
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePreparative separation of structural isomeric pentacyclic triterpene oleanolic acid and ursolic acid from natural products by pH-zone-refining countercurrent chromatography†
Chaoyue Wang ,
Xiang Wang,
Shanshan Zhao,
Wenyu Sun and
Shengqiang Tong*
,
Xiang Wang,
Shanshan Zhao,
Wenyu Sun and
Shengqiang Tong*
College of Pharmaceutical Science, Zhejiang University of Technology, Chaowang Road 18, Chaohui No. 6 District, Hangzhou, China. E-mail: sqtong@zjut.edu.cn; Fax: +86 571 88320984; Tel: +86 571 88320984
First published on 27th November 2019
Abstract
In this work, pH-zone-refining countercurrent chromatography was investigated in the preparative separation of two bioactive components, oleanolic acid and ursolic acid, from three different natural products, Aralia chinensis, apple peels and Eriobotrya japonica Thunb. Oleanolic acid and ursolic acid are structurally isomeric pentacyclic triterpene acids that are widely distributed in many natural products. However, it was difficult to separate these components with high purity by conventional methods. A biphasic solvent system composed of n-hexane–dichloromethane–methanol–water (7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3:2:8, v/v) was selected, in which an optimized concentration of 10 mmol L−1 trifluoroacetic acid was added in the upper phase as the retainer and 10 mmol L−1 ammonia (with 25–28% NH3) was added in the aqueous phase as the eluter. Consequently, 38.56 mg of oleanolic acid with 99.01% purity was separated from 100 mg of the crude extract of Aralia chinensis, while 65.6 mg of a mixture of ursolic acid (90.98%) and oleanolic acid (6.51%) and 46.6 mg of a mixture of ursolic acid (74.35%) and oleanolic acid (23.61%) were separated from 100 mg of the crude extract of apple peels and 100 mg of the crude extract of Eriobotrya japonica Thunb., respectively, by pH-zone-refining countercurrent chromatography using the above selected biphasic solvent system. The results showed that pH-zone-refining countercurrent chromatography is an efficient method for the preparative separation of pentacyclic triterpene acids from natural products.
:3:2:8, v/v) was selected, in which an optimized concentration of 10 mmol L−1 trifluoroacetic acid was added in the upper phase as the retainer and 10 mmol L−1 ammonia (with 25–28% NH3) was added in the aqueous phase as the eluter. Consequently, 38.56 mg of oleanolic acid with 99.01% purity was separated from 100 mg of the crude extract of Aralia chinensis, while 65.6 mg of a mixture of ursolic acid (90.98%) and oleanolic acid (6.51%) and 46.6 mg of a mixture of ursolic acid (74.35%) and oleanolic acid (23.61%) were separated from 100 mg of the crude extract of apple peels and 100 mg of the crude extract of Eriobotrya japonica Thunb., respectively, by pH-zone-refining countercurrent chromatography using the above selected biphasic solvent system. The results showed that pH-zone-refining countercurrent chromatography is an efficient method for the preparative separation of pentacyclic triterpene acids from natural products.
Introduction
Oleanolic acid (3β-hydroxy-olea-12-en-oic acid, OA) and ursolic acid (3β-hydroxy-urs-12-en-28-oic acid, UA) are pentacyclic triterpene acids (Fig. 1). They widely exist in various plant resources and their presence has been demonstrated in diverse well-known medicinal plants as well as the pulps and rinds of many fresh fruits.1,2 OA and UA are considered a criterion in the quality control of many Chinese herbal medicines, nutraceuticals and health products.3–5 The only difference in the chemical structure between the two components is the position of a methyl group. Therefore, they share numerous common pharmacological properties, including anti-allergy properties,6 anti-inflammation properties,7,8 anti-diabetic properties,9 anti-tumor properties,10,11 a calcium balance,12 immunomodulatory properties,13 the prevention of skin diseases, and protection of the liver.14 According to recent research, both OA and UA possess the ability to inhibit the growth of a variety of cancer cells, which brings the gospel to mankind.15 Mixtures of the two triterpene acids are more effective than a single ingredient in producing the desired biological effects, and they are commonly used in oral drugs and clinical treatments.16 Hence, in many pharmacological experiments, the effect of the combination of OA and UA were usually studied.12,17–19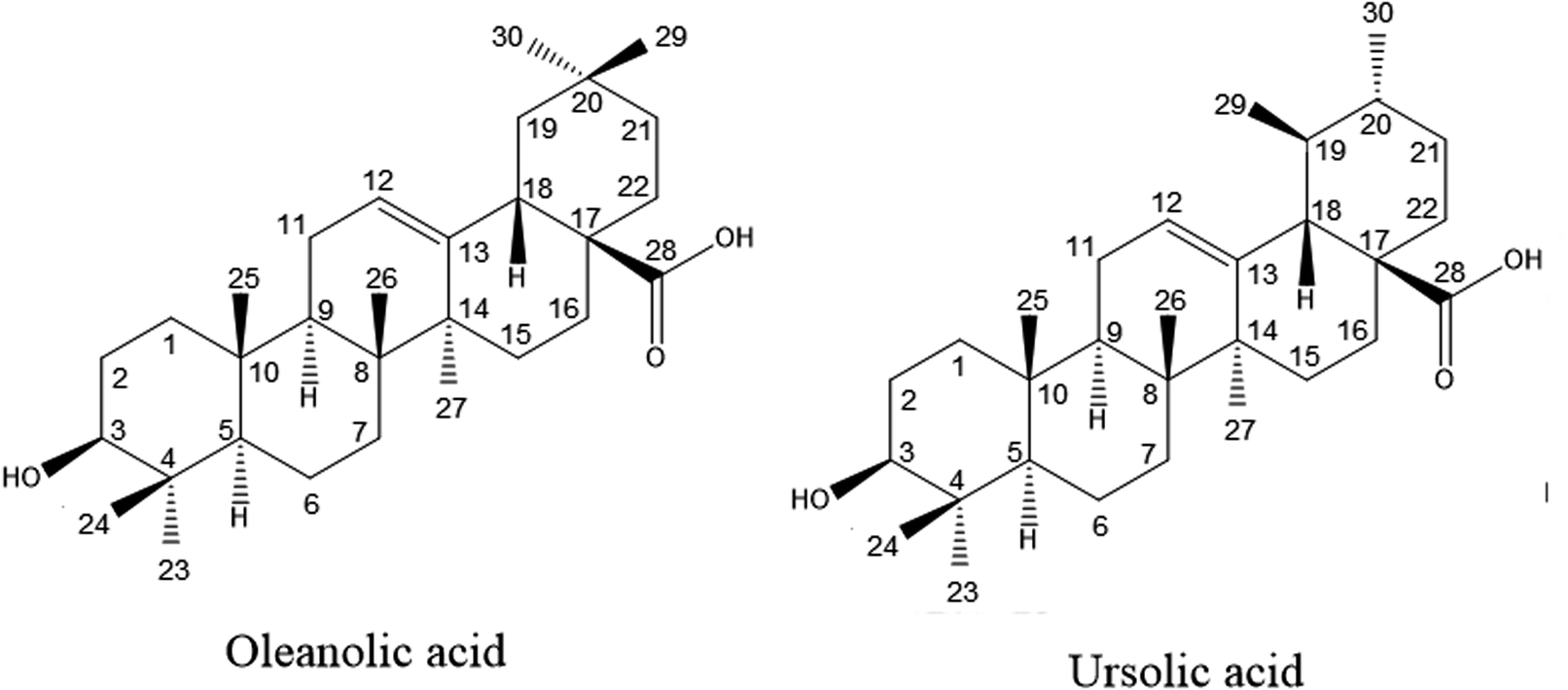 | ||
| Fig. 1 Chemical structures of oleanolic acid and ursolic acid. | ||
OA and UA are constitutional (structural) isomers with only one difference in the substitutional position of the methyl group in their chemical structure as shown in Fig. 1. It was demonstrated that both components exist together in most plants.1,20 Although, there are several analytical methods available in literature for the separation of positional isomers,21,22 it is difficult to separate OA and UA on the preparative scale due to their similar chemical properties. Several studies have been reported on the separation of OA and UA from natural products using silica gel column chromatography,23–26 which is a time- and solvent-consuming procedure. Meanwhile, in order to achieve complete separation of the stereoisomers OA and UA by silica gel chromatography, a prechromatographic derivatization of OA and UA is necessary. Furthermore, it is still very challenging to isolate the two components from crude extracts of the natural products because of the complex composition resulting from the pentacyclic triterpenes derivatives. Therefore, it is important to establish an efficient method for the preparative separation of OA and UA in light of the above pharmacological activities as well as the difficulty in separation by traditional methods.
Countercurrent chromatography is a support-free liquid–liquid distribution technique that avoids the problems of irreversible adsorption, deactivation and denaturation of the sample.27 pH-zone-refining countercurrent chromatography is a relatively large-scale preparative chromatography technique for ionizable analytes developed from conventional countercurrent chromatography.28 The sample injection amount for pH-zone-refining countercurrent chromatography could be ten-fold that for conventional countercurrent chromatography, and the sample amount could be injected at the gram level.29,30 Moreover, pH-zone-refining countercurrent chromatography can be easily used for the separation of the ionizable analytes without UV absorption using a pH detector. This method has been gradually developed into a useful technique for the preparative separation and purification of organic acids and alkaloids from natural medicinal plants.29 However, very limited number of papers have been published on the preparative separation of triterpene acids by pH-zone-refining countercurrent chromatography due to the complexity of the chemical constituents and their difficulty in UV detection.31–33 The simultaneous separation of triterpenoid saponins and flavonoid glycosides from the roots of Glycyrrhiza uralensis Fisch. by pH-zone-refining countercurrent chromatography was reported by Kong's group,32 in which the triterpenoid, glycyrrhizinic acid, was successfully separated. Five terpenoids, including two new ones, were purified by pH-zone-refining countercurrent chromatography and conventional countercurrent chromatography from olibanum by Wang's group.33 To the best of our knowledge, no paper on pH-zone-refining countercurrent chromatography for separating OA or UA from natural products has been published. In our present work, we wanted to report our recent work on the selection of a solvent system for the preparative separation of constitutional isomers, OA and UA, from natural products by pH-zone-refining countercurrent chromatography.
Experimental
Apparatus
The countercurrent chromatographic apparatus used in the present study was a multilayer coil planet centrifuge designed in our laboratory (Zhejiang University of Technology, Hangzhou, China), which has been described in previous research.34 The apparatus was equipped with multilayer coils. The total capacity of the preparative column was 105 mL. The β values of the preparative columns ranged from 0.28 to 0.55. The rotation speed of the column coils was modulated with a speed controller in the range from 0 to 600 rpm. The solvent was pumped into the column with a model MP0106 constant-flow pump (Shanghai Sanotac Scientific Instruments Co., Ltd, Shanghai, China). A model UVD-200 detector and a Jinda Biotechnology workstation (Shanghai Jinda Biotechnology, Shanghai, China) were employed to record the chromatogram. The column temperature was room temperature and the detection wavelength was determined at 254 nm. The effluents were collected by a Model BSZ-100 automatic component collector (Shanghai Huxi technology Co. Ltd, Shanghai, China) and the pH value of each fraction collected was manually determined using a portable Delta 320-s pH meter (Mettler-Toledo, Zurich, Switzerland).HPLC analysis was performed on a Shimadzu LC-20AT system (Shimadzu, Kyoto, Japan) equipped with a Shimadzu LC pump, a Shimadzu SPD-20Avp UV detector, a Shimadzu SCL-10ASvp controller and LC-solution software.
Reagents and materials
Aralia chinensis and Eriobotrya japonica Thunb. were purchased from Huadong Wulin drugstore. Commercial Red Fuji Malus domestica was bought in the local fruit supermarket in July. Methanol and acetonitrile used for HPLC analysis was of chromatographic grade. Trifluoroacetic acid (TFA, neat) and aqueous ammonia (with 25–28% NH3) were of reagent grade. All organic solvents used for countercurrent chromatography were of analytical grade. Ethanol used for the extraction was of analytical grade. All organic reagents were purchased from Huipu Chemical Company, Hangzhou, China. Water was redistilled for the mobile phase of HPLC. The ammonium acetate added in the mobile phase of HPLC was analytical grade with a purity of 99.0% and was purchased from Aladdin Biochemical Technology Co., Ltd, Shanghai, China. Analytical grade sulfuric acid, sodium hydroxide and activated carbon were purchased from Huipu Chemical Company, Hangzhou, China.Preparation of crude sample
Preparation of biphasic solvent system and sample solution
The biphasic solvent system consisting of n-hexane–dichloromethane–methanol–water (7:3:2:8, v/v) was placed in a 2 L separatory funnel. After shaking vigorously and a thorough equilibration, the solution was divided into two phases. TFA was added to the upper phase to obtain a final concentration of 10 mol L−1 (stationary phase) and ammonia (25–28% NH3) was added to the lower phase to obtain a final concentration of 10 mol L−1 (mobile phase). The crude sample was prepared by dissolving 100 mg of the crude sample in 5 mL of stationary phase with retainer and 5 mL of mobile phase without eluter.
Separation procedure
The coil column was first filled with the upper organic phase containing 10 mmol L−1 of TFA at a flow rate of 10 mL min−1. Then, the crude sample solution was injected through a six-way valve. The lower aqueous phase containing 10 mmol L−1 ammonia (25–28% NH3) was then pumped into the column at 1.5 mL min−1, while the apparatus was rotated at 600 rpm. Each effluent was collected in a 10 mL test tube using an automatic fraction collector at 4 min intervals. The pH of each fraction collected was determined with a pH meter. After the separation experiment was completed, the retention of the stationary phase was measured by forcing the remaining contents in the separation column into a graduated cylinder with pressurized gas. The whole experimental procedure was shown in Fig. 2. | ||
| Fig. 2 A pictorial diagram of the overall procedure. | ||
Analysis of crude sample and the separated fractions
The crude sample and the collected fractions from pH-zone-refining countercurrent chromatography was performed on an Agilent 5 HC-C18 column (250 mm × 4.6 mm, 5 μm) and detection wavelength was set at 205 nm. The mobile phase was composed of 0.5% ammonium acetate, acetonitrile and methanol (21:67:21, v/v/v). The flow rate was 1 mL min−1 with isocratic elution. The column temperature was 25 °C.
Results and discussion
Selection of solvent systems for pH-zone-refining countercurrent chromatography
The successful separation of OA and UA from natural plants was strongly influenced by the selection of an appropriate biphasic solvent system, which had a suitable partition coefficient as well as a good solubility for the compounds.28 The Kacid (the partition coefficient K value in the solvent system added with an acidic retainer) should be much larger than 1 while the Kbase (the partition coefficient K value in the solvent system added with a basic eluter) should be much smaller than 1. As OA and UA were organic acids with small polarity, biphasic solvent systems with low polarity were investigated. Several biphasic solvent systems at different volume ratios were investigated as follows: n-hexane–ethyl acetate–methanol–water, methyl tert-butyl ether–tetrahydrofuran–water, methyl tert-butyl ether–acetonitrile–water, chloroform–methanol–water, chloroform–methanol–n-butanol–water and n-hexane–dichloromethane–methanol–water. The partition performance of OA and UA in the studied solvent systems is presented in Table 1 in the ESI.† The results demonstrated that the solvent system of n-hexane–dichloromethane–methanol–water (7:3:2:8, v/v) produced an ideal Kacid and Kbase value (Kbase was 0.0726 and Kacid was over 35.492 for OA; Kbase was 0.0938 and Kacid was over 47.841 for UA). Meanwhile, the high solubility of the crude sample in the above solvent system was determined with the help of ultrasonic dissolution. Therefore, the biphasic solvent system composed of n-hexane–dichloromethane–methanol–water (7:3:2:8, v/v) was selected, in which an optimized concentration of 10 mmol L−1 trifluoroacetic acid was added in the upper phase as the retainer and a 10 mmol L−1 ammonia (with 25–28% NH3) was added in the aqueous phase as the eluter.
Preparative separation of OA and UA by pH-zone-refining countercurrent chromatography
Fig. 3 is a typical chromatogram for the analysis of a crude sample of Araliae chinensis, apple peels and Eriobotrya japonica Thunb. by HPLC. As shown in Fig. 3, a different content for the two pentacyclic triterpene acids, OA (peak 1) and UA (peak 2), was found in the three crude samples. OA represented a high content of 6.63% and UA had only a 0.29% content in the crude sample of Araliae chinensis, while in other two crude samples, the content of UA was predominant.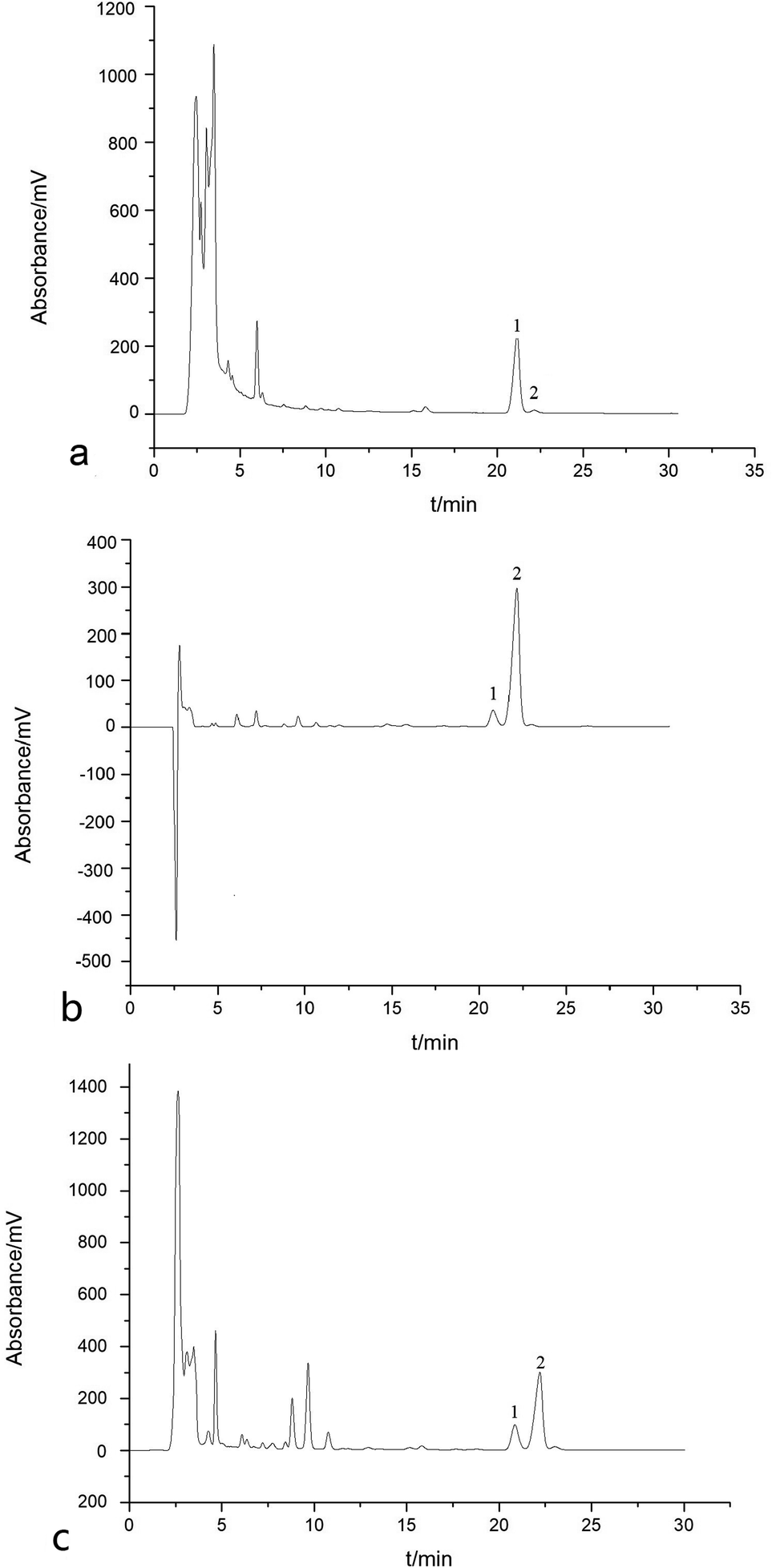 | ||
| Fig. 3 Chromatogram for the analysis of three crude samples by HPLC. (a) Crude sample 1 of Aralia chinensis. (b) Crude sample 2 of apple peels. (c) Crude sample 3 of Eriobotrya japonica Thunb. Peak 1: oleanolic acid; peak 2: ursolic acid; HPLC column: Agilent 5 HC-C18 (250 mm × 4.6 mm, 5 μm); mobile phase: 0.5% ammonium acetate:acetonitrile:methanol (21:67:21, v/v) with an isocratic elution; flow rate: 1 mL min; UV wavelength: 205 nm; column temperature: 25 °C. | ||
Fig. 4 shows three typical chromatograms for the preparative separation of OA and UA from three crude samples by pH-zone-refining countercurrent chromatography with the selected biphasic solvent system composed of n-hexane–dichloromethane–methanol–water (7:3:2:8, v/v), in which the upper organic phase was dissolved with 10 mmol L−1 of TFA and the lower aqueous phase contained 10 mmol L−1 ammonia (25–28% NH3). The results showed that 38.56 mg of OA with a purity of 99.01% was obtained from 100 mg of crude sample 1 as shown in the shadow area of Zone I in Fig. 4(a). The retention of the stationary phase was up to 80.7% and the recovery of OA was 85.2%. A pH-zone platform was observed by measuring the pH value of each collected fraction, which further indicated the successful separation of OA.
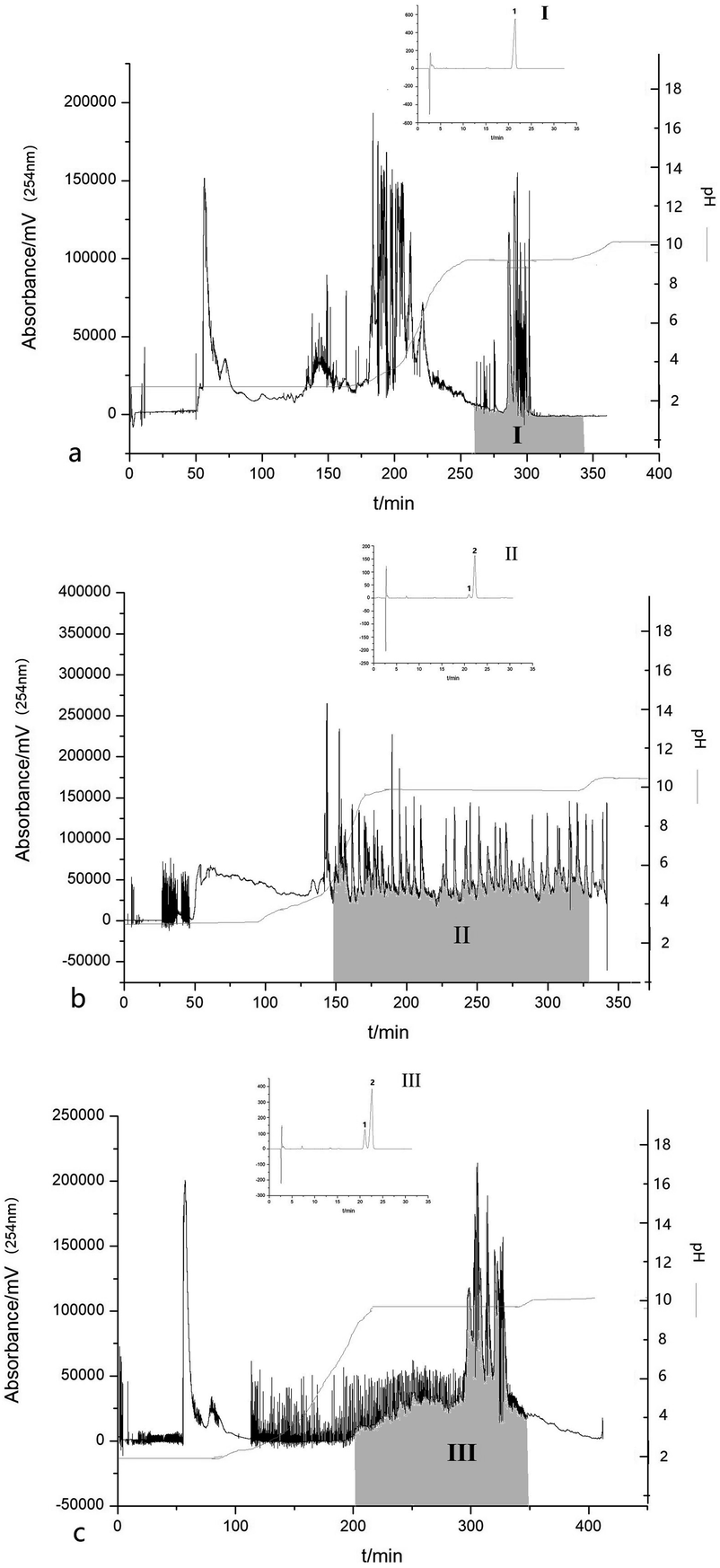 | ||
| Fig. 4 Chromatogram for the preparative separation of ursolic acid and oleanolic acid from 100 mg of crude sample by pH-zone-refining countercurrent chromatography. (a) Crude sample 1 of Aralia chinensis. (b) Crude sample 2 of apple peels. (c) Crude sample 3 of Eriobotrya japonica Thunb. Zone I: oleanolic acid. Zone II: a mixture of ursolic acid and oleanolic acid. Zone III: a mixture of ursolic acid and oleanolic acid. Solvent system: n-hexane–dichloromethane–methanol–water (7:3:2:8, v/v); 10 mmol L−1 TFA in upper organic stationary phase, 10 mmol L−1 ammonia (with 25–28% NH3) in lower aqueous mobile phase; revolution speed: 600 rpm; flow-rate: 1.5 mL min; sample size: 100 mg; UV detection wavelength: 254 nm. | ||
The same method was used to separate OA and UA from apple peels as shown in Fig. 4(b) and from Eriobotrya japonica Thunb. as shown in Fig. 4(c). Unfortunately, OA and UA eluted together with peak overlapping. The shadow area of Zone II showed approximately 65.6 mg of a mixture of OA and UA, which was obtained from the crude extract of apple peels. The content of UA was 90.98% and the content of OA was 6.51%. Meanwhile, the shadow area of Zone III showed approximately 46.6 mg of a mixture of OA and UA, which was obtained from the crude exact of Eriobotrya japonica Thunb. The content of UA was 74.35% and that of OA was 23.61%. As shown in Fig. 4(a) and (b), two target triterpene acids eluted as rectangular peaks in Zone II and Zone III, whereas other impurities were concentrated and eluted at the boundaries of the rectangular peaks. The HPLC analysis of each eluted fraction from the countercurrent chromatography indicated that UA eluted first with a small amount of OA, and then was followed by a large content of OA. A certain peak resolution could be observed, but no complete resolution was achieved. A series of other two-phase solvent systems were also investigated in our work. However, no successful separation was obtained for OA and UA. The only difference in the chemical structure between OA and UA was the position of the methyl group. The two methyl groups of OA were substituted at the 20-position while the two methyl groups of UA were substituted at the 19- and 20-position. Therefore, OA and UA shared a quite similar hydrophobicity and acidity, resulting in very similar chemical properties. Therefore, it was still difficult to get a complete separation for the two components using the above solvent system. Nevertheless, the two compounds were highly concentrated with short retention time after a one-step separation by pH-zone-refining countercurrent chromatography, which provided an alternative method for the preparative separation of OA and UA from natural products.
According to reports in literature, the mixture of OA and UA had a higher pharmacological activity compared with the activity observed with either OA or UA alone.16,35–37 The interaction of OA and UA may have taken place chemically and biologically to produce an ideal effect.38 Therefore, pharmacological research on the mixture of OA and UA was conducted by numerous researchers.39–41 The combination of OA and UA has been widely used in clinical practices.42 pH-zone-refining countercurrent chromatography provided a rapid and simple method for the enrichment of a mixture of OA and UA, which might be useful for the clinical use of OA and UA. The most significant advantage of this method was that a large load capacity and an effective separation of OA and UA from natural products were achieved in a short time.
Compared with previously reported methods for the separation of OA and UA from natural products, our methodology is more feasible and practical. According to the experimental results in the reference,23–26 for the preparative separation of OA and UA from natural products by silica gel column chromatography and dextran gel column chromatography, a more complicated experimental procedure was necessary in which the high consumption of organic solvents, a long separation time, irreversible sample adsorption and a low recovery of the solute would always be involved. For example, in the research by Huang'group,24 silica gel column chromatography, dextran gel column chromatography and preparative HPLC were successively used to isolate and purify OA and UA. The mobile phase for the dextran gel column chromatography was composed of chloroform and methanol (v/v, 1:1), which would not only cause harm to people's health, but would also cause air pollution. Furthermore, only 12 mg of OA and 11 mg of UA were obtained from 28.12 g of the crude extract from the roots of Ilex kudingcha after a series of complex experimental procedures. In our present work, all impurities were concentrated and eluted outside the rectangular peak of the triterpene acids, OA and UA. The two target components and impurities were completely separated by pH-zone-refining countercurrent chromatography using the selected biphasic solvent system. Therefore, the two ionizable triterpene acids were well concentrated and enriched with a high content from the crude extract of natural products in a very convenient and efficient way.
Conclusions
A useful method for the separation of two structural isomeric triterpenic acids, OA and UA, from Aralia chinensis, apple peels and Eriobotrya japonica Thunb., was established using pH-zone-refining countercurrent chromatography. A biphasic solvent system composed of n-hexane–dichloromethane–methanol–water (7:3:2:8, v/v) containing 10 mmol L−1 trifluoroacetic acid in the upper phase as a retainer and 10 mmol L−1 ammonia (with 25–28% NH3) in the aqueous phase as an eluter was screened after investigating the partition coefficients of the target components in several different solvent systems. OA and UA were well separated and enriched from the crude sample. pH-zone-refining countercurrent chromatography is a practical and efficient method for the preparative separation of bioactive triterpene acids from natural products.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (21978266).Notes and references
- S. Qi, L. Ding, K. Tian, X. Chen and Z. Hu, Novel and simple nonaqueous capillary electrophoresis separation and determination bioactive triterpenes in Chinese herbs, J. Pharm. Biomed. Anal., 2006, 40, 35–41 CrossRef CAS PubMed.
- J. Sun, S. Dong, Y. Wu, H. Zhao, X. Li and W. Gao, Oleanolic acid and ursolic acid as potential inhibitors of human salivary α-amylase: insights from in vitro assays and in silico simulations, J. Mol. Model., 2017, 23, 248 CrossRef PubMed.
- T. Cui, J.-Z. Li, H. Kayahara, L. Ma, L.-X. Wu and K. Nakamura, Quantification of the polyphenols and triterpene acids in Chinese hawthorn fruit by high-performance liquid chromatography, J. Agric. Food Chem., 2006, 54, 4574–4581 CrossRef CAS PubMed.
- P. Kaur, R. C. Gupta, A. Dey and D. K. Pandey, Simultaneous quantification of oleanolic acid, ursolic acid, betulinic acid and lupeol in different populations of five Swertia species by using HPTLC-densitometry: comparison of different extraction methods and solvent selection, Ind. Crop. Prod., 2019, 130, 537–546 CrossRef CAS.
- T. P. D. Vu, T. Q. Khong, T. M. N. Nguyen, Y. H. Kim and J. S. Kang, Phytochemical profile of Syzygium formosum (Wall.) Masam leaves using HPLC-PDA-MS/MS and a simple HPLC-ELSD method for quality control, J. Pharm. Biomed. Anal., 2019, 168, 1–12 CrossRef PubMed.
- H. Tan, T. Sonam and K. Shimizu, The potential of triterpenoids from loquat leaves (Eriobotrya japonica) for prevention and treatment of skin disorder, Int. J. Mol. Sci., 2017, 18, 1030 CrossRef PubMed.
- D. Kashyap, A. Sharma, H. S. Tuli, S. Punia and A. K. Sharma, Ursolic acid and oleanolic acid: pentacyclic terpenoids with promising anti-inflammatory activities, Recent Pat. Inflammation Allergy Drug Discovery, 2016, 10, 21–33 CrossRef CAS PubMed.
- N. Sultana and Z. S. Saify, Naturally occurring and synthetic agents as potential anti-inflammatory and immunomodulants, Anti-Inflammatory Anti-Allergy Agents Med. Chem., 2012, 11, 3–19 CrossRef CAS PubMed.
- Z. Kalaycloglu, S. Uzasci, T. Dirmenci and F. B. Erim, α-Glucosidase enzyme inhibitory effects and ursolic and oleanolic acid contents of fourteen Anatolian Salvia species, J. Pharm. Biomed. Anal., 2018, 155, 284–287 CrossRef PubMed.
- L. Sheng, J. Huang, C. Liu, J. Zhang and K. Cheng, Synthesis of oleanolic acid/ursolic acid/glycyrrhetinic acid-hydrogen sulfide donor hybrids and their antitumor activity, Med. Chem. Res., 2019, 28, 1212–1222 CrossRef CAS.
- Y.-Y. Han, X.-W. Xue, Z.-M. Shi, P.-Y. Wang, X.-R. Wu and X.-J. Wang, Oleanolic acid and ursolic acid inhibit proliferation in transformed rat hepatic oval cells, World J. Gastroenterol., 2014, 20, 1348–1356 CrossRef PubMed.
- S. Cao, X.-L. Tian, W.-X. Yu, L.-P. Zhou, X.-L. Dong, M.-J. Favus and M.-S. Wong, Oleanolic acid and ursolic acid improve bone properties and calcium balance and modulate vitamin D metabolism in aged female rats, Front. Pharmacol., 2018, 9, 1435 CrossRef CAS PubMed.
- L. Saaby, A. K. Jager, L. Moesby, E. W. Hansen and S. B. Christensen, Isolation of immunomodulatory triterpene acids from a standardized rose hip powder (Rosa cannina L.), Phytother. Res., 2011, 25, 195–201 CAS.
- J. Liu, Y. Liu, Q. Mao and C. D. Klaassen, The effect of 10 triterpenoid compounds on experimental liver injury in mice, Fundam. Appl. Toxicol., 1994, 22, 34–40 CrossRef CAS PubMed.
- Y. S. Gao, Y. Yuan, G. Song and S. Q. Lin, Inhibitory effect of ursolic acid and oleanolic acid from Eriobotrya fragrans on A549 cell viability in vivo, Genet. Mol. Res., 2016, 15, 1–8 Search PubMed.
- H. Liu, Oleanolic acid and ursolic acid: research perspectives, J. Ethnopharmacol., 2005, 100, 92–94 CrossRef PubMed.
- H. Ding, X. Hu, X. Xu, G. Zhang and D. Gong, Inhibitory mechanism of two allosteric inhibitors, oleanolic acid and ursolic acid on α-glucosidase, Int. J. Biol. Macromol., 2018, 107, 1844–1855 CrossRef CAS PubMed.
- M. Hamza, M. Nadir, N. Mehmood and A. Farooq, In vitro effectiveness of triterpenoids and their synergistic effect with antibiotics against Staphylococcus aureus strains, Indian J. Pharmacol., 2016, 48, 710–714 CrossRef CAS PubMed.
- H. Ali, P. J. Houghton and A. Soumyanath, Alpha-amylase inhibitory activity of some Malaysian plants used to treat diabetes; with particular reference to Phyllanthus amarus, J. Ethnopharmacol., 2006, 107, 449–455 CrossRef PubMed.
- Y. M. Hao, Analysis of liquid microextraction in traditional Chinese medicine oleanolic acid and ursolic acid and its application to protein binding, Shanxi Medical University, Taiyuan, Shanxi, China, 2013 Search PubMed.
- S. Saha, R. Mistri and B. C. Ray, Rapid and sensitive method for simultaneous determination of six carcinogenic aromatic amines in mainstream cigarette smoke by liquid chromatography/electrospray ionization tandem mass spectrometry, J. Chromatogr. A, 2009, 1216, 3059–3063 CrossRef CAS PubMed.
- S. Saha, R. Mistri and B. C. Ray, Determination of pyridine, 2-picoline, 4-picoline and quinoline from mainstream cigarette smoke by solid-phase extraction liquid chromatography/electrospray ionization tandem mass spectrometry, J. Chromatogr. A, 2010, 1217, 307–311 CrossRef CAS PubMed.
- A. Singh, K. N. Venugopala, M. A. Khedr, M. Pillay, K. U. Nwaeze, Y. Coovadia, F. Shode and B. Odhav, Antimycobacterial, docking and molecular dynamic studies of pentacyclic triterpenes from Buddleja saligna leaves, J. Biomol. Struct. Dyn., 2017, 35, 2654–2664 CrossRef CAS PubMed.
- Y. Huang, J. Zheng, G. Yang, M. Lai, Y. Huang and B. Liu, Chemical constituents in roots of Ilex kudingcha, Chinese traditional and herbal drug, 2015, 16, 2371–2376 Search PubMed.
- M. D. Awouafack, P. Tane and H. Morita, Tricalycoside, a new cerebroside from Tricalysia coriacea (Rubiaceae), Chem. Biodivers., 2018, 15, e1700472 CrossRef PubMed.
- N. Das, A. G. Atanasov, P. K. Deb, A. Mocan, S. M. Nabavi, R. Ghosh and B. Dinda, Hepatoprotective Naphthalene Diglucoside from Neanotis wightiana Aerial Parts, Phytomedicine, 2017, 33, 14–20 CrossRef CAS PubMed.
- Y. Ito, Golden rules and pitfalls in selecting optimum conditions for high-speed counter-current chromatography, J. Chromatogr. A, 2005, 1065, 145–168 CrossRef CAS PubMed.
- Y. Ito and Y. Ma, pH-zone-refining countercurrent chromatography, J. Chromatogr. A, 1996, 753, 1–36 CrossRef CAS PubMed.
- Y. Ito, pH-zone-refining counter-current chromatography: origin, mechanism, procedure and applications, J. Chromatogr. A, 2013, 1271, 71–85 CrossRef CAS PubMed.
- C. Sun, F. Liu, J. Sun, J. Li and X. Wang, Optimisation and establishment of separation conditions of organic acids from Usnea longissima Ach. by pH-zone-refining counter-current chromatography: discussion of the eluotropic sequence, J. Chromatogr. A, 2016, 1427, 96–101 CrossRef CAS PubMed.
- H. Song, J. Lin, X. Zhu and Q. Chen, Developments in high-speed countercurrent chromatography and its applications in the separation of terpenoids and saponins, J. Sep. Sci., 2016, 39, 1574–1591 CrossRef CAS PubMed.
- J. Xu, J. Luo and L. Kong, Simultaneous separation of triterpenoid saponins and flavonoid glycosides from the roots of Glycyrrhiza uralensis Fisch. by pH-zone-refining counter-current chromatography, J. Sep. Sci., 2013, 36, 3295–3301 CAS.
- J. Yu, H. Zhao, D. Wang, X. Song, L. Zhao and X. Wang, Extraction and purification of five terpenoids from olibanum by ultrahigh pressure technique and high-speed countercurrent chromatography, J. Sep. Sci., 2017, 40, 2732–2740 CrossRef CAS PubMed.
- C. Wang, W. Sun, X. Wang, Y. Jin, S. Zhao, M. Luo and S. Tong, Large-scale separation of baicalin and wogonoside from Scutellaria baicalensis Georgi by the combination of pH-zone-refining and conventional counter-current chromatography, J. Chromatogr. A, 2019, 1601, 266–273 CrossRef CAS PubMed.
- C. Lu, C. Zhang, F. Zhao, D. Li and W. Lu, Biosynthesis of ursolic acid and oleanolic acid in saccharomyces cerevisiae, AIChE J., 2018, 64, 3794–3802 CrossRef CAS.
- C. Soica, C. Oprean, F. Borcan, C. Danciu, C. Trandafirescu, D. Coricovac, Z. Crainiceanu, C. A. Dehelean and M. Munteanu, The synergistic biologic activity of oleanolic and ursolic acids in complex with hydroxypropyl-γ-cyclodextrin, Molecules, 2014, 19, 4924–4940 CrossRef CAS PubMed.
- G. A. Gutierrez-Rebolledo, G. A. Siordia-Reyes, M. Meckes-Fischer and A. Jimenez-Arellanes, Hepatoprotective properties of oleanolic and ursolic acids in antitubercular drug-induced liver damage, Asian Pac. J. Trop. Med., 2016, 9, 644–651 CrossRef CAS PubMed.
- A. Jimenez-Arellanes, J. Luna-Herrera, J. Cornejo-Garrido, S. Lopez-Garcia, M. E. Castro-Mussot, M. Meckes-Fischer, D. Mata-Espinosa, B. Marquina, J. Torres and R. Hernandez-Pando, Ursolic and oleanolic acids as antimicrobial and immunomodulatory compounds for tuberculosis treatment, BMC Complem. Altern. M., 2013, 13, 258 CrossRef PubMed.
- M. M. De Los Reyes, G. G. Oyong, V. A. S Ng, C.-C. Shen and C. Y. Ragasa, Cytotoxic compounds from Wrightia pubescens (R. Br.), Pharmacogn. Res., 2018, 10, 9–15 CrossRef CAS.
- H. L. Alvarado, A. C. Calpena, M. L. Garduno-Ramirez, R. Ortiz, C. Melguizo, J. C. Prados and B. Clares, Nanoemulsion Strategy for Ursolic and Oleanic Acids Isolates from Plumeria obtusa Improves Antioxidant and Cytotoxic Activity in Melanoma Cells, Anti-Cancer Agent Me., 2018, 18, 847–853 CrossRef CAS PubMed.
- S. Ceballos, A. Guillen, D. L. Munoz, A. Castano, L. F. Echeverri, S. Acin and N. Balcazar, Immunometabolic regulation by triterpenes of Eucalyptus tereticornis in adipose tissue cell line models, Phytomedicine, 2018, 50, 109–117 CrossRef CAS PubMed.
- M. Zhang and Y. Shen, Research advances in pharmacologic actions of ursolic acid and oleanolic acid in resistance to skin cancer and leukemia, Anti Infect. Pharm., 2012, 9, 177–181 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra06082k |
| This journal is © The Royal Society of Chemistry 2019 |