
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAtomic layer deposition with rotary reactor for uniform hetero-junction photocatalyst, g-C3N4@TiO2 core–shell structures†
Eunyong Janga,
Won Jun Kimb,
Dae Woong Kimb,
Seong Hwan Hongb,
Ijaz Alib,
Young Min Park *c and
Tae Joo Park*ab
*c and
Tae Joo Park*ab
aDepartment of Advanced Materials Engineering, Hanyang University, Ansan 15588, Korea. E-mail: tjp@hanyang.ac.kr
bDepartment of Materials Science & Chemical Engineering, Hanyang University, Ansan 15588, Korea
cSurface Technology Group, Korea Institute of Industrial Technology, Incheon 31056, Korea. E-mail: youngmin@kitech.re.kr
First published on 17th October 2019
Abstract
A heterojunction of TiO2 grown on g-C3N4 particles is demonstrated using atomic layer deposition (ALD), equipped with a specifically designed rotary reactor for maintaining stable mechanical dispersion of g-C3N4 particles during ALD. The photocatalytic activity of the g-C3N4@ALD-TiO2 core–shell composites was examined using the degradation of rhodamine B dye under visible light irradiation. The optimal composite with 5 ALD cycles of TiO2 exhibited the highest photocatalytic reaction rate constant among the composites with a range of ALD cycles from 2 to 200 cycles, which was observed to be 3 times higher than that of pristine g-C3N4 and 2 times higher than that of g-C3N4@TiO2 composite prepared using a simple impregnation method. The ALD-TiO2 were well-dispersed on the g-C3N4 surface, while TiO2 nanoparticles were agglomerated onto the g-C3N4 in the g-C3N4@TiO2 composite prepared by the impregnation method. This created uniform and stable heterojunctions between the g-C3N4 and TiO2, thus, enhancing the photocatalytic activity.
1. Introduction
As various environmental issues from pollutants and alternative energy resources have emerged, research to identify solutions that address these problems has received much attention. Among a variety of suggested solutions, photocatalysts have attracted intensive interest due to their potential for photodecomposition of organic pollutants and hydrogen generation from solar light. Ever since Wang et al. used graphitic carbon nitride (g-C3N4) as a metal-free photocatalyst to generate hydrogen from water utilizing solar radiation in 2009,1 g-C3N4 has been widely studied as a promising photocatalyst. g-C3N4 is a two-dimensional, π-conjugated polymeric semiconductor which exhibits photocatalytic activity from visible light with an energy bandgap of ∼2.7 eV.2 g-C3N4 is physicochemically stable, environmentally friendly,3 and easily synthesized using cheap precursors.4 The photocatalytic activity of g-C3N4, however, is limited by the rapid recombination of photogenerated electron–hole pairs.5,6 To enhance the photocatalytic performance of g-C3N4, various methods have been investigated, such as nanostructure design,7,8 crystal structure engineering,9 doping,10 and coupling with other semiconductors.11 Among these methods, coupling with other semiconductor has been identified as one of the most efficient methods to lower the recombination rate of photogenerated charge carriers. As a result, previous researchers have coupled g-C3N4 with other semiconductors, including Bi2WO6,12 BiVO4,13 TaON,14 CdS,15 ZnO,16 and TiO2,17 and their results showed improved photocatalytic activity owing to efficient charge separation. This semiconductor heterostructure design has been proved as a promising strategy for photocatalytic dye degradation through a number of publications.18–30In the early stages of research on g-C3N4, we demonstrated the potential of a high-efficiency photocatalyst using a g-C3N4@TiO2 heterojunction composite with chemical stability and non-toxicity.31–33 Various attempts to coat TiO2 on g-C3N4 are documented in the literature; synthesis methods such as ball-milling,34 the impregnation method,35 hydrolysis,36 and solvothermal techniques37 have been employed to form g-C3N4@TiO2 composites. However, when utilizing the solid-state mixing and impregnation methods, it is difficult to make a uniform heterojunction between pre-synthesized g-C3N4 and TiO2. For the hydrolysis method, it is hard to achieve the formation of well-distributed TiO2 particles on the surface of g-C3N4 due to the rapid hydrolysis reaction.36 Therefore, the solvothermal method has been widely used to produce a uniform coating by utilizing in situ TiO2 growth on the surface of g-C3N4. However, practical problems also exist in liquid phase synthesis processes like the solvothermal method; it is difficult to precisely control the size and concentration of TiO2 particles in the coating. Furthermore, still problematic are common issues of solvothermal method such as residual impurities and time-consuming process. Cleaning, centrifugation, and drying steps must be performed after the main synthesis process.
In this work, atomic layer deposition (ALD) was introduced to resolve the aforementioned problems in the synthesis of g-C3N4 based heterojunction composites. ALD is a vapor deposition method consisting of self-limiting surface chemical reactions. Alternating pulses of a precursor and an oxidant (denoted as A, B sequence, respectively) are supplied into the ALD reactor, and surface reactions occur on the surface of the substrate. This AB reaction cycle runs repeatedly in a sequence,38 which can achieve ultrathin and uniform film deposition by controlling the number of AB cycles.39 Recently, reports on particle coating via ALD have increased.40 Longrie et al. proved a pinhole-free and conformal Al2O3 film coating on ZnO nanoparticles.41 Hakim et al. reported the conformal Al2O3 coating on TiO2 to suppress the phototoxicity of TiO2 in sunscreen applications.42–44 Here, introduction of an ALD to form a TiO2 coating on the surface of g-C3N4 (g-C3N4@ALD-TiO2) allows for precise size control of the TiO2 nanoparticles, low concentrations of impurity residues, and a simple synthesis process. However, the general ALD system is designed for the deposition process on fixed Si wafer. Therefore, the ALD system used in this work is equipped with specially designed rotary reactor, which provides stable mechanical dispersion of g-C3N4 particles in the reactor for uniform coating of TiO2.16 As a result, the photocatalytic activity of the synthesized composite was enhanced by producing stable, uniform, and intimate heterojunctions between g-C3N4 and TiO2. A photocatalytic degradation experiment of rhodamine b (RhB) was utilized to probe the enhancement in the photocatalytic activity of g-C3N4@ALD-TiO2 composite, and various physicochemical analyses were conducted on the composite. A g-C3N4@TiO2 composite synthesized using a simple impregnation method was also prepared to be used as a control group.
2. Experimental
2.1. Preparation of g-C3N4
Bulk g-C3N4 was synthesized using a simple pyrolysis method.36 10 g of urea was ground in a mortar and put into a crucible covered with aluminum foil. The crucible was calcined at 550 °C for 4 h at a heating rate of 10 °C per min in a muffle furnace. The yellow product that was obtained was ground into a powder for further use.2.2. Preparation of g-C3N4@TiO2 composite
The g-C3N4@TiO2 composites were prepared using ALD with a specially designed rotary reactor. The pristine g-C3N4 powder was loaded into the rotary reactor (Atomic shell, CN-1 Co. Ltd., Korea), which was connected to ALD equipment. TiO2 was grown on the g-C3N4 using titanium isopropoxide (Ti(OCH(CH3)2)4, TTIP) and deionized (DI) water as the metal precursor and oxygen source, respectively. The temperature of the reactor was kept at 250 °C. The rotation speed of the rotary reactor was maintained at 60 rpm. N2 gas at 200 sccm was used for the purge process. The sequence of an ALD cycle is as follows: TTIP pulse, reactor isolation, N2 purge, H2O pulse, reactor isolation, and N2 purge. We introduced the reactor isolation step to enhance the surface reaction on g-C3N4. g-C3N4@TiO2 composites with 2, 5, 20, 50, and 200 cycles of TiO2 ALD were prepared. The prepared g-C3N4@TiO2 composites were calcined at 400 °C for 1 h in an atmosphere using a muffle furnace. For comparison, g-C3N4@TiO2 composites were also prepared using a simple impregnation method.45 In a typical procedure, 0.3 g of g-C3N4 was added to methanol (80 mL), and the solution was sonicated for 30 min. Then, 0.0375 g of TiO2 nanoparticles (P25) was added to the solution and stirred in a fume hood for 24 h. Next, the solution was dried in a drying oven at 60 °C for 24 h. We denote pristine g-C3N4 as GCN; the g-C3N4@TiO2 composites obtained by ALD are denoted as GT2, GT5, GT20, GT50, and GT200 (GTs) according to the number of ALD cycles; and the composites obtained from the simple impregnation method are denoted as GTC.2.3. Materials characterizations
The morphology of the composites was observed using energy dispersive X-ray spectroscopy (EDS) attached to a field emission-scanning electron microscope (FESEM, HITACHI-S4800) and a high-resolution-transmission electron microscope (HRTEM, JEM-2100F). Nitrogen adsorption–desorption isotherms were obtained on a Micrometrics ASAP 2010 system to measure the Brunauer–Emmett–Teller (BET) specific surface area and porosity of the composites. The crystalline structure of the composites was examined with an X-ray diffractometer (XRD, Rigaku-Dmax 2500) using Cu-Kα radiation (λ = 0.15405 nm, 40 kV, 100 mA). Fourier transform-infrared (FT-IR) spectra of the composites were collected using an attenuated total reflectance FT-IR spectrometer (Thermo Fisher Scientific-NICOLET iS10). UV-Vis diffuse reflectance spectroscopy (DRS) analysis of the composites was carried out on a UV-Vis spectrophotometer (JASCO V-650) using BaSO4 as a reference. Photoluminescence (PL) spectra of the composites were acquired using HORIBA-LabRAM HR to analyze the recombination behavior of the photo-generated carriers. An excitation wavelength of 320 nm was used, and PL emission spectra were obtained under λ = 350–700 nm.2.4. Evaluation of photocatalytic activity
The photocatalytic activities of the samples were evaluated using the degradation of RhB under visible light irradiation to examine the effect of ALD TiO2. RhB (0.0072 g) was added to 1 L DI water and stirred in the dark for 1 h. 30 mg of a photocatalyst were transferred into 200 mL of the aqueous RhB solution and stirred in the dark for 1 h to establish an adsorption–desorption equilibrium. Then, the reaction slurry was irradiated under visible light for 2 h using a 100 W halogen lamp with UV-absorbing glass filter (HI-Spot 95) obtained from Osram Sylvania Inc. Luminous intensity is 80 klux (∼11.7 mW cm−2 or 7400 foot-candle), which was measured with a lux meter (TES-1339 Light Meter Pro) at several points. Each suspension was collected at 20 minute intervals. We recorded the changes in concentration of the suspension versus irradiation time by observing the maximum absorption spectra using a JASCO V-550 UV-Vis spectrophotometer. To determine the active species during the photocatalytic degradation of the RhB dye, benzoquinone (BQ, 1 mM), EDTA-2Na (1 mM), and isopropyl alcohol (IPA, 5 mM) were used to trap the superoxide radical (O2˙−), the hole (h+) scavenger, and the hydroxyl radical (OH˙) scavenger, respectively.3. Results and discussion
SEM images of GCN, GT200, and GTC are shown in Fig. 1a, b, and c, respectively. Morphological changes between GT200 and GCN were difficult to observe with SEM due to resolution limits. However, the GTC prepared with the simple impregnation method in Fig. 1c exhibited agglomerates of TiO2 particles on the g-C3N4 surface. SEM-EDS point analyses further confirmed the presence of agglomerated TiO2 particles in the GTC samples (Fig. S1†). Fig. 1d, e, and f show the SEM images and corresponding EDS elemental (N and Ti) mapping results for GT50, respectively. The coincidence of the g-C3N4 particle shape (or N mapping results) with the Ti mapping results suggests a homogeneous coating of ALD TiO2 on g-C3N4. The concentration of TiO2 on the g-C3N4 can be estimated using the Ti/N ratio, which was measured with SEM-EDS and TEM-EDS and plotted as a function of the number of ALD cycles, as shown in Fig. 1g. For both cases the Ti/N wt% ratio increased linearly with the number of TiO2 ALD cycles, which suggests that ALD TiO2 was successfully grown on g-C3N4.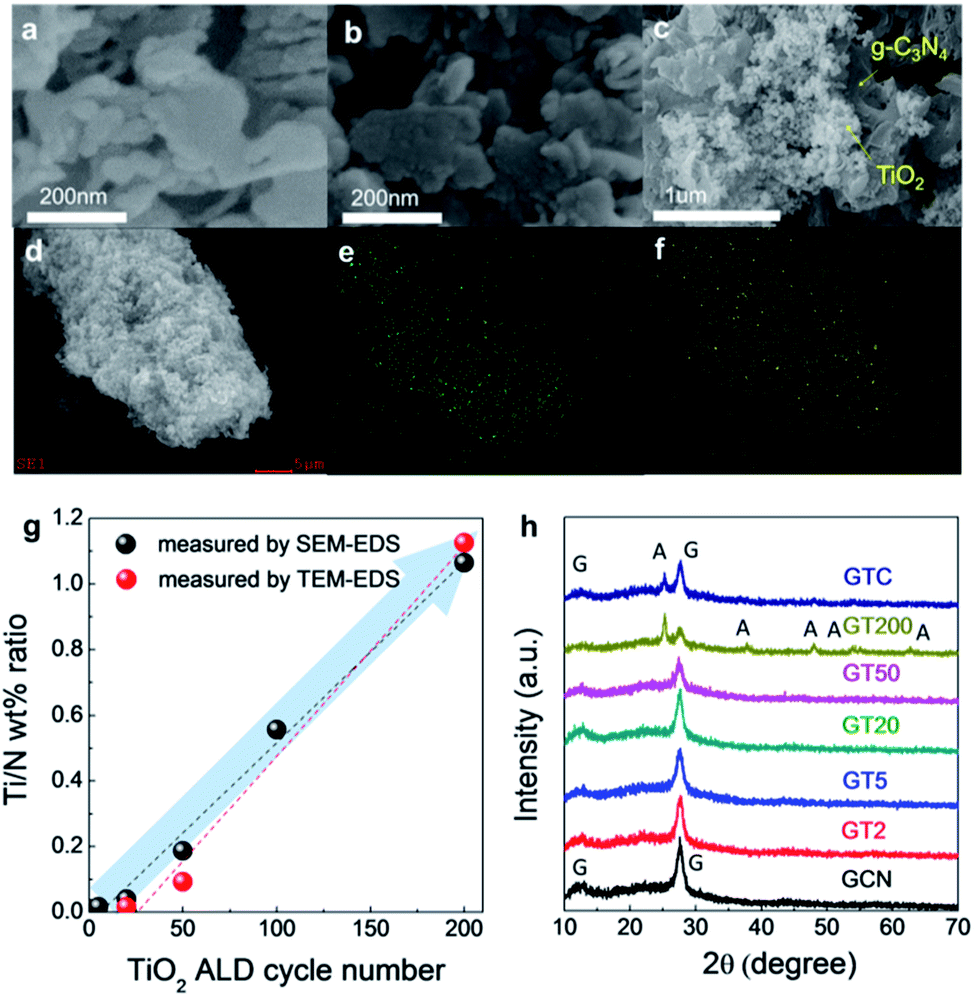 | ||
| Fig. 1 SEM images of (a) GCN, (b) GT200, and (c) GTC. (d) A SEM image and elemental mapping of (e) N and (f) Ti in GT50. (g) Ti/N ratio as a function of the number of TiO2 ALD cycles, and (h) the XRD patterns of GCN, GTs, and GTC. | ||
Fig. 1h shows the XRD patterns of GCN, GTs, and GTC. GCN exhibits two characteristic diffraction peaks which are 13.1°, 27.6°, corresponding to the in-plane ordering of the tri-s-triazine unit for the (001) plane and the interlayer-stacking of the aromatic system for the (002) plane of g-C3N4, respectively.46 When more than 20 TiO2 ALD cycles were used, we observed a gradual decrease in the intensity of the g-C3N4 (001) plane peak, and anatase TiO2 (101), (004), (200), (105), (211), and (204) peaks clearly appeared at GT200. The TiO2 crystalline structure in the GTs with <200 ALD cycles was hardly detected by XRD because ALD TiO2 exhibits nanocrystalline particle-like growth on the g-C3N4, and layer growth only occurs at the edge of the g-C3N4, which is discussed in more detail with the HRTEM analysis. For GTC, the peaks of g-C3N4 and anatase TiO2 were clearly observed.
The HRTEM image in Fig. 2a shows the smooth surface of GCN. The amorphous structure of GCN was confirmed using a magnified image in the inset of Fig. 2a. Although TiO2 particles are scarce in the GT50 shown in Fig. 2b, nano-crystalline structures with several nm in size were observed in the magnified HRTEM image in Fig. 2c, where the d-spacing is 0.23 nm and corresponds to the (001) plane in anatase TiO2. In Fig. 2d, it is apparent that the conformal TiO2 layer is coated along the edge of the g-C3N4 in the GT200 sample, and the TiO2 nanoparticles are well distributed on the g-C3N4 surface. Furthermore, the magnified HRTEM image obtained from the area marked with a yellow box in Fig. 2d (Fig. 2e) confirmed that the interplanar distance of TiO2 coated on the surface and along the edge of the g-C3N4 was also 0.23 nm, which coincides with the d-spacing of the (001) plane of anatase TiO2. The fringes on the g-C3N4 with a lattice spacing of 0.34 nm in the orange dashed circle are consistent with that of the (101) plane of anatase TiO2. Since the average thickness of the TiO2 layer on the edge of the g-C3N4 is ∼3.1 nm and the average diameter of the TiO2 nanoparticles on the surface of the g-C3N4 is ∼6.3 nm, the growth rate of ALD TiO2 was calculated to be 0.15 Å per cycle, which is similar to reported growth rates for ALD TiO2 thin film grown with TTIP and H2O at 250 °C (0.15–0.3 Å per cycle).47 EDS line scan was performed downward through the red line in Fig. 2d (Fig. 2f), which confirmed the growth behavior of ALD TiO2 on the g-C3N4 substrate: layer growth at the edge of g-C3N4 and particle-like growth on the g-C3N4 surface. This growth behavior takes place because the growth of ALD TiO2 only occurs at chemically active defect sites, such as at a few surface defects and line defects at the edge of the g-C3N4. A similar phenomenon was also observed in various ALD studies on carbon nanotubes and graphene. Sun et al. examined ALD of TiO2 on graphene surfaces with very low reactivity. Their results were similar to our work; the surface defects of graphene acted as TiO2 nucleation sites.48 Additionally, Cavanagh et al. coated Al2O3 on multi-walled carbon nanotubes (MWCNT) using ALD and found that Al2O3 nucleation only occurred at a few defects and active chemical species on the surface of the MWCNT.49 g-C3N4 has many defects consisting of NH and NH2 functional groups,37,50 which facilitate nucleation during the ALD process. Furthermore, the reaction between C–NH2 and H2O produces OH functional groups.51,52 Therefore, the nucleation of TiO2 occurs on the NH, NH2, and OH functional groups at the defect sites on the surface and edge of g-C3N4, as shown in Fig. 2g. Furthermore, IR spectra of the prepared samples indicate that the change of defect site on g-C3N4 occurred with increasing number of ALD TiO2 cycles as shown in Fig. 2h. The broad absorption band in the region of 3000–3400 cm−1 corresponds to the stretching mode of the terminal NH2, NH, and OH surface active species.37 The absorption peaks between 1200 and 1630 cm−1 are assigned to the stretching modes of CN heterocycles. The peak at 805 cm−1 is attributed to the characteristic mode of the triazine unit.53 In all cases, these peaks confirm the presence of g-C3N4. Note that the peak intensity in the region of 3000–3400 cm−1 corresponding to the surface active species decreased with an increasing number of ALD cycles, which suggests that the surface active species were consumed by ALD TiO2 growth, as discussed above.
 | ||
| Fig. 2 TEM images of (a) GCN, (b and c) GT50, (d and e) GT200, and (f) TEM-EDS line scanning result of GT200. (g) Schematics of ALD TiO2 growth on g-C3N4, and (h) FT-IR spectra of the prepared composites. | ||
The band gap energy of GCN, GT2, GT5, GT50, and GT200 were obtained based on UV-Vis diffuse reflectance spectra, and the results were similar (∼2.75 eV) regardless of number of ALD cycle (Fig. S2†). Since the band gap energy of TiO2 (∼3.21 eV) is larger than that of g-C3N4 (∼2.75 eV), TiO2 loading on the g-C3N4 has a minimal effect on the observed band gap energy of g-C3N4@TiO2 composite.
Fig. 3a shows the degradation of the RhB dye concentration in the presence of GCN, GTs, GTC, and pure TiO2 (P25) as a function of time exposed to visible light irradiation. All the samples exhibited photocatalytic activity under visible light irradiation. The photolysis of RhB dyes in the absence of the photocatalysts under visible light is also included, which was negligible. For GCN, 71% of the RhB was decomposed under visible light irradiation after 2 h. Even though it is generally known that pure TiO2 is not capable of absorbing visible light, the pure TiO2 (P25) that we used for comparison removed 32% of RhB dye molecules due to dye sensitization of the RhB adsorbed on the TiO2 surface, which is in agreement with previous studies.54 RhB dye can generate e−–h+ pairs by absorbing light. Then, the photo-generated electrons are injected into the TiO2 conduction band. The injected electrons reduce O2 to superoxide O2˙−, which contributes to the decomposition of the RhB dye molecules. For quantitative analysis of the photocatalytic activity, a pseudo-first-order kinetics model was employed according to Langmuir–Hinshelwood using the following equation:
| ln(C0/C) = kt |
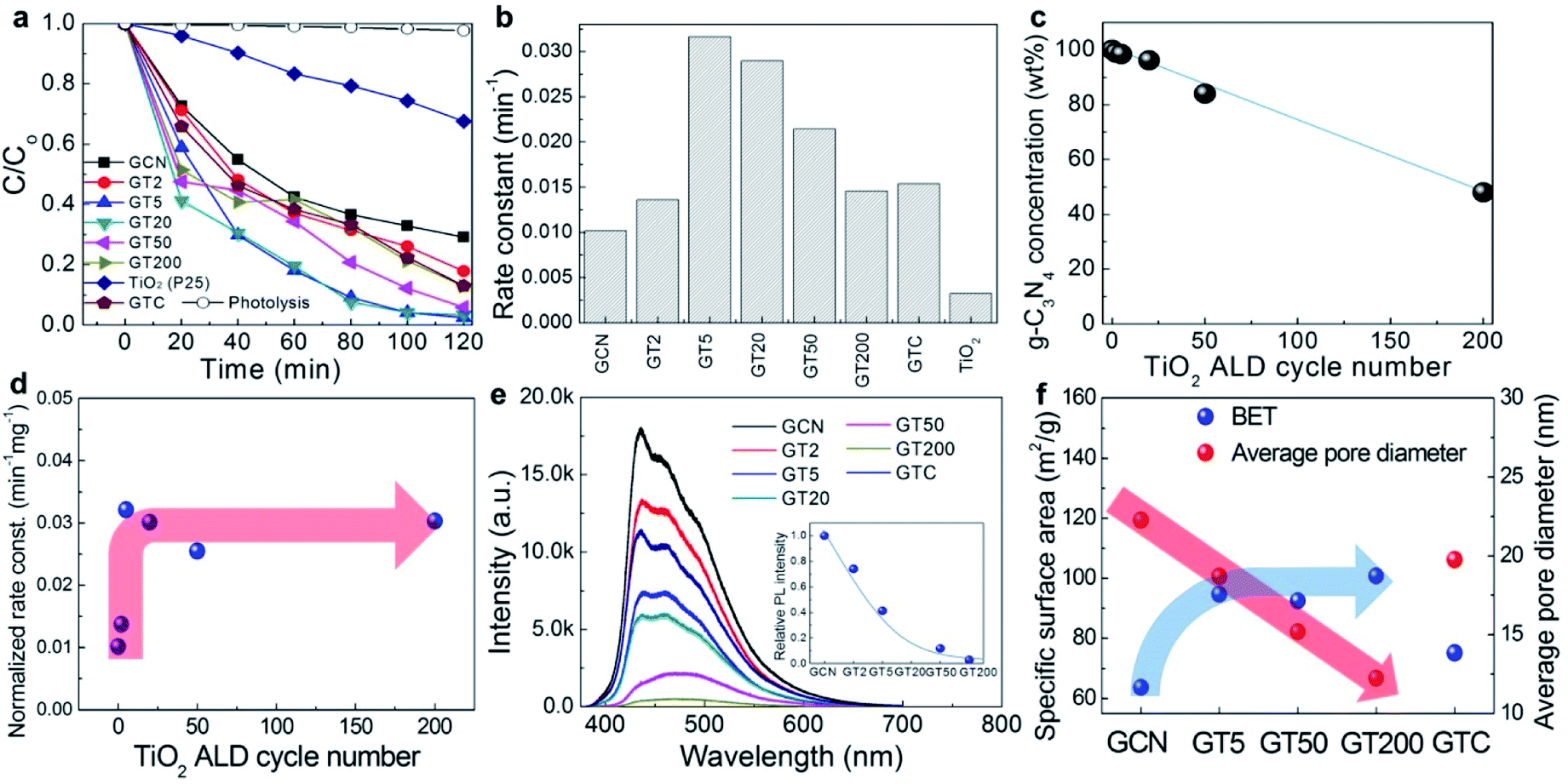 | ||
| Fig. 3 (a) The photocatalytic degradation of RhB in the presence of GCN, GTs, GTC, and pure TiO2 (P25) under visible light irradiation and (b) the corresponding reaction kinetics. (c) The weight concentration of g-C3N4 in GTs, and (d) the normalized photocatalytic reaction rate constant of GTs as a function of the number of ALD cycles. (e) PL emission results, and (f) the specific surface area and average pore diameter of GCN, GTs, and GTC. | ||
For a better comparison, however, it is necessary to control the concentration of g-C3N4 in the GTs to be identical or normalized because bare TiO2 rarely absorbs visible light due to its large energy band gap. Fig. 3c shows the weight concentration of g-C3N4 in the GTs as a function of the number of ALD cycles based on the N/Ti values measured by SEM-EDS. In the figure, the g-C3N4 concentration decreased from 98.4% to 48.1% when the number of ALD cycles increased from 2 (GT2) to 200 (GT200). Therefore, the photocatalytic reaction rate constant was normalized to the weight concentration of g-C3N4 in the GTs, as shown in Fig. 3d. The saturation trend of the normalized rate constant was observed after 5 ALD cycles. Such an important observation suggests that only 5 cycles of TiO2 ALD are required to reach to the saturated photocatalytic reaction rate constant. The origin of such a behavior was examined as follows. The photocatalytic reaction rate is mainly determined by the charge recombination behavior and specific surface area of the GTs. First, the charge recombination behavior was examined using the photoluminescence (PL) in Fig. 3e. Since PL emissions result from the recombination of excited e−–h+ pairs, PL is generally used to study the state of photo-generated carriers. The higher the recombination of the charge carriers occurred, the higher the PL emission intensity was observed. GCN exhibited the highest PL intensity, suggesting that the charge recombination rate is the largest, and thus, GCN has the lowest photoactivity. The decrease in the PL intensity of the GTs compared to GCN indicates that photo-induced electrons from g-C3N4 are transferred to TiO2, which leads to efficient charge separation.55,56 The PL intensity decreased abruptly when the number of ALD cycles increased from 2 (GT2) to 5 (GT5), and gradually saturated over 5 ALD cycles. Therefore, 5 cycles of ALD with TiO2 are enough to form intimate heterojunctions with g-C3N4 for efficient charge separation. Secondly, as shown in Fig. 3f, the specific surface area (BET) of the GTs increased when TiO2 was coated on the g-C3N4 surface in a three-dimensional manner, but it was saturated over 5 ALD cycles because the average pore diameter in the GTs decreased with an increasing number of TiO2 ALD cycles. Consequently, the PL emission and BET results in Fig. 3e and f, respectively, coincide with the normalized photocatalytic activity results in Fig. 3d; the reaction rate constant was saturated over 5 ALD cycles. On the other hand, GTC exhibited a specific surface area similar to that of GCN and showed intermediate PL intensity because the TiO2 particles were mechanically agglomerated onto the g-C3N4 without intimate contact to form heterojunctions. As a result, GTC displayed poor photocatalytic activity (the g-C3N4 concentration in GTC is ∼89%). Meanwhile, the saturation behaviour of photocatalytic activity might be also associated with charge carrier recombination at the interface under the overload of ALD TiO2 exceeding the optimal value.26
The photocatalytic reaction mechanism was examined using active species trapping (scavenger) experiments. BQ,57 EDTA-2Na,57 and IPA58 were used to trap O2˙−, h+ and ˙OH, respectively. Fig. 4a and b present the results of the scavenger experiments associated with GCN and GT5, respectively. The slowest degradation rate of RhB was observed in the presence of BQ for GCN, suggesting that O2˙− plays a key role for photodegradation. The degradation rate of RhB in the presence of IPA was 69%, similar to the results observed without the scavenger, implying that ˙OH has a minor role for RhB dye degradation. On the other hand, in the presence of EDTA-2Na, the degradation of the RhB dye reached 99% within 100 min. In this case, it is expected that EDTA-2Na promoted charge separation in the g-C3N4 by trapping h+ which has minimal involvement in the decomposition reaction of RhB, thus enhancing the degradation of RhB.57,59
 | ||
| Fig. 4 The photocatalytic degradation of RhB in the presence of (a) GCN and (b) GT5 under visible light irradiation with the addition of various scavengers, including BQ, IPA, and EDTA-2Na, and (c) the photocatalytic degradation mechanism. | ||
In Fig. 4b, the lowest photocatalytic activity of GT5 in the presence of BQ confirmed that O2˙− was a major active species for RhB dye degradation, similar to GCN. However, in the presence of IPA, the photocatalytic activity of GT5 decreased significantly, while the change in the photocatalytic activity of GCN with IPA was negligible. Such a result suggests that the contribution of ˙OH to the photocatalytic reaction increased when ALD TiO2 was adopted on g-C3N4. This result can be explained with the behaviors of the active species and the charge separation in the heterojunctions. g-C3N4, with an energy band gap of 2.87 eV, is capable of absorbing visible light with a wavelength over 450 nm, but TiO2 does not typically utilize visible light due to its wide energy band gap. Therefore, electrons are photo-generated in g-C3N4 to form the superoxide radical (O2˙−) by reacting with O2 (−0.28 V) according to reaction (1), which is the main active species for the photocatalytic degradation of both GCN and GTs as discussed above.
| e− + O2 → O2˙− (−0.28 V) | (1) |
For GTs, the lowest unoccupied molecular orbital (LUMO) and highest occupied molecular orbital (HOMO) levels (−1.27 and 1.55 eV) of g-C3N4 are more negative than the conduction band and valence band edge (−0.5 and 1.99 eV) of TiO2, which produces the type II heterojunction, as shown in Fig. 4c. Photo-generated electrons in g-C3N4 are injected from the LUMO level of g-C3N4 to the conduction band of TiO2, while photo-generated holes remain in the g-C3N4 (charge separation). Therefore, carrier recombination is suppressed, and more superoxide radicals (O2˙−) are generated compared to GCN, which contribute to the formation of the hydroxyl radical (˙OH), another active species, through reactions (2) and (3) in a sequence (0.305 and 0.67 V, respectively). The redox potential of all the reactions [(1)–(3)] are more positive than the conduction band of TiO2, which suggests that these reactions are thermodynamically favorable. Therefore, the hydroxyl radical became a secondary active species for RhB degradation in the GTs.
| O2˙− + 2H+ → H2O2 (0.305 V) | (2) |
| H2O2 + e− → OH− + ˙OH (0.67 V) | (3) |
Photo-generated holes in g-C3N4 have minimal involvement in photocatalytic reactions due to similar HOMO levels between g-C3N4 (1.55 V) and RhB (1.43 V).60 Hydroxyl radical formation from the reaction of the holes with H2O is also implausible, because the redox potential of OH−/˙OH (1.99 V) is more positive than the HOMO level of g-C3N4.
4. Conclusions
In summary, TiO2 was coated on g-C3N4 using ALD to control the recombination rate of photo-generated charge carriers in g-C3N4 (g-C3N4@ALD-TiO2). g-C3N4@TiO2 composites synthesized by a simple impregnation method were also prepared and used for comparison against the proposed method. When using ALD, TiO2 nanoparticles with a diameter of ∼6 nm were homogeneously distributed on the g-C3N4 surface. On the other hand, the impregnation method resulted in TiO2 agglomerates on the surface of the g-C3N4. The photocatalytic activity of the g-C3N4@ALD-TiO2 composites was estimated using the photo-degradation of RhB under visible light irradiation. Five cycles of TiO2 ALD was enough to form intimate, stable, and uniform heterojunctions on g-C3N4 and enhance the photocatalytic activity, as demonstrated by the saturation of the normalized photocatalytic reaction rate constant. With respect to the surface area of a composite, 5 is also an optimal number of ALD cycles. The photocatalytic reaction rate constant was 3 times higher than that of the bare g-C3N4 and 2 times higher than that of the composite prepared by the simple impregnation method.Conflicts of interest
TP and YP conceived and designed the experiments. EJ and WK conducted most of the experiments. DK, SH and IA performed the data analysis. TP, YP and EJ co-wrote the paper. All authors discussed the results and commented on the manuscript.Acknowledgements
This work was supported by the Future Semiconductor Device Technology Development Program (No. 10067739) funded by MOTIE (Ministry of Trade, Industry & Energy) and KSRC (Korea Semiconductor Research Consortium).References
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76 CrossRef CAS.
- G. Mamba and A. Mishra, Appl. Catal., B, 2016, 198, 347–377 CrossRef CAS.
- X. Dong and F. Cheng, J. Mater. Chem. A, 2015, 3, 23642–23652 RSC.
- Z. Zhao, Y. Sun and F. Dong, Nanoscale, 2015, 7, 15–37 RSC.
- L. Huang, H. Xu, Y. Li, H. Li, X. Cheng, J. Xia, Y. Xu and G. Cai, Dalton Trans., 2013, 42, 8606–8616 RSC.
- X. Wang, S. Blechert and M. Antonietti, ACS Catal., 2012, 2, 1596–1606 CrossRef CAS.
- X. Zhang, X. Xie, H. Wang, J. Zhang, B. Pan and Y. Xie, J. Am. Chem. Soc., 2012, 135, 18–21 CrossRef.
- K. Sridharan, P. Sreekanth, T. J. Park and R. Philip, J. Phys. Chem. C, 2015, 119, 16314–16320 CrossRef CAS.
- K. Schwinghammer, M. B. Mesch, V. Duppel, C. Ziegler, J. r. Senker and B. V. Lotsch, J. Am. Chem. Soc., 2014, 136, 1730–1733 CrossRef CAS.
- S. Yan, Z. Li and Z. Zou, Langmuir, 2010, 26, 3894–3901 CrossRef CAS.
- C. Pan, J. Xu, Y. Wang, D. Li and Y. Zhu, Adv. Funct. Mater., 2012, 22, 1518–1524 CrossRef CAS.
- L. Ge, C. Han and J. Liu, Appl. Catal., B, 2011, 108, 100–107 CrossRef.
- C. Li, S. Wang, T. Wang, Y. Wei, P. Zhang and J. Gong, Small, 2014, 10, 2783–2790 CrossRef CAS PubMed.
- S. Yan, S. Lv, Z. Li and Z. Zou, Dalton Trans., 2010, 39, 1488–1491 RSC.
- J. Fu, B. Chang, Y. Tian, F. Xi and X. Dong, J. Mater. Chem. A, 2013, 1, 3083–3090 RSC.
- W. Yu, D. Xu and T. Peng, J. Mater. Chem. A, 2015, 3, 19936–19947 RSC.
- X. Chen, J. Wei, R. Hou, Y. Liang, Z. Xie, Y. Zhu, X. Zhang and H. Wang, Appl. Catal., B, 2016, 188, 342–350 CrossRef CAS.
- Y.-H. Chiu, T.-F. M. Chang, C.-Y. Chen, M. Sone and Y.-J. Hsu, Catalysts, 2019, 9, 430 CrossRef CAS.
- Y.-C. Pu, H.-Y. Chou, W.-S. Kuo, K.-H. Wei and Y.-J. Hsu, Appl. Catal., B, 2017, 204, 21–32 CrossRef CAS.
- W.-H. Lin, Y.-H. Chiu, P.-W. Shao and Y.-J. Hsu, ACS Appl. Mater. Interfaces, 2016, 8, 32754–32763 CrossRef CAS.
- Y.-C. Chen, T.-C. Liu and Y.-J. Hsu, ACS Appl. Mater. Interfaces, 2015, 7, 1616–1623 CrossRef CAS.
- Y.-C. Pu, W.-H. Lin and Y.-J. Hsu, Appl. Catal., B, 2015, 163, 343–351 CrossRef CAS.
- A. T. Nguyen, W.-H. Lin, Y.-H. Lu, Y.-D. Chiou and Y.-J. Hsu, Appl. Catal., A, 2014, 476, 140–147 CrossRef CAS.
- M.-Y. Chen and Y.-J. Hsu, Nanoscale, 2013, 5, 363–368 RSC.
- J. L. Guo, Y. D. Chiou, W. I. Liang, H. J. Liu, Y. J. Chen, W. C. Kuo, C. Y. Tsai, K. A. Tsai, H. H. Kuo and W. F. Hsieh, Adv. Mater., 2013, 25, 2040–2044 CrossRef CAS.
- Y.-F. Lin and Y.-J. Hsu, Appl. Catal., B, 2013, 130, 93–98 CrossRef.
- Y.-C. Chen, Y.-C. Pu and Y.-J. Hsu, J. Phys. Chem. C, 2012, 116, 2967–2975 CrossRef CAS.
- W.-T. Chen and Y.-J. Hsu, Langmuir, 2009, 26, 5918–5925 CrossRef.
- T.-T. Yang, W.-T. Chen, Y.-J. Hsu, K.-H. Wei, T.-Y. Lin and T.-W. Lin, J. Phys. Chem. C, 2010, 114, 11414–11420 CrossRef CAS.
- Y.-C. Pu, Y.-C. Chen and Y.-J. Hsu, Appl. Catal., B, 2010, 97, 389–397 CrossRef CAS.
- K. Sridharan, E. Jang and T. J. Park, Appl. Catal., B, 2013, 142, 718–728 CrossRef.
- M. R. Hoffmann, S. T. Martin, W. Choi and D. W. Bahnemann, Chem. Rev., 1995, 95, 69–96 CrossRef CAS.
- A. Fujishima, T. N. Rao and D. A. Tryk, J. Photochem. Photobiol., C, 2000, 1, 1–21 CrossRef CAS.
- J. Zhou, M. Zhang and Y. Zhu, Phys. Chem. Chem. Phys., 2015, 17, 3647–3652 RSC.
- C. Miranda, H. Mansilla, J. Yáñez, S. Obregón and G. Colón, J. Photochem. Photobiol., A, 2013, 253, 16–21 CrossRef CAS.
- Y. Li, J. Wang, Y. Yang, Y. Zhang, D. He, Q. An and G. Cao, J. Hazard. Mater., 2015, 292, 79–89 CrossRef CAS.
- L. Gu, J. Wang, Z. Zou and X. Han, J. Hazard. Mater., 2014, 268, 216–223 CrossRef CAS.
- R. L. Puurunen, J. Appl. Phys., 2005, 97, 9 CrossRef.
- S. George, A. Ott and J. Klaus, J. Phys. Chem., 1996, 100, 13121–13131 CrossRef CAS.
- D. M. King, X. Liang and A. W. Weimer, Powder Technol., 2012, 221, 13–25 CrossRef CAS.
- D. Longrie, D. Deduytsche, J. Haemers, K. Driesen and C. Detavernier, Surf. Coat. Technol., 2012, 213, 183–191 CrossRef CAS.
- L. F. Hakim, D. M. King, Y. Zhou, C. J. Gump, S. M. George and A. W. Weimer, Adv. Funct. Mater., 2007, 17, 3175–3181 CrossRef CAS.
- K. Sridharan, E. Jang, Y. M. Park and T. J. Park, Chem. –Eur. J., 2015, 21, 19136–19141 CrossRef CAS.
- E. Jang, K. Sridharan, Y. M. Park and T. J. Park, Chem. –Eur. J., 2016, 22, 12022–12026 CrossRef CAS PubMed.
- L. Zhang, D. Jing, X. She, H. Liu, D. Yang, Y. Lu, J. Li, Z. Zheng and L. Guo, J. Mater. Chem. A, 2014, 2, 2071–2078 RSC.
- M. Groenewolt and M. Antonietti, Adv. Mater., 2005, 17, 1789–1792 CrossRef CAS.
- D. M. King, X. Liang, Y. Zhou, C. S. Carney, L. F. Hakim, P. Li and A. W. Weimer, Powder Technol., 2008, 183, 356–363 CrossRef CAS.
- X. Sun, M. Xie, G. Wang, H. Sun, A. S. Cavanagh, J. J. Travis, S. M. George and J. Lian, J. Electrochem. Soc., 2012, 159, A364–A369 CrossRef CAS.
- A. S. Cavanagh, C. A. Wilson, A. W. Weimer and S. M. George, Nanotechnology, 2009, 20, 255602 CrossRef.
- Y. Wang, X. Wang and M. Antonietti, Angew. Chem., Int. Ed., 2012, 51, 68–89 CrossRef CAS.
- B. Zhu, P. Xia, W. Ho and J. Yu, Appl. Surf. Sci., 2015, 344, 188–195 CrossRef CAS.
- J. Aarik, A. Aidla, T. Uustare, M. Ritala and M. Leskelä, Appl. Surf. Sci., 2000, 161, 385–395 CrossRef CAS.
- F. Dong, L. Wu, Y. Sun, M. Fu, Z. Wu and S. Lee, J. Mater. Chem., 2011, 21, 15171–15174 RSC.
- T. Wu, G. Liu, J. Zhao, H. Hidaka and N. Serpone, J. Phys. Chem. B, 1998, 102, 5845–5851 CrossRef CAS.
- H. Li, L. Zhou, L. Wang, Y. Liu, J. Lei and J. Zhang, Phys. Chem. Chem. Phys., 2015, 17, 17406–17412 RSC.
- R. Hao, G. Wang, H. Tang, L. Sun, C. Xu and D. Han, Appl. Catal., B, 2016, 187, 47–58 CrossRef CAS.
- Y. Tian, B. Chang, J. Lu, J. Fu, F. Xi and X. Dong, ACS Appl. Mater. Interfaces, 2013, 5, 7079–7085 CrossRef CAS.
- L. Ye, J. Liu, Z. Jiang, T. Peng and L. Zan, Appl. Catal., B, 2013, 142–143, 1–7 CAS.
- S. Hu, L. Ma, J. You, F. Li, Z. Fan, F. Wang, D. Liu and J. Gui, RSC Adv., 2014, 4, 21657–21663 RSC.
- X. Ma, Y. Lv, J. Xu, Y. Liu, R. Zhang and Y. Zhu, J. Phys. Chem. C, 2012, 116, 23485–23493 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra05958j |
| This journal is © The Royal Society of Chemistry 2019 |