
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEpoxy resin composites with commercially available graphene: toward high toughness and rigidity
Jianxiang Suna,
Jingqi Jia,
Zhigeng Chen *ab,
Shumei Liuab and
Jianqing Zhao*ab
*ab,
Shumei Liuab and
Jianqing Zhao*ab
aSchool of Materials Science and Engineering, South China University of Technology, Guangzhou 510641, P. R. China. E-mail: chenzg@scut.edu.cn; psjqzhao@scut.edu.cn
bThe Key Laboratory of Polymer Processing Engineering, Ministry of Education, Guangzhou 510641, P. R. China
First published on 16th October 2019
Abstract
Some commercially available graphene (CG) samples, actually graphite nanoplates, are difficult to be applied in polymers due to their large thickness, even though their fabrication is more cost-effective. In this work, a CG sample with a thickness of about 25 nm is pre-treated via a facile acid soaking technique, and then the acid-treated CG is introduced into epoxy resin (EP) to improve the toughness while maintaining the high rigidity of the EP. The thickness of the acid-treated CG is dramatically decreased to about 1 nm, well-dispersing CG in the EP matrix. The tensile strength and the fracture energy of EP composite with 0.2 wt% acid-treated CG are increased to 85.2 MPa and 3.6 kJ m−2 from 74.3 MPa and 2.3 kJ m−2 of pure EP, respectively. In addition, the tensile-fractured surfaces of EP composites reveal that the toughening mechanism is mainly attributed to the crack deflection and crack pinning caused by the firmly-embedded CG nanosheets in the EP matrix. Besides, the glass transition temperature of EP composites is increased to 170.5 °C from 163.9 °C of pure EP. The present study provides a valuable approach for making use of the cost-effective graphite nanoplates to achieve a similar performance as graphene in EP.
Introduction
Epoxy resin (EP) has been widely used in many fields such as electrical and automotive industries due to its low curing shrinkage, high adhesion, good chemical stability, high strength and ease of processing.1,2 However, the traditional EP usually shows poor toughness, high brittleness and easy cracking since the high crosslinking density, which has to be improved for enabling EP to meet higher requirements in some high-tech fields.3,4 In previous studies,5 incorporating rubber elastomers or thermoplastic polymers into EP was able to effectively improve the toughness while sacrificing EP's rigidity and thermal stability. Recently, different nanofillers such as nanoclay,6 carbon nanotubes,7 and graphene8 were used to overcome the above problems. Among these nanofillers, graphene-based materials have attracted considerable attention due to their excellent physical and mechanical properties.Wang et al.9 investigated the strengthening and toughening effects of graphene on polymer composites. The microcrack, resulting from interfacial debonding between graphene and matrix as well as breakage and pulling out of graphene, was proposed to be the main toughening mechanism. Besides, the prevention of crack propagation was also contributed to the improved mechanical properties. However, graphene is prone to agglomerate because of the strong van der Waals forces and π–π interactions,10 leading to the deterioration of mechanical properties for graphene/polymer composites. Thus, surface modification of graphene is usually executed to improve the dispersion of graphene in polymer matrix viz. covalent modification and non-covalent modification of graphene.
For the way of covalent modification, graphene oxide (GO) is considered to be the most promising graphene-based materials for its abundant reactive oxygen-containing groups on the nanosheets.11 Ramezanzadeh et al.12 modified GO with p-phenylenediamine (GO–PPDA), and prepared GO–PPDA/EP composites. It revealed that the interactions between GO–PPDA and EP matrix was increased with the increase of p-phenylenediamine, which was favorable to avoid the agglomeration of GO–PPDA nanosheets. In addition, the tensile strength and the elongation at break of GO–PPDA/EP composites were increased to 53 MPa and 4.0% from 28 MPa and 3.1% of pure EP, respectively. Chhetri et al.13 prepared monolayer or multilayer N-methyl-2-pyrrolidone/graphene (NMPG) by one-step hydrothermal reduction. The tensile strength of NMPG/EP composite with 0.2 wt% NMPG was increased to 58.3 MPa from 45.7 MPa of pure EP. Meanwhile, the fracture toughness (critical intensity factor) and fracture energy of NMPG/EP composites were improved by 100% and 240%, respectively.
Furthermore, for the way of non-covalent modification, it seems more conducive to obtain the well-dispersed graphene-based materials.14,15 Saha et al.16 employed polyvinylpyrrolidone as intercalant to exfoliate the reduced GO, promoting the dispersion of the reduced GO in EP matrix effectively. Wan et al.17 fabricated EP composites with highly dispersed graphene of a thickness of about 2 nm via a facile surfactant-assisted process. The tensile strength and the elongation at break of EP composite with 0.1 wt% graphene were increased to 83.4 MPa and 4.3% from 53.0 MPa and 2.6% of pure EP, respectively. Notably, it was proved that the monolayer graphene (with thickness about 1 nm or below) and the multilayer graphene (consisting of 2–10 graphene layers) were able to be well dispersed in EP matrix, which was potential to achieve the enhanced rigidity and toughness simultaneously.18
Nowadays, the mass production of commercially available graphene (CG) at low manufacturing costs does not appear to be a problem. However, Kauling et al.19 analyzed CG samples from 60 companies all over the world, and it was found that most of them contained less than 10% monolayer graphene. Actually, the thickness of some CG samples was more than 20 nm, which should have been called graphite nanoplates according to the definition of graphene.20 Since the fabrication of graphite nanoplates is more cost-effective than that of monolayer or multilayer graphene, it is meaningful to propose a facile method to make use of graphite nanoplates to achieve high toughness and rigidity for EP.
In the present work, a CG sample with about 25 nm thickness is soaked in the mixture of nitric acid and sulfuric acid under an ice bath for promoting the exfoliation of CG. The structure evolution of CG via acid soaking technique is characterized by Fourier-transform infrared spectroscopy (FT-IR), Raman spectroscopy, X-ray diffraction (XRD), X-ray photoelectron spectroscopy (XPS), and atomic force microscope (AFM). The acid-treated CG is then introduced into EP for improving the mechanical properties. And the corresponding toughening mechanism is studied based on the scanning electron microscopy (SEM) images of the tensile-fractured surfaces of EP composites.
Experimental
Chemicals and materials
Epoxy (DGEBA, epoxy value = 0.51 mol/100 g) was supplied by SINOPEC Assets Management Corporation Baling Petrochemical Branch, Beijing, China. 4,4′-Methylenedianiline (DDM) was provided by Shanghai Titan Scientific Co., Ltd., Shanghai, China. The commercially available graphene (CG) sample was purchased from Angstron Materials Inc., America. Graphite flakes (∼150 μm flakes) is purchased from Alfa Aesar (China) Chemical Co., Ltd. and used for characterization. All other reagents used in this study were purchased from Aladdin Chemical Reagent Co., Ltd. and used as received unless otherwise specified.Preparation of acid-treated CG
Based on Cui's study,21 CG sample (0.1 g) was immersed in concentrated nitric acid (30 mL) under an ice bath, and concentrated sulfuric acid (60 mL) was slowly added under stirring. The mixture was kept for 12 h before dilution with plenty of cold water. The resulting black solution was filtered and washed by plenty of water. The residue was re-dispersed in ethanol solution (100 mL) containing polyvinylpyrrolidone (PVP, 0.2 g) through ultrasonic treatment for 30 min. Finally, the black homogeneous dispersion of acid-treated CG with PVP (ATCG–PVP) was obtained.Preparation of EP composites
Typically, DEGBA and ATCG–PVP dispersion were mixed in the flask and stirred at 120 °C for 3 h to remove the ethanol. When the mixture was cooled down to 80 °C, DDM with the equivalent protons to epoxy groups of DEGBA was added, and the mixture was stirred until the complete dissolution of DDM. Then, the mixture was poured into a preheated mold, and degassed in a vacuum oven at 80 °C for 30 min. Finally, the mixture was cured in the following steps: 80 °C for 2 h, 110 °C for 1 h, 150 °C for 2 h, 180 °C for 2 h, 200 °C for 2 h. The formulations of EP composites were listed in Table 1. For comparison, CG–PVP/EP composite with 0.2 wt% CG and PVP/EP with 0.4 wt% PVP were also prepared.| Samples | ATCG or CG (wt%) | PVP (wt%) |
|---|---|---|
| a The contents of DEGBA and DDM for all EP composites were 25 g and 6.3 g, respectively. | ||
| Pure EP | 0 | 0 |
| ATCG–PVP/EP-0.1 | 0.1 | 0.2 |
| ATCG–PVP/EP-0.2 | 0.2 | 0.4 |
| ATCG–PVP/EP-0.5 | 0.5 | 1.0 |
| CG–PVP/EP | 0.2 | 0.4 |
| PVP/EP | 0 | 0.4 |
Characterization techniques
The structural differences between CG and ATCG powders were characterized by FT-IR, Raman, XRD and XPS. FT-IR spectra ranged from 500 to 4000 cm−1 were recorded in the transmission mode on a Vertex70 spectrometer (Bruker, Germany). Raman spectra ranged from 800 to 3000 cm−1 were acquired on a Lab RAM Aramis Micro-Raman spectrometer (HORIBACJOBIN YVON Co., France) with a He laser at an excitation wavelength of 632.8 nm. XRD patterns in the range of 0–50° were taken by a D8 Advance X-ray diffractometer (Bruker, Germany) using Cu Kα irradiation (λ = 1.54184 Å) at the step length of 0.02°. XPS was carried out on an Axis Ultra-DLD X-ray photoelectron spectrometer (Kratos, UK) with Al Kα radiation (1486.6 eV) as the X-ray source for excitation.CG and ATCG were dispersed in ethanol with the help of PVP as surfactant, respectively. And they were dropped onto mica sheets for AFM measurement on a DI/MultiMode (Veeco, USA) instrument with tapping mode under ambient condition. The CG–PVP and ATCG–PVP dispersions were further filtered and then dried for thermogravimetry analysis (TGA), which was carried out on a Netzsch 2209F1 thermogravimetric analyzer with the temperature range of 30–800 °C and a heating rate of 10 °C min−1 under a nitrogen atmosphere.
The tensile properties of EP composites were carried out according to ASTM D638-08 using an Instron-5967 universal testing machine at the room temperature and a crosshead speed of 2 mm min−1. Differential scanning calorimeter (DSC) analysis was performed on a Netzsch DSC 200F3A01 thermal analyser (Selb, Germany) at a heating rate of 10 °C min under a nitrogen atmosphere. The mid-point between onset and offset of the inflectional tangent on the measured curve was defined as glass transition temperature. EP Samples were sliced into ultra-thin sections and placed on copper meshes for transmission electron microscopy (TEM) measurement on a JEM2010-HR (JEOL, Japan) instrument. SEM images of the tensile-fractured surfaces were acquired on a Nova Nano SEM430 (FEI, Eindhoven, The Netherlands) equipped with EDS. The sample surfaces were coated with a thin layer of gold–palladium alloy prior to the experiment.
Results and discussion
Characterization of ATCG
At the beginning, CG was ultrasonically treated directly in ethanol with or without PVP. And the optical photographs of CG/ethanol and CG–PVP/ethanol dispersions are given in the left of Fig. 1. It is found that two dispersions exhibit clear stratification. After high-speed centrifugation and gravimetric analysis,22 there is almost no solid left in the upper supernatants of CG/ethanol and CG–PVP/ethanol dispersions, which indicates that CG sample used here is difficult to be directly applied in EP composites. Comparatively, the optical photographs of ATCG/ethanol and ATCG–PVP/ethanol dispersions are shown in the right of Fig. 1. The dispersion of ATCG/ethanol exhibits only slightly stratification. Furthermore, the dispersion of ATCG–PVP/ethanol exhibits the uniformity and stability on account of the intercalation of PVP into the graphene layers to prevent effectively their aggregation. | ||
| Fig. 1 Optical photographs of CG/ethanol, CG–PVP/ethanol, ATCG/ethanol and ATCG–PVP/ethanol dispersions. | ||
For further analysis, graphite, CG and ATCG were characterized by FT-IR, the spectra are shown in Fig. 2. Compared to graphite, CG contains some oxygen groups, arose from the production of CG via oxidation–reduction method. Moreover, there is an emerging absorption peak around 1729 cm−1 belonging to the stretching vibration of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O for carboxyl in the spectrum of ATCG. Besides, a broad peak around 3000–3400 cm−1 corresponding to the stretching vibration of –OH is observed. FT-IR results imply that CG is further oxidized to obtain ATCG via acid soaking technique.
O for carboxyl in the spectrum of ATCG. Besides, a broad peak around 3000–3400 cm−1 corresponding to the stretching vibration of –OH is observed. FT-IR results imply that CG is further oxidized to obtain ATCG via acid soaking technique.
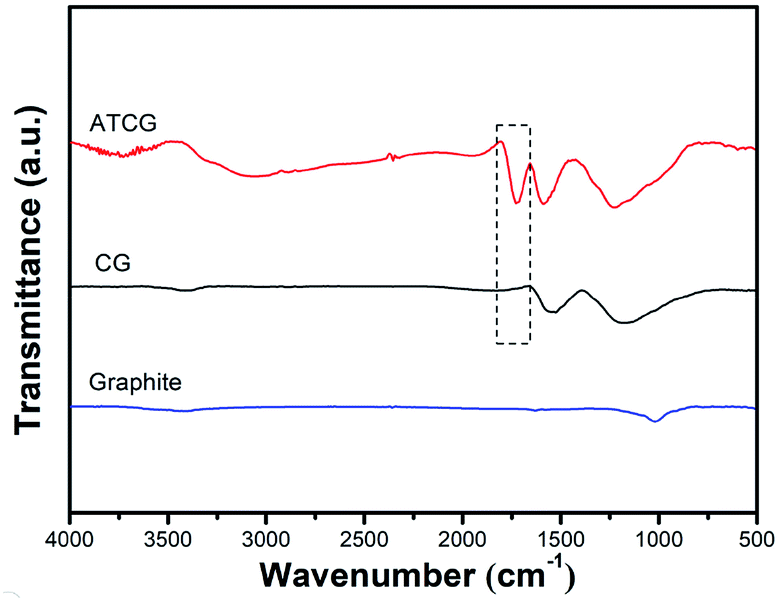 | ||
| Fig. 2 FT-IR spectra of graphite, CG and ATCG. | ||
The contents of O element on the surfaces of CG and ATCG nanosheets were figured out by XPS. From XPS spectrum of CG in Fig. 3a, there is a small amount of O element, and the molar ratio of C/O is about 17.7. Comparatively, the intensity of O element of ATCG in Fig. 3b is higher than that of CG, and the molar ratio of C/O is decreased to nearly 4.5. Compared to the high-resolution C1s spectra of CG in Fig. 3c, besides the peak at 286.6 eV of C–O bond, one additional peak at 289.0 eV of O–CO bond is found in the high-resolution C1s spectra of ATCG in Fig. 3d. The XPS results are consistent with the results of FT-IR.
 | ||
| Fig. 3 XPS spectra of (a) CG and (b) ATCG; high-resolution C1s spectra of (c) CG and (d) ATCG. | ||
The structural disorder of CG and ATCG were evaluated by the intensity ratio of D peak and G peak (ID/IG) from Raman spectra. As shown in Fig. 4, in addition to the G peak around 1580 cm−1 ascribed to the sp2 hybridized carbon atom, the Raman spectrum of CG has an emerging D peak around 1300 cm−1 originated from the disordered edge region of graphene-based materials,23 which means that defect sites are introduced into CG during the mass production. From the Raman spectrum of ATCG, the intensity of D peak is slightly higher than that of CG, and the ID/IG value of ATCG is only increased to 0.97 from 0.92 of CG, implying that few defects are introduced into graphene nanosheets through acid soaking technique.
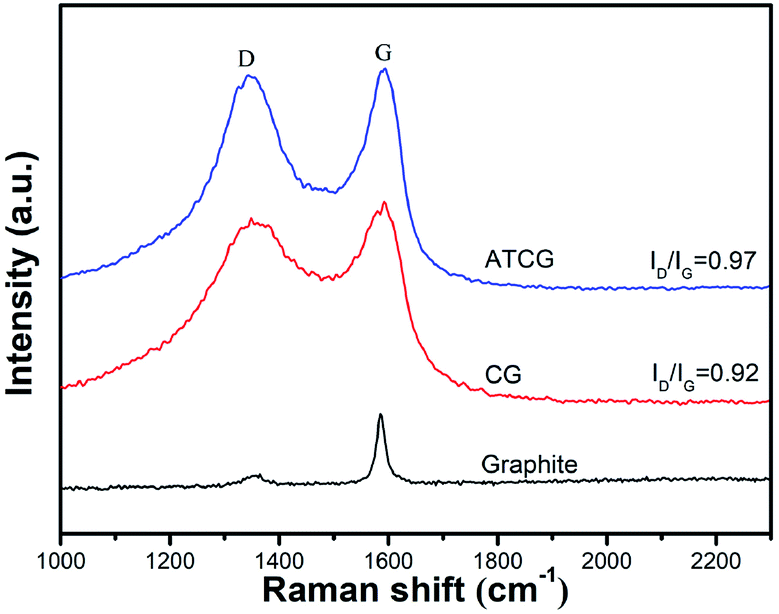 | ||
| Fig. 4 Raman spectra of graphite, CG and ATCG. | ||
The structure of CG and ATCG was further investigated by XRD, and the spectra are shown in Fig. 5. A sharp diffraction peak of graphite appears at about 26.7°, which indicates the high crystallinity of this material. Based on Braggs law, the d-spacing of graphite layer is 0.34 nm. The diffraction peaks of CG and ATCG are much weaker and broader than that of graphite, suggesting the regular stack structure of graphite is destroyed to some extent. From the enlarged view of the spectra, the diffraction peak of CG is at about 25.6°, which means a slightly increase in d-spacing (0.01 nm). Instead, since the introduction of oxygen groups via acid soaking technique, the diffraction peak of ATCG is further decreased to 23.6°, meaning a larger d-spacing of 0.38 nm. According to previous studies,24 graphene with larger d-spacing is more advantageous to be exfoliated into monolayer.
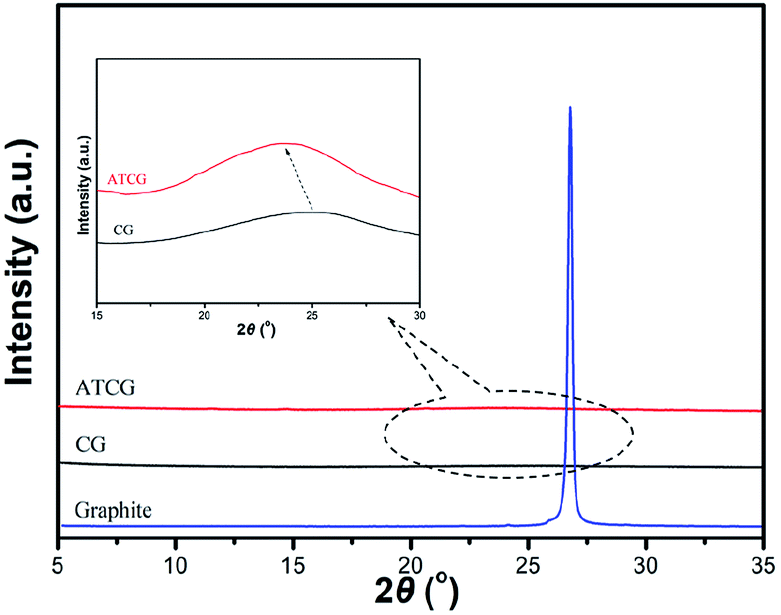 | ||
| Fig. 5 XRD patterns of graphite, CG and ATCG. | ||
AFM images with the height profiles of CG–PVP and ATCG–PVP dispersions are presented in Fig. 6. From Fig. 6a, it is apparently found that CG is granular and agglomerated. The average thickness of CG is about 25 nm after ultrasonic treatment (seen in Fig. 6c), indicating there is barely any monolayer nanosheet existed in CG. Even with the help of PVP as intercalant, CG is still difficult to be peeled off. As mentioned in the results of XRD, the d-spacing of ATCG is larger than that of CG, thus it is more conducive to the intercalation of PVP. As shown in Fig. 6b, ATCG displays relatively even lamellar morphology. And the height profile of ATCG in Fig. 6d shows that the average thickness is reduced to about 1 nm, revealing that monolayer nanosheets are obtained.25
 | ||
| Fig. 6 AFM images of (a) CG–PVP and (b) ATCG–PVP; the height profiles of (c) CG–PVP and (d) ATCG–PVP. | ||
In addition, the CG–PVP and ATCG–PVP dispersions are further filtered and then dried for TGA characterization. Fig. 7 shows TGA curves of CG, ATCG, PVP, CG–PVP and ATCG–PVP. Compared to CG, a more significant weight loss occurs from 100–800 °C for ATCG, which is attributed to the removal of the oxygen groups on ATCG nanosheets, corresponding to the oxidation of CG via acid soaking technique. Besides, CG–PVP and ATCG–PVP exhibit a major weight loss stage in the range of 400–500 °C due to the decomposition of PVP, even though the dispersions have been filtered to remove PVP surfactant in ethanol. The results indicate that CG and ATCG are wrapped by PVP surfactant, which can improve the dispersity of CG and ATCG in EP matrix.26
 | ||
| Fig. 7 TGA curves of CG, ATCG, PVP, CG–PVP and ATCG–PVP. | ||
Performance of EP composites
As mentioned above, CG is exfoliated into monolayer, which make it possible for being applied in EP. The dispersion of CG and ATCG nanosheets in EP composites was investigated via TEM measurement, and the results are shown in Fig. 8. Undoubtedly, agglomerated morphology is found in Fig. 8a for a CG–PVP/EP composite, revealing the poor dispersion of CG in EP matrix. In contrast, some black lines are observed in Fig. 8b for ATCG–PVP/EP composite with 0.2 wt% ATCG. The black lines are the images of the monolayer/multilayer ATCG nanosheets, which is consistent with those of graphene/polymer composites reported in the literature.27 Besides, it can be seen that the well-dispersed ATCG nanosheets are curved and interconnected with each other, implying the possibility of improving performance of EP composites.28 However, when the loading of ATCG is further increased to 0.5 wt%, it is found that some ATCG nanosheets are agglomerated together (seen in Fig. 8c), which may deteriorate the performance of EP composites. | ||
| Fig. 8 TEM images of the ultrathin section of (a) CG–PVP/EP, (b) ATCG–PVP/EP-0.2 and (c) ATCG–PVP/EP-0.5. | ||
In addition to the dispersion of graphene nanosheets in polymer matrix, the interactions between graphene nanosheets and polymer matrix is another key factor for the mechanical properties of composites. Natarajan et al.29 revealed that the strong interactions between graphene nanosheets and polymer matrix were beneficial to the increase in glass transition temperature (Tg). Besides, the well-dispersed graphene nanosheets in polymer matrix decreased the Tg value considerably. In the present work, Tg of EP composites was characterized by DSC, and the results are shown in Fig. 9. When the loading of ATCG is 0.2 wt%, Tg of the EP composite is increased to 170.5 °C from 163.9 °C of pure EP, implying the strong interactions between ATCG nanosheets and EP matrix.
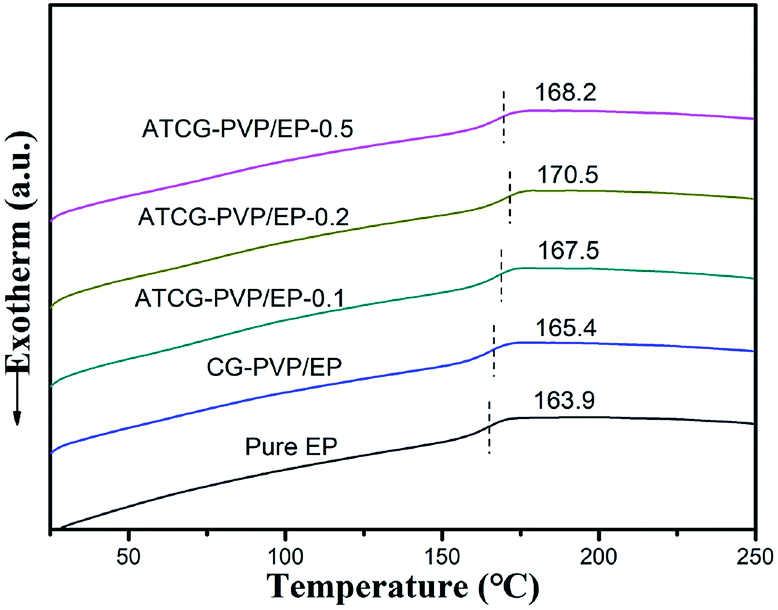 | ||
| Fig. 9 DSC curves of pure EP, CG–PVP/EP and ATCG–PVP/EP composites. | ||
The well-dispersed ATCG nanosheets and the strong interactions between ATCG nanosheets and EP matrix tempt us to explore the mechanical properties of EP composites. The tensile test results are presented in Fig. 10 and summarized in Table 2. Since the poor dispersion of CG in EP, the tensile properties of CG–PVP/EP composite with 0.2 wt% CG are almost unchanged compared with those of pure EP. Instead, the tensile strength of ATCG–PVP/EP composite with 0.2 wt% ATCG is up to 85.2 MPa (14.7% higher than that of pure EP). However, excessive loading leads to the poor dispersion and partially aggregation of ATCG, reducing the effective reinforcement. In addition, it is interesting to find that the tensile modulus of EP composites increases slightly, despite the good dispersion of ATCG in EP. According to Anwer's study,26 the addition of PVP surfactant might reduce the interfacial adhesion between ATCG nanosheets and EP matrix, which probably inhibits the stress transfer from EP matrix to ATCG nanosheets, resulting the almost invariable tensile modulus.
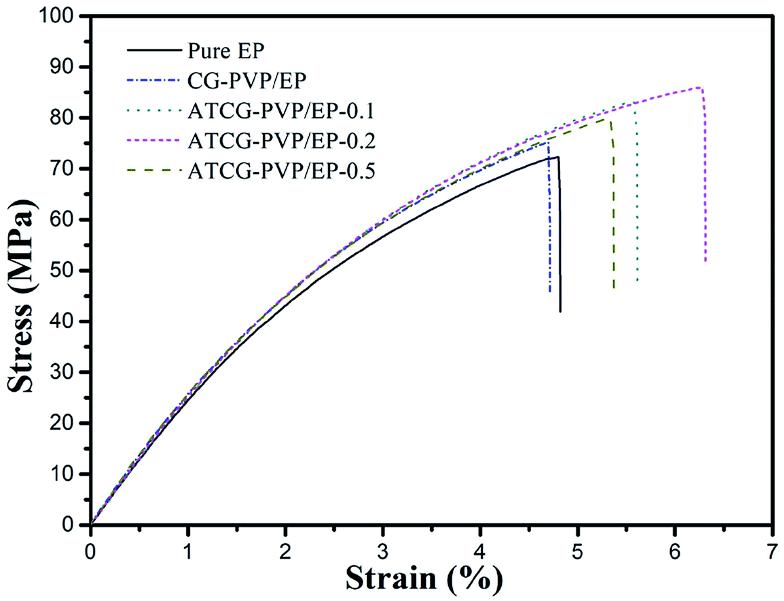 | ||
| Fig. 10 Stress–strain curves of pure EP, CG–PVP/EP and ATCG–PVP/EP composites. | ||
| Sample name | Tensile strength (MPa) | Tensile modulus (GPa) | Elongation at break (%) | Fracture energy (kJ m−2) |
|---|---|---|---|---|
| Pure EP | 74.3 ± 2.6 | 2.85 ± 0.07 | 5.0 ± 0.3 | 2.3 ± 0.2 |
| ATCG–PVP/EP-0.1 | 81.0 ± 2.7 | 3.01 ± 0.20 | 5.8 ± 0.6 | 3.0 ± 0.4 |
| ATCG–PVP/EP-0.2 | 85.2 ± 2.8 | 3.02 ± 0.19 | 6.4 ± 0.4 | 3.6 ± 0.3 |
| ATCG–PVP/EP-0.5 | 79.4 ± 4.9 | 3.04 ± 0.27 | 5.5 ± 0.5 | 2.8 ± 0.5 |
| CG–PVP/EP | 75.9 ± 6.0 | 3.03 ± 0.13 | 5.2 ± 1.2 | 2.5 ± 0.8 |
| PVP/EP | 74.2 ± 2.2 | 2.84 ± 0.19 | 5.4 ± 0.4 | 2.6 ± 0.3 |
Notably, the ATCG–PVP/EP-0.2 composite shows the higher elongation at break of 6.4%, which indicates 28% improvement as compared with that of pure EP. Furthermore, the fracture energy calculated from the area under the stress–strain curves is employed to study the toughness of EP composites.30 The fracture energy of ATCG–PVP/EP-0.2 composite is increased to 3.6 kJ m−2 (56.5% higher than the corresponding value for pure EP), indicating the remarkable improvement in toughness. Dose the PVP surfactant play the role of toughening agent in EP composites? In fact, the fracture energy of PVP/EP rises slightly, which is probably due to the inadequate loading of PVP.31 Therefore, the increase in the toughness of ATCG–PVP/EP composites is mainly ascribed to the well-dispersed ATCG, which is further investigated by SEM.
SEM images of tensile-fractured surface of pure EP are shown in Fig. 11. From Fig. 11a, the tensile-fractured surface of pure EP shows typical cleavage patterns with a series of parallel steps, and the smooth morphology of the crack is seen at high magnification in Fig. 11b, indicating the brittleness of pure EP. After the introduction of the poor-dispersed CG, a number of dimples (the red circles in Fig. 12a) are observed in the tensile-fractured surface of CG–PVP/EP composite. Besides, from the high magnification in Fig. 12b, a CG aggregate is found to cause stress concentration, which is deleterious to the mechanical properties of EP composites.
 | ||
| Fig. 11 (a) SEM image of tensile-fractured surface of pure EP; (b) the magnified image of (a). | ||
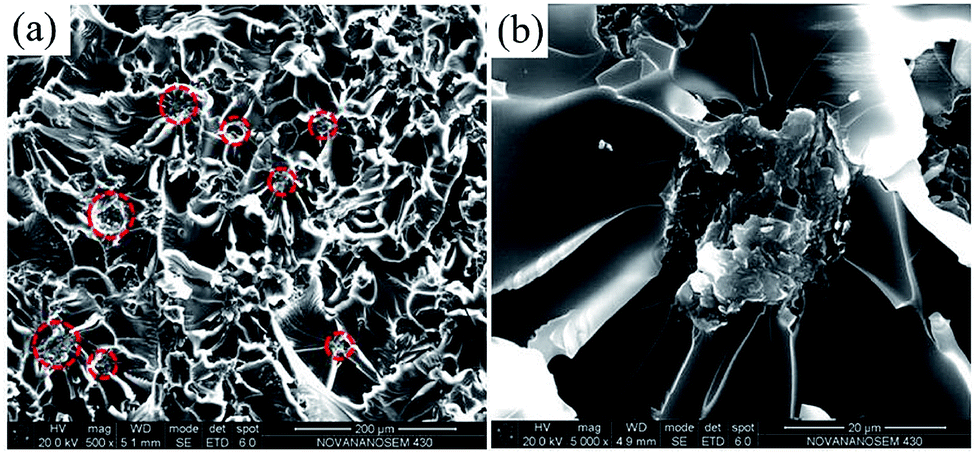 | ||
| Fig. 12 (a) SEM image of tensile-fractured surface of CG–PVP/EP; (b) the magnified image of (a). | ||
Since the well dispersion of ATCG in EP matrix, there is seldom aggregates in the tensile-fractured surface of ATCG–PVP/EP-0.2 composite (shown in Fig. 13a), and many fine ridges are found at high magnification in Fig. 13b, in favor of improving tensile properties. Furthermore, the introduction of the firmly-embedded ATCG nanosheets in EP prevents the crack propagation through inducing crack deflection and crack pinning (shown in Fig. 13c and d). Crack deflection means that ATCG nanosheets deviate the crack from its main plane resulting in an increase in the surface area of the crack and the energy to propagate such a crack. Crack pinning means that the crack growth is prevented by ATCG nanosheets, and extra energy is needed for crack propagation.9 Thus, the fracture energy of ATCG–PVP/EP composites is improved accordingly. In a word, CG with about 25 nm thickness is exfoliated into monolayer by a facile acid soaking technique, and is able to be applied in EP for improving the toughness while maintaining the high rigidity.
 | ||
| Fig. 13 (a) SEM image of tensile-fractured surface of ATCG–PVP/EP-0.2; (b) the magnified image of (a); SEM images of (c) crack deflection and (d) crack pinning caused by ATCG on the tensile-fractured surface. | ||
In addition, the tensile properties of EP composites are compared with those from some literatures (summarized in Table 3). Although the tensile strength and tensile modulus of ATCG–PVP/EP composites (increased by 14.7% and 6% compared to those of pure EP) are somewhat below those from the literature, the fracture energy of ATCG–PVP/EP composites (increased by 56.5% compared to that of pure EP) is competitive.
| Reference | Properties of neat EP | Properties of composite | % increase in σa | % increase in Eb | % increase in δc | % increase in fracture energy |
|---|---|---|---|---|---|---|
| a σ: tensile strength.b E: tensile modulus.c δ: elongation at break. | ||||||
| The present work | σ = 74.3 MPa | σ = 85.2 MPa | 14.7 | 6.0 | 28 | 56.5 |
| E = 2.85 GPa | E = 3.02 GPa | |||||
| δ = 5.0% | δ = 6.4% | |||||
| 12 | σ = 28 MPa | σ = 53 MPa | 89.3 | — | 29 | — |
| δ = 3.1% | δ = 4.0% | |||||
| 13 | σ = 45.7 MPa | σ = 58.3 MPa | 27.6 | 17.8 | 19.2 | 100 |
| E = 1.29 GPa | E = 1.52 GPa | |||||
| δ = 5.2% | δ = 6.2% | |||||
| 17 | σ = 53 MPa | σ = 83.4 MPa | 57.4 | 8.2 | 65.4 | — |
| E = 2.91 GPa | E = 3.15 GPa | |||||
| δ = 2.6% | δ = 4.3% | |||||
| 28 | σ = 46.5 MPa | σ = 68.3 MPa | 46.9 | 47.3 | 23.9 | — |
| E = 1.82 GPa | E = 2.68 GPa | |||||
| δ = 4.6% | δ = 5.7% | |||||
| 32 | σ = 33.9 MPa | σ = 70.9 MPa | 109.1 | 134.2 | 78.6 | — |
| E = 0.79 GPa | E = 1.85 GPa | |||||
| δ = 3.6% | δ = 6.43% | |||||
| 33 | σ = 48.4 MPa | σ = 51.2 MPa | 5.8 | 6.7 | — | — |
| E = 1.50 GPa | E = 1.60 GPa | |||||
| 34 | σ = 49.2 MPa | σ = 51.1 MPa | 3.8 | 28.0 | −31.3 | 50.4 |
| E = 1.28 GPa | E = 1.64 GPa | |||||
| δ = 12.8% | δ = 8.8% | |||||
| 35 | σ = 70.5 MPa | σ = 77.3 MPa | 9.6 | 7.5 | 34.5 | 40.9 |
| E = 2.80 GPa | E = 3.01 GPa | |||||
| δ = 3.25% | δ = 4.37% | |||||
| 36 | σ = 47.5 MPa | σ = 59.0 MPa | 24.2 | 5.0 | 126.8 | |
| E = 2.62 GPa | E = 2.75 GPa | |||||
| δ = 1.57% | δ = 3.56% | |||||
| 37 | σ = 69.5 MPa | σ = 79.8 MPa | 14.9 | 32.6 | 9.6 | 30.8 |
| E = 2.70 GPa | E = 3.58 GPa | |||||
| δ = 2.61% | δ = 2.86% | |||||
Conclusions
A CG sample with about 25 nm thickness was exfoliated into monolayer by a facile acid soaking technique, which was then incorporated into EP to construct graphene/EP composites. Thanks to the well dispersion of ATCG nanosheets and the strong interactions between ATCG nanosheets and EP matrix, the mechanical strength of EP composites was enhanced. Moreover, due to the prevention of crack propagation causing by ATCG nanosheets, a remarkable improvement in toughness of EP composites was achieved. This work gave reference for making use of the cost-effective graphite nanoplates to achieve the almost same performance as graphene in EP.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research is supported by the National Natural Science Foundation of China (Grant No. U1201243), the Project for Science and Technology of Guangdong (Grant No. 2019B010929001), the Opening Project of Key Laboratory of Polymer Processing Engineering (South China University of Technology), Ministry of Education (Grant No. KFKT1802).References
- R. Atif, I. Shyha and F. Inam, Polymers, 2016, 8, 281 CrossRef PubMed.
- H. B. Gu, C. Ma, J. W. Gu, J. Guo, X. R. Yan, J. N. Huang, Q. Y. Zhang and Z. H. Guo, J. Mater. Chem. C, 2016, 4, 5890 RSC.
- A. Hussein, S. Sarkar, K. Lee and B. Kim, Composites, Part B, 2017, 129, 133 CrossRef CAS.
- J. Cha, G. H. Jun, J. K. Park, J. C. Kim, H. J. Ryu and S. H. Hong, Composites, Part B, 2017, 129, 169 CrossRef CAS.
- K. P. Unnikrishnan and E. T. Thachil, Des. Monomers Polym., 2006, 9, 129 CrossRef CAS.
- Y. Rostamiyan, A. Fereidoon, M. Rezaeiashtiyani, A. H. Mashhadzadeh and A. Salmankhani, Mater. Des., 2015, 69, 96 CrossRef CAS.
- A. Montazeri, J. Javadpour, A. Khavandi, A. Tcharkhtchi and A. Mohajeri, Mater. Des., 2010, 31, 4202 CrossRef CAS.
- M. Sahu and A. M. Raichur, Composites, Part B, 2019, 168, 15 CrossRef CAS.
- J. F. Wang, C. H. Li, X. M. Zhang, L. C. Xia, X. L. Zhang, H. Wu and S. Y. Guo, Chem. Eng. J., 2017, 325, 474 CrossRef CAS.
- C. N. R. Rao, A. K. Sood, K. S. Subrahmanyam and A. Govindaraj, Angew. Chem., Int. Ed., 2009, 48, 7752 CrossRef CAS.
- R. K. Singh, R. Kumar and D. P. Singh, RSC Adv., 2016, 6, 64993 RSC.
- B. Ramezanzadeh, G. Bahlakeh, M. H. M. Moghadam and R. Miraftab, Chem. Eng. J., 2018, 335, 737 CrossRef CAS.
- S. Chhetri, N. C. Adak, P. Samanta, N. C. Murmu and T. Kuila, Polym. Test., 2017, 63, 1 CrossRef CAS.
- H. B. Kulkarni, P. Tambe and G. M. Joshi, Compos. Interfaces, 2018, 25, 381 CrossRef CAS.
- H. Bai, Y. X. Xu, L. Zhao, C. Li and G. Q. Shi, Chem. Commun., 2009, 13, 1667 RSC.
- M. Saha, P. Tambe and S. Pal, Compos. Interfaces, 2016, 23, 255 CrossRef CAS.
- Y. J. Wan, L. C. Tang, D. Yan, L. Zhao, Y. B. Li, L. B. Wu, J. X. Jiang and G. Q. Lai, Compos. Sci. Technol., 2013, 82, 60 CrossRef CAS.
- G. Shi, S. Araby, C. T. Gibson, Q. S. Meng, S. M. Zhu and J. Ma, Adv. Funct. Mater., 2018, 28, 1706705 CrossRef.
- A. P. Kauling, A. T. Seefeldt, D. P. Pisoni, R. C. Pradeep, R. Bentini, R. V. B. Oliveira, K. S. Novoselov and A. H. C. Neto, Adv. Mater., 2018, 30, 1803784 CrossRef.
- M. V. D. Z. Park, E. A. J. Bleeker, W. Brand, F. R. Cassee, M. V. Elk, I. Gosens, W. H. D. Jong, J. A. J. Meesters, W. J. G. M. Peijnenburg, J. T. K. Quik, R. J. Vandebriel and A. J. A. M. Sips, ACS Nano, 2017, 11, 9574 CrossRef CAS.
- W. J. Cui, J. Chen, C. Xie, Y. Z. Jin and J. Y. Zhang, Front. Sci., 2011, 5, 40 Search PubMed.
- Y. Hernandez, V. Nicolosi, M. Lotya, F. M. Blighe, Z. Y. Sun, S. De, I. T. McGovern, B. Holland, M. Byrne, Y. K. Gun’ko, J. J. Boland, P. Niraj, G. Duesberg, S. Krishnamurthy, R. Goodhue, J. Hutchison, V. Scardaci, A. C. Ferrari and J. N. Coleman, Nat. Nanotechnol., 2008, 3, 563 CrossRef CAS.
- Z. G. Chen, H. H. Huang, S. J. Yan, Z. P. Zheng, S. M. Liu, Y. C. Yuan, J. Q. Zhao and Y. Fu, Ind. Eng. Chem. Res., 2017, 56, 9926 CrossRef CAS.
- Y. Y. Xu, H. Z. Cao, Y. Q. Xue, B. Li and W. H. Cai, Nanomaterials, 2018, 8, 942 CrossRef.
- Z. G. Chen, S. M. Liu, S. J. Yan, X. Shu, Y. C. Yuan, H. H. Huang and J. Q. Zhao, Mater. Des., 2017, 117, 150 CrossRef CAS.
- M. A. S. Anwer, J. Wang and H. E. Naguib, Ind. Eng. Chem. Res., 2019, 58, 8131 CrossRef CAS.
- T. Q. Li, S. Y. He, A. Stein, L. F. Francis and F. S. Bates, Macromolecules, 2016, 49, 9507 CrossRef CAS.
- W. Zheng, W. G. Chen, Q. Zhao, S. X. Ren and Y. Q. Fu, Polymer, 2019, 163, 171 CrossRef CAS.
- B. Natarajan, Y. Li, H. Deng, L. C. Brinson and L. S. Schadler, Macromolecules, 2013, 46, 2833 CrossRef CAS.
- M. Harada, M. Hirotani and M. Ochi, J. Appl. Polym. Sci., 2019, 136, 47891 CrossRef.
- J. S. Lee, N. Y. Ko, N. H. Kwak, W. B. Ying and B. Lee, J. Appl. Polym. Sci., 2019, 136, 48178 CrossRef.
- L. Wei, X. Chen, K. Hong, Z. Yuan, L. Wang, H. Wang, Z. Qiao, X. Wang, Z. Li and Z. Wang, Compos. Interfaces, 2019, 26, 141 CrossRef.
- M. K. Shukla, A. Kumar, A. Yadav and K. Sharma, Mater. Today: Proc., 2019, 11, 679 CAS.
- D. Galpaya, M. Wang, G. George, N. Motta, E. Waclawik and C. Yan, J. Appl. Phys., 2014, 116, 053518 CrossRef.
- M. M. Kooshki and A. Jalali-Arani, Polym. Compos., 2018, 39, E2497 CrossRef.
- R. Aradhana, S. Mohanty and S. K. Nayak, Polymer, 2019, 166, 215 CrossRef CAS.
- U. R. Hashim and A. Jumahat, Mater. Res. Express, 2019, 6, 025303 CrossRef.
| This journal is © The Royal Society of Chemistry 2019 |