
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePlatinum and rhodium complexes ligated by imidazolium-substituted phosphine as efficient and recyclable catalysts for hydrosilylation
Magdalena Jankowska-Wajda *a,
Olga Bartlewicza,
Andrea Szpechta,
Adrian Zajacb,
Marcin Smiglakb and
Hieronim Maciejewskiab
*a,
Olga Bartlewicza,
Andrea Szpechta,
Adrian Zajacb,
Marcin Smiglakb and
Hieronim Maciejewskiab
aLaboratory of Chemistry and Technology of Inorganic Polymers, Faculty of Chemistry, Adam Mickiewicz University in Poznan, Uniwersytetu Poznanskiego 8, 61-614 Poznan, Poland. E-mail: magdajw@amu.edu.pl
bPoznan Science and Technology Park of Adam Mickiewicz University Foundation, Rubiez 46, 61-612 Poznan, Poland
First published on 17th September 2019
Abstract
Phosphine ligands functionalized with imidazolium salt were prepared and used for the synthesis of two new ionic Pt(0) complexes and four Rh(I) complexes. The catalysts show very good catalytic activity in hydrosilylation reaction of olefins of different polarities (1-octene and allyl glycidyl ether) with 1,1,1,3,5,5,5-heptamethyltrisiloxane. Their insolubility in the reaction medium facilitated their isolation and permitted their multiple use in subsequent catalytic runs. In hydrosilylation of nonpolar olefins, all the catalysts showed similar activity, while in hydrosilylation of polar olefins the catalysts containing the bromide anion showed higher activity. The results permitted identification of the most effective catalysts for hydrosilylation of olefins of different polarities. The most active complexes did not lose their activity even after 10 catalytic runs, thereby providing a very good alternative to the commonly used homogeneous catalysts.
Introduction
The hydrosilylation process is widely applied in the synthesis of industrially employed organofunctional silicon compounds (silanes, polysiloxanes and silsesquioxanes) and also in the synthesis of important reagents for organic synthesis.1–4 It is also commonly used as a commercially important method of crosslinking of silicone rubbers as well as organic polymers containing vinyl or allyl groups with siloxanes and polysiloxanes with Si–H functionality.5,6 In most cases the catalysts used in the process are transition metal complexes, particularly compounds and complexes of platinum and rhodium. Although both rhodium and platinum complexes are highly active catalysts, they are homogeneous. High prices of the above metals and inadmissibility of their presence (for many applications) even in trace amounts in the final product, imply a continuous search for methods of their recycling and reuse. Classical methods based on distillation (rectification) of post-reaction mixtures followed by catalyst regeneration are very costly and often difficult to perform because of high viscosity and boiling point of reaction product (particularly the polymeric products). Thus much effort is devoted to the search for new active catalysts that would combine the advantages of homogeneous and heterogeneous catalysis. An example of such a solution is the application of ionic liquids that make it possible to conduct processes in biphasic liquid–liquid systems.7,8 The use of ionic liquids as solvents for chemical reactions is well documented in literature and has been for a long time the area of interest.9 Due to their unique properties; mainly high polarity, extremely low volatility, high thermal stability, ionic liquids allow realization of chemical processes in new pressure and temperature regimes, often acting not only as solvents but also catalysts, catalyst immobilizers or initiators.10–13 ILs (ionic liquids) have been employed in most of catalytic processes in the field of organic chemistry, but as yet their use in the synthesis of organosilicon compounds has been still poorly perceptible.14–19 There are barely a few literature reports on the application of ILs to processes of hydrosilylation with polyhydrosiloxanes.15–17 Studies on the addition of hydrosiloxanes to functional olefins, catalyzed by diverse platinum complexes in the presence of imidazolium ILs, have been among the first in this area.15,18 The contribution of our research group to this field has concerned the processes of hydrosilylation of diverse olefins catalyzed both by rhodium and platinum complexes in the medium of imidazolium,20,21 phosphonium,22,23 morpholinium,24 pyrilium25 and ammonium ionic liquids.26,27 In most cases, ILs can well dissolve and immobilize metal complexes and, at the same time, they are insoluble in reactants thus enabling performance the processes in biphasic systems and easy isolation of products (after the reaction) offering the possibility of reusing the catalytic system. Our research has enabled a selection of suitable ionic liquids and complexes of rhodium or platinum that formed very active catalytic systems, however, they have not provided us with the information on the kind of metal complex and ionic liquid interactions and on the structure of the actual catalyst. It should be mentioned that this is a formidable task because of the complexity of the systems and a large number of ions. An alternative way for obtaining heterogenized metal complexes in ILs media is the application of metal complexes in which ionic liquids act as ligands. As indicated above the use of ionic liquids as solvent media for carrying out the catalytic reactions is well documented in literature. Taking into the account the possible tuning of the physical properties of ionic liquids through changes in their structure, it is surprising that the research on the use of ionic liquids as structural components of the complexing ions has been much less explored. For example, diphenylphosphine functionalized phosphonium ionic liquids have been presented by Moores et al., as an excellent example of using ionic liquids as rhodium complex stabilizer in the hydroformylation reaction.28 In a similar fashion Jin et al. have prepared chiral pyrrolidinodiphosphine ligands attached directly to the imidazolium cation and used in the catalytic asymmetric hydrogenation reaction.29 In another example, Chen et al. have presented the synthesis and use of novel ionic complex, bis-[1-butyl-2-diphenylphosphanyl-3-methylimidazolium]-tetrachloridorhodium(III) hexafluorophosphate, which was used in the hydroformylation reaction.30 Taking into consideration that the best effects have been observed for complexes immobilized in imidazolium and phosphonium liquids, we have decided to modify the ionic liquid so that it could be employed as a ligand in the synthesis of complexes.The aim of the present study was to synthesize and characterize rhodium and platinum complexes containing these kinds of ligands, as well as to evaluate their catalytic activity for the process of olefin hydrosilylation. For this purpose we synthesized phosphonium ligands functionalized with imidazolium salt. Similar syntheses have been presented earlier by Consorti31 and Lai.32 At the next stage we applied the obtained phosphines as ligands for the synthesis of rhodium and platinum complexes. Since most of hydrosilylation catalysts are based on platinum and rhodium compounds, we also determined their catalytic activity and the possibility of multiple use in subsequent reaction runs.
Experimental
Materials
All reagents applied in the synthesis of imidazolium salts, synthesis of phosphine ligands and catalytic measurements and i.e., dibromobutane, allyl chloride, diphenylphosphine (HPPh2), 2,2′-azobis(2-methylpropionitrile) (AIBN), potassium diphenylphosphide solution (KPPh2, 0.5 M in THF), silver nitrate solution (AgNO3, 0.1 M), 1-octene, allyl glycidyl ether, n-decane and 1,1,1,3,5,5,5-heptamethyltrisiloxane (HMTS) were purchased from Sigma Aldrich and used as received. Also metal precursors [Pt2{(CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHSiMe2)2O}3], [RhCl(PPh3)3], [{Rh(μ-Cl)(cod)}2], used for the synthesis of new complexes were supplied by Sigma Aldrich. The ionic liquid 1,2-dimethylimidazole and aqueous solution of lithium bis(trifluoromethanesulfonyl)amide (LiNTf2, 80%) was purchased from Iolitec GmbH, Germany. Acetonitrile HPLC grade (CH3CN) was purchased form Sigma Aldrich and used as received, as well as all other solvents i.e., dichloromethane (DCM), tetrahydrofuran (THF), ethyl acetate (AcOEt), diethyl ether (Et2O). Deuterated solvents dimethyl sulfoxide (DMSO-d6), chloroform (CDCl3) and acetonitrile (CD3CN) were purchased from Sigma Aldrich.
CHSiMe2)2O}3], [RhCl(PPh3)3], [{Rh(μ-Cl)(cod)}2], used for the synthesis of new complexes were supplied by Sigma Aldrich. The ionic liquid 1,2-dimethylimidazole and aqueous solution of lithium bis(trifluoromethanesulfonyl)amide (LiNTf2, 80%) was purchased from Iolitec GmbH, Germany. Acetonitrile HPLC grade (CH3CN) was purchased form Sigma Aldrich and used as received, as well as all other solvents i.e., dichloromethane (DCM), tetrahydrofuran (THF), ethyl acetate (AcOEt), diethyl ether (Et2O). Deuterated solvents dimethyl sulfoxide (DMSO-d6), chloroform (CDCl3) and acetonitrile (CD3CN) were purchased from Sigma Aldrich.
General methods
All used reagents and solvents of high purity were commercially available, purchased and used as received. All reactions for synthesis of phosphine ligands and respective complexes were carried out under a dry argon atmosphere using Schlenk glassware and vacuum-line techniques. Before use the solvents were dried and degassed by standard methods.1H and 31P NMR spectra were measured using a Bruker BioSpin spectrometer operating at 400, 500 and 600 MHz in commercially available deuterated solvents. Chemical shifts for 1H are given in ppm relative to the residual proton signal of either CDCl3 (δ 7.24), CD3CN (δ 2.10) and for DMSO-d6 (δ 2.49). 31P NMR chemical shifts were specified relative to 85% H3PO4 as an external standard. The yield of a product of a given reaction was determined using a Clarus 680 gas chromatograph (Perkin Elmer) equipped with a 30 m capillary column Agilent VF-5ms and TCD detector, using the temperature program: 60 °C (3 min), 10 °C min−1, 290 °C (5 min). Thermogravimetric analysis (TGA) was carried out using an analyzer TA Instruments model TG Q50 at a linear heating rate of 10 °C min−1 under synthetic air (50 mL min−1). The tested samples were placed in a platinum pan and the weight of the samples was kept within 9–10 mg. The experimental error was 0.5% for weight and 1 °C for temperature. Melting points were measured on a Melting Point M-565 instrument (Buchi) equipped with a video camera. Temperature gradient: 10 °C min−1. FT-IR in situ measurements were performed using a Mettler Toledo ReactIR 15 instrument. For selected samples, the spectra were recorded with 256 scans for 1 at 30 second intervals with a resolution of 1 cm−1. Intensity change of the band at 903 cm−1, characteristic of Si–H bond was recorded by using an ATR probe with a diamond window. ICP-MS analysis of post-reaction mixture was carried out on a Perkin Nexion 300D inductively coupled mass spectrometer. The reaction mixture after the hydrosilylation reaction, was cooled and removed from the system using a syringe with the needle. The samples subjected to quantitative analysis, due to the high concentration of the standard samples, were diluted 10 and 100 times. In order to determine the concentration of rhodium and platinum in the analysed samples, a four-point calibration curve was made and the concentration of the analysed elements was determined on its basis.
Synthesis of imidazolium salts
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :40), and the purified product was dried again overnight at 60 °C, under high vacuum. Yield: 85%.
:40), and the purified product was dried again overnight at 60 °C, under high vacuum. Yield: 85%.1H NMR (400 MHz, DMSO-d6): δ = 7.81–7.46 (m, 2H, imidazolium –CH), 4.16 (t, J = 6.7 Hz, 2H, CH2), 3.74 (s, 3H, CH3), 3.56 (s, 2H), 2.58 (s, 3H, CH3), 1.83 (dd, J = 6.2 Hz, J = 3.1 Hz, 4H, CH2–CH2).
1H NMR (400 MHz, DMSO-d6): δ = 7.84 (d, 3J = 2.0 Hz, 1H, imidazolium –CH), 7.77 (d, J = 2.0 Hz, 1H, imidazolium –CH), 5.95 (ddt, J = 17.1 Hz, J = 10.7 Hz, J = 5.2 Hz, 1H, vinyl –CH), 5.25 (dd, J = 10.4 Hz, J = 1.0 Hz, 1H, vinyl –CH–H), 5.14 (dd, J = 17.2 Hz, 2J = 1.1 Hz, 1H, vinyl –CH–H), 4.91 (d, J = 5.5 Hz, 2H, –CH2–), 3.81 (s, 3H, CH3), 2.59 (s, 3H, CH3).
1H NMR (500 MHz, CDCl3): δ = 7.14 (d, J = 2.0 Hz, 1H, imidazolium –CH), 7.10 (d, J = 2.0 Hz, 1H, imidazolium –CH), 5.81 (ddt, J = 16.2 Hz, 3J = 10.7 Hz, J = 5.6 Hz, 1H, vinyl –CH), 5.28 (d, J = 10.4 Hz, 1H, vinyl –CH–H), 5.13 (d, J = 17.1 Hz, 1H, vinyl –CH–H), 4.58 (d, J = 5.6 Hz, 2H, –CH2–), 3.67 (s, 3H, CH3), 2.45 (s, 3H, CH3).
Synthesis of phosphine ligands
1H NMR (600 MHz, DMSO-d6): δ = 7.79 (m, 4H, Ar–H), 7.63 (d, J = 2.0 Hz, 2H, imidazolium –CH), 7.58 (m, 6H, Ar–H), 4.23 (t, J = 7.1 Hz, 2H, –CH2–), 3.72 (s, 3H, CH3), 2.54 (s, 3H, CH3), 2.45 (m, 2H, –CH2–), 1.90 (m, 2H, –CH2–).
31P NMR (242 MHz, DMSO-d6): δ = −17.53.
1H NMR (600 MHz, DMSO-d6): δ = 7.60 (d, J = 2.4 Hz, 2H, imidazolium –CH), 7.38 (m, 6H, Ar–H), 7.20 (m, 4H, Ar–H), 4.13 (dt, J = 14.6 Hz, J = 7.2 Hz, 2H, CH2), 3.73 (s, 3H, CH3), 2.55 (d, J = 2.7 Hz, 2H. CH2), 1.85 (dt, J = 14.7 Hz, J = 7.3 Hz, 2H, CH2), 1.69 (s, 3H, CH3), 1.37 (m, 2H, CH2).
31P NMR (242 MHz, DMSO-d6): δ = −17.54.
Synthesis of transition-metal based complexes
1H NMR (CDCl3) ppm: 9.05 and 8.74 (s, 2H, imidazolium –CH), 7.80–7.18 (m, 40H, Ar–H), 4.15 (dt, 2H, N–CH2), 3.72 (s, 3H, N–CH3), 2.55 (d, J = 3.7 Hz, 2H, CH2), 2.52 (dt, J = 15.7 Hz, J = 8.2 Hz, 2H, CH2), 2.3 (s, 3H, CH3), 1.2 (m, 4H, J = 7.5, CH2).
13C NMR (CDCl3) ppm: 138 (NC(CH3)N), 135, 134, 133, 132, 131, 130, 129, 128, 127 (Ar–C), 123 (CHCH), 121 (CHCH), 49 (N–CH3), 35 (N–CH2), 32, 28 (CH2), 13 (CH3).
31P(CDCl3) ppm: 28.35 (P–Ar).
1H NMR (CDCl3) ppm: 8.57 and 8.16 (s, 2H, imidazolium –CH), 7.85–7.18 (m, 10H, Ar–H), 5.45 (m, 2H, J = 7.5, –CH), 4.34 (dt, 2H, N–CH2), 4.24 (m, 4H, CH–), 3.71 (s, 3H, N–CH3), 3.1 (m, 2H, –CH), 2.6 (d, J = 3.4 Hz, 2H, CH2), 2.2 (dt, J = 13.5, 2H, CH2), 1.8 (s, 3H, CH3), 1.4 (m, 4H, J = 7.1, CH2).
13C NMR (CDCl3) ppm: 139 (NC(CH3)N), 135, 132, 131, 129 (Ar–C), 122 (–CHCH–), 121 (–CHCH), 101 (cod, CH–), 64.8, 64 (cod, –CH), 47 (N–CH3), 34 (N–CH2), 33, 31 (cod, CH2), 30, 29 (CH2), 14 (CH3).
31P(CDCl3) ppm: 26 (P–Ar).
1H NMR (CDCl3) ppm: 8.98 and 8.75 (s, 2H, imidazolium –CH), 7.85–7.18 (m, 10H, Ar–H), 5.9 (d, 2H, CHCH2), 5.3 (d, 1H, CHCH2), 4.25 (dt, 2H, N–CH2), 3.71 (s, 3H, N–CH3), 2.6 (d, 2H, CH2), 2.5 (dt, J = 13.4, J = 7.8, 2H, CH2), 2.1 (s, 3H, CH3), 1.9 (m, 4H, J = 6.9, CH2), 0.34 (s, 6H, SiCH3), −0.25 (s, 6H, SiCH3).
13C NMR (CDCl3) ppm: 137 (NC(CH3)N), 135, 132, 131, 130 (Ar–C), 50 (N–CH3), 40.3 (–CHCH2), 37 (N–CH2), 33.5 (CHCH2), 32, 31 (CH2), 13 (CH3), 1.4 (SiCH3), −1.9 (SiCH3).
31P(CDCl3) ppm: 31 (P–Ar).
1H NMR (CDCl3) ppm: 8.89 and 8.88 (s, s, 2H, imidazolium –CH), 8.15–7.18 (m, 40H, Ar–H), 4.32 (t, J = 6.4, 2H, N–CH2), 3.92 (s, 3H, N–CH3), 2.6 (s, 3H, CH3), 2.52 (m, 2H, CH2), 1.2 (m, 4H, J = 7.5, CH2).
13C NMR (CDCl3) ppm: 138 (NC(CH3)N), 135, 132, 131, 130, 129, 128, 127 (Ar–C), 123 (–CHCH–), 121 (CHCH), 49 (N–CH3), 35 (N–CH2), 32, 31 (CH2), 12 (CH3).
31P(CDCl3) ppm: 28.34 (P–Ar).
1H NMR (CDCl3) ppm: 9.05 and 8.74 (s, 2H, imidazolium –CH), 7.85–7.18 (m, 10H, Ar–H), 4.45 (m, 2H, J = 6.8 Hz, –CH), 4.30 (t, 2H, N–CH2), 4.14 ( m, 4H, CH–), 3.70 (s, 3H, N–CH3), 3.35 (m, 2H, –CH), 2.64 (s, 3H CH3), 2.55 (m, 2H, CH2), 2.1 (m, 4H, CH2), 1.5 (m, 4H, J = 6.7, CH2).
13C NMR (CDCl3) ppm: 140 (NC(CH3)N), 135, 132, 131, 130, (Ar–C), 123 (CHCH), 122 (CHCH), 90 (cod, CH–), 64.9, 64.3 (cod, –CH), 46 (N–CH3), 35 (N–CH2), 33, 32 (cod CH2), 30, 28 (CH2), 12 (CH3).
31P(CDCl3) ppm: 27 (P–Ar).
1H NMR (CDCl3) ppm: 9.01 and 8.72 (s, 2H, imidazolium –CH), 7.85–7.18 (m, 10H, Ar–H), 5.9 (d, 1H, CHCH2), 5.3 (d, 2H, CHCH2), 4.25 (t, 2H, N–CH2), 3.70 (s, 3H, N–CH3), 2.5 (s, 3H, CH3), 2.5 (m, 2H, CH2), 1.91 (m, 2H, J = 7.1, CH2), 0.34 (s, 6H, SiCH3), −0.25 (s, 6H, SiCH3).
13C NMR (CDCl3) ppm: 138 (NC(CH3)N), 135, 133, 132, 130 (Ar–C), 122 (CHCH), 121 (CHCH), 51 (N–CH3), 41 (CHCH2), 37 (N–CH2), 33.5 (CHCH2), 32, 30 (CH2), 13 (–CH3), 1.4 (SiCH3), −1.9 (SiCH3).
31P(CDCl3) ppm: 32 (P–Ar).
General procedure for catalytic tests
The catalytic activity of the obtained platinum and rhodium complexes was determined in the reaction of hydrosilylation of 1-octene or allyl glycidyl ether with 1,1,1,3,5,5,5-heptamethyltrisiloxane (HMTS). For this purpose, 10−5 mol of catalyst molar mass per 1 mol of Si–H, 3.68 mmol of 1-octene or 4.41 mmol of allyl glycidyl ether and 3.68 mmol of HMTS were used. In the system with allyl glycidyl ether, which is more problematic reagent, to avoid formation of side products and obtain full conversion of Si–H, 1.2 equivalent of olefin to 1 equivalent of HMTS was added. Moreover, 1 mmol of n-decane as an internal standard was added. The reaction was carried out in a reaction vessel in the presence of air at 110 °C for 1 h, without stirring. Then the reaction mixture was cooled down and subjected to GC analysis to determine the reaction yield, which in both cases, was calculated based on the HMTS area peak. Due to the very small amount of catalyst the products were taken entirely with a syringe with the needle. The catalyst remaining in the flask was not washed or regenerated in any way. After complete removal of the products from the reaction system, another, same portion of substrates was added, and the reaction was carried out in the same way. The above operation was repeated until 10 catalytic cycles were carried out. The product was isolated and subjected to NMR analyses.29Si NMR (CDCl3) ppm: −2.19 (–O–Si–O–), 6.75 (OSi(CH3)3).
13C NMR (CD3CN) ppm: −1.0 (–Si–CH3), 0.37–1.39 (–Si(CH3)3); 12.77 (–Si–C–); 23.16 (–Si–C–C–); 43.56 (–C–O–C–); 50.71 (–C–O–C–); 71.41 (–O–C–C–); 73.67 (–C–C–O–).
29Si NMR (CD3CN) ppm: −20.52 (–O–Si–O–), 8.07 (OSi(CH3)3).
Results and discussion
Synthesis of ionic liquids
The first step was the synthesis of initial nitrogen-based ionic liquids. 1,2-Dimethylimidazole was treated with two different alkylating agents, one being 1,4-dibromobutane and the other one allyl chloride (Scheme 1).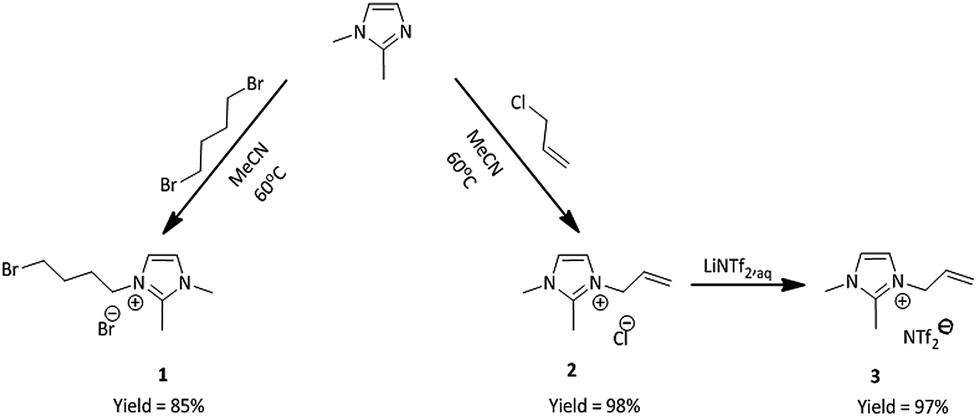 | ||
| Scheme 1 Synthesis of 1,2-dimethylimidazolium based ionic liquids 1 and 3. | ||
The product of the first quaternization reaction was 3-(4-bromobutyl)-1,2-dimethylimidazolium bromide (1), while the product of the second reaction was 3-allyl-1,2-dimethylimidazolium chloride (2). These reactions provided us with two different ionic liquids bearing modifiable substituent, in the form of unsaturated group or bromoalkyl group in quantitative yield. Next, compound (2) was treated with aqueous solution of lithium bis(trifluoromethylsulfonyl)amide (LiNTf2) in two-phase system at room temperature to furnish pure 1,2-dimethylimidazolium bis(trifluoromethylsulfonyl)amide (3) in quantitative yield. Both ionic liquids have been previously obtained31,33 and their properties have been confirmed. In the next step, these ionic liquids were used as substrates in the synthesis of phosphine-based IL's.
Synthesis of phosphine ligands
Previously obtained IL's (1) and (3), allowed us to use two different synthesis approaches for obtaining phosphine ligands (Scheme 2). | ||
| Scheme 2 Synthesis of phosphine ligands 4 and 5. | ||
1,2-Dimethylimidazolium bis(trifluoromethylsulfonyl)amide ionic liquid bearing allyl group (3) underwent the radical chain addition of diphenylphosphine (HPPh2), assisted by AIBN (2,2′-azobis(2-methylpropionitrile)) at 80 °C.31 The reaction was carried out entirely in the Schlenk flask, without any solvent, with constant argon flow, in order to avoid oxidation of both diphenylphosphine and the product. Diphenylphosphine was used in excess and after the reaction time, its excess was evaporated under vacuum. while heating the mixture for 24 h at 60 °C obtained. Properties of the phosphine ligand obtained by our group are compatible with those reported in literature.31 3-(3-(Diphenylphosphanyl)propyl-1,2-dimethylimidazolium)bis(trifluoromethanesulfonyl)amide (4) was obtained in almost quantitative yield.
The second approach involved nucleophilic substitution of bromoalkyl-derivative (1) with potassium diphenylphosphide in THF at 50 °C and led to the formation of desired phosphinated product. The synthesis was carried out according to literature33 with one main difference, which was to not oxidize the phosphine ligand to the phosphine oxide using hydrogen peroxide. It was imperative to stop at this moment in order to obtain phosphine ligand with diphenylphosphinyl group as one of the ligands of the new complexes. 3-(4-(Diphenylphosphanyl)butyl)-1,2-dimethylimidazolium bromide (5) was obtained in almost quantitative yield. The product was obtained with the use of Schlenk line and Schlenk flasks, upon continuous argon flow to also avoid any oxidation. The phosphine ligand obtained in this method has been reported in literature33 and the properties are compatible.
Synthesis of rhodium and platinum complexes
The new Rh(I) and Pt(0) complexes, of the general formula 6–11 were synthesized using a diphenylphosphine group connecting the ionic liquid (4 or 5) as a ligand (Scheme 3 and 4): | ||
| Scheme 3 Synthesis of complexes 6, 7 and 8. | ||
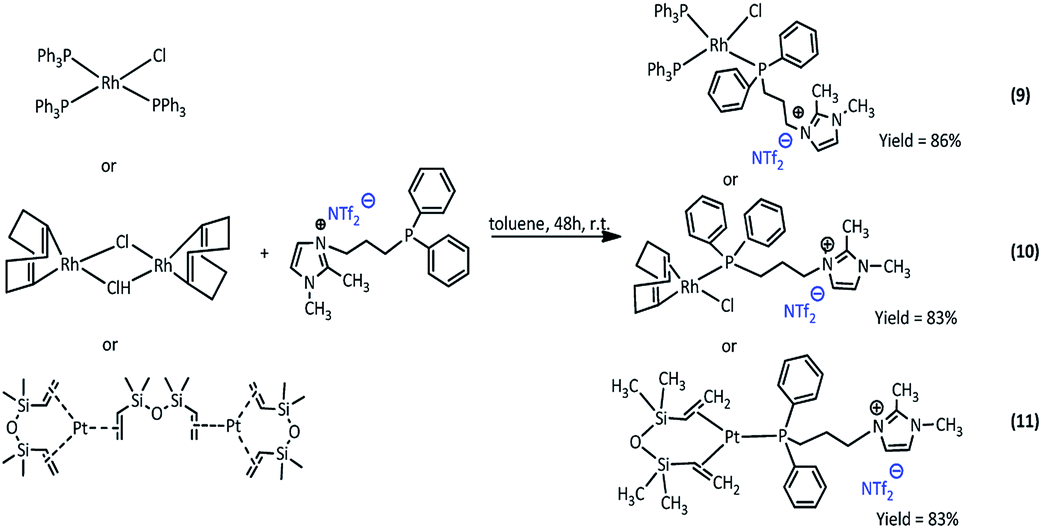 | ||
| Scheme 4 Synthesis of complexes 9, 10 and 11. | ||
The rhodium(I) complexes were obtained in the reaction of 3-(4-(diphenylphosphanyl)butyl)-1,2-dimethylimidazolium bromide (5) or 3-(3-(diphenylphosphanyl)propyl-1,2-dimethylimidazolium)-bis(trifluoromethanesulfonyl)amide (4) with dimer rhodium(I) complex or Wilkinson's catalyst in toluene. In the same way, the synthesis of platinum(0) complexes was carried out, using the Karstedt complex as platinum precursor. All complexes obtained in the reaction with the ligand containing Br− anion were solid but those with the ligand containing NTf2− anion were liquids. The complexes produced were isolated with very high yields in the range of 83–91%. The obtained complexes were characterized by 1H, 31P and 13C NMR analysis, which confirmed their assumed structures. In the 31P NMR spectra, a visible difference in the chemical shift values of the phosphorus atom signal present in the imidazolium ligand was observed, which indicates the coordination of the ligand to the rhodium or platinum atom. The signal shifts from −17 ppm to +26–32 ppm depending on the type of precursor used. For the solid complexes their melting points were determined (Table 1).
| Complex | Melting point [°C] | Decomposition temperature [°C] |
|---|---|---|
| 6 | 148 | 211.16 |
| 7 | 141 | 206.07 |
| 8 | 178 | 222.98 |
| 9 | Oil | 213.17 |
| 10 | Oil | 235.88 |
| 11 | Oil | 272.80 |
The melting points certainly affects the type of anion, because as mentioned above only the complexes with the bromide anion were solids, while the complexes with the NTf2− anion were liquid at room temperature. When comparing the melting points of solid complexes, it can be seen that rhodium complexes have lower melting points than platinum complex. In addition, the temperatures of both rhodium complexes are very similar. A similar relationship can be observed for the thermal stability of the above complexes.
The thermal stability was evaluated by thermogravimetric analysis (TGA) and the results are shown in Fig. 1. Decomposition temperatures were estimated by taking temperature at 10% of weight loss (Table 1). Such a value of weight loss was chosen to discriminate between decomposition temperature and water desorption temperature.
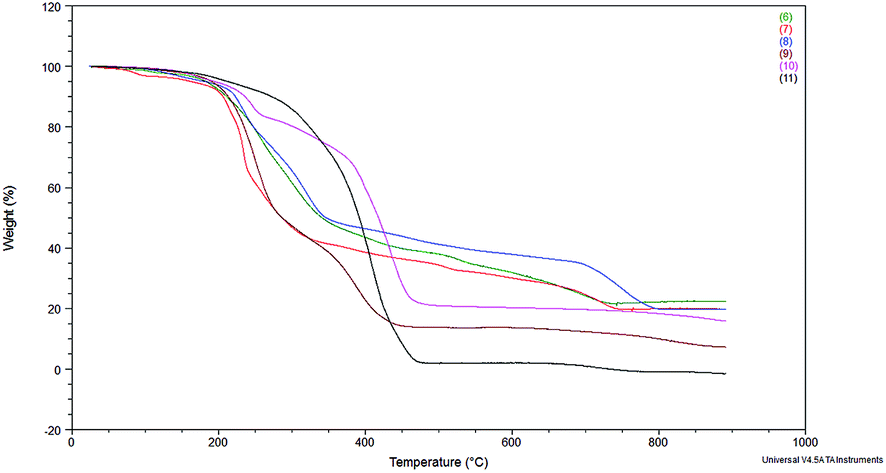 | ||
| Fig. 1 Thermogravimetric curves recorded for rhodium and platinum complexes. | ||
The results in Table 1 show that the platinum complexes (8 and 11) have higher decomposition temperatures than the rhodium complexes, among the complexes with bromide anion and NTf2− anion. Thus, divinyltetramethyldisiloxane as a chelating ligand (present in complexes 8 and 11) can be supposed to better stabilize the complexes than triphenylphosphine or cyclooctadiene ligands present in the other complexes. In addition, the complexes with the NTf2− anion (9–11) showed higher decomposition temperatures. Nevertheless, all complexes have decomposition temperatures above 200 °C, so they should be considered thermally stable. This observation is important from the point of view of their use in catalytic processes carried out at elevated temperature and the possibility of their recycle and reuse.
Catalysis
All the synthesized platinum and rhodium complexes were employed as catalysts for olefin hydrosilylation. The reactants were 1-octene, which is nonpolar and hydrophobic, and allyl glycidyl ether, which is polar and hydrophilic. Both olefins were subjected to hydrosilylation with 1,1,1,3,5,5,5-heptamethyltrisiloxane (HMTS) (Scheme 5). | ||
| Scheme 5 Model reaction of hydrosilylation process of 1-octene/allyl glycidyl ether with 1,1,1,3,5,5,5-heptamethyltrisiloxane. | ||
Chromatographic analysis of post-reaction mixtures showed that in each case only the product of β-addition was formed. No other side products, e.g. α-adducts or products of olefin isomerization were observed. To determine the catalytic activity and optimize the reaction conditions, the FT-IR in situ analysis was performed for the reaction of 1-octene (or allyl-glycidyl ether) hydrosilylation. Real-time monitoring of the reaction catalyzed by the rhodium and platinum complexes using an in situ FT-IR probe enabled determination of particular reaction profiles and differences in their activity. Fig. 2 presents the change in the 1-octene hydrosilylation product yield as a function of time, as observed for particular reactions catalyzed by phosphine rhodium and platinum complexes (at the concentration 10−5 mol Pt or Rh/mol Si–H), conducted at 80 °C.
 | ||
| Fig. 2 The change in the product yield as a function of time for the reaction of 1-octene hydrosilylation catalyzed by phosphine rhodium and platinum complexes in the FT-IR in situ reaction. | ||
These results showed that the complexes with bromide anions (6–8) permitted almost full conversion of reactants already within the first minutes of the reaction, whereas the complexes with amide anion (except 10) were characterized by a longer reaction time of about 30–35 minutes.
The catalysts are characterized by a high activity and they do not dissolve in the reagents. For this reason they could be easily separated from the reaction mixture and reused in a subsequent reaction run. To verify this possibility, we conducted 10 reaction runs using this same catalyst portion. Fig. 3 shows the product yield in subsequent catalytic reaction runs.
 | ||
| Fig. 3 Yields of the product of hydrosilylation of 1-octene with 1,1,1,3,5,5,5-heptamethyltrisiloxane as determined for 10 subsequent reaction runs catalyzed by the same catalyst portion. | ||
Also in this case, the complexes with bromide anion (6–8) were characterized by higher activities and reproducibilities in subsequent runs. However, the most effective catalyst was the rhodium complex 10 which enabled obtaining the product with the same yield of 96% in each of 10 runs. The performance of platinum complex 11 containing imide anion was poorer, because the yield dropped to 69% after the fourth run, however, the same yield was maintained throughout subsequent runs. Turnover numbers (TON) for each catalyst after 10 reaction runs are shown in Table 2 to easily compare the catalytic activity.
| Catalyst | Yield of product in subsequent cyclesb [%] | Total TON |
|---|---|---|
| a [HSi]:[CHCH]:[cat] = 1:1:10−5; T = 110 °C; t = 1 h.b Yields were determined on the basis of GC analysis with n-decane as internal standard. |
||
| 6 | 95 (95……95) | 95000 |
| 7 | 98 (96, 96, 96, 96, 95, 95, 95, 89, 89) | 94500 |
| 8 | 99 (94, 94, 94, 94, 93, 90, 88, 87, 87) | 92000 |
| 9 | 90 (89, 89, 89, 89, 89, 89, 89, 88, 75) | 87600 |
| 10 | 96 (96, 96, 96, 96, 96, 96, 96, 96, 96) | 96000 |
| 11 | 90 (87, 87, 75, 69, 69, 66, 66, 66, 66) | 74100 |
| Catalyst | Yield of product in subsequent cyclesb [%] | Total TON |
|---|---|---|
| a [HSi]:[CHCH]:[cat] = 1:1.2:10−5; T = 110 °C; t = 1 h.b Yields were determined on the basis of GC analysis with n-decane as internal standard. |
||
| 6 | 79 (79……79) | 79000 |
| 7 | 81 (81, 81, 81, 81, 81, 81, 79, 79, 79) | 80400 |
| 8 | 77 (77……77) | 77000 |
| 9 | 83 (83, 72, 12, 8, 7, 4, 3, 2, 0) | 27400 |
| 10 | 85 (85, 53, 20, 0) | 24300 |
| 11 | 81 (81, 19, 7, 5, 4, 4, 4, 3, 2) | 22100 |
The differences in the activities are not large but one can notice some advantage of rhodium complexes, in particular complex 6 and 10 for which the yield in the next 10 cycles was stable and very high (95 or 96% respectively). It is worth mentioning that the catalytic measurements were finished after 10 runs, although these catalysts were still highly active.
The catalyst effectiveness also depends on the kind of olefin used in the reaction of hydrosilylation. In the reaction of allyl glycidyl ether hydrosilylation, greater differences are visible in the activity of the studied complexes.
Fig. 4 presenting the change in the product yield as a function of time, shows that the reaction proceeds with the highest rate in the presence of platinum and rhodium complexes with bromide anion, however, the reaction time extends and falls in the range between 7 and 45 minutes. Moreover, the rhodium complex with bromide anion (complex 6) is characterized by one of the longest reaction time. The product yields obtained in the presence of the studied complexes are about 80%. Furthermore, complex 10 which in the reaction with octene was characterized by one of the shorter reaction times, also in this case showed high activity, albeit in the first cycle.
 | ||
| Fig. 4 The change in the product yield as a function of time for the reaction of hydrosilylation of allylglycidyl ether, catalyzed by phosphine rhodium and platinum complexes in the FT-IR in situ reaction. | ||
Catalytic activity in subsequent reaction runs (see Fig. 5) is even more diversified because the complexes with imide anion lose their activity after the third run, while all complexes with bromide anion maintain constant activity in 10 subsequent runs and permit obtaining the product with 80% yield after each run.
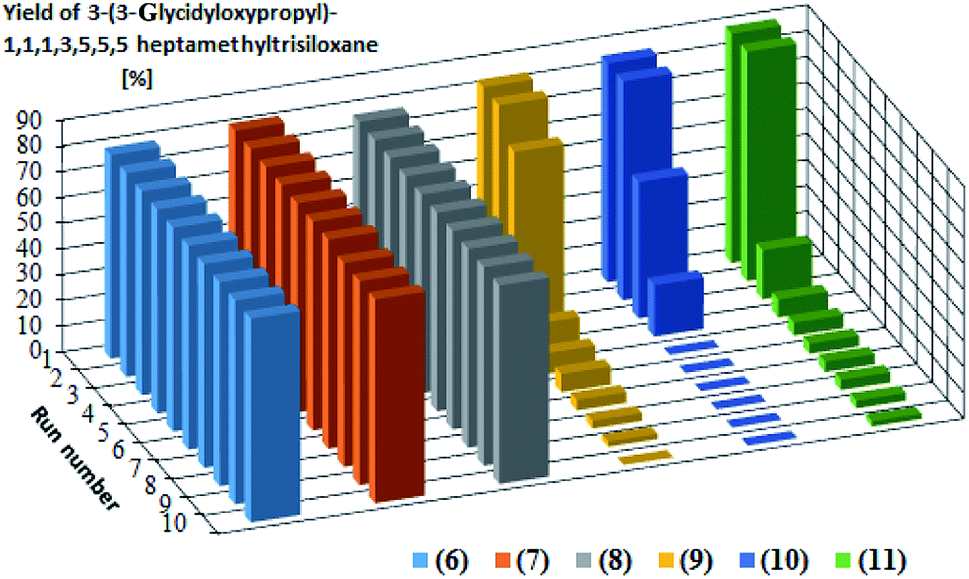 | ||
| Fig. 5 Yields of the product of hydrosilylation of allyl glycidyl ether with 1,1,1,3,5,5,5-heptamethyltrisiloxane in 10 subsequent reaction runs catalyzed by the same catalyst portion. The yields in the subsequent catalytic cycles and turnover numbers after 10 cycles are presented in Table 3. | ||
The presented catalytic data confirm the high catalytic activity of all obtained complexes, however in the first cycle. The catalytic activity of the complexes containing NTf2− anion starts to decrease drastically in subsequent catalytic cycles, which is related to the nature of the anion present in the complex but also to the properties of the olefin used in the hydrosilylation reaction. One of the reasons for decreasing yield is the catalyst leaching by the reagents, which results in a decrease in the catalysts concentration after the isolation of reaction products. The complexes with weak and delocalized charged NTf2− anion are more susceptible to leaching by polar compounds. Since allyl glycidyl ether is such a compound, there in its hydrosilylation a successive leaching of this type of complexes takes place, which was confirmed by ICP-MS analysis (Table 4). The complexes containing the Br− anion, both in the reaction with 1-octene and with allyl glycidyl ether, are much more resistant to leaching and thus remain catalytically active for longer time than the complexes containing NTf2− anion. Comparing the results obtained for individual olefins it can be concluded that the polarity of olefins plays a key role in the above process. In the system with non-polar 1-octene, the yields were at a similar level in subsequent catalytic cycles. In turn, in the systems with polar allyl glycidyl ether, catalyst efficiency drastically decreases between the first and the last catalytic cycle.
| Run no. | Complex 6 | Complex 11 | ||||||
|---|---|---|---|---|---|---|---|---|
| Rh content in the mixture after the reaction with | Pt content in the mixture after the reaction with | |||||||
| 1-Octene | Allyl glycidyl ether | 1-Octene | Allyl glycidyl ether | |||||
| [ppm] | [%] | [ppm] | [%] | [ppm] | [%] | [ppm] | [%] | |
| 1 | 0.037 | 0.80 | 0.079 | 1.69 | 0.039 | 0.72 | 0.21 | 3.87 |
| 5 | 0.001 | 0.02 | 0.004 | 0.09 | 0.0047 | 0.09 | 0.0007 | 0.01 |
Conclusions
Functionalization of imidazolium ionic liquids with diphenyl phosphine allowed obtaining the compounds that were used as ligands in the synthesis of rhodium and platinum complexes. In reactions with various metal precursors, new ionic complexes (four Rh(I) complexes and two Pt(0) complexes) were obtained with high yields (83–96%). All complexes obtained are stable in the air and thermally stable (decomposition temperature above 200 °C). The complexes thus formed were used as catalysts for the hydrosilylation of olefins of different polarity (1-octene and allyl glycidyl ether) with 1,1,1,3,5,5,5-heptamethyltrisiloxane. Due to their insolubility in the reaction medium, their easy isolation and reuse in subsequent catalytic runs were possible. The most active complexes did not lose their activity even after 10 catalytic runs. In general, higher activity and repeatability was demonstrated by rhodium complexes (I). However, the activity of the complexes also depends on the type of olefin and the products formed. In the case of nonpolar products, the activity of all catalysts was similar. However, in the case of polar products, higher stability was demonstrated by bromide anion complexes, which can be explained by their lower susceptibility to leaching by polar reagents. The obtained results made it possible to select the most effective catalysts for hydrosilylation of olefins of different polarities. In addition, the possibility of their recycle and repeated use makes them a very good alternative to commonly used homogeneous rhodium and platinum catalysts. The design of effective catalysts and their application in the synthesis of organofunctional silicon compounds have brought the following advantages:(i) Economical – the possibility of catalyst recycling (the catalyst price is usually high) will reduce the cost of raw materials and easiness of final product isolation will eliminate additional operations (e.g. distillation, precipitation, extraction, etc.). The latter advantage will also shorten the time of desirable product preparation which can be reflected by an increase in the production capacity.
(ii) Ecological – the elimination of classical solvents and waste-generating operations (washing, precipitation, distillation) will result in a considerable reduction in the emission of volatile compounds and waste generation.
(iii) Scientific – the obtained results significantly broaden our knowledge in the field of coordination chemistry, catalysis and new directions in the application of ionic liquids.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Financial support from the National Science Center (Poland), grant OPUS UMO-2014/15/B/ST5/04257, is gratefully acknowledged.References
- B. Marciniec, H. Maciejewski, C. Pietraszuk and P. Pawluć, Hydrosilylation. A Comprehensive Review on Recent Advances, Springer, 2009 Search PubMed.
- D. Trogel and J. Strohrer, Coord. Chem. Rev., 2011, 255, 1440–1459 CrossRef.
- Y. Nakajima and S. Shimada, RSC Adv., 2015, 5, 20603–20616 RSC.
- B. Marciniec, H. Maciejewski, C. Pietraszuk and P. Pawluć, in Applied Homogeneous Catalysis with Organometallic Compounds, ed. B. Cornils, W.A. Hermmann, M. Belier and R. Pawelo, Wiley, 2017, ch. 8, pp. 569–620 Search PubMed.
- R. J. Hoffmann, M. Vlatkovie and F. Wiesbrock, Polymers, 2017, 9, 534–571 CrossRef PubMed.
- B. Marciniec, H. Maciejewski and P. Pawluć, in Organosilicon Compounds, ed. V.Y. Lee, Academic Press, 2017, ch. 5, pp. 169–218 Search PubMed.
- Ionic Liquids – Industrial Applications to Green Chemistry, ed. R. D. Roger and K. R. Seddon, ACS, Washington, 2002 Search PubMed.
- Ionic Liquids in Synthesis, ed. P. Wasserscheid and T. Welton, Wiley-VCH, Weinheim, 2003 Search PubMed.
- D. J. Adams, P. J. Dyson and S. I. Travener, Chemistry in Alternative Reaction Media, Wiley, Hoboken, 2004 Search PubMed.
- P. J. Dyson and T. J. Geldbach, Metal Catalysed Reactions in Ionic Liquids, Springer, Dordrecht, 2005 Search PubMed.
- C. Hardacacre and V. Parvulescu, Catalysis in Ionic Liquids. From Catalyst Synthesis to Application, RS Chemistry, 2014 Search PubMed.
- Ionic Liquids in Organometallic Catalysis, ed. J. Dupont and L. Kollar, Springer, 2015 Search PubMed.
- R. L. Vekariya, J. Mol. Liq., 2017, 227, 44–60 CrossRef CAS.
- A. Behr and N. Toslu, Chem. Eng. Technol., 2000, 23, 122–125 CrossRef CAS.
- T. J. Geldbach, D. Zhao, N. C. Castillo, G. Laurenczy, B. Weyershausen and P. J. Dyson, J. Am. Chem. Soc., 2006, 128, 9773–9780 CrossRef CAS PubMed.
- N. Hofmann, A. Bauer, T. Auer, V. Stanjek, P. Schulz, N. Tanardi and P. Wasserscheid, Adv. Synth. Catal., 2008, 35, 2599–2609 CrossRef.
- N. Taccardi, M. Fekete, M. E. Berger, V. Stanjek, P. S. Schulz and P. Wasserscheid, Appl. Catal., A, 2011, 399, 69–74 CrossRef CAS.
- B. Weyershausen, K. Hell and U. Hesse, Green Chem., 2005, 7, 283–287 RSC.
- T. Schulz and T. Strassner, J. Organomet. Chem., 2013, 744, 113–118 CrossRef CAS.
- J. Peng, J. Li, Y. Bai, H. Qiu, K. Jiang, J. Jiang and G. Lai, Catal. Commun., 2008, 9, 2236–2238 CrossRef CAS.
- H. Maciejewski, A. Wawrzyńczak, M. Dutkiewicz and R. Fiedorow, J. Mol. Catal. A: Chem., 2006, 257, 141–148 CrossRef CAS.
- W. Zieliński, R. Kukawka, H. Maciejewski and M. Śmiglak, Molecules, 2016, 21, 1115–1126 CrossRef PubMed.
- H. Maciejewski, K. Szubert, B. Marciniec and J. Pernak, Green Chem., 2009, 11, 1045–1051 RSC.
- M. Jankowska-Wajda, R. Kukawaka, M. Śmiglak and H. Maciejewski, New J. Chem., 2018, 42, 5229–5236 RSC.
- H. Maciejewski, M. Jankowska-Wajda, I. Dabek and R. Fiedorow, RSC Adv., 2018, 8, 26922–26927 RSC.
- J. Pernak, A. Swierczynska, M. Kot, F. Walkiewicz and H. Maciejewski, Tetrahedron Lett., 2011, 52, 4342–4345 CrossRef CAS.
- H. Maciejewski, K. Szubert, R. Fiedorow, R. Giszter, M. Niemczak and J. Pernak, Appl. Catal., A, 2013, 451, 168–175 CrossRef CAS.
- K. L. Luska, K. Z. Demmans, S. A. Stratton and A. Moores, Dalton Trans., 2012, 41, 13533–13540 RSC.
- X. Jin, X.-F. Xu and K. Zhao, Tetrahedron: Asymmetry, 2012, 23(14), 1058–1067 CrossRef CAS.
- S. J. Chen, Y. Y. Wang, W. M. Yao, X. L. Zhao, G. Vo-Thanh and Y. Liu, J. Mol. Catal. A: Chem., 2013, 378, 293–298 CrossRef CAS.
- C. S. Consorti, G. L. P. Aydos, G. Ebeling and J. Dupont, Org. Lett., 2008, 10, 237–240 CrossRef CAS PubMed.
- J. Li, J. Peng, D. Wang, Y. Bai, J. Jiang and G. Lai, J. Organomet. Chem., 2011, 696, 263–268 CrossRef CAS.
- J. A. Vincente, A. Mlonka, H. Q. N. Gunarante, M. Swadźba- Kwaśny and P. Nockemann, Chem. Commun., 2012, 48, 6115–6117 RSC.
| This journal is © The Royal Society of Chemistry 2019 |