
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLaccase-immobilized tannic acid-mediated surface modification of halloysite nanotubes for efficient bisphenol-A degradation†
Liting Zhang ac,
Wen Tangb,
Tonghao Mac,
Lina Zhouc,
Chenggong Huia,
Xiaoli Wangc,
Ping Wangd,
Changai Zhang*a and
Chao Chen*c
ac,
Wen Tangb,
Tonghao Mac,
Lina Zhouc,
Chenggong Huia,
Xiaoli Wangc,
Ping Wangd,
Changai Zhang*a and
Chao Chen*c
aZhejiang Province Key Laboratory of Recycling and Eco-treatment of Waste Biomass, Zhejiang University of Science and Technology, Hangzhou, 310023, China. E-mail: zca2006@sina.com
bState Key Laboratory of Bioreactor Engineering, New World Institute of Biotechnology, East China University of Science and Technology, No. 130 Meilong Road, Shanghai 200237, People's Republic of China
cState Key Laboratory of Bioreactor Engineering, Biomedical Nanotechnology Center, School of Biotechnology, East China University of Science and Technology, Shanghai 200237, People's Republic of China. E-mail: chaochen@ecust.edu.cn
dDepartment of Bioproducts and Biosystems Engineering, University of Minnesota, St Paul, MN 55108, USA
First published on 27th November 2019
Abstract
Halloysite nanotubes (HNTs) have been pursued as promising carriers for enzyme immobilization, but the lack of functional groups severely limits their applications. Herein, we reported a simple tannic acid (TA)-mediated surface modification strategy for the fabrication of HNT-based efficient enzyme immobilization supports. Particularly, TA was first self-polymerized and deposited onto the surface of HNTs to form a thin active film via a mussel-inspired method, and the model enzyme laccase was directly conjugated via the Michael addition and/or Schiff base condensation between quinone groups on poly(tannic acid) layer surfaces and exposed amine groups on laccase surfaces. Under the optimum conditions, this newly fabricated support retained good enzyme-loading and activity recovery properties with 197.9 mg protein per gram of support and 55.4% of activity recovery being achieved. In addition, this immobilized laccase was less influenced by pH, temperature, and inhibitor changes and exhibited higher storage stability than free laccases as more than 70% of initial activity was retained by the immobilized laccase, while less than 30% was retained for free laccase after one-month storage at 4 °C. Finally, a higher bisphenol-A (BPA) removal efficiency and more reuse cycles were demonstrated for immobilized laccases. As a result, this TA-mediated surface modification is a simple and green method for biological macromolecule immobilization on HNTs in one step.
Introduction
In recent years, the accumulation and spread of persistent organic contaminants in water environment has received considerable attention as it is a great threat to environment and human health.1–3 Particularly, phenolic compounds, most of which are poorly degradable and could impose teratogenic, mutagenic, carcinogenic and toxic effects on humans and animals even at low concentrations, are listed as top pollutants by the US Environmental Protection Agency.4 Till date, many researchers have made great endeavors for phenolic pollutant removal or degradation, among which enzyme-based treatment has been one of the most sustainable and promising strategies due to the green treatment process and high efficiency in the removal of the pollutants.5–7Laccases are multi-copper oxidases produced by numerous fungi, bacteria and plants, which can oxidize a wide range of phenolic compounds with simultaneous reduction of molecular oxygen to water.8,9 Moreover, due to their high catalytic activity and wide substrate specificity, laccases have received great attention for the treatment of phenol-contaminated wastewater. However, free laccases are not widely used in practical applications due to their low stability, and the lack of reusability induced high cost.10 Previous studies have revealed that high-density immobilization of laccases has been proved to be an effective and the most straightforward strategy to address these deficiencies along with extra advantages, such as continuous treatment.4,11
Till date, laccases have been immobilized on many different kinds of supports, including inorganic, organic and polymeric materials.12,13 Halloysite nanotube (HNT)-based immobilization systems have received great attention due to their tubular structure, low cost, good biocompatibility and environmentally friendly nature.14,15 However, the absence of active reactive groups and intrinsic inorganic property of HNTs lead to weak noncovalent bonding between HNTs and enzymes. Additionally, HNTs tend to form agglomerates or bundles, resulting in reduced effective specific surface area for enzyme immobilization.16 To address these issues, biomimetic modification of HNTs provides a facile and promising approach. Recently, tannic acid (TA)-based enzyme immobilization supports have been demonstrated to be versatile platforms for enzyme immobilization.17,18 Tannic acid, as a kind of natural water-soluble polyphenol, can self-polymerize and hence, they can be coated onto support surfaces.19–21 The formed poly(tannic acid) layer possesses abundant quinone groups, which could efficiently bind with nucleophilic functional groups such as thiol and amine groups via the Michael addition and/or Schiff base reactions in one step.17,20,22 As a result, enzyme molecules with thiol and amine groups can be directly immobilized on HNTs via covalent bonding.
Based on the above-mentioned considerations, we introduced a tannic acid (TA)-mediated surface modification strategy for the fabrication of HNT-based enzyme immobilization supports. Laccase was chosen as the model enzyme to demonstrate the functionalities of our designed support for enzyme immobilization. Meanwhile, the enzyme properties including catalytic activity and environmental tolerance were explored. Furthermore, preliminary molecular dynamic simulation was adapted to explain the relationships between enzyme activity, stability and structure changes after immobilization. Finally, a potential industrial application for the removal of BPA was also investigated.
Material and methods
Materials
Tannic acid (TA) was purchased from Adamas Reagent Co., Ltd (Shanghai, China). 2,2′-Azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) (ABTS) and BPA (99%) were supplied by Aladdin Reagent Co., Ltd (Shanghai, China). Freeze-dried laccase obtained from Trametes versicolor (protein content is higher than 98% and specific activity is 1.07 U mg−1 solid) was provided by Yuanye Biotech. Co., Ltd (Shanghai, China). Halloysite nanotubes (HNTs) were purchased from Sigma-Aldrich (Shanghai, China). Other chemicals were of analytical grade and used without further purification.Preparation of tannic acid-functionalized halloysite nanotubes (HNTs-TA)
Halloysite nanotubes (HNTs) were modified with tannic acid by a mussel-inspired method. Specifically, 40 mg of HNTs was dispersed into 40 mL Tris-HCl solution (100 mM, pH 8.5) containing 500 μg mL−1 tannic acid (TA). After being ultrasonicated for 30 min, the mixture was shaken for 24 h at room temperature. Finally, the TA-modified HNTs (denoted as HNTs-TA) were separated from the mixture by centrifugation at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 100 rpm for 10 min.
100 rpm for 10 min.
Immobilization of laccases on HNTs-TA
In the typical enzyme immobilization process, a certain amount of laccase was added into 10 mL of PBS solution (100 mM, pH 7.5) containing 100 mg of HNT-TA in each flask. After immobilization for 6 h, the immobilized laccase was collected by centrifugation and then washed several times by PBS solution. Subsequently, the amount of laccase immobilized on HNT-TA was calculated by determining the initial and final concentrations of laccase in liquid solution, using a typical Bradford protein assay method.23 For the influence of immobilization time on enzyme loading and apparent activity of the immobilized enzyme, 3 mg mL−1 enzyme and 10 mg mL−1 HNT-TA were added, respectively, into 10 mL of PBS solution in the flask. The enzyme loading and apparent activity of immobilized laccase were measured every 2 h. The relative activity was calculated from the ratio between the apparent activity and the optimized apparent activity.Assays of free and immobilized laccase activity
The enzyme activity of free and immobilized laccase was measured by monitoring the oxidation of 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) (ABTS) used as a substrate according to a previous study.24 Specifically, 10 μL of pre-diluted free or immobilized laccase solution was added into sodium acetate buffer (100 mM, pH 5.0) containing 1 mM of ABTS for 5 min. The oxidation of ABTS to its cation radical (ABTS+) was measured using a U-5100 UV-vis spectrometer (Hitachi, Japan) at 420 nm. One activity unit (U) of laccase was defined as the amount of enzyme required to oxidize 1 μmol of substrate per minute.25Molecular dynamic simulation of free and immobilized laccase
In order to explore the structural change of laccase before and after immobilization, molecular dynamic (MD) simulation of free and immobilized enzyme was conducted according to the method reported previously with some modifications.26,27 In brief, the crystal structure of laccase (PDB ID: 1KYA) was first download from Protein Data Bank (http://www.rcsb.org/). Subsequently, GROMACS 5.1.5 and AmberTool 18 with ff14SB as well as gaff force field were used to study the enzyme conformation change before and after immobilization. The free and immobilized laccases were solvated with TIP3P water, and Na+ was used to neutralize the system charge. Then, the steepest descent and conjugate gradient energy minimization were employed to optimize the system. Finally, 20 ns MD simulation of free and immobilized laccases was performed after NPT optimizations. After 20 ns MD simulation, the visual analysis of the enzyme structure was carried out using PyMOL-edu.Properties of free and immobilized laccases
In order to compare the stability of free and immobilized laccases in different pH and temperature environments, the free and immobilized laccases were incubated in buffers of different pH values ranging from 3 to 9 at room temperature for a certain time, or in PBS buffer at different temperatures (20–80 °C), respectively. Then the residual activity was determined and calculated from the ratio between activity and the optimized activity at predetermined time. Besides, the storage stability at 4 °C was further investigated in PBS (100 mM, pH 7.4), and the relative activity was calculated from the ratio between residual activity and the initial activity before storage. To investigate the influence of activators/inhibitors (metal ions, EDTA, SDS and organic solvents) on the laccase activity, free and immobilized laccases were pre-incubated in PBS (100 mM, pH 5.0) for a certain time in the presence of 0.5 mM Cu2+, Mn2+, EDTA, SDS or 20% (v/v) of acetonitrile, and DMSO, respectively. Finally, the percent relative activity in each case represents the enzyme activity relative to the control (without any activators/inhibitors), which was taken as 100%.Bisphenol-A removal experiments
To determine and compare the removal efficiency of bisphenol-A (BPA) catalyzed by free and immobilized laccase, experiments were conducted in acetate buffer (100 mM, pH 6.0) solution containing 140 μM bisphenol-A. An equal amount of free or immobilized laccase was added into the buffer, and the reaction was conducted at 30 °C in an oven for 18 h with magnetic stirring at 500 rpm, using HNT-TA as the control to eliminate the interference induced by abiotic BPA adsorption. The samples were taken from the obtained mixture solution at regular intervals, and the BPA concentrations were quantitatively analyzed by HPLC according to previous studies.28 For the reusability evaluation, the reaction was continued for 18 h and was catalyzed by the same batch of immobilized laccases, which was repeated for 9 cycles. The laccase re-collected after centrifugation was washed with acetate buffer for three times before use in the next cycle, and the final concentration of BPA was recorded by HPLC.Characterization
The morphology and structure of HNTs and HNT-TA was characterized using a transmission electron microscope (JEOL, JEM-1400, Japan). A RINT2000 vertical goniometer (Rigaku, Japan) equipped with a Cu Kα irradiation source was used to conduct X-ray diffraction (XRD) analysis. The chemical structure of the modified HNTs was analyzed using a Fourier transform infrared spectrometer (Bruker, Switzerland). Thermogravimetric analysis (TGA) of HNTs and HNT-TA was performed using a TGA-50 thermogravimetry analyzer (Shimadzu, Japan). ESCALAB 250Xi (Thermo, MA) was applied to perform X-ray photoelectron spectroscopy (XPS) of HNTs and HNT-TA. The secondary structure of laccases before and after immobilization was analyzed using a Chirascan-plus system (Applied Photophysics, UK).Statistical analysis
All experiments were repeated thrice, and all statistical data were expressed as means ± standard errors. One-way analysis of variance (ANOVA) was used to determine the difference of each group in this study. In all cases, a p-value less than 0.05 was statistically significant.Results and discussion
Preparation and characterization of tannic acid-modified HNTs
The strategies of tannic acid (TA)-mediated surface modification of halloysite nanotubes (HNTs) and laccase immobilization are illustrated in Scheme 1A. TA was first polymerized and coated onto the surface of HNTs, and the formed polymer can be used as a reactive surface coating for laccase immobilization via the Schiff base formation and/or the Michael addition reaction between quinone groups on poly(tannic acid) layer surfaces and exposed amine groups on laccase surfaces, as shown in Scheme 1B.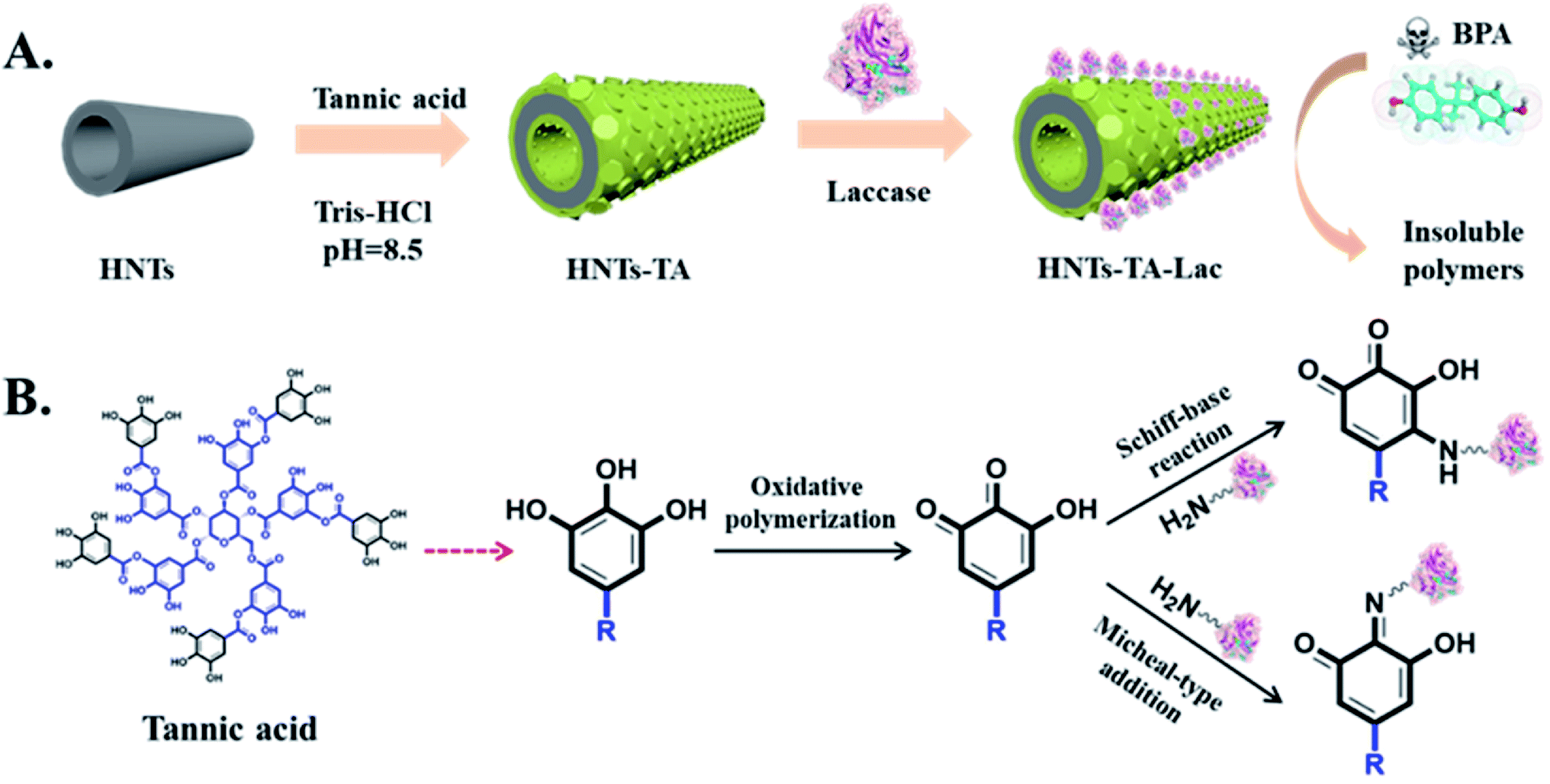 | ||
| Scheme 1 Schematic of (A) fabrication process of HNT-TA-Lac for the removal of BPA and (B) the mechanism of laccase immobilization. | ||
The structural morphology of original HNTs and TA-modified HNTs (HNT-TA) was first characterized by TEM. Specifically, Fig. 1A and B displays that pristine HNTs have cylindrical-shaped tubular morphology and open-ended lumen along the nanotubes, and the walls are smooth and clean. The lengths of the HNTs range from 100 to 5000 nm, the inner diameters range from 20 to 30 nm, and the shell thickness is about 15–20 nm. After coating with TA, the walls of HNTs changed rough and indistinct, and an organic layer with light gray color and about 20 nm in thickness was formed and located on the outer surfaces of HNTs (Fig. 1C and D). This phenomenon could be the most direct evidence for the presence of TA layer on the nanotube surfaces. Meanwhile, the zeta potential values were changed after TA modification. This indicated that a TA polymer layer had successfully modified the surface of HNTs (Fig. S1†).
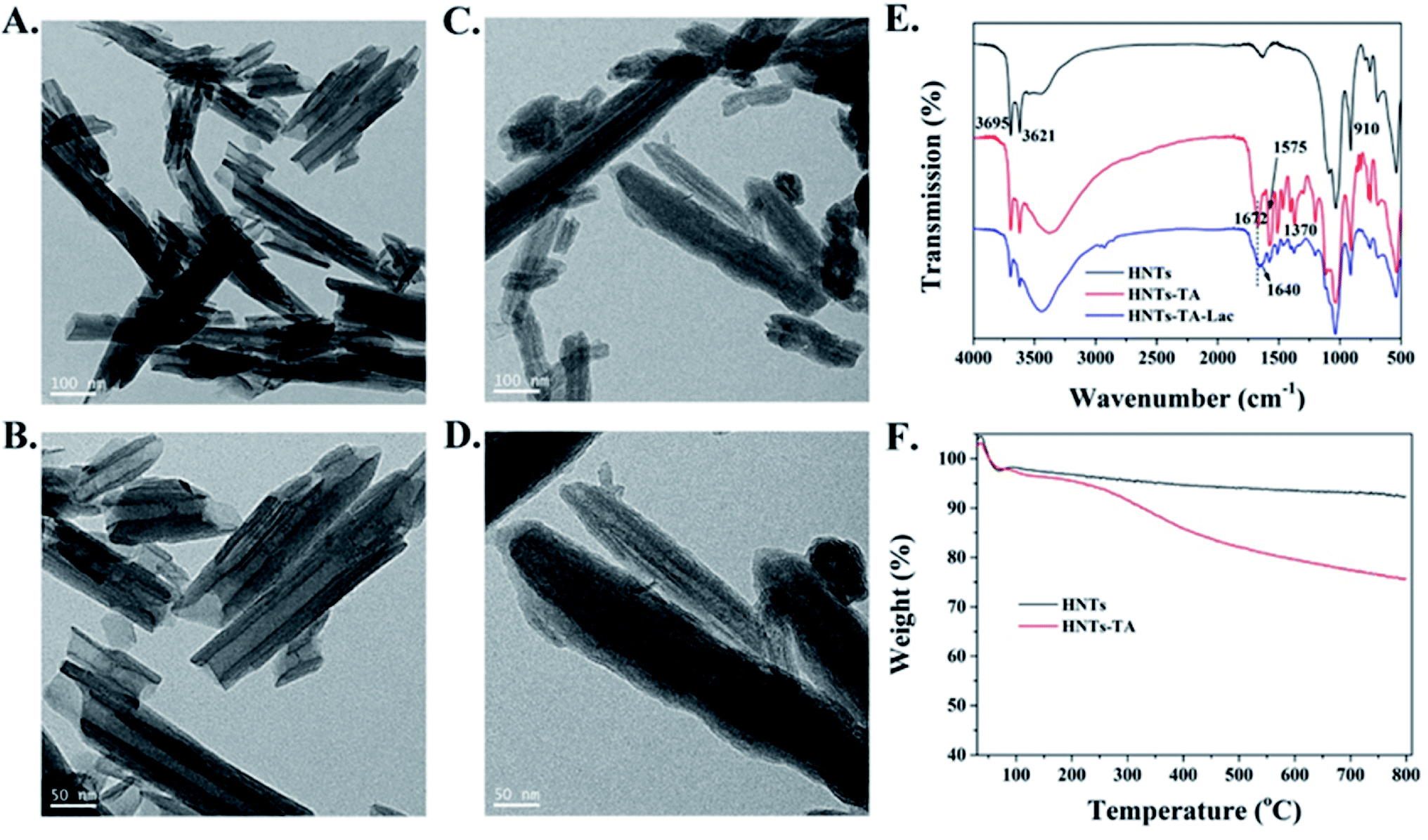 | ||
| Fig. 1 TEM images of the (A and B) HNTs and (C and D) HNTs-TA. (E) FTIR spectra of HNTs, HNT-TA and HNT-TA-Lac. (F) TGA curves of HNTs and HNT-TA. | ||
Furthermore, FT-IR spectroscopy was performed to characterize the chemical structure of both samples. As shown in Fig. 1E, pristine HNTs displayed two bands at 433 and 1087 cm−1, which correspond to the vibration modes of the Si–O network. Besides, the absorption bands at 3695 and 3621 cm−1 are ascribed to –OH groups. The characteristic bands at 910 cm−1 are attributed to Al–OH stretching vibrations. After the modification of HNTs with TA, a new absorption signal at 1672 cm−1 belongs to the absorbance of –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups of HNT-TA. The peaks at 1370 cm−1 in the spectrum of HNT-TA belong to phenol groups, and the absorption bands between 1600 and 1400 cm−1 are related to aromatic –CC– bonds. These results gave a very strong corroboration regarding the successful modification of TA on the surface of HNTs. After laccase immobilization, the changed relative peaks of HNT-TA-Lac appeared between 1000 and 1700 cm−1 region due to protein-tannate complex formation between laccase and some phenolic groups of tannin.17 Furthermore, the new absorption band at 1640 cm−1 is attributed to the formation of C = N in the Schiff base reaction between –CO of HNT-TA and amino groups of laccases, which directly indicated that the laccase was conjugated on the surface of HNT-TA.29 Furthermore, the XRD patterns of HNTs before and after modification with TA are shown in Fig. S2.† Both of them are in good agreement with previously published patterns for HNTs. Meanwhile, TGA analysis was also performed to characterize the process of surface modification of HNTs with TA (Fig. 1F). The weight loss for HNTs was about 7.7% of total weight, which was attributed to the loss of possible water residue. After modification with TA, an obvious weight loss of about 24.3% of HNT-TA can be observed, which can be attributed to the physically adsorbed water in HNTs, bounded water in hydrophilic TA layer and the degradation of TA layer. All results indicated that a novel immobilization support, HNT-TA, was successfully fabricated. Then, the cytotoxicity of the synthesized HNT-TA was examined by an MTT assay to examine whether “secondary” pollutant problems exist. From Fig. S3,† it can be observed that more than 90% cell viability of L-02 cells was retained for HNT-TA and higher than 80% for HNTs at the tested concentrates, ranging from 0 to 200 μg mL−1. This demonstrated good biocompatibility of HNTs and even better for HNT-TA.
O groups of HNT-TA. The peaks at 1370 cm−1 in the spectrum of HNT-TA belong to phenol groups, and the absorption bands between 1600 and 1400 cm−1 are related to aromatic –CC– bonds. These results gave a very strong corroboration regarding the successful modification of TA on the surface of HNTs. After laccase immobilization, the changed relative peaks of HNT-TA-Lac appeared between 1000 and 1700 cm−1 region due to protein-tannate complex formation between laccase and some phenolic groups of tannin.17 Furthermore, the new absorption band at 1640 cm−1 is attributed to the formation of C = N in the Schiff base reaction between –CO of HNT-TA and amino groups of laccases, which directly indicated that the laccase was conjugated on the surface of HNT-TA.29 Furthermore, the XRD patterns of HNTs before and after modification with TA are shown in Fig. S2.† Both of them are in good agreement with previously published patterns for HNTs. Meanwhile, TGA analysis was also performed to characterize the process of surface modification of HNTs with TA (Fig. 1F). The weight loss for HNTs was about 7.7% of total weight, which was attributed to the loss of possible water residue. After modification with TA, an obvious weight loss of about 24.3% of HNT-TA can be observed, which can be attributed to the physically adsorbed water in HNTs, bounded water in hydrophilic TA layer and the degradation of TA layer. All results indicated that a novel immobilization support, HNT-TA, was successfully fabricated. Then, the cytotoxicity of the synthesized HNT-TA was examined by an MTT assay to examine whether “secondary” pollutant problems exist. From Fig. S3,† it can be observed that more than 90% cell viability of L-02 cells was retained for HNT-TA and higher than 80% for HNTs at the tested concentrates, ranging from 0 to 200 μg mL−1. This demonstrated good biocompatibility of HNTs and even better for HNT-TA.
The changes in the surface chemical compositions of the HNT-TA were determined by XPS analysis. Fig. 2A and B shows that both HNTs and HNT-TA contain Al, Si, C and O elements. Furthermore, the increase in the C element content indirectly reflected that TA was successfully formed on the surface of HNTs. Besides, Fig. 2C exhibits that the peaks in the C 1s spectra at 290.5, 288.7, 286.2, 285.6 and 284.9 eV corresponded to CO–O, CO, C–O, C–OH and C–C, respectively. In the O 1s peak, two peaks were observed at 532.8 and 531.8 eV (Fig. 2D), which were attributed to the O atoms of polymerized TA in the form of catechol and quinone groups. According to the above-mentioned analysis, it is reasonable to conclude that a large number of enzyme-binding sites have been generated on the surface of HNT-TA.
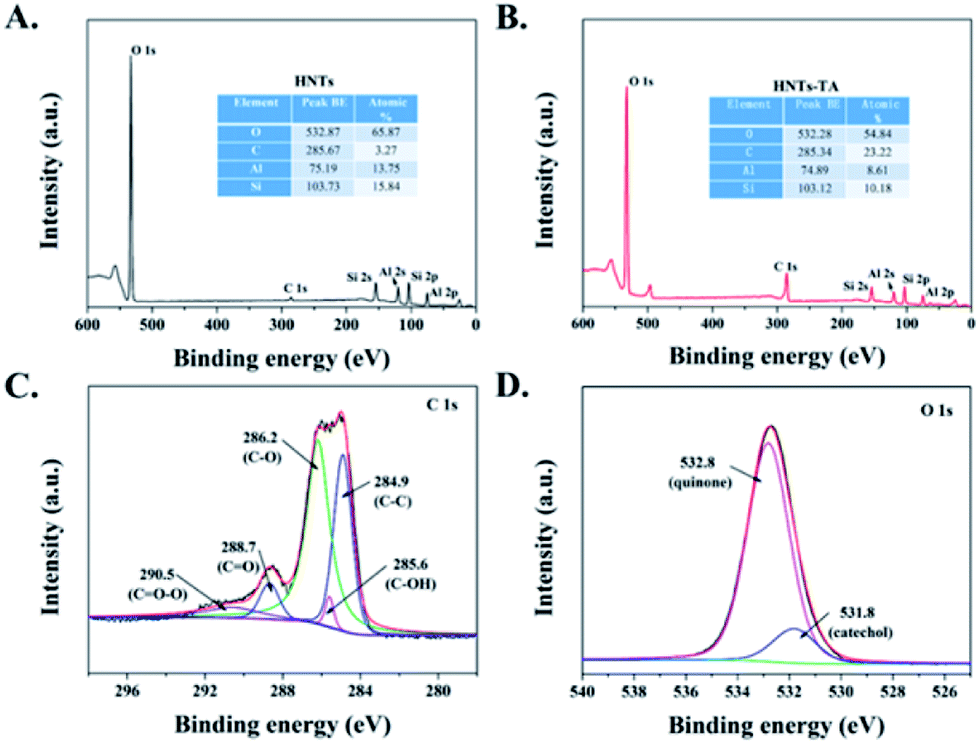 | ||
| Fig. 2 XPS survey spectra of (A) HNTs and (B) HNTs-TA. High-resolution XPS spectra of (C) C 1s and (D) O 1s peaks for HNTs-TA. | ||
Optimization of laccase immobilization
The concentration of enzymes used in the immobilization process could influence the enzyme loading and apparent activity on the support. Fig. 3A displays that the enzyme loading of HNT-TA is enhanced with the increasing concentration of laccase at 1–3 mg mL−1 range, where the amount of HNT-TA was kept at 10 mg mL−1. However, the apparent activity of immobilized laccase gradually decreased when the added laccase exceeded 300 mg g−1 support even when the enzyme loading still slightly increased. As shown in Fig. 3B, similar results were also observed in the immobilization time optimization that both enzyme loading and apparent activity of immobilized laccase improved with the time extension at the beginning 6–8 h, while the apparent activity decreased after 8 h. This phenomenon is similar to the previous research.12,30 The increase in the relative activity could be caused by the increase in enzyme density on the HNT-TA surface, while the decrease in relative activity could be attributed to the overcrowded enzyme hindering the substrate diffusion into the binding pocket.11 As a result, 300 mg g−1 support of added enzyme with 8 h of conjugation process was an optimum combination for laccase immobilization on HNTs-TA. Meanwhile, the enzyme loading and activity recovery of immobilized laccase on HNTs and HNT-TA were further compared in Table 1. Under this optimal condition, the enzyme loading of laccase on HNT-TA reached 197.9 mg g−1 support, which was 12.9 times as high as that on HNTs (15.4 mg g−1 support). Besides, the enzyme activity recovery on HNT-TA support (55.4%) was also higher than that on the HNT support (42.7%). These results indicated that TA-functionalized HNTs retained more advantages for enzyme immobilization, which could be explained by that pure HNTs tend to form agglomerates or bundles, resulting in reduced effective specific surface area for enzyme immobilization, while modification of tannic acid on HNTs could overcome the problem of pure HNTs and introduce abundant quinone groups to effectively immobilize laccase.31 Therefore, tannic acid-modified HNTs were promising supports for enzyme immobilization via covalent binding, exhibiting high loading capacity and activity recovery.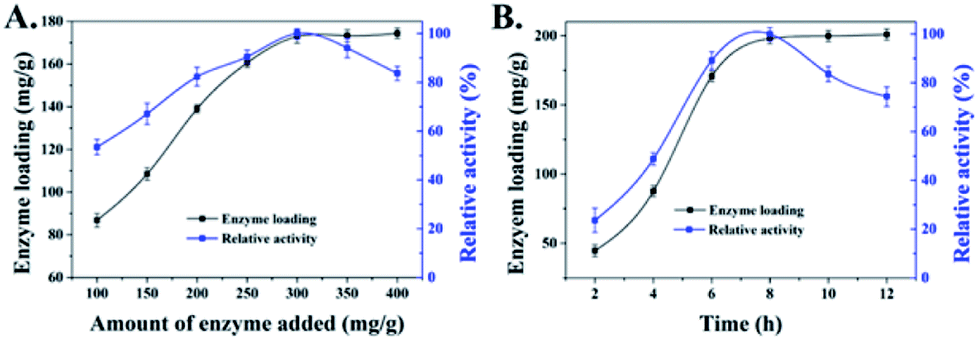 | ||
| Fig. 3 Dependence of the enzyme loading (black line) and relative activity (blue line) of laccase immobilized on HNT-TA on (A) different concentrations of enzyme added and (B) immobilization time. Data are represented as mean ± SD (n = 3). | ||
| Samples | Amount of laccase added (mg g−1) | Enzyme loading (mg g−1) | Immobilization efficiency (%) | Activity recovery (%) |
|---|---|---|---|---|
| HNTs | 300 | 15.4 | 5.1 | 42.7 |
| HNT-TA | 300 | 197.9 | 66.0 | 55.4 |
Enzymatic properties of free and immobilized laccases
The catalytic activity of the enzyme is easily affected by the microenvironment, especially under extreme conditions, which usually leads to enzyme inactivation.32 In this study, the effect of solution pH and temperature on the stability of free and immobilized laccases was further examined within the pH range from 3 to 9 (Fig. 4A) and at a temperature between 20 and 80 °C (Fig. 4B). For both free and immobilized laccases, the optimal pH was 6 and the optimal temperature was 50 °C, and the immobilized laccases are more stable with less influence from pH and temperature changes. Both observations might be due to the fact that the immobilized laccases would stabilize the biologically active conformation, which was consistent with some previous studies on immobilized laccases.33,34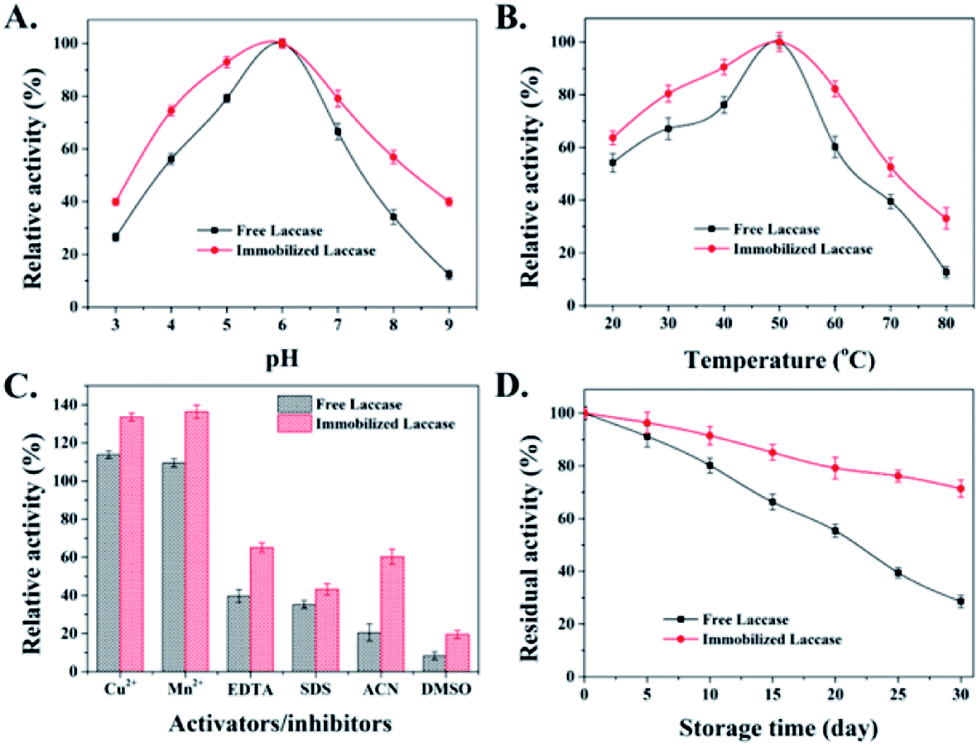 | ||
| Fig. 4 Effects of different (A) pH values and (B) temperatures on the enzyme activity of free and immobilized laccases. (C) Effects of activators/inhibitors (metal ions, EDTA, SDS and organic solvents) on the catalytic activity of free and immobilized laccases. The percent relative activity in each case represents the enzyme activity relative to the control (without any activators/inhibitors) which was taken as 100%. (D) The storage stability at 4 °C of free and immobilized laccases. Data are represented as mean ± SD (n = 3). | ||
The effects of different activators and inhibitors, such as metal ions, chelating agent (EDTA), detergent (SDS) and organic solvent, on free and immobilized laccase activity were also evaluated. As shown in Fig. 4C, free and immobilized laccases were activated by Cu2+ and Mn2+, which might be owing to the stabilization of the active conformation that stimulates the laccase activity.35 Besides, compared with the free enzyme, immobilized laccases exhibited lower inhibitory effects when exposed to EDTA, SDS, ACN and DMSO, which might be attributed to the theory that the restricted mobility and compacted conformation of the protein chain after enzyme immobilization could effectively alleviate the cofactor loss and protein unfolding under unfavorable conditions.35,36 Considering that the storage stability of an enzyme is also an important aspect for practical application, the storage stability of free and immobilized laccases was also investigated at 4 °C. As depicted in Fig. 4D, downward trends are presented for both free and immobilized laccases during 30 days of storage. However, the residual activity of immobilized laccases is much higher than that of free laccases. After 30 days, the residual activity of immobilized laccases was around 71.4%, whereas the activity of free laccases was only 28.6% of its initial activity. Taken together, it is reasonable to conclude that immobilized laccases exhibit enhanced stability and better tolerance on environmental changes.
Structural analysis of free and immobilized laccases
The overall secondary structure of laccases before and after immobilization was analyzed by circular dichroism (CD) spectra to probe the protein structure change caused by HNT-TA immobilization. According to the CD spectra in Fig. 5A, there exists a positive peak at around 198 nm and a negative peak at around 218 nm correlated with the β-sheet content of laccase. After immobilization, a decreased circular dichroism value of positive and negative peaks was observed, indicating that the native β-sheet content is lost to some extent. Such a phenomenon is common in reported enzyme immobilizations,37,38 while the overall secondary structure of immobilized laccases have not changed a lot. Additionally, the free and immobilized laccases were simulated by a molecular dynamic (MD) simulation for 20 ns to analyze the three-dimensional structural changes. In order to verify the convergence of simulations, the time-evolution of root-mean-square deviation (RMSD) of the simulation system is plotted in Fig. S4 and S5.† It can be found that a 20 ns MD simulation was sufficient to simulate the laccase immobilization as the RMSD reached equilibrium after 5 ns. As shown in Fig. 5B, there exist abundant β-sheets in free laccases. After laccase immobilization on HNT-TA, a sheet-loop transition was observed, which was consistent with the results in the CD spectra of immobilized laccases. Meanwhile, the reduced β-sheet was near to the binding pocket of laccases that might affect the substrate binding, thus leading to a declined specific activity after immobilization. Besides, the conformation changes of binding pockets in laccases covalently immobilized on HNTs-TA were further examined by RMSD (Fig. 5C). When laccases covalently immobilized on HNT-TA, the RMSD value of laccases reduced from 0.17 nm to 0.1 nm, which means that laccases bound to the surface of HNT-TA suffer less structural deviations of binding pockets than free laccases in the solution. This could be due to the interactions between the support and the enzyme causing constraints on the enzyme and leading to the alteration of the enzyme hydration shell and/or the reduction of enzyme flexibility.39,40 Therefore, the laccase immobilized on TA-functionalized HNTs basically maintains its native structure of binding pockets but it is more rigid. This simulation result may provide a proper explanation for the enzyme stabilization by covalent binding on the HNT-TA surface.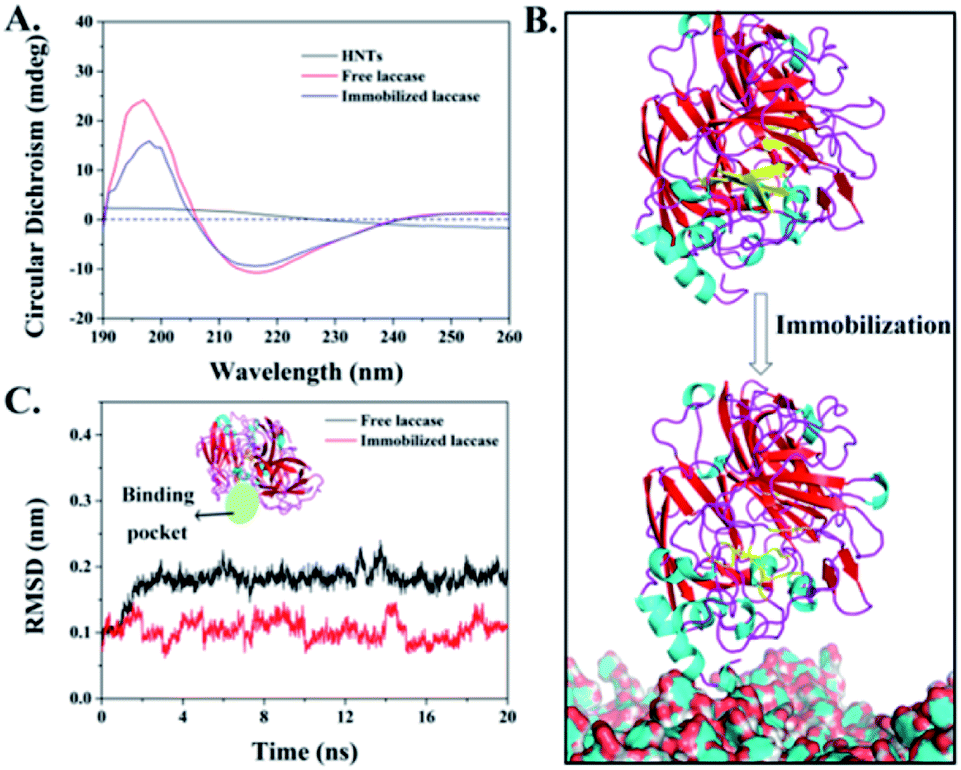 | ||
| Fig. 5 (A) CD spectra of free and immobilized laccase; (B) simulated structure of free and immobilized laccases at 20 ns. The absence of β-sheet (in yellow) in immobilized laccases compared with free laccases (in yellow) at the same position. (C) Time-evolution of RMSD of binding pockets in free and immobilized enzymes. | ||
Immobilized laccase-mediated BPA removal
In order to examine the feasibility of immobilized laccase used in practical phenol treatment, BPA was selected as a model contaminant, which can be oxidized by laccase in the presence of O2 to BPA radicals, which will further be self-polymerized to form insoluble macromolecular polymers and it can be easily precipitated and removed from the reaction solution by filtration or sedimentation (Fig. S6†).41–43 Fig. 6A shows that immobilized laccases exhibited higher BPA removal capacity with respect to free laccases that 80.2% and 60.4% of BPA were eliminated at the beginning 6 h, respectively. Meanwhile, the control HNT-TA also presented an obvious BPA decrease that about 8.9% of substrate was quickly absorbed under the experimental condition, which indicated a synergistic effect between the BPA enrichment on HNT-TA and laccase catalysis. The enhanced BPA removal efficiency might also be contributed by the high enzyme density on the HNT-TA support surface, and the ability of HNT-TA support protecting the laccase from changes in the microenvironment.44 It is worth to mention that unlike immobilized enzyme, free laccases are challenging to be recovered from the reaction solution to be reused, that will make application cost higher. As a result, the reusability of immobilized laccase was further investigated and the removal efficiency of the first reaction cycle was defined as 100%. Although Fig. 6B shows that the removal efficiency of BPA decreased gradually during the repeated use, which might be due to the activity loss and the precipitation of polymeric reaction products formed through the catalytic reaction, which gradually blocked the substrate diffusion channel.45 After nine cycles of reuse, the immobilized laccase still retained more than 60% of its initial activity, indicating a good potential application value. Thus, these results clearly displayed that immobilizing laccases on HNT-TA could successfully eliminate phenolic substances from water and further bring chance for laccases to be used in continuous reactors.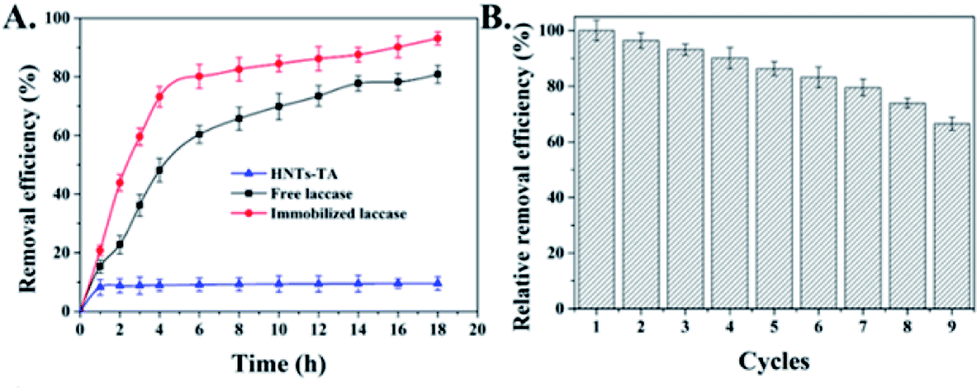 | ||
| Fig. 6 (A) Removal efficiency of bisphenol-A (BPA) by HNT-TA, and free and immobilized laccases with time. (B) Reusability of immobilized laccase in the removal of bisphenol-A (BPA). Data are represented as mean ± SD (n = 3). | ||
Conclusions
In summary, we have fabricated a novel-based enzyme immobilization support by a TA-mediated surface modification strategy. Various physicochemical characterizations confirmed that HNT-TA supports retained abundant quinone groups on their surface, and the model enzyme laccase can be directly immobilized on it via the Michael addition and/or Schiff base reaction. Under optimum conditions, laccases could covalently conjugate on such supports with 197.9 mg g−1 of support enzyme loading and 55.4% of activity recovery. Compared with the free laccase, the pH, temperature, activator/inhibitor and storage stability of immobilized laccases were enhanced. Besides, CD analysis and MD simulation further revealed the secondary structure changes and potential stability enhancement mechanism of immobilized laccase. In addition, immobilized laccase exhibited good catalytic efficiency and reusability in the removal of BPA. Therefore, this simple and green modification method opened a new gate for inert supports to be used in practical enzyme immobilizations.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Science and Technology Department of Zhejiang Province (2019C02053 and 2017C03010), National Natural Science Foundation of China (21908059, 21636003 and 41907318), Shanghai Sailing Program (19YF1410900), the China Postdoctoral Science Foundation (2019M651419) and the Shanghai “Super Postdoctoral” Incentive Plan. The authors thank Research Center of Analysis and Test of East China University of Science and Technology for the help on the characterizations.Notes and references
- O. M. L. Alharbi, A. A. Basheer, R. A. Khattab and I. Ali, J. Mol. Liq., 2018, 263, 442–453 CrossRef CAS.
- X. Cao, J. Luo, J. M. Woodley and Y. Wan, ACS Appl. Mater. Interfaces, 2016, 8, 30511–30522 CrossRef CAS PubMed.
- M. Bilal, T. Rasheed, F. Nabeel, H. M. N. Iqbal and Y. P. Zhao, J. Environ. Manage., 2019, 234, 253–264 CrossRef CAS PubMed.
- Y. Y. Liu, Z. T. Zeng, G. M. Zeng, L. Tang, Y. Pang, Z. Li, C. Liu, X. X. Lei, M. S. Wu, P. Y. Ren, Z. F. Liu, M. Chen and G. X. Xie, Bioresour. Technol., 2012, 115, 21–26 CrossRef CAS PubMed.
- J. H. Lin, L. Fan, R. L. Miao, X. Y. Le, S. Chen and X. H. Zhou, Int. J. Biol. Macromol., 2015, 78, 1–8 CrossRef CAS PubMed.
- Y. R. Dai, J. Yao, Y. H. Song, S. Y. Wang and Y. Yuan, Environ. Sci.: Nano, 2016, 3, 857–868 RSC.
- L. Y. Jun, L. S. Yon, N. Mubarak, C. H. Bing, S. Pan, M. K. Danquah, E. Abdullah and M. Khalid, J. Environ. Chem. Eng., 2019, 102961 CrossRef CAS.
- A. A. Kadam, J. Jang and D. S. Lee, ACS Appl. Mater. Interfaces, 2017, 9, 15492–15501 CrossRef CAS PubMed.
- C. Chen, W. Sun, H. Y. Lv, H. Li, Y. B. Wang and P. Wang, Chem. Eng. J., 2018, 350, 949–959 CrossRef CAS.
- N. Jaiswal, V. P. Pandey and U. N. Dwivedi, Int. J. Biol. Macromol., 2016, 86, 288–295 CrossRef CAS PubMed.
- F. Zheng, B. K. Cui, X. J. Wu, G. Meng, H. X. Liu and J. Si, Int. Biodeterior. Biodegrad., 2016, 110, 69–78 CrossRef CAS.
- F. P. Ran, Y. L. Zou, Y. X. Xu, X. Y. Liu and H. X. Zhang, Chem. Eng. J., 2019, 375, 121947 CrossRef CAS.
- E. H. Wu, Y. X. Li, Q. Huang, Z. K. Yang, A. Y. Wei and Q. Hu, Chemosphere, 2019, 233, 327–335 CrossRef CAS PubMed.
- C. Chao, B. Zhang, R. Zhai, X. Xiang, J. D. Liu and R. F. Chen, ACS Sustainable Chem. Eng., 2014, 2, 396–403 CrossRef CAS.
- H. B. Wang, X. P. Zhao, S. F. Wang, S. Tao, N. Ai and Y. Wang, J. Chromatogr. A, 2015, 1392, 20–27 CrossRef CAS PubMed.
- Y. Wu, Z. L. Du, H. B. Wang and X. Cheng, J. Appl. Polym. Sci., 2016, 133, 43949 Search PubMed.
- K. Atacan and M. Ozacar, Colloids Surf., B, 2015, 128, 227–236 CrossRef CAS PubMed.
- S. Altun, B. Cakiroglu, M. Ozacar and M. Ozacar, Colloids Surf., B, 2015, 136, 963–970 CrossRef CAS PubMed.
- S. H. Zhang, Z. Y. Jiang, X. L. Wang, C. Yang and J. F. Shi, ACS Appl. Mater. Interfaces, 2015, 7, 19570–19578 CrossRef CAS PubMed.
- S. A. Abouelmagd, F. F. Meng, B. K. Kim, H. Hyun and Y. Yeo, ACS Biomater. Sci. Eng., 2016, 2, 2294–2303 CrossRef CAS PubMed.
- T. S. Sileika, D. G. Barrett, R. Zhang, K. H. A. Lau and P. B. Messersmith, Angew. Chem., Int. Ed., 2013, 52, 10766–10770 CrossRef CAS PubMed.
- K. Atacan, B. Cakiroglu and M. Ozacar, Colloids Surf., B, 2017, 156, 9–18 CrossRef CAS PubMed.
- M. M. Bradford, Anal. Biochem., 1976, 72, 248–254 CrossRef CAS.
- R. Xu, Q. J. Zhou, F. T. Li and B. R. Zhang, Chem. Eng. J., 2013, 222, 321–329 CrossRef CAS.
- B. Karagoz, G. Bayramoglu, B. Altintas, N. Bicak and M. Y. Arica, Bioresour. Technol., 2011, 102, 6783–6790 CrossRef CAS.
- H. Zhou, Y. Y. Qu, C. L. Kong, D. X. Li, E. Shen, Q. Ma, X. W. Zhang, J. W. Wang and J. T. Zhou, Colloids Surf., B, 2014, 116, 365–371 CrossRef CAS PubMed.
- D. E. B. Gomes, R. D. Lins, P. G. Pascutti, C. H. Lei and T. A. Soares, J. Phys. Chem. B, 2010, 114, 531–540 CrossRef CAS PubMed.
- Y. R. Dai, J. Yao, Y. H. Song, X. L. Liu, S. Y. Wang and Y. Yuan, J. Hazard. Mater., 2016, 317, 485–493 CrossRef CAS.
- C. Hou, H. Zhu, Y. F. Li, Y. J. Li, X. Y. Wang, W. W. Zhu and R. D. Zhou, Appl. Microbiol. Biotechnol., 2015, 99, 1249–1259 CrossRef CAS.
- X. Liu, L. Lei, Y. F. Li, H. Zhu, Y. J. Cui and H. Y. Hu, Biochem. Eng. J., 2011, 56, 142–149 CrossRef CAS.
- C. Chao, J. D. Liu, J. T. Wang, Y. W. Zhang, B. Zhang, Y. T. Zhang, X. Xiang and R. F. Chen, ACS Appl. Mater. Interfaces, 2013, 5, 10559–10564 CrossRef CAS.
- J. Navarro-Sanchez, N. Almora-Barrios, B. Lerma-Berlanga, J. J. Ruiz-Pernia, V. A. Lorenz-Fonfria, I. Tunon and C. Marti-Gastaldo, Chem. Sci., 2019, 10, 4082–4088 CAS.
- J. Z. Wang, G. H. Zhao, Y. F. Li, X. M. Peng and X. Y. Wang, Chem. Eng. J., 2015, 263, 471–478 CrossRef CAS.
- M. Khoobi, S. F. Motevalizadeh, Z. Asadgol, H. Forootanfar, A. Shafiee and M. A. Faramarzi, Mater. Chem. Phys., 2015, 149, 77–86 CrossRef.
- M. Asgher, S. Noreen and M. Bilal, Int. J. Biol. Macromol., 2017, 95, 54–62 CrossRef CAS.
- M. Asgher, A. Wahab, M. Bilal and H. M. N. Iqbal, Waste Biomass Valorization, 2018, 9, 2071–2079 CrossRef CAS.
- A. Kumar, G. D. Park, S. K. S. Patel, S. Kondaveeti, S. Otari, M. Z. Anwar, V. C. Kalia, Y. Singh, S. C. Kim, B. K. Cho, J. H. Sohn, D. R. Kim, Y. C. Kang and J. K. Lee, Chem. Eng. J., 2019, 359, 1252–1264 CrossRef CAS.
- Y. Masuda, S. Kugimiya, Y. Kawachi and K. Kato, RSC Adv., 2014, 4, 3573–3580 RSC.
- F. Secundo, Chem. Soc. Rev., 2013, 42, 6250–6261 RSC.
- J. Liu, G. B. Yu and J. Zhou, Chem. Eng. Sci., 2015, 121, 331–339 CrossRef CAS.
- J. W. Hou, G. X. Dong, Y. Ye and V. Chen, J. Membr. Sci., 2014, 469, 19–30 CrossRef CAS.
- S. Beck, E. Berry, S. Duke, A. Milliken, H. Patterson, D. L. Prewett, T. C. Rae, V. Sridhar, N. Wendland, B. W. Gregory and C. M. Johnson, Int. Biodeterior. Biodegrad., 2018, 127, 146–159 CrossRef CAS.
- J. Zdarta, K. Antecka, R. Frankowski, A. Zgola-Grzeskowiak, H. Ehrlich and T. Jesionowski, Sci. Total Environ., 2018, 615, 784–795 CrossRef CAS PubMed.
- M. Asgher, S. Noreen and M. Bilal, Chem. Eng. Res. Des., 2017, 119, 1–11 CrossRef CAS.
- J. W. Hou, G. X. Dong, B. Luu, R. G. Sengpiel, Y. Ye, M. Wessling and V. Chen, Bioresour. Technol., 2014, 169, 475–483 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra06171a |
| This journal is © The Royal Society of Chemistry 2019 |