DOI:
10.1039/C9RA06188F
(Paper)
RSC Adv., 2019,
9, 33834-33842
Retracted Article: Long non-coding RNA PCAT1 facilitates cell growth in multiple myeloma through an MTDH-mediated AKT/β-catenin signaling pathway by sponging miR-363-3p†
Received
9th August 2019
, Accepted 10th October 2019
First published on 21st October 2019
Abstract
Multiple Myeloma (MM) is a plasma cell myeloma. Long non-coding RNA (lncRNA) prostate cancer associated transcript 1 (PCAT1) has been identified as being associated with various types of cancer. However, the mechanism of PCAT1 was still undefined in MM. In the current study, PCAT1 and metadherin (MTDH) were up-regulated and miR-363-3p was down-regulated in MM bone marrow plasma and cells. Furthermore, PCAT1 knockdown inhibited cell proliferation, cell cycle progression, and the protein kinase B (AKT) and β-catenin signaling pathway, but induced cell apoptosis by regulating MTDH. PCAT1 was verified to sponge miR-363-3p, and MTDH was identified as a candidate target of miR-363-3p. Meanwhile, PCAT1 positively modulated MTDH expression by sponging miR-363-3p. In addition, PCAT1 knockdown blocked xenograft tumor growth in vivo. Thus, we concluded that PCAT1 facilitated cell growth in multiple myeloma by activating the AKT/β-catenin signaling pathway through the miR-363-3p/MTDH axis.
Introduction
Multiple Myeloma (MM, also known as plasma cell myeloma) is a rare malignancy of the blood originating from post-germinal center B-cells and accounting for about 10% of all hematological malignancies.1–3 Despite improvements in diagnosis and therapy, the overall survival rate remains low and MM patients often suffer a high rate of relapse.4–6 But the mechanism of MM has rarely been documented.
Long non-coding RNAs (lncRNAs) are a type of long RNA (>200 nt) without translation capacity, which affect target gene expression at the post-transcriptional stage.7 Many lncRNAs have been identified as being implicated in the pathological processes of MM. For example, MALAT1 was significantly increased in MM blood samples and cells, and also promoted autophagy in vitro.8,9 Meanwhile, other lncRNAs were also dysregulated in MM or regulated cell behaviors in MM, like CCAT1,10 CRNDE,11 and H19.12
Prostate cancer associated transcript 1 (PCAT1), an lncRNA ∼2 kb in length located on 8q24.21, was first found in prostate cancer.13 Emerging evidence indicated that PCAT1 regulated cell growth and was associated with the clinical stage in diverse cancers.14–16 Moreover, PCAT1 was also reported to be up-regulated in MM.17 However, the mechanism of PCAT1 remained unclear in MM.
MicroRNAs (miRNAs) are a series of small RNA (about ∼22 nt) with no ability of translation and with blocked target gene expression.18 Also, the aberrant expression of miRNAs was also found in MM. For instance, many miRNAs, like miR-203,19 miR-342,20 and miR-29b,21 all apparently decreased in MM and further affected MM progression. In addition, Gowda et al. reported that miR-363 modulates cell behavior in MM.20 Metadherin (MTDH, also named LYRIC, AEG-1 or 3D3) is a gene located on chromosome 8q22.1 (ref. 22) and has been identified as an oncogene to regulate cell behavior,23,24 angiogenesis,25 and chemoresistance22 in various cancers including MM.26,27 However, the molecular mechanisms of miR-363-3p and MTDH have barely been reported in MM. In this research, we focused mainly on the roles of PCAT1 and MTDH in MM.
Materials and methods
Specimen collection
Thirty-six MM bone marrow blood samples were collected according to standard procedures from The First Affiliated Hospital of Xi'an Jiaotong University, as well as twenty-two normal blood samples. The centrifuged plasmas were stored in RNase-free tubes at −80 °C. The study was allowed by the Ethics Committee of The First Affiliated Hospital of Xi'an Jiaotong University and performed on the basis of Declaration of Helsinki Principles. All patients in this study provided written informed consent.
Quantitative real-time polymerase chain reaction (qRT-PCR)
The extraction of RNA was carried out using a miScript RT Kit (TaKaRa, Dalian, China). Then random primers (TaKaRa) were utilized to synthesize cDNA. Quantitative PCR was performed using an SYBR Green PCR Master Mix (Applied Biosystems, Foster City, USA). The relative expression levels of PCAT1, MTDH were normalized with glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and miR-363-3p was normalized with small nuclear RNA U6, and then calculated according to the 2−ΔΔCt method. The primers were obtained from Songon (Shanghai, China) and are listed in Table 1.
Table 1 Oligonucleotide sequences used in this researcha
| Gene |
Sequences |
| Abbreviation: PCAT1, prostate cancer associated transcript 1; MTDH, metadherin; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; U6, small nuclear RNA U6; F, forward; R, reverse. |
| PCAT1-F |
5′-TTGTGGAAGCCCCGCAAGGCCTGAA-3′ |
| PCAT1-R |
5′-TGTGGGGCCTGCACTGGCACTT-3′ |
| miR-363-3p-F |
5′-ACACTCCAGCTGGGGAATTGCACGGTATCCA-3′ |
| miR-363-3p-R |
5′-TGGTGTCGTGGAGTCG-3′ |
| MTDH-F |
5′-TGCCTCCTTCACAGACCAA-3′ |
| MTDH-R |
5′-TCGGCTGCAGATGAGATAG-3′ |
| GAPDH-F |
5′-TGTTCGTCATGGGTGTGAAC-3′ |
| GAPDH-R |
5′-ATGGCATGGACTGTGGTCAT-3′ |
| U6-F |
5′-CTCGCTTCGGCAGCACA-3′ |
| U6-R |
5′-AACGCTTCACGAATTTGCGT-3′ |
Cell culture and transfection
Three MM cell lines, RPMI-8226, OPM2 and U266B1, were bought from Huzhen (Shanghai, China), another MM cell line, MM1S, was purchased from Tongpai (Shanghai, China) and neural progenitor cells (nPCs) were obtained from Hopstem (Hangzhou, China). All cells were cultivated in Dulbecco's modified Eagle's medium (DMEM; Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS; Thermo Fisher Scientific, Rockville, MD, USA) in a 5% CO2 incubator at 37 °C. Short hairpin RNA (shRNA) against PCAT1 (sh-PCAT1), shRNA targeting MTDH (sh-MTDH), PCAT1 overexpression vector (PCAT1), MTDH overexpression plasmid (MTDH), miR-363-3p mimics (miR-363-3p), miR-363-3p inhibitor (anti-miR-363-3p), and their negative controls (sh-NC, pcDNA, miR-NC, or anti-NC) were all obtained from GeneChem (Shanghai, China). The transfection was carried out using Lipofectamine 2000 (Invitrogen).
3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide (MTT) assay
MTT (Sigma-Aldrich, Louis, MO, USA) was used to monitor the cell viability of OPM2 and U266B1 cells. Briefly, 180 μL of OPM2 and U266B1 cells (4 × 103 per well) were first cultured for 24 h. Following transfection, the cells were further cultivated for another 24 h, 48 h, or 72 h. Then 10 μL of MTT was added into each well and incubated for 4 h at 37 °C. After removing the supernatant, 110 μL of dimethyl sulfoxide (DMSO, Sigma-Aldrich) was injected into each well and oscillated at low speed for 10 min to dissolve the formazan. The absorbance at 490 nm was tested with a Multiscan Spectrum (Petenov, Beijing, China).
Flow cytometry analysis
A cell cycle kit (Beyotime, Shanghai, China) was utilized to detect the cell cycle arrest. The OPM2 and U266B1 cells (1 × 106 per tube) were first fixed in 500 μL of precooled ethanol for 2 h, then re-suspended in 100 μL of RNase A and incubated for 30 min at 37 °C. Subsequently, the OPM2 and U266B1 cells were stained with 400 μL of propidium iodide (PI) for 30 min at 4 °C in the dark. The cell phase fraction was assessed through flow cytometrics (Agilent, Beijing, China).
An Annexin V-fluorescein isothiocyanate (FITC)/PI apoptosis detection kit (Solarbio, Beijing, China) was used to assess the apoptotic rate. 100 μL of OPM2 and U266B1 cells (1 × 105 per tube) were re-suspended in binding buffer, and then incubated with 5 μL of Annexin V-FITC for 10 min, and 5 μL of PI solution for 5 min at 37 °C under dark conditions. Then phosphate buffer solution (PBS) was added into the tube to 500 μL. The cell apoptotic rate was evaluated using flow cytometrics (Agilent).
Western blot assay
Equal samples of 30 μg of extracted protein were separated by sodium dodecyl sulfonate-polyacrylamide gel electrophoresis (SDS-PAGE), and then transferred onto a polyvinylidene fluoride (PVDF) membrane. The membrane was firstly blocked into skim milk for 4 h, then incubated with primary antibody at 4 °C overnight. Following 2 h of incubation with secondary antibody, the chemiluminescence intensity of the membrane was evaluated using a Clarity™ Western ECL Substrate Kit (Bio-Rad, Shanghai, China). The cyclin D1 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1000), c-Myc (1:1000), B-cell lymphoma 2 (Bcl-2, 1:1000), Bcl2-associated X (Bax, 1:1000), GAPDH (1:10000), MTDH (1:1000), AKT (1:500), p-AKT (1:500), β-catenin (1:5000), and p-β-catenin (1:500) rabbit primary antibodies and goat anti-rabbit secondary antibodies (1:10000) were purchased from Abcam (Cambridge, MA, USA).
:1000), c-Myc (1:1000), B-cell lymphoma 2 (Bcl-2, 1:1000), Bcl2-associated X (Bax, 1:1000), GAPDH (1:10000), MTDH (1:1000), AKT (1:500), p-AKT (1:500), β-catenin (1:5000), and p-β-catenin (1:500) rabbit primary antibodies and goat anti-rabbit secondary antibodies (1:10000) were purchased from Abcam (Cambridge, MA, USA).
Dual-luciferase reporter assay
Through the miRcode (http://mircode.org/) and starBase v3.0 (http://starbase.sysu.edu.cn/) online databases, potential candidate targets of PCAT1 or MTDH were searched for. The wild type and mutant sequences of PCAT1, MTDH 3′-untranslated regions (3′UTR) were inserted into pmirGLO vector (Promega, Madison, WI, USA), namely WT-PCAT1, MUT-PCAT1, MTDH 3′UTR-WT, or MTDH 3′UTR-MUT. The co-transfection of luciferase reporter and miR-NC or miR-363-3p was conducted using Lipofectamine 2000 (Invitrogen). The transfected OPM2 and U266B1 cells were lysed by cell lysis buffer for 5 min on ice. Then the cell lysis samples were incubated with firefly luciferase substrate or renilla luciferase substrate. A Dual-Lucy Assay Kit (Solarbio) was utilized to assess the luciferase activity. The firefly luciferase activity was normalized by renilla luciferase activity.
Mice xenograft models
The experiments on nude mice were performed according to the criteria approved by the Animal Care Committee of The First Affiliated Hospital of Xi'an Jiaotong University. All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of Xi'an Jiaotong University and the experiments were approved by the Animal Ethics Committee of The First Affiliated Hospital of Xi'an Jiaotong University. Six-week-old nude mice (n = 5 per group) were purchased from Shanghai Laboratory Animal Center (Shanghai, China). Then the U266B1 cells (5 × 106) transfected with sh-PCAT1 or sh-NC were subcutaneously injected into the right back of the nude mice. After inoculation, the tumor volumes were calculated every 7 d for 35 d according to the formula: volume (mm3) = width2 × length/2. Then the xenograft tumor samples were excised for weight measurement and further analysis.
Statistical analysis
The results were obtained with GraphPad Prism 7 (GraphPad Inc., La Jolla, CA, USA). All experiments were repeated at least three times and presented as mean ± standard deviation (SD). Comparison between two groups was processed via Student's t-test, while comparison among multiple groups was evaluated by one-way analysis of variance (ANOVA). A P value less than 0.05 was considered a significant difference.
Results
PCAT1 and MTDH were strikingly up-regulated in MM bone marrow plasma
To investigate the effects of PCAT1 and MTDH in MM, the levels of PCAT1 and MTDH were firstly studied in MM bone marrow plasma. As shown in Fig. 1A and B, the relative expression levels of PCAT1 and MTDH were both significantly enhanced in MM bone marrow plasma compared to those in normal healthy patients. A scatter diagram shows that the level of MTDH was positively linearly correlated with the level of PCAT1 (Fig. 1C). In addition, the Kaplan–Meier curves exhibited that high levels of PCAT1 and MTDH were associated with a low overall survival rate, while low levels were related to a high overall survival rate (Fig. 1D and E). These results indicated that PCAT1 and MTDH may play vital roles in MM.
 |
| | Fig. 1 PCAT1 and MTDH were strikingly up-regulated in MM bone marrow plasma. (A and B) The levels of PCAT1 and MTDH in MM or healthy patients' bone marrow plasma were measured by qRT-PCR. (C) The correlation between MTDH and PCAT1 was processed by the Pearson test. (D and E) The overall survival rate in high or low expression of PCAT1 and MTDH group is displayed. *P < 0.05. | |
PCAT1 knockdown repressed cell proliferation by inducing cell cycle arrest and promoted cell apoptosis in OPM2 and U266B1 cells
Also the level of PCAT1 was apparently up-regulated in MM cells MM1S, RPMI-8226, OPM2 and U266B1 in contrast to the level in nPCs cells (Fig. 2A). To further explore the functions of PCAT1 in MM, sh-PCAT1 was transfected into OPM2 and U266B1 cells. As shown in Fig. 2B, the knockdown efficiency was confirmed, as indicated by the obvious decline in PCAT1 level in OPM2 and U266B1 cells transfected with sh-PCAT1. Furthermore, the MTT assay indicated that cell viability was remarkably suppressed in OPM2 and U266B1 cells transfected with sh-PCAT1 relative to that in the sh-NC group (Fig. 2C and D). The cells in the G1 phase fraction were markedly elevated in OPM2 and U266B1 cells transfected with sh-PCAT1, while the cells in the S phase fraction were significantly decreased (Fig. 2E and F). However, the transfection of sh-PCAT1 evidently facilitated the apoptotic rate in OPM2 and U266B1 cells (Fig. 2G). Since cyclin D1, c-Mycaspro-proliferation factors, Bax, and Bcl-2 act as apoptosis-related markers, we next detected the levels of these proteins in OPM2 and U266B1 cells. The western blot assay indicated that the protein levels of cyclin D1, c-Myc and Bcl-2 were all conspicuously reduced, while Bax was effectively increased in OPM2 and U266B1 cells transfected with sh-PCAT1 (Fig. 2H and I). Taken together, PCAT1 silencing constrained cell proliferation and cell cycle progression, but boosted apoptosis in OPM2 and U266B1 cells.
 |
| | Fig. 2 PCAT1 knockdown repressed cell proliferation by inducing cell cycle arrest and promoted cell apoptosis in OPM2 and U266B1 cells. (A) The level of PCAT1 in nPCs, MM1S, RPMI-8226, OPM2 and U266B1 cells was tested by qRT-PCR. (B–I) The OPM2 and U266B1 cells were transfected with sh-PCAT1 or sh-NC. (B) The level of PCAT1 in transfected cells was detected via qRT-PCR. (C and D) The cell viability of transfected OPM2 and U266B1 cells was monitored by MTT assay. (E and F) The G1 and S phase fractions in transfected cells were evaluated through flow cytometry. (G) The apoptotic rate in transfected cells was analyzed via flow cytometry. (H and I) The protein levels of cyclin D1, c-Myc, Bax and Bcl-2 were detected by western blot assay. *P < 0.05. | |
MTDH depletion blocked cell proliferation and cell cycle progression, as well as promoting apoptosis in OPM2 and U266B1 cells
Based on the above results, we further explored whether the functions of MTDH were similar to those of PCAT1. Also, the protein level of MTDH was obviously increased in MM cells MM1S, RPMI-8226, OPM2 and U266B1 compared with that in nPCs cells (Fig. 3A). The western blot assay showed that the protein level of MTDH was dramatically down-regulated in OPM2 and U266B1 cells transfected with sh-MTDH in comparison with that in the sh-NC group (Fig. 3B). Furthermore, the transfection of sh-MTDH notably decreased cell viability in OPM2 and U266B1 cells (Fig. 3C and D). Whereas the G1/S ratio was greatly accelerated in the sh-MTDH group (Fig. 3E and F). Also, the apoptotic rate was drastically speeded up in OPM2 and U266B1 cells transfected with sh-MTDH (Fig. 3G). In addition, the introduction of sh-MTDH resulted in a significant decline in cyclin D1, c-Myc, and Bcl-2 protein levels, as well as a striking increase in the Bax protein level in sh-MTDH-transfected OPM2 and U266B1 cells (Fig. 3H and I). To sum up, MTDH knockdown retarded cell proliferation and cell cycle progression, but accelerated apoptosis in OPM2 and U266B1 cells.
 |
| | Fig. 3 MTDH depletion blocked cell proliferation, as well as promoting G1/S ratio and cell apoptosis in OPM2 and U266B1 cells. (A) The protein level of MTDH in nPCs, MM1S, RPMI-8226, OPM2 and U266B1 cells was tested by western blot assay. (B–I) The OPM2 and U266B1 cells were transfected with sh-MTDH or sh-NC. (B) The protein level of MTDH in transfected cells was detected via western blot assay. (C and D) The cell viability of transfected OPM2 and U266B1 cells was monitored by MTT assay. (E and F) The G1 and S phase fractions in transfected cells were evaluated through flow cytometry. (G) The apoptotic rate in transfected cells was analyzed via flow cytometry. (H and I) The protein levels of cyclin D1, c-Myc, Bax, and Bcl-2 were detected by western blot assay. *P < 0.05. | |
PCAT1 silencing impeded cell proliferation and cell cycle progression, but facilitated apoptosis in OPM2 and U266B1 cells by regulating MTDH
To explore whether PCAT1 regulated cell behavior in MM cells was mediated by MTDH, sh-PCAT1 and MTDH were transfected into OPM2 and U266B1 cells. Firstly, the mRNA and protein levels of PCAT1 were low expressed in sh-PCAT1-transfected OPM2 and U266B1 cells, but obviously elevated in OPM2 and U266B1 cells transfected with PCAT1 (Fig. 4A and B). As exhibited in Fig. 4C and D, the transfection of MTDH alleviated the suppressive impact on cell viability in OPM2 and U266B1 cells retarded by sh-PCAT1. Meanwhile, the facilitated effect on G1/S ratio induced by sh-PCAT1 was weakened by MTDH in OPM2 and U266B1 cells, (Fig. 4E and F). Also, the apoptotic rate was mitigated by the introduction of MTDH in OPM2 and U266B1 cells, facilitated by PCAT1 silencing (Fig. 4G). In addition, the transfection of sh-PCAT1 counteracted the inhibitory impact on the protein levels of cyclin D1, c-Myc, and Bcl-2 and the accelerated effect on the protein level of Bax in OPM2 and U266B1 cells caused by sh-PCAT1 (Fig. 4H and I). These data revealed that PCAT1 depletion confined cell proliferation while promoting cell cycle arrest and apoptosis in OPM2 and U266B1 cells by regulating MTDH.
 |
| | Fig. 4 PCAT1 silencing impeded cell proliferation and cell cycle progression, but facilitated cell apoptosis in OPM2 and U266B1 cells by positively regulating MTDH. (A and B) The OPM2 and U266B1 cells were transfected with sh-NC, sh-PCAT1, pcDNA, or PCAT1. (A) The level of MTDH in transfected cells was measured by qRT-PCR. (B) The protein level of MTDH in transfected cells was detected by western blot assay. (C–I) The OPM2 and U266B1 cells were transfected with sh-NC, sh-PCAT1, sh-PCAT1 + pcDNA, or sh-PCAT1 + MTDH. (C and D) The cell viability of transfected cells was monitored via MTT assay. (E and F) The G1 and S phase fractions in transfected cells were evaluated through flow cytometry. (G) The apoptotic rate in transfected cells was analyzed through flow cytometry. (H and I) The protein levels of cyclin D1, c-Myc, Bax, and Bcl-2 in transfected cells were detected by western blot assay. *P < 0.05. | |
PCAT1 up-regulated MTDH expression in MM cells by sponging miR-363-3p
Interestingly, we found that miR-363-3p was notably down-regulated in MM bone marrow plasma and cells (Fig. 5A and B). In addition, the level of miR-363-3p was negatively linearly correlated with PCAT1 and MTDH (Fig. 5C and D). To illustrate the mechanisms of PCAT1 and MTDH in MM, miRcode and starBase v3.0 online databases were utilized to search for miRNAs which had complementary base pairing with PCAT1 and MTDH. The results implied that miR-363-3p had complementary sequences with PCAT1 or MTDH 3′UTR (Fig. 5E and F). A dual-luciferase reporter assay demonstrated that the transfection of miR-363-3p contributed to the prominent decrease in luciferase activity of WT-PCAT1 or MTDH 3′UTR-WT reporter compared with that in the miR-NC group, while the luciferase activity of MUT-PCAT1 or MTDH 3′UTR-MUT reporter showed no effective fluctuation in any group (Fig. 5G–J). In addition, the level of miR-363-3p was dramatically enhanced in sh-PCAT1-transfected OPM2 and U266B1 cells, but substantially declined in the PCAT1 group (Fig. 5K). Moreover, the mRNA and protein levels of MTDH were both obviously down-regulated in OPM2 and U266B1 cells transfected with miR-363-3p, but partially retained in OPM2 and U266B1 cells co-transfected with miR-363-3p and PCAT1 (Fig. 5L and M). These data disclosed that PCAT1 positively regulated MTDH expression in OPM2 and U266B1 cells via miR-363-3p.
 |
| | Fig. 5 MiR-363-3p was a direct target of PCAT1 and MTDH, and PCAT1 positively regulated MTDH expression by sponging miR-363-3p. (A and B) The level of miR-363-3p in MM bone marrow plasma and cells. (C and D) The correlations between miR-363-3p and PCAT1 or MTDH were analyzed by the Pearson test. (E and F) The complementary binding sites between miR-363-3p and PCAT1 or MTDH 3′UTR are shown, as well as the sequences of a corresponding negative control. (G–J) The OPM2 and U266B1 cells were transfected with miR-363-3p or miR-NC. The luciferase activities of WT-PCAT1, MUT-PCAT1, MTDH 3′UTR-WT, or MTDH 3′UTR-MUT reporter were assessed via a dual-luciferase reporter assay. (K) The level of miR-363-3p in OPM2 and U266B1 cells transfected with sh-NC, sh-PCAT1, pcDNA or PCAT1 was examined by qRT-PCR. (L and M) The OPM2 and U266B1 cells were transfected with miR-NC, miR-363-3p, miR-363-3p + pcDNA, or miR-363-p + PCAT1. (L) The level of MTDH in transfected cells was monitored via qRT-PCR. (M) The protein level of MTDH in transfected cells was evaluated by western blot assay. *P < 0.05. | |
PCAT1 silencing modulated miR-363-3p and MTDH expression in OPM2 and U266B1 cells via the AKT/β-catenin signaling pathway
To explore whether the AKT/β-catenin signaling pathway was implicated in the MM progression, the levels of p-AKT and p-β-catenin were tested in OPM2 and U266B1 cells. A western blot assay showed that the emergence of anti-miR-363-3p or MTDH partly restored the protein levels of p-AKT and p-β-catenin in OPM2 and U266B1 cells constrained by PCAT1 knockdown (Fig. 6A and B). These results elucidated that PCAT1 regulated miR-363-3p and MTDH expression in MM cells by activating the AKT/β-catenin signaling pathway.
 |
| | Fig. 6 PCAT1 silencing modulated miR-363-3p and MTDH expression in OPM2 and U266B1 cells via the AKT/β-catenin signaling pathway. (A and B) The protein levels of AKT, p-AKT, β-catenin, and p-β-catenin in OPM2 and U266B1 cells transfected with sh-NC, sh-PCAT1, sh-PCAT1 + anti-NC, sh-PCAT1 + anti-miR-363-3p, sh-PCAT1 + pcDNA, or sh-PCAT1 + MTDH were detected via western blot assay. *P < 0.05. | |
PCAT1 depletion retarded xenograft tumor growth in vivo
Based on the above results, we found that PCAT1 knockdown inhibited MM cell proliferation, but promoted cell cycle arrest and cell apoptosis in vitro. To further confirm the PCAT1 functions on MM progression in vivo, the U266B1 cells stably transfected with sh-PCAT1 were inoculated into nude mice. As presented in Fig. 7A–C, the volume and weight of xenograft tumor were both dramatically reduced in the sh-PCAT1 group in comparison with those in the sh-NC group. Furthermore, the levels of PCAT1 and MTDH both declined remarkably in the sh-PCAT1 group, while miR-363-3p was distinctly enhanced in the sh-PCAT1 group (Fig. 7D). In addition, the protein level of MTDH was also notably decreased in the sh-PCAT1 group (Fig. 7E). These data revealed that the xenograft tumor growth was blocked by PCAT1 silencing in vivo.
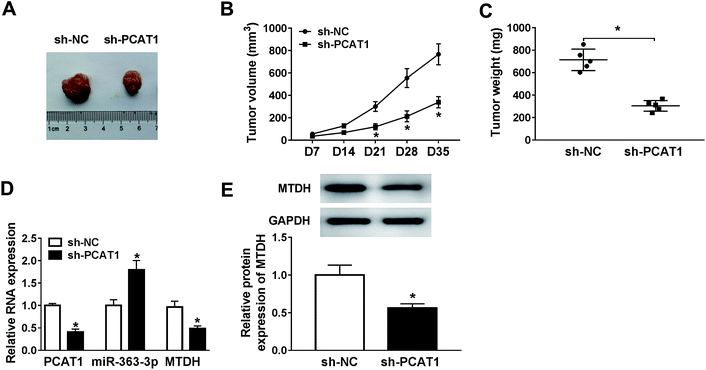 |
| | Fig. 7 PCAT1 depletion retarded xenograft tumor growth in vivo. (A–E) Nude mice were injected with U266B1 cells transfected with sh-PCAT1 or sh-NC. (A–C) The tumor volume and weight were measured. (D) The levels of PCAT1, miR-363-3p, and MTDH in xenograft tumors were assessed via qRT-PCR. (E) The protein level of MTDH was detected by western blot assay. *P < 0.05. | |
Discussion
MM ranks as the second most common type of blood cancer to leukemia. Convincing data indicated that PCAT1 plays a crucial role in tumor progression. In this study, we mainly explored the molecular mechanisms of PCAT1 and MTDH in MM.
Mounting evidence showed that the aberrant expression of PCAT1 was related to multiple types of cancers. For instance, a study of endometrial carcinoma (EC) showed that PCAT1 was distinctly enhanced in EC tissues and cells; cell growth and metastasis were blocked, while cell apoptosis was enhanced by PCAT1 silencing.28 Another report of oesophageal squamous cell carcinoma (ESCC) demonstrated the high expression of PCAT1 in ESCC promoted cell growth and cell cycle progression.29 In this research, a high expression of PCAT1 was found in MM bone marrow plasma and cells. This result was consistent with that reported by Shen et al.17 Furthermore, the depletion of PCAT1 suppressed cell proliferation and the AKT/β-catenin signaling pathway, but accelerated cell cycle arrest and cell apoptosis in OPM2 and U266B1 cells. In addition, PCAT1 knockdown restrained xenograft tumor growth in vivo. These results revealed that PCAT1 promoted cell growth in MM through the AKT/β-catenin signaling pathway.
Accumulating data showed that MTDH was implicated in tumor progression, including MM. For example, Chen et al. suggested that the promoted impacts on cell proliferation, metastasis and the repressive impact on cell apoptosis in pancreatic cancer were caused by the overexpression of MTDH.30 Similar results were also documented in non-small cell lung cancer.31 Currently, MTDH is up-regulated in MM bone marrow plasma and cells. MTDH silencing constrains cell proliferation by inducing cell cycle arrest, while boosting cell apoptosis in MM cells. Furthermore, MTDH overexpression attenuates the inhibitory impacts on cell proliferation, cell cycle progression and the AKT/β-catenin signaling pathway, as well as facilitating the impact on cell apoptosis in MM cells caused by PCAT1 knockdown. Previous reports were in agreement with our data.26,27 These data elucidated that PCAT1 positively modulates MTDH expression to promote cell growth in MM through the AKT/β-catenin signaling pathway.
Recent explorations have revealed that miR-363-3p is also associated with cancer development, including MM. For example, miR-363-3p retarded colorectal cancer progression by targeting EZH2.32 Also, miR-363-3p overexpression blocked non-small cell lung cancer development through regulating NEDD9 and SOX4.33 At present, miR-363-3p is down-regulated in MM bone marrow plasma and cells, and negatively correlated with PCAT1 or MTDH. Although miR-363-3p has not been reported in MM, a study of MM demonstrated that low expression of its analogue miR-363 accelerated cancer development through the AKT/β-catenin signaling pathway in MM by modulating Runx2.20 Furthermore, miR-363-3p was validated as a candidate target of PCAT1 and MTDH. In addition, PCAT1 enhanced MTDH expression in MM cells by sponging miR-363-3p. These data unraveled that PCAT1 positively modulated MTDH expression to promote cell growth in MM through the AKT/β-catenin signaling pathway by sponging miR-363-3p.
Conclusions
In conclusion, PCAT1 and MTDH were up-regulated, while miR-363-p was down-regulated in MM. The functional and mechanistic experiments demonstrated that PCAT1 promoted cell growth through the AKT/β-catenin signaling pathway in MM, partly via the miR-363-3p/MTDH axis. This novel regulatory network may shed light on the mechanism of MM and may provide a therapeutic target for MM patients.
Funding
This work was supported by Key R&D Program Projects of Shaanxi Province (no. 2018SF-045).
Abbreviations
| MM | Multiple Myeloma |
| lncRNA | Long non-coding RNA |
| PCAT1 | Prostate cancer associated transcript 1 |
| miRNA | MicroRNA |
| MTDH | Metadherin |
| qRT-PCR | Quantitative real-time polymerase chain reaction |
| GAPDH | Glyceraldehyde 3-phosphate dehydrogenase |
| MTT | 3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide |
| nPCs | Neural progenitor cells |
| DMEM | Dulbecco's modified Eagle's medium |
| FBS | Fetal bovine serum |
| DMSO | Dimethyl sulfoxide |
| FITC | Fluorescein isothiocyanate |
| PI | Propidium iodide |
| PBS | Phosphate buffer solution |
| SDS-PAGE | Sodium dodecyl sulfonate-polyacrylamide gel electrophoresis |
| PVDF | Polyvinylidene fluoride |
| AKT | Protein kinase B |
Conflicts of interest
There are no conflicts to declare.
References
- S. V. Rajkumar and S. Kumar, Mayo Clin. Proc., 2016, 91, 101–119 CrossRef.
- S. V. Rajkumar, Am. J. Hematol., 2014, 89, 998–1009 CrossRef.
- S. V. Rajkumar, M. A. Dimopoulos, A. Palumbo, J. Blade, G. Merlini, M. V. Mateos, S. Kumar, J. Hillengass, E. Kastritis and P. Richardson, Lancet Oncol., 2014, 15, e538–e548 CrossRef.
- E. Hatzimichael, A. Dasoula, L. Benetatos, N. Syed, G. Dranitsaris, T. Crook and K. Bourantas, Leuk. Lymphoma, 2010, 51, 2270–2274 CrossRef CAS.
- A. J. Greenberg, S. V. Rajkumar and C. M. Vachon, Blood, 2012, 119, 5359–5366 CrossRef CAS.
- R. Kyle and S. V. Rajkumar, Leukemia, 2009, 23, 3–9 CrossRef CAS.
- G. Yang, X. Lu and L. Yuan, Biochim. Biophys. Acta, 2014, 1839, 1097–1109 CrossRef CAS PubMed.
- S. F. Cho, Y. C. Chang, C. S. Chang, S. F. Lin, Y. C. Liu, H. H. Hsiao, J. G. Chang and T. C. Liu, BMC Cancer, 2014, 14, 809 CrossRef PubMed.
- D. Gao, A. e. Lv, H. P. Li, D. H. Han and Y. P. Zhang, J. Cell. Biochem., 2017, 118, 3341–3348 CrossRef CAS.
- L. Chen, N. Hu, C. Wang, H. Zhao and Y. Gu, Cell Cycle, 2018, 17, 319–329 CrossRef CAS.
- Y. B. Meng, X. He, Y. F. Huang, Q. N. Wu, Y. C. Zhou and D. J. Hao, Oncol. Res., 2017, 25, 1207–1214 CrossRef.
- Y. Pan, H. Chen, X. Shen, X. Wang, S. Ju, M. Lu and H. Cong, Clin. Chim. Acta, 2018, 480, 199–205 CrossRef CAS.
- J. R. Prensner, M. K. Iyer, O. A. Balbin, S. M. Dhanasekaran, Q. Cao, J. C. Brenner, B. Laxman, I. A. Asangani, C. S. Grasso and H. D. Kominsky, Nat. Biotechnol., 2011, 29, 742 CrossRef CAS.
- B. Zhao, X. Hou and H. Zhan, Int. J. Clin. Exp. Med., 2015, 8, 18482–18487 CAS.
- L. Liu, Y. Liu, C. Zhuang, W. Xu, X. Fu, Z. Lv, H. Wu, L. Mou, G. Zhao and Z. Cai, Tumor Biol., 2015, 36, 7685–7689 CrossRef CAS.
- W. h. Shi, Q. q. Wu, S. q. Li, T. x. Yang, Z. h. Liu, Y. s. Tong, L. Tuo, S. Wang and X. F. Cao, Tumor Biol., 2015, 36, 2501–2507 CrossRef CAS.
- X. Shen, Y. Zhang, X. Wu, Y. Guo, W. Shi, J. Qi, H. Cong, X. Wang, X. Wu and S. Ju, Cancer Biomarkers, 2017, 18, 257–263 CAS.
- M. Esteller, Nat. Rev. Genet., 2011, 12, 861–874 CrossRef CAS.
- N. Gupta, R. Kumar, T. Seth, B. Garg, H. C. Sati and A. Sharma, J. Cancer Res. Clin. Oncol., 2019, 145, 1601–1611 CrossRef CAS.
- P. S. Gowda, B. J. Wildman, T. N. Trotter, X. Xu, X. Hao, M. Q. Hassan and Y. Yang, Mol. Cancer Res., 2018, 16, 1138–1148 CrossRef CAS.
- H. Wang, Q. Ding, M. Wang, M. Guo and Q. Zhao, Hematology, 2019, 24, 32–38 CrossRef CAS.
- G. Hu, R. A. Chong, Q. Yang, Y. Wei, M. A. Blanco, F. Li, M. Reiss, J. L.-S. Au, B. G. Haffty and Y. Kang, Cancer Cell, 2009, 15, 9–20 CrossRef CAS.
- C. Yu, K. Chen, H. Zheng, X. Guo, W. Jia, M. Li, M. Zeng, J. Li and L. Song, Carcinogenesis, 2009, 30, 894–901 CrossRef CAS PubMed.
- W. Chen, Z. Ke, H. Shi, S. Yang and L. Wang, Neoplasma, 2010, 57, 522–529 CrossRef CAS.
- L. Emdad, S.-G. Lee, Z. Z. Su, H. Y. Jeon, H. Boukerche, D. Sarkar and P. B. Fisher, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 21300–21305 CrossRef CAS.
- C. Gu, L. Feng, H. Peng, H. Yang, Z. Feng and Y. Yang, Oncotarget, 2016, 7, 4559–4569 Search PubMed.
- B. Zhu, H. Chen, X. Zhang, Y. Pan, R. Jing, L. Shen, X. Wang, S. Ju, C. Jin and H. Cong, Int. J. Oncol., 2018, 53, 2131–2144 CAS.
- X. Zhao, Y. Fan, C. Lu, H. Li, N. Zhou, G. Sun and H. Fan, Bosnian J. Basic Med. Sci., 2019, 19, 274–281 Search PubMed.
- L. Huang, Y. Wang, J. Chen, Y. Wang, Y. Zhao, Y. Wang, Y. Ma, X. Chen, W. Liu and Z. Li, Cell Death Dis., 2019, 10, 513 CrossRef.
- Z. Chen, Y. Ma, Y. Pan, H. Zhu, C. Yu and C. Sun, Mol. Cell. Probes, 2018, 40, 19–26 CrossRef CAS.
- Q. Lu, S. Shan, Y. Li, D. Zhu, W. Jin and T. Ren, FASEB J., 2018, 32, 3957–3967 CrossRef CAS.
- J. Xie, W. Li, X. Li, W. Ye and C. Shao, J. Biol. Regul. Homeostatic Agents, 2019, 33, 331–343 CAS.
- J. Chang, F. Gao, H. Chu, L. Lou, H. Wang and Y. Chen, J. Cell. Physiol., 2019 DOI:10.1002/jcp.29099.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra06188f |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *b
*b