, 43, 5469–5470 CrossRef CAS.
- For Review:
(a) M. Fochi, L. Caruana and L. Bernardi, Synthesis, 2014, 46, 135–157 CrossRef;
(b) V. V. Kouznetsov, Tetrahedron, 2009, 65, 2721–2750 CrossRef CAS;
(c) P. Buonora, J.-C. Olsen and T. Oh, Tetrahedron, 2001, 57, 6099–6138 CrossRef CAS;
(d) H. Posson, J.-P. Hurvois and C. Moinet, Synlett, 2000, 2000, 209–212 CrossRef ; selected references for Povarov cyclization: ;
(e) Q. Gao, S. Liu, X. Wu and A. Wu, Org. Lett., 2014, 16, 4582–4585 CrossRef CAS;
(f) H. Richter and O. García Mancheño, Org. Lett., 2011, 13, 6066–6069 CrossRef CAS;
(g) J. Liu, Y. Wang, L. Yu, C. Huo, X. Wang and X. Jia, Adv. Synth. Catal., 2014, 356, 3214–3218 CrossRef CAS;
(h) C. Huo, Y. Yuan, M. Wu, X. Jia, X. Wang, F. Chen and J. Tang, Angew. Chem., Int. Ed., 2014, 53, 13544–13547 CrossRef CAS;
(i) K. V. Sashidhara, G. R. Palnati, L. R. Singh, A. Upadhyay, S. R. Avula, A. Kumar and R. Kant, Green Chem., 2015, 17, 3766–3770 RSC;
(j) X. Wu, X. Geng, P. Zhao, J. Zhang, X. Gong, Y.-d. Wu and A.-x. Wu, Org. Lett., 2017, 19, 1550–1553 CrossRef CAS;
(k) J. Liu, F. Liu, Y. Zhu, X. Ma and X. Jia, Org. Lett., 2015, 17, 1409–1412 CrossRef CAS;
(l) G. Dagousset, J. Zhu and G. Masson, J. Am. Chem. Soc., 2011, 133, 14804–14813 CrossRef CAS PubMed;
(m) P. M. Khaja Mohinuddin, R. Dada, A. I. Almansour, N. Arumugam and S. Yaragorla, Tetrahedron Lett., 2019, 60, 1043–1048 CrossRef CAS;
(n) D. A. Powell and R. A. Batey, Tetrahedron Lett., 2003, 44, 7569–7573 CrossRef CAS;
(o) T. R. M. Rezende, J. O. S. Varejão, A. L. L. d. A. Sousa, S. M. B. Castañeda and S. A. Fernandes, Org. Biomol. Chem., 2019, 17, 2913–2922 RSC;
(p) M. Xie, X. Chen, Y. Zhu, B. Gao, L. Lin, X. Liu and X. Feng, Angew. Chem., Int. Ed., 2010, 49, 3799–3802 CrossRef CAS;
(q) O. Ghashghaei, C. Masdeu, C. Alonso, F. Palacios and R. Lavilla, Drug Discovery Today: Technol., 2018, 29, 71–79 CrossRef ; selected references for copper catalyzed Povarov cyclization:;
(r) H. Wang, C. Wang, K. Huang, L. Liu, W. Chang and J. Li, Org. Lett., 2016, 18, 2367–2370 CrossRef CAS;
(s) I. Muthukrishnan, P. Vinoth, T. Vivekanand, S. Nagarajan, C. U. Maheswari, J. C. Menéndez and V. Sridharan, J. Org. Chem., 2016, 81, 1116–1124 CrossRef CAS;
(t) H. Huang, H. Jiang, K. Chen and H. Liu, J. Org. Chem., 2009, 74, 5476–5480 CrossRef CAS;
(u) S. Ramesh and R. Nagarajan, J. Chem. Sci., 2014, 126, 1049–1054 CrossRef CAS.
-
(a) N. Lezana, M. Matus-Pérez, A. Galdámez, S. Lühr and M. Vilches-Herrera, Green Chem., 2016, 18, 3712–3717 RSC;
(b) A. I. Almansour, N. Arumugam, R. Suresh Kumar, J. Carlos Menéndez, H. A. Ghabbour, H.-K. Fun and R. Ranjith Kumar, Tetrahedron Lett., 2015, 56, 6900–6903 CrossRef CAS;
(c) M. Chen, N. Sun and Y. Liu, Org. Lett., 2013, 15, 5574–5577 CrossRef CAS;
(d) A. A. Kudale, D. O. Miller, L. N. Dawe and G. J. Bodwell, Org. Biomol. Chem., 2011, 9, 7196–7206 RSC;
(e) H. Twin and R. A. Batey, Org. Lett., 2004, 6, 4913–4916 CrossRef CAS PubMed;
(f) M. Toyota, C. Komori and M. Ihara, J. Org. Chem., 2000, 65, 7110–7113 CrossRef CAS.
- S. Desrat and P. van de Weghe, J. Org. Chem., 2009, 74, 6728–6734 CrossRef CAS.
-
(a) W. Dong, B. Hu, X. Gao, Y. Li, X. Xie and Z. Zhang, J. Org. Chem., 2016, 81, 8770–8776 CrossRef CAS PubMed;
(b) Y. Wang, F. Peng, J. Liu, C. Huo, X. Wang and X. Jia, J. Org. Chem., 2015, 80, 609–614 CrossRef CAS.
- S. Hati, U. Holzgrabe and S. Sen, Beilstein J. Org. Chem., 2017, 13, 1670–1692 CrossRef CAS.
-
(a) K. C. Nicolaou, C. J. N. Mathison and T. Montagnon, Angew. Chem., Int. Ed., 2003, 42, 4077–4082 CrossRef CAS;
(b) C. de Graaff, L. Bensch, M. J. van Lint, E. Ruijter and R. V. A. Orru, Org. Biomol. Chem., 2015, 13, 10108–10112 RSC.
-
(a) H. Hussain, I. R. Green and I. Ahmed, Chem. Rev., 2013, 113, 3329–3371 CrossRef CAS PubMed;
(b) L. V. Desai, H. A. Malik and M. S. Sanford, Org. Lett., 2006, 8, 1141–1144 CrossRef CAS.
-
(a) Q. Zhou, T. B. Vu Ngoc, G. Leszczynska, J.-L. Stigliani and G. Pratviel, Biomolecules, 2018, 8, 145 CrossRef;
(b) A. Armstrong, Angew. Chem., Int. Ed., 2004, 43, 1460–1462 CrossRef CAS.
- A. S. Kumar, K. Praneeth, P. Srihari and J. S. Yadav, Tetrahedron Lett., 2017, 58, 509–511 CrossRef.
- J. Panteleev, R. Y. Huang, E. K. J. Lui and M. Lautens, Org. Lett., 2011, 13, 5314–5317 CrossRef CAS.
- Rotameric mixture was observed in NMR of compounds 3c–3o due to restricted rotation of –NCO– bond.
-
(a) E. Schwartz, P. Bodis, M. Koepf, J. J. L. M. Cornelissen, A. E. Rowan, S. Woutersen and R. J. M. Nolte, Chem. Commun., 2009, 4675–4677 RSC;
(b) D. D. S. Sharley and J. M. J. Williams, Chem. Commun., 2017, 53, 2020–2023 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra06212b |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a and
M.
Muthukrishnan
a and
M.
Muthukrishnan