
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Essential oil from halophyte Lobularia maritima: protective effects against CCl4-induced hepatic oxidative damage in rats and inhibition of the production of proinflammatory gene expression by lipopolysaccharide-stimulated RAW 264.7 macrophages
Anis Ben Hsouna ab,
Sabah Dhibi*c,
Wissal Dhifid,
Rania Ben Saadb,
Faical Brinic,
Najla Hfaidhc and
Wissem Mnifef
ab,
Sabah Dhibi*c,
Wissal Dhifid,
Rania Ben Saadb,
Faical Brinic,
Najla Hfaidhc and
Wissem Mnifef
aDepartment of Life Sciences, Faculty of Sciences of Gafsa, Zarroug 2112, Gafsa, Tunisia
bLaboratory of Biotechnology and Plant Improvement, Centre of Biotechnology of Sfax, Tunisia
cUnit of Macromolecular Biochemistry and Genetics, Faculty of Sciences of Gafsa, Sidi Ahmed Zarrouk, 2112, Gafsa, Tunisia. E-mail: sabahdhibi7@gmail.com
dLR17-ES03 Physiopathology, Food and Biomolecules, Higher Institute of Biotechnology of Sidi Thabet, Biotechpole Sidi Thabet, 2020, Ariana, Tunisia
eDepartment of Chemistry, Faculty of Sciences and Arts in Balgarn, University of Bisha, Bisha 61922, P. O. Box 199, Saudi Arabia
fUniversity of Manouba, ISBST, BVBGR-LR11ES31, Biotechpole Sidi Thabet, 2020, Ariana, Tunisia
First published on 11th November 2019
Abstract
The present study evaluates the chemical profiling of the essential oil of a halophyte, L. maritima (LmEO), and its protective potential against CCl4-induced oxidative stress in rats. Forty compounds have been identified in LmEO. The major components are α-pinene (3.51%), benzyl alcohol (8.65%), linalool (22.43%), pulegone (3.33%), 1-phenyl butanone (7.33%), globulol (4.32%), γ-terpinene (6.15%), terpinen-4-ol (4.31%), α-terpineol (3.9%), ledol (3.59%), epi-α-cadinol (3.05%) and α-cadinol (4.91%). In comparison with the CCl4-intoxicated group, LmEO treatment resulted in decreased liver serum marker enzymes, decreased lipid peroxidation and increased antioxidant enzyme levels, with overall further amelioration of oxidative stress. The administration of LmEO to CCl4-treated rats at a dose of 250 mg kg−1 body weight significantly reduced the toxic effects and the oxidative stress on the liver, thus validating the traditional medicinal claim of this plant. Moreover, the anti-inflammatory activity of LmEO was evaluated in lipopolysaccharide-stimulated murine RAW 264.7 cells. Our oil could modulate the inflammatory mode of the macrophages by causing reduction in iNOS and COX2 enzymes as well as in IL-1β, IL-6, and TNF-α cytokine levels. These findings suggest that LmEO exerts anti-inflammatory effects by regulating the expression of inflammatory cytokines.
Introduction
The liver is a very important organ which is responsible for metabolic and secretor activities. It is a sensitive target site for substances that modulate biotransformation. Furthermore, it plays a key role in body detoxification from exogenous and endogenous challenges, such as xenobiotics, drugs, viral infections and chronic alcoholism.1 The carbon tetrachloride (CCl4)-induced hepatotoxicity model is extensively used to evaluate the antioxidant effects of drugs and plant extracts.2 CCl4 accumulates in hepatic parenchyma cells and is metabolized into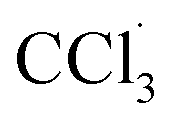 radicals by liver cytochrome P450-dependent monooxygenases.3 It causes oxidative stress and accumulation of reactive oxygen species (ROS). ROS, including superoxide anions (O2˙−), hydrogen peroxide (H2O2), hydroxyl radicals
radicals by liver cytochrome P450-dependent monooxygenases.3 It causes oxidative stress and accumulation of reactive oxygen species (ROS). ROS, including superoxide anions (O2˙−), hydrogen peroxide (H2O2), hydroxyl radicals 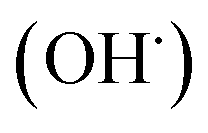 and peroxyl radical
and peroxyl radical 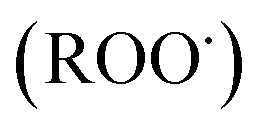 , have been recognized to stimulate tissue oxidative damage and cause several diseases, such as atherosclerosis, diabetes mellitus, cancer, and neurodegenerative diseases, as well as ageing processes.4 In addition to these conditions, oxidative stress is involved in liver pathologies and mostly results in progressive evolution of fatty liver, necrosis, fibrosis, cirrhosis and hepatocellular carcinoma.
, have been recognized to stimulate tissue oxidative damage and cause several diseases, such as atherosclerosis, diabetes mellitus, cancer, and neurodegenerative diseases, as well as ageing processes.4 In addition to these conditions, oxidative stress is involved in liver pathologies and mostly results in progressive evolution of fatty liver, necrosis, fibrosis, cirrhosis and hepatocellular carcinoma.
Oxidative stress can result from excess ROS production and/or deficient antioxidant capacity. Several chemical drugs have been implicated in the aetiology of liver oxidative-stress diseases. In fact, CCl4 is a toxic substance used to induce liver damage in rats.5 CCl4 catabolic radicals induce lipid peroxidation and damage liver cell membranes and organelles, causing swelling and necrosis. Therefore, CCl4, a hepatotoxin used to evaluate the hepatoprotective potential of plant extracts, is commonly used to induce liver damage by producing free radical intermediates (malondialdehyde and 4-hydroxy-2-nonenal). These radicals quickly add molecular oxygen to form highly reactive trichloromethyl peroxy radicals, CCl3OO˙, which react with proteins and lipids.3 They remove hydrogen atoms from unsaturated lipids; this initiates lipid peroxidation, which has the ability to cause structural and functional injury to cell membranes and thus increase membrane permeability, leading to leakage of hepatic enzymes (ALT, AST, ALP, and LDH).6 Biochemically, hepatocellular damage occurs due to high levels of cytosolic enzymes, such as AST, ALT and ALP, in the blood circulation.7
In addition to oxidative stress, inflammation is a common biological mechanism contributing to compromised liver function. Inflammation is a physiological body response to stimuli, including infections and tissue injury. Macrophages, which are ubiquitously distributed in tissues via the mononuclear phagocyte system, play an essential role in innate immunity and are also involved in both host defense mechanism and inflammation.8 They support homeostasis and host defense against intracellular parasitic bacteria and pathogenic protozoa.9 When activated, macrophages are responsible for excessive secretion of proinflammatory enzymes, such as inducible NO synthase (iNOS) and cyclooxygenase (COX-2).10 However, overproduction of these mediators can cause harmful effects to tissues. These effects have been reported to be associated with the pathogenesis of various inflammatory-related diseases, such as rheumatoid arthritis, diabetes, inflammatory bowel disease, atherosclerosis, and cancer.11
Lipopolysaccharide (LPS), a component of the outer membrane of Gram-negative bacteria, stimulates macrophage activation.12 Activated macrophages play an important role in inflammatory response by producing cytokines, including interleukin-1 beta (IL-1β), interleukin-6 (IL-6), and tumor necrosis factor-alpha (TNF-α).9 Inflammatory response is a crucial protective attempt of the host defense to remove injurious stimuli and initiate healing. Despite this protective role, the overproduction of inflammatory mediators can be a major cause of tissue damage, sepsis, and cancer.13
ROS overproduction and cell redox imbalance play key roles in the pathophysiology of the inflammatory response.14 Substantial evidence implicates oxidative stress as an important pathogenic factor in inflammatory response.15 ROS are intracellularly generated from several sources, including mitochondrial respiration, cytochrome P450, and the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase system.16 Phagocyte NADPH oxidase 2 (NOX2) is the major source of ROS generation in the macrophage response to LPS.17 Therefore, LPS may induce an inflammatory response through NADPH oxidase activation-driven oxidative stress. Likewise, the inhibition of ROS production using inhibitors of NADPH oxidase has been proposed as an alternative approach to conventional antioxidant therapies against LPS-induced inflammation. In ROS-induced liver diseases, exogenous antioxidative compounds must be delivered to maintain balance between oxidants and antioxidants in order to prevent subsequent pathologies. However, conventional and synthetic drugs used in treating liver diseases are inadequate to some extent and may cause serious adverse effects.18 For this reason, a considerable number of liver patients prefer safer approaches, such as using more effective natural antioxidants. Accordingly, plant extracts and their derived metabolites, such as phenolic compounds, offer an opportunity in this respect.19 The use of natural antioxidants has been proposed as a therapeutic regime and as drug co-adjutants in liver damage treatments.
Plant secondary metabolites are reputed for their anti-inflammatory properties.20,21 Among these metabolites, essential oils (EOs) are well-known for their antioxidant properties. These oils have been widely used in traditional medicine to treat inflammatory diseases.22,23 They are also exploited as natural additives or ingredients in foods which claim beneficial health properties.24
Lobularia maritima (Alyssum martima, Brassicaceae), commonly known as sweet alyssum, is an annual ornamental halophyte that is commonly found along the Tunisian seashore. It can tolerate salinity up to 400 mM NaCl.25
To our knowledge, the chemical composition and biological activities of the halophyte L. maritima have not been reported. Moreover, the use of this plant to alleviate the oxidative damage induced by CCl4 has not been previously explored. This work is the first investigation dealing with the (i) chemical identification of the bioactive compounds extracted from L. maritima using gas chromatography-mass spectrometry (GC-MS); (ii) the investigation of the in vitro antioxidant effects of LmEO in several in vitro systems; (iii) the anti-inflammatory mechanism of LmEO in LPS-induced murine macrophage cells; and (iv) the exploration of the possible protective effects of LmEO against liver oxidative damage following intraperitoneal administration of CCl4 by assessing the oxidative stress profile and some serum biochemical parameters.
Materials and methods
Plant material
The aerial parts of the halophyte Lobularia maritima (L. maritima) (flowering stage) were collected in April 2017 in Chebba, Mahdia in Tunisia. The aerial parts were air-dried at room temperature in shade. After that, they were finely ground and maintained in a sealed bag in a cold, dry place until they were used.Essential oil isolation
The oil extraction was performed on 1 kg of fresh L. maritima leaves by steam distillation for 3 h using a Clevenger-type apparatus. The aqueous phase was extracted with dichloromethane (3 × 50 mL) and dried with anhydrous sodium sulphate. The resulting LmEO was stored at 4 °C prior to further analysis.26 LmEO was solubilized in n-hexane for GC-MS analysis. The oil extraction yield was calculated according to the following formula: oil (% v/w) = volume of LmEO (mL)/weight of raw material (g).Gas chromatography-mass spectrometry (GC-MS)
GC/MS analysis was carried out using a Shimadzu QP2010SE 15A operating at 70 eV ionization energy, equipped with a Rtx-5MS column (phenyl methyl siloxane 30 m × 0.25 mm, 0.25 μm film thickness) with He as the carrier gas at a flow rate of 0.9 mL min−1 and a split ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20. The acquisition mass range was 35 to 300 and the scan time was 0.5 s/scan. Retention indices were specified using retention times of n-alkanes that were injected after the oil under the same chromatographic conditions. The retention indices for all components were specified according to the Van Den Dool method using n-alkanes as a standard. The compounds were identified by comparison of their retention indices (RI, Rtx-5MS) with those reported in the literature and by comparison of their mass spectra with the Wiley and NIST libraries or with published mass spectra.26
:20. The acquisition mass range was 35 to 300 and the scan time was 0.5 s/scan. Retention indices were specified using retention times of n-alkanes that were injected after the oil under the same chromatographic conditions. The retention indices for all components were specified according to the Van Den Dool method using n-alkanes as a standard. The compounds were identified by comparison of their retention indices (RI, Rtx-5MS) with those reported in the literature and by comparison of their mass spectra with the Wiley and NIST libraries or with published mass spectra.26
Antioxidant testing assays
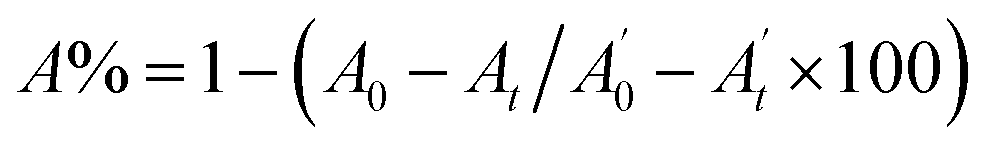 , where A0 and
, where A0 and 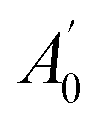 are the absorbances of the sample and the blank, respectively, measured at zero time, and At and
are the absorbances of the sample and the blank, respectively, measured at zero time, and At and 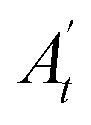 are the absorbances of the sample and the blank, respectively, measured after 2 h. All tests were carried out in triplicate.
are the absorbances of the sample and the blank, respectively, measured after 2 h. All tests were carried out in triplicate.In vivo antioxidant properties
Experimental design
Rats were randomly assigned to four groups of eight animals each. Animals in the first group receiving distilled water and a standard laboratory diet served as controls (C). The second group (CCl4), the hepatoxicity model, was given a single dose of CCl4 (1 mL kg−1 in 1% olive oil i.p.) on the 14th day.6,26 These doses were selected on the basis of previous studies which did not reveal any toxic effects in adult rats and were effective against toxicity. Animals in the third group (LmEO) were given a daily i.p. injection of LmEO at 250 mL kg−1 b.w. and distilled water as sole beverage for 15 days. The fourth group (LmEO + CCl4) was pretreated with LmEO and intoxicated with CCl4 on the 14th day.6,26 The CCl4 dose was selected according to the chronic oral reference dose recommended for CCl4 (CASRN 56-23-5).30 During the 2 weeks of the experimental period, all animals survived.Organ sampling
At the end of the experimental period (15 days), 24 h after the administration of CCl4, control and treated rats were anesthetized with chloral hydrate by intra-abdominal injection. The body weights of the rats were recorded, and blood samples from the brachial artery were collected in heparin tubes. At the end of the experimental period, the animals in the different groups were killed by cervical decapitation to avoid animal stress. Plasma samples were obtained from blood after centrifugation at 2500 × g for 15 min to estimate selected serum biochemical parameters. The samples were maintained at −20 °C until analysis. All samples were analyzed in triplicate.The livers of the rats were collected, cleaned and weighed. Some samples were homogenized (1:2, w/v) in 50 mmol L−1 Tris buffer (pH 7.4) containing 150 mmol L−1 of NaCl using an ultra-Turrax device. The homogenates were centrifuged at 5000 × g for 25 min at 4 °C, and aliquots of the supernatant were maintained at −20 °C until the analyses.
In parallel, portions of the livers were immediately fixed in Bouin solution (saturated picric acid added to 37% to 40% formaldehyde and glacial acetic acid, 75:25:5 v/v) for histological studies.6,26
Serum parameters
Serum samples were obtained by centrifugation of blood at 2700 × g for 15 min at 4 °C and were then divided into Eppendorf tubes. Isolated sera were stored at −20 °C until they were used for further analyses. The levels of serum ALT, AST, ALP, and lactate dehydrogenase (LDH) were measured using commercial kits according to the manufacturer's directions (Biolabo, Maizy, France) on an automatic biochemistry analyzer (Vitalab Flexor E, Diamond Diagnostics, Holliston, MA).Biochemical assays
The nitrite concentration in the cultured media was measured as an indicator of nitric oxide (NO) production based on the Griess reaction.37 The nitrite concentration was calculated using a sodium nitrite standard curve. The percentage of NO inhibition in the treated cells was compared to LPS-only treated cells (100%), and the half NO inhibitory concentrations (IC50) for various LmEO concentrations were determined.
The levels of IL-1β, IL-6, IL-10, and TNF-α in the culture media were quantitated by ELISA (R&D Systems) in accordance with the manufacturer's instructions.
The results of the western blot analysis were quantified by measuring the relative intensity compared to the control and are represented by relative intensities. Mouse β-actin was simultaneously detected as an internal control to monitor the intensity. The bands for the iNOS, COX-2, and β-actin antibodies were recognized at ∼135 kDA, ∼72 kDA, and ∼45 kDa, respectively.
Results
LmEO chemical constitution
The hydrodistillation of L. maritima aerial parts generated a yellow oil (yield of 2.4%, v/w). Upon GC/MS analysis, LmEO was found to contain 40 constituents, with 90.55% identified constituents (Table 1). The volatile oil contained 74.40% oxygenated monoterpenes and 16.15% monoterpene hydrocarbons. The major components were α-pinene (3.51%), benzyl alcohol (8.65%), linalool (22.43%), α-terpineol (3.9%), pulegone (3.33%), 1-phenyl butanone (7.33%), ledol (3.59%), globulol (4.32%) and α-cadinol (4.91%). It is important to note that the oxygenated monoterpene fraction was present in a relatively high amount (>74.40%).| No. | Compounda | KIb | %c |
|---|---|---|---|
| a Identification of components based on GC-MS Wiley version 7.0 library data and National Institute of Standards and Technology 05 MS (NIST) library data.b KI: Kovats indices on an HP-5MS capillary column in reference to C10–C22 n-alkanes injected under the same conditions.c %: percentages are the means of two runs and were obtained from electronic integration measurements using a selective mass detector. | |||
| 1 | Furfural | 800 | 0.18 |
| 2 | α-Thujene | 883 | 0.12 |
| 3 | α-Pinene | 938 | 3.51 |
| 4 | Sabinene | 976 | 2.13 |
| 5 | Myrcene | 947 | 0.15 |
| 8 | α-Phellandrene | 1003 | 0.37 |
| 9 | δ-3-Carene | 1016 | 0.15 |
| 10 | Benzyl alcohol | 1040 | 8.65 |
| 11 | γ-Terpinene | 1052 | 6.15 |
| 12 | Acetophenone | 1065 | 0.15 |
| 13 | Z-Linalool oxide (furanoid) | 1074 | 0.16 |
| 14 | Linalool | 1082 | 22.43 |
| 15 | 1-Terpineol | 1143 |
5.6 |
| 16 | Terpinen-4-ol | 1175 | 4.31 |
| 17 | α-Terpineol | 1176 | 3.9 |
| 18 | Pulegone | 1238 | 3.33 |
| 19 | δ-Elemene | 1338 | 0.22 |
| 20 | Isoledene | 1376 | 0.37 |
| 21 | α-Copaene | 1379 | 0.22 |
| 22 | β-Bournonen | 1380 | 0.26 |
| 23 | β-Cubebene | 1384 | 0.24 |
| 24 | α-Gurjunene | 1406 | 0.22 |
| 25 | 1-Phenyl butanone | 1425 | 7.33 |
| 26 | α-Humulene | 1455 | 0.15 |
| 27 | Germacrene D | 1462 | 0.42 |
| 28 | β-Selinene | 1484 | 0.24 |
| 29 | β-Sesquiphellandrene | 1501 | 0.17 |
| 30 | Germacrene B | 1535 | 0.34 |
| 31 | Ledol | 1561 | 3.59 |
| 32 | Germacrene D-4-ol | 1573 | 0.12 |
| 33 | Spathulenol | 1576 | 0.47 |
| 34 | Globulol | 1590 | 4.32 |
| 35 | 1.10-di-epi-Cubenol | 1627 | 1.67 |
| 36 | 10-epi-γ-Eudesmol | 1635 | 0.27 |
| 37 | epi-α-Cadinol | 1643 | 3.05 |
| 38 | α-Cadinol | 1672 | 4.91 |
| 39 | Z-Methyl epijasmonate | 1675 | 0.14 |
| 40 | Z-α-Bisabolene epoxide | 1680 | 0.54 |
| Monoterpene hydrocarbons | — | — | 16.15 |
| Oxygenated monoterpenes | — | — | 74.40 |
| Total (%) | 90.55 | ||
In vitro antioxidant properties
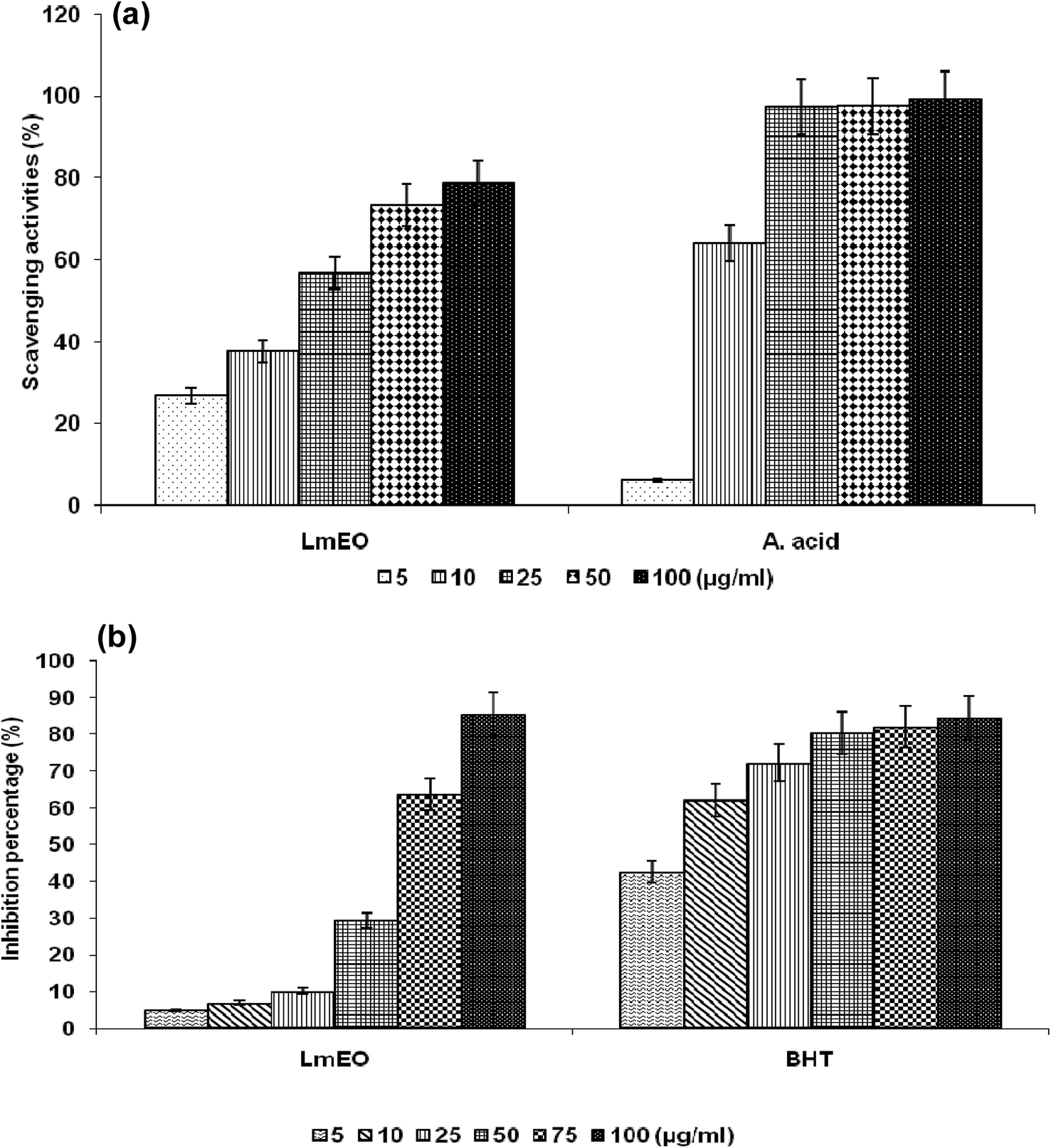 | ||
| Fig. 1 (a) Scavenging effects of LmEO at different concentrations of 5, 10, 25, 50 and 100 μg mL−1 on the stable 1,1-diphenyl-2-picrylhydrazyl radical (DPPH). Results are expressed as percentage of decrement of absorbance at 517 nm with respect to the control. Ascorbic acid was used as a standard. (b) Antioxidant activities of LmEO at different concentrations of 5, 10, 25, 50, 75 and 50 μg mL−1 measured by the β-carotene bleaching method. Butylhydroxytoluene (BHT) was used as a standard. The results presented are representative of three independent experiments. Values are expressed as mean ± SEM (n = 3). | ||
For the β-carotene bleaching method, the inhibitory effects of LmEO on lipid peroxidation were determined by the β-carotene/linoleic acid bleaching test. Fig. 1b shows various degrees of linoleic acid oxidation and subsequent β-carotene bleaching after addition of LmEO and BHT used as a positive control at different concentrations. This antioxidant activity was dose dependent, as found in the DPPH tests.
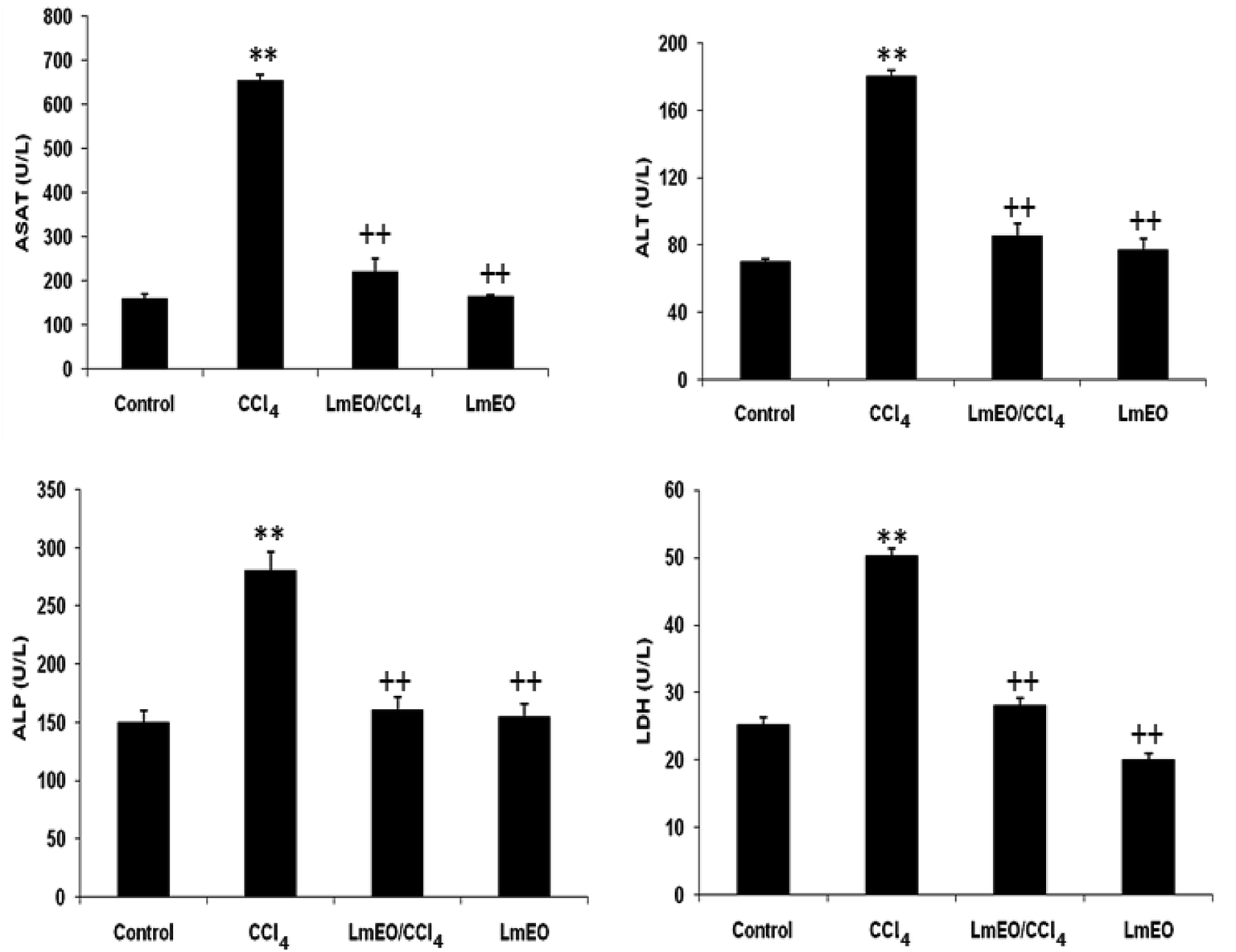 | ||
| Fig. 2 Plasma levels of bio-indices of liver functions in adult rats treated with CCl4 alone or concomitantly with LmEO for 15 days; C: control; CCl4: carbon tetrachloride; (CCl4 + LmEO): rats pre-treated with LmEO and intoxicated with CCl4 at 14 days; aspartate aminotransferase (AST); alanine aminotransferase (ALT); alkaline phosphatase (ALP); lactate dehydrogenase (LDH). Values are expressed as mean ± SEM of eight animals in each group. One-way ANOVA followed by Tukey test for comparison between groups: comparison between CCl4 and control groups: **P < 0.01; comparison between CCl4 + LmEO and CCl4 groups: ++P < 0.01. | ||
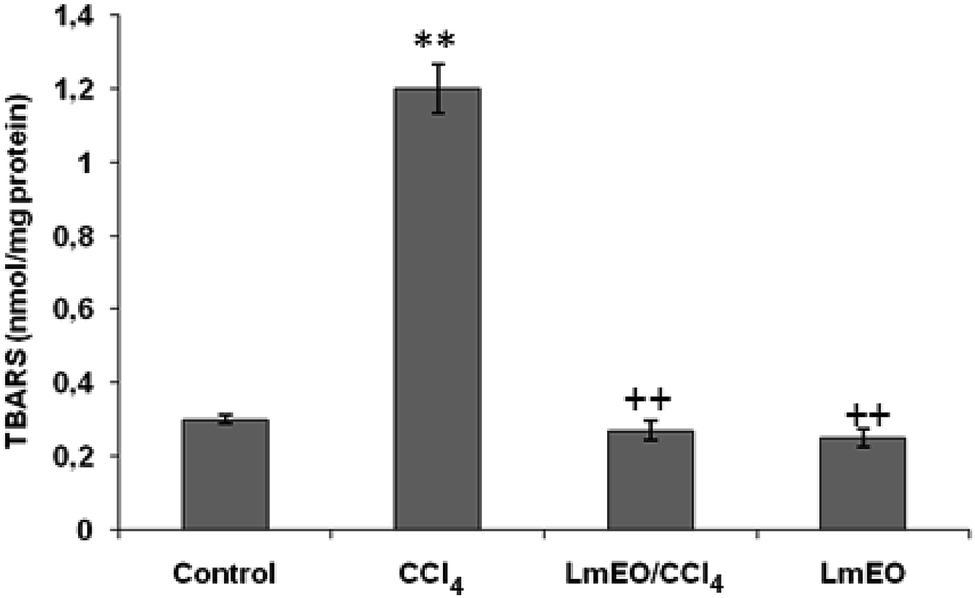 | ||
| Fig. 3 Effects of CCl4, LmEO and their combinations (LmEO/CCl4) on hepatic TBARS of control and experimental rats. Values are expressed as mean ± SEM of eight animals in each group. One-way ANOVA followed by Tukey test for comparison between groups: comparison between CCl4 and control groups: **P < 0.01; comparison between CCl4 + LmEO and CCl4 groups: ++P < 0.01. | ||
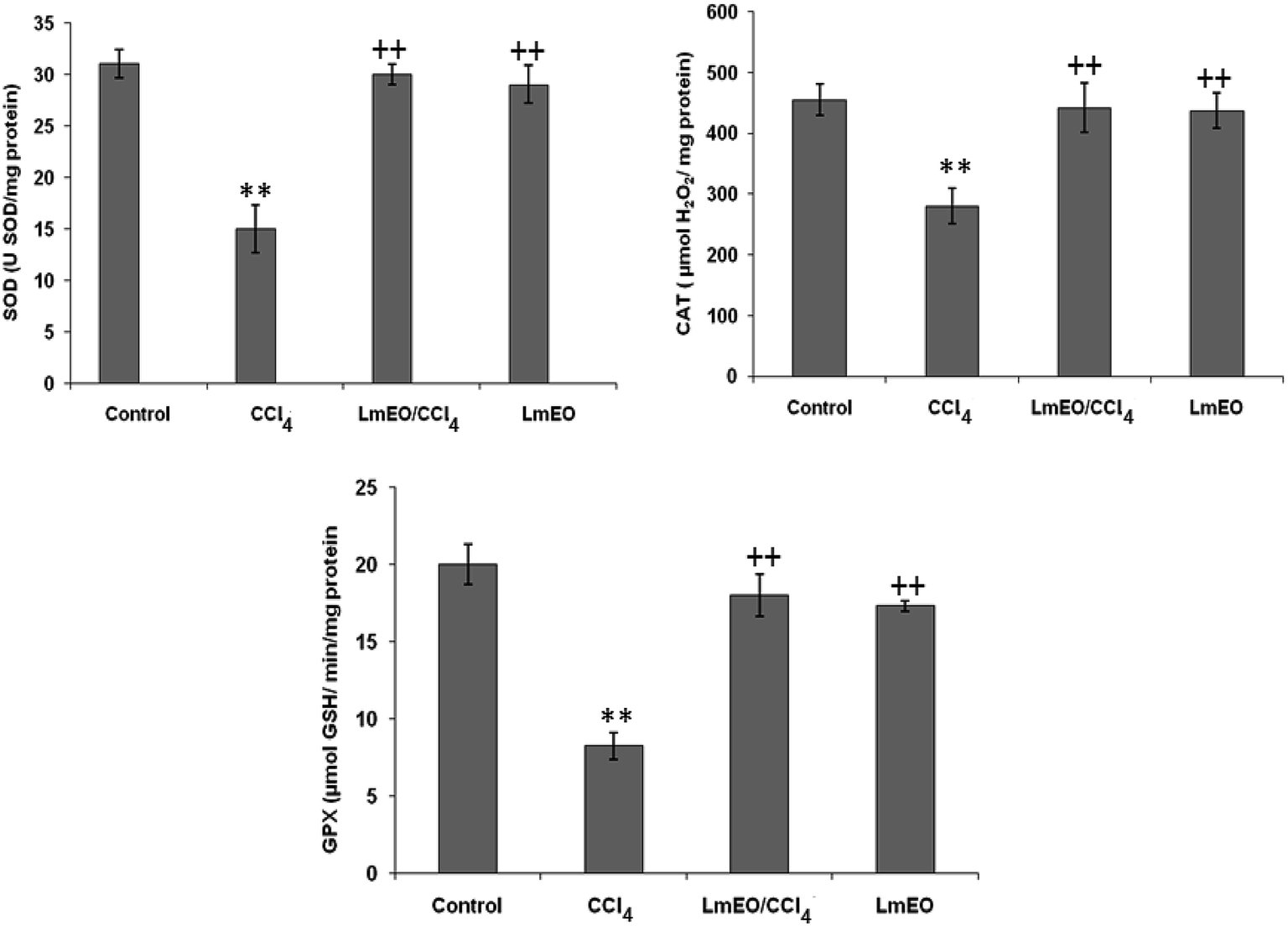 | ||
| Fig. 4 Effects of CCl4, LmEO and their combination LmEO/CCl4 on the activities of enzymatic antioxidants in the livers of control and experimental rats. C: control; CCl4: carbon tetrachloride; (CCl4 + LmEO): rats pre-treated with LmEO and intoxicated with CCl4 at 14 days; SOD: superoxide dismutase (U SOD per mg protein); CAT: catalase (mmol per mg protein); GPx: glutathione peroxidase (nmol per mg protein). Values are expressed as mean ± SEM of eight animals in each group. One-way ANOVA followed by Tukey test for comparison between groups: comparison between CCl4 and control groups: **P < 0.01; comparison between CCl4 + LmEO and CCl4 groups: ++P < 0.01. | ||
Histopathological findings
The treatment with CCl4 caused excessive necrosis associated with neutrophilic infiltration, which is frequently observed in the case of swelling of liver cells, as well as neutrophilic infiltration (arrows) and several ballooning degenerations of hepatocytes (Fig. 5). However, according to the microscopic examinations, the severe hepatic lesions induced by CCl4 were considerably reduced by the administration of LmEO (250 mg kg−1 BW) (Fig. 5). These data are well correlated with those of the serum biochemical parameters and oxidative stress markers. Necrosis was markedly reduced and minimized by pre-treatment with 250 mg kg−1 of LmEO.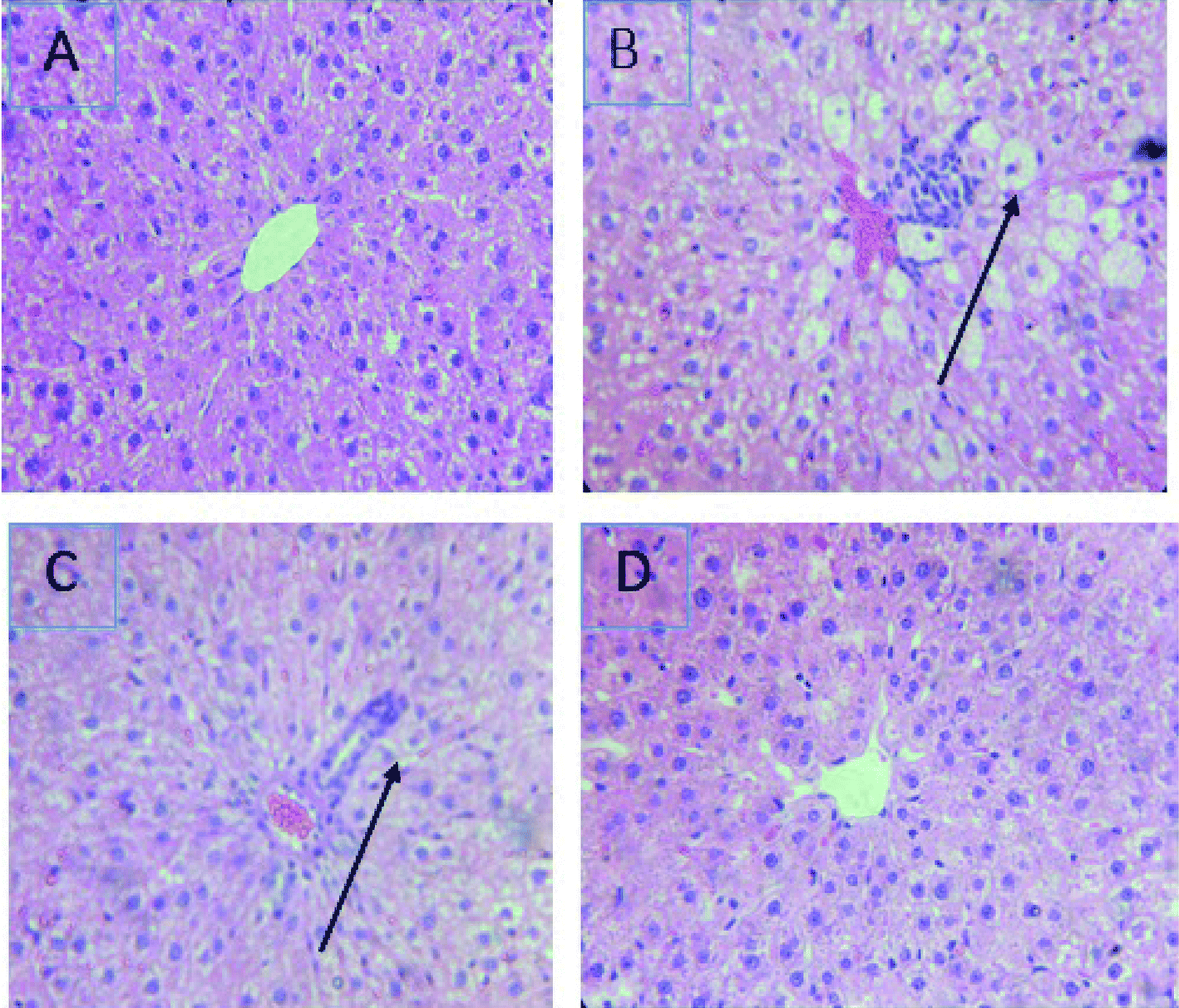 | ||
| Fig. 5 Effects of LmEO on the histological morphology of rat liver fibrosis with Masson staining (×100). (A) Control group; (B) LmEO group; (C) CCl4-treated group; (D) CCl4 + LmEO group. | ||
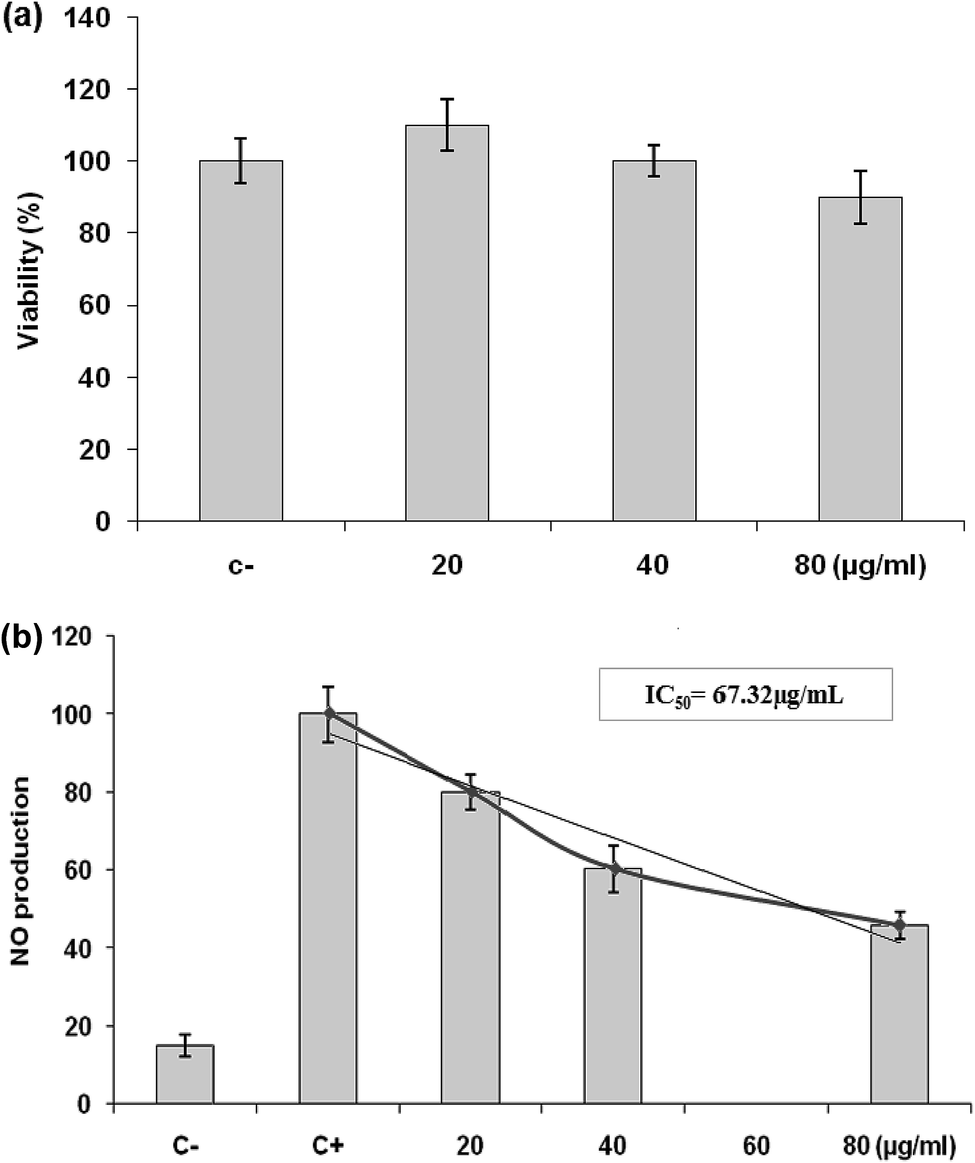 | ||
| Fig. 6 (a) Effects of LmEO on the viability of RAW 264.7 cells determined by MTT assay. The cells were treated with different concentrations of LmEO for 24 h. (C−): negative control values were obtained in the absence of components. None of the tested concentrations of LmEO showed significant detrimental effects. (b) Effects of LmEO on nitric oxide production (NO) in lipopolysaccharide-stimulated macrophages. Cells were treated with lipopolysaccharide and different concentrations of LmEO for 24 h. The supernatants of the cultured cells were examined for NO levels by the Griess method. (C−): negative control values were obtained in the absence of lipopolysaccharide and the essential oil. (C+): the positive control was lipopolysaccharide-only treated cells. Data represent mean ± SEM of three independent experiments.*P < 0.05 compared with the LPS-only treatment group, **P < 0.01 vs. the positive control. Ctrl = control. | ||
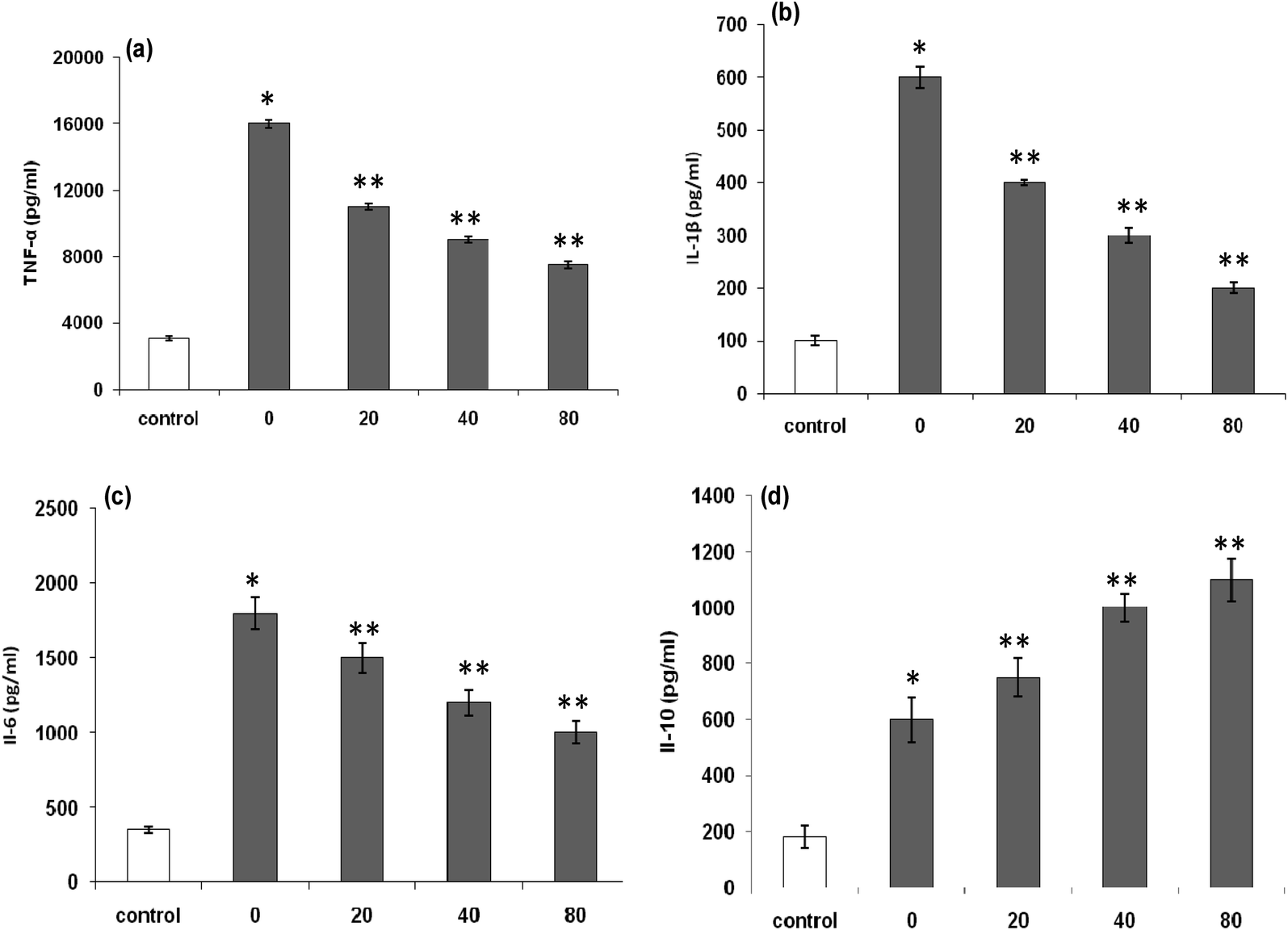 | ||
| Fig. 7 (a) Effects of LmEO on lipopolysaccharide (LPS)-induced tumor necrosis factor (TNF)-α production of RAW 264.7 macrophages; (b) effects of LmEO on LPS-induced interleukin (IL)-1β production of RAW 264.7 macrophages; (c) effects of LmEO on LPS-induced IL-6 production of RAW 264.7 macrophages; and (d) effects of LmEO on LPS-induced IL-10 production of RAW 264.7 macrophages. Values are expressed as mean ± SEM (n = 3). *P < 0.05 compared with the control group. **P < 0.01 compared with the LPS only treatment group. | ||
The increased levels of TNF-α (Fig. 7a), IL-1β (Fig. 7b), and IL-6 (Fig. 7c) in RAW 264.7 cells by LPS stimulation remarkably decreased in a dose-dependent manner after exposure of the cells to LmEO (p < 0.05). In contrast, the levels of anti-inflammatory cytokine IL-10 significantly increased in a dose-dependent manner after the cells were exposed to LmEO (p < 0.05; Fig. 7d).
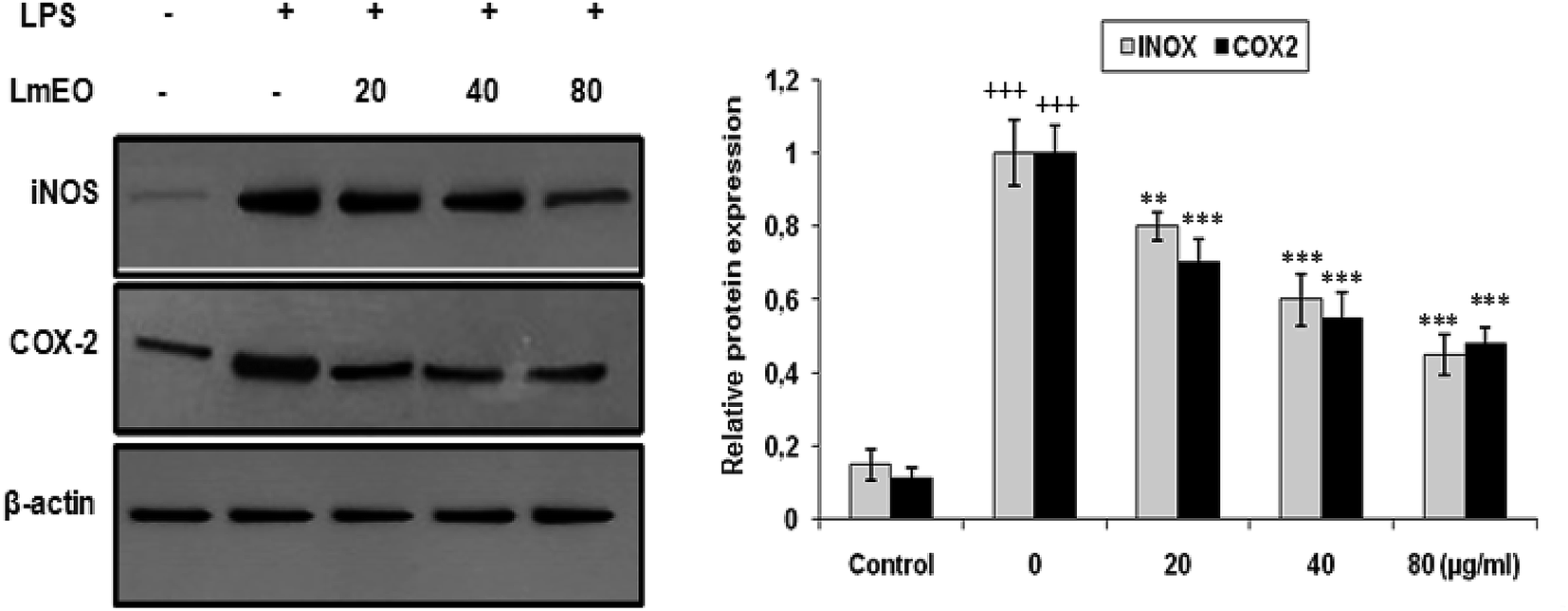 | ||
| Fig. 8 Inhibitory effects of LmEO on protein expression of iNOS and COX-2 in LPS-stimulated RAW 264.7 cells. The results presented are representative of three independent experiments. Values are expressed as mean ± SEM (n = 3). +++P < 0.001 compared with the control group. ** and ***P < 0.01 and +++P < 0.001, respectively, compared with the LPS-only treatment group. | ||
In these experiments, LPS-activated macrophages increased the protein expression levels of COX-2 and iNOS compared with those in the untreated control group. In contrast, LmEO treatment down-regulated the expression of these LPS-stimulated proteins in a concentration-dependent manner (p < 0.05).
Discussion
In the current study, the role of L. maritima leaf EO on the oxidative stress induced by CCl4 hepatotoxicity was investigated in male rats. The dose of CCl4 was selected based on the literature.38 The selected dose of LmEO was based on the acute toxicity studies. LmEO is mainly composed of oxygenated monoterpenes and monoterpene hydrocarbons in respective amounts of 74.4% and 16.5%. The major component is linalool, which accounts for 22.43% of the whole oil. To ascertain the LmEO antioxidant potential, we chose the DPPH method because the stable DPPH radical is widely used to evaluate the free radical-scavenging activity of many plants. Antioxidants are able to scavenge the radical by hydrogen donation, thus inducing a decrease of the DPPH absorbance at 517 nm. The assessment of the LmEO antioxidant activity shows that it is a free radical scavenger and may act as a primary antioxidant. This activity can be ascribed to the chemical composition of LmEO. LmEO monoterpenes may act as radical scavenging agents. It appears to be a general trend that LmEO, which contains monoterpene hydrocarbons, oxygenated monoterpenes and/or sesquiterpenes, has great antioxidative properties.39 Monoterpene hydrocarbons, particularly terpinolene, α-terpinene and γ-terpinene, may also account for the antioxidant activity. However, this activity is less important than that of the oxygenated monoterpenes. On the other hand, low antioxidant activity is associated with sesquiterpene hydrocarbons and their oxygenated derivatives.40 Numerous reports clearly demonstrate the importance of medicinal plants in the treatment of oxidative stress-induced cell death.41 The present study was undertaken to study the possible hepatoprotective role of L. maritima leaf EO in a CCl4-induced liver toxicity rat model. High serum levels of hepatic enzymes (ALT, AST, ALP, and LDH) constitute a sensitive indicator of liver cell injury and are very helpful in recognizing hepatic diseases.42 Hence, free radical scavenging helps protect against CCl4-induced oxidative injury. In the present study, the protective effects of LmEO against CCl4-induced hepatotoxicity and oxidative stress were investigated. According to our results, the pre-treatment with LmEO attenuated the increased serum levels of liver enzymes (AST, ALT, ALP and LDH) induced by CCl4 toxicity with values similar to those of control rats; this was indicated by the structural and functional integrity of hepatic parenchyma cells.In this investigation, CCl4 induced an increase in hepatic LPO in the experimental group II animals, expressed in terms of high TBARS level in the liver tissue of these animals. Compared to the control animals, hepatocellular damage was indicated as previously described.43 Pre-treatment with the LmEO exhibited a significant decrease in TBARS level in the liver tissue, indicating an inhibitory role of LmEO against lipid peroxidation. Consequently, we noted a decrease of CCl4-induced hepatic damage. The prevention of lipid peroxidation can be attributed to the ability of LmEO to scavenge ROS.
ROS, such as superoxide anions and H2O2, are produced during normal aerobic metabolism, and their intracellular concentration depends on the rate of their production and their removal by various antioxidants. The antioxidant system in mammalian cells is mainly represented by three enzymes: superoxide dismutase (SOD), catalase (CAT) and glutathione peroxidase (GPx). These enzymes act in a synergic way to detoxify the superoxide anions and H2O2 in cells. In our study, the SOD, CAT and GPx activities significantly decreased in the liver tissue of CCl4-treated rats (group II) compared to the control group. The reduced activity of these enzymes may be due to enhanced lipid peroxidation or inactivation of the antioxidative enzymes.44 Intra-peritoneal injection of LmEO for 14 days in group IV rats abolished the CCl4-induced reduction in the SOD, CAT and GPx activities, protecting liver tissue against the oxidative insults of CCl4. The histological data basically supported the results relative to the serum enzyme assays. We noticed massive fatty changes, such as necrosis, infiltration of lymphocytes, and several ballooning degenerations of hepatocytes, in the livers of CCl4-intoxicated rats; meanwhile, for rats pre-treated with LmEO and subsequently given CCl4, the liver histopathological data revealed more or less normal architectures.
These results suggest that LmEO reduces oxidative stress by preventing the generation of free radicals. The reduced oxidative stress and lipid peroxidation occurring in the LmEO-treated animals can be attributed to the antioxidant potential of LmEO. LmEO antioxidants are able to decompose free radicals by quenching ROS and trapping radicals before they reach their cellular targets. The antioxidant activity of LmEO can also be assigned to its monoterpenes. Moreover, the measured antioxidant activities may be due to the synergistic effects of two or more LmEO constituents. In this context, many authors have reported that most natural antioxidative compounds act synergistically to produce a broad spectrum of antioxidative properties that create an effective defence system against free radicals.45,46
As natural volatile substances from plants, EOs may represent an alternative source of anti-inflammatory agents. These oils consist of mixtures containing many bioactive compounds that are biodegradable into nontoxic products and potentially suitable for use in integrated management programs.47 In the present study, we also investigated the anti-inflammatory effects of LmEO on LPS-stimulated RAW 264.7 macrophage cells. To our knowledge, this study is the first to assess the chemical composition and bioactivity of the genus Lobularia.
Inflammation is a bodily response to harmful stimuli such as injury and infection.48 Various inflammatory models allow evaluation of test compounds and provide further understanding of the inflammatory process. In many studies, anti-inflammatory compounds have been investigated for their potential inhibitory effects in vitro using LPS-stimulated RAW 264.7 macrophages. LPS, a component of the outer membrane of Gram-negative bacteria, can activate murine macrophages; this induces oversecretion of various inflammatory and toxicity-mediating molecules, such as TNF-α, IL-6, eicosanoids, and NO.49 At concentrations of 20–80 μg mL−1, LmEO did not show any cytotoxic effects on cells. It suppressed NO production in the LPS-treated cells in a dose-dependent manner. Ko et al. investigated the potential anti-inflammatory effects of Lindera erythrocarpa EO (LEO) through the evaluation of NO production in LPS-stimulated RAW 264 cells.50 They reported that NO production was substantially higher in LPS-treated cells than in untreated cells. According to the same authors, LEO suppressed NO production in LPS-treated cells in a dose-dependent manner.
In our case, LPS-activated macrophages increased the protein expression levels of COX-2 and iNOS compared with those in the untreated control group. In contrast, LmEO treatment down-regulated the expression of these LPS-stimulated proteins in a concentration-dependent manner. Similar facts have been reported by Ko et al., who used western blot analyses to determine whether the inhibitory activity of LEO on the production of NO and PGE2 was related to the expression levels of iNOS and COX-2.50 Ko et al. noted increased expression levels of iNOS and COX-2 in LPS-stimulated cells in comparison with untreated controls.50 Furthermore, they reported that LEO inhibited an LPS-induced increase in iNOS and COX-2 levels in a dose-dependent manner. These results are consistent with the inhibitory effects of LEO on the production of NO and PGE2. Additionally, EO extracted from Hibiscus sabdariffa was characterized by excellent anti-inflammatory activity in LPS-stimulated macrophage RAW 264.7 cells. It induced NO inhibition, which reached 67.46% at an EO concentration of 200 μg mL−1.51
To evaluate the anti-inflammatory mechanism mediated by L. maritima, we investigated the effects of LmEO on LPS-induced cytokine production, including proinflammatory cytokines such as IL-1β and IL-6 as well as TNF-α and the anti-inflammatory cytokine IL-10, which are regarded as crucial anti-inflammatory targets.52,53
Proinflammatory cytokines, such as TNF-α, IL-1β, and IL-6, are primarily produced by activated monocytes or macrophages. IL-6 is characterized as an inflammatory factor because it synergistically consolidates the inflammatory actions of IL-1 in human synovial cells.54 According to our results, the treatment of RAW 264.7 cells with LPS alone resulted in a significant increase in proinflammatory cytokine production compared with that in the control group (p < 0.001). In contrast, LmEO significantly reduced these cytokine levels (p < 0.001; Fig. 7). IL-10 is generally considered as an anti-inflammatory and immunosuppressive cytokine. Its inhibitory effects on the production of inflammatory cytokines, including TNF-α, IL-1β, and IL-6, have been reported in numerous studies.55 In the current study, LmEO significantly increased the levels of anti-inflammatory cytokine IL-10 (Fig. 7d). These results are in concordance with those of Ko et al., who reported that IL-6 and TNF-α production was considerably increased in LPS-stimulated RAW 264.7 cells but significantly inhibited by LEO in a dose-dependent manner.50 Similar results have been reported for curcumin. Compared to LPS-stimulated controls, curcumin led to a dose-dependent reduction in the levels of proinflammatory cytokines. LPS-stimulated production of TNF-α was reduced in a dose-dependent manner by cells exposed to curcumin. TNF-α is a major cytokine involved in inflammation.56 Curcumin supplementation also resulted in inhibition of LPS-induced IL-10 and IFN-γ. Moreover, curcumin downregulated IL-6 and TNF-α production.56 Therefore, the regulation of cytokines observed in this study may reflect one of the mechanisms underlying the anti-inflammatory effects of LmEO.
Conclusion
In vivo investigations were performed to explore the therapeutic role of a halophyte plant in CCl4-induced liver injury for the first time. The main results of the current in vivo study revealed that LmEO has hepato-protective effects against CCl4-induced oxidative stress in rats, as evidenced by the decreased TBARS levels in liver tissue and the decrease of liver marker enzyme levels in serum to their normal values. Moreover, LmEO exerted potent anti-inflammatory effects through down-regulating the expression of multiple inflammatory cytokines and related mediators, including IL-1, IL-6, TNF-α, NO, iNOS, and COX-2, in LPS-induced macrophages. These significant findings not only support the traditional use of some medicinal plants in the treatment of various inflammation-associated diseases, but also provide evidence that LmEO can be exploited to develop new potential therapeutic agents for inflammatory diseases. As a continuation of the present study, isolation and investigation of the LmEO constituents responsible for these hepatoprotective and anti-inflammatory effects should be undertaken in order to confirm and elucidate the mechanisms behind these activities.Conflicts of interest
The authors declare that there are no conflicts of interest.Acknowledgements
This study was supported by a grant from the Ministry of Higher Education and Scientific Research of Tunisia (contract programme 2019–2022).References
- A. Roy, D. Bhoumik, R. K. Sahu and J. Dwivedi, UK J. Pharm. Biosci., 2014, 2, 23–33 CrossRef.
- J. Avijeet, S. Manish, D. Lokesh, J. Anurekha, S. P Rout, V. B Gupta and L. K. Krishna, J. Ethnopharmacol., 2008, 115, 61–66 CrossRef.
- D. Zhou, J. Ruan, Y. Cai, Z. Xiong, W. Fu and A. Wei, J. Ethnopharmacol., 2010, 129, 232–237 CrossRef.
- B. N. Ames, M. K. Shigenaga and T. M. Hagen, Proc. Natl. Acad. Sci. U. S. A., 1993, 7915–7922 CrossRef CAS.
- A. R. Opoku, I. M. Ndlovu, S. E. Terblanche and A. H. Hutchings, S. Afr. J. Bot., 2007, 73, 372–377 CrossRef.
- B. H. Anis, G. Manel, D. Wissal, B. S. Rania, S. Naima, M. Wissem and S. Walid, Environ. Toxicol., 2019, 34(4), 388–400 CrossRef PubMed.
- B. Singh, A. K. Saxena, B. K. Chandan, K. K. Anand, O. P. Suri, K. A. Suri and N. K. Satti, Fitoterapia, 1998, 69, 135–140 CAS.
- S. K. Mitra, M. V. Venkataranganna, R. Sundaram and S. Gopumadhavan, Phytother. Res., 1998, 12, 114–117 CrossRef.
- S. Ran and K. E. Montgomery, Cancers, 2012, 4, 618–657 CrossRef CAS.
- J. H. Choi, Y. N. Park, Y. Li, M. H Jin, J. Lee, Y. Lee, J. K. Son, H. W. Chang and E. Lee, Immune Network, 2010, 10, 145e52 CrossRef.
- Y. Yang, T. Yu, Y. G. Lee, W. S. Yang, J. Oh, D. Jeong, S. Lee, T. W. Kim, Y. C. Park, G. H. Sung and J. Y. Cho, J. Ethnopharmacol., 2013, 145, 598e607 Search PubMed.
- S. J. Heo, W. J. Yoon, K. N. Kim, C. Oh, Y. U. Choi, K. T. Yoon, D. H. Kang, Z. J. Qian, I. W. Choi and W. K. Jung, Food Chem. Toxicol., 2012, 50, 3336–3342 CrossRef CAS.
- M. Rossol, H. Heine, U. Meusch, D. Quandt, C. Klein, M. J. Sweet and S. Hauschildt, Crit. Rev. Immunol., 2011, 31, 379–446 CrossRef CAS.
- E. Ayroldi, A. Bastianelli, L. Cannarile, M. G. Petrillo, D. V. Delfino and A. Fierabracci, Curr. Pharm. Des., 2011, 17, 3176–3190 CrossRef CAS.
- A. A. Alfadda and R. M. Sallam, J. Biomed. Biotechnol., 2012, 936486 Search PubMed.
- A. U. Khan and A. H. Gilani, Phytother. Res., 2011, 25, 577–583 CrossRef CAS.
- B. Benedek and B. Kopp, Wien. Med. Wochenschr., 2007, 157, 312–314 CrossRef.
- K. Wolf, X. Wu and R. H. Liu, J. Agric. Food Chem., 2003, 51, 609–614 CrossRef.
- J. R. Wallace, Proc. Nutr. Soc., 2004, 63, 621–629 CrossRef.
- T. Sergent, N. Piront, J. Meurice, O. Toussaint and Y. J. Schneider, Chem.-Biol. Interact., 2010, 188(3), 659–667 CrossRef CAS.
- P. Zorrilla, A. Rodriguez-Nogales and F. Algieri, et al., J. Funct. Foods, 2014, 11, 449–459 CrossRef CAS.
- F. Bakkali, S. Averbeck, D. Averbeck and M. Idaomar, Food Chem. Toxicol., 2008, 46(2), 446–475 CrossRef CAS.
- J. Sharifi-Rad, A. Sureda and G. C. Tenore, et al., Molecules, 2017, 22(1), 70 CrossRef.
- D. D. Jayasena and C. Jo, Trends Food Sci. Technol., 2013, 34(2), 96–108 CrossRef CAS.
- F. X. Picó and J. Retana, Bot. J. Linn. Soc., 2003, 142, 273–280 CrossRef.
- A. B. Hsouna, S. Dhibi, W. Dhifi, W. Mnif, H. B. Nasr and N. Hfaiedh, RSC Adv., 2019, 9, 3777–3787 RSC.
- A. J. Kirby and R. J. Schmidt, J. Ethnopharmacol., 1997, 56(2), 103–108 CrossRef CAS.
- D. E. Pratt, Natural antioxidants of soybean and other oil-seeds, in Autoxidation in food andbiological system, ed. M. G. Simic and M. Karel, Plenum Press, New York, 1980, pp. 283–292 Search PubMed.
- Council of European Communities, Official Journal of the European Union, 2010, 276, 1–33 Search PubMed.
- J. V. Bruckner, W. F. MacKenzie, S. Muralidhara, R. Luthra, M. Kyle and D. Acosta, Fundam. Appl. Toxicol., 1986, 6, 16–34 CrossRef CAS.
- O. H. Lowry, et al., Biol. Chem., 1951, 193, 265–275 CAS.
- K. Yagi, Biochem. Med., 1976, 15, 212–216 CrossRef CAS.
- W. F. Beyer and I. Fridovich, Anal. Biochem., 1987, 161, 559–566 CrossRef CAS.
- H. Aebi, Methods Enzymol., 1984, 105, 121–126 CAS.
- L. Floke and R. Gunzler, Methods Enzymol., 1984, 105, 114–121 Search PubMed.
- D. E. Kleiner, et al., J. Hepatol., 2005, 41(6), 1313–1321 CrossRef.
- V. M. Dirsch, H. Stuppner and A. M. Vollmar, Planta Med., 1998, 64, 423e6 CrossRef.
- C. Girish, B. C. Koner, S. Jayanthi, K. R. Rao, B. Rajesh and S. C. Pradhan, J. Exp. Biol., 2009, 47, 257–263 CAS.
- B. Tepe, D. Daferera, M. Sökmen, M. Polissiou and A. Sökmen, J. Agric. Food Chem., 2004, 52(5), 1132–1137 CrossRef CAS.
- B. H. Anis, H. Naceur, B. H. Nihed and A. Slim, J. Oleo Sci., 2013, 62, 763–772 CrossRef.
- M. Jung, M. Park, H. C. Lee, Y. K. Kang, E. S. Kang and S. K. Kim, Curr. Med. Chem., 2006, 13(10), 1203–1218 CrossRef CAS.
- D. H. Abdelaziz and S. A. Ali, J. Ethnopharmacol., 2014, 155(1), 736–743 CrossRef.
- G. Pérez-Trueba, C. Ramos-Guanche, B. Martínez-Sánchez, I. Márquez-Hernández, A. Giuliani and G. Martínez-Sánchez, Redox Rep., 2003, 8(4), 215–221 CrossRef.
- A. B. Hsouna, M. Saoudi, M. Trigui, K. Jamoussi, T. Boudawara, S. Jaoua and A. El Feki, Food Chem. Toxicol., 2011, 49, 3183–3191 CrossRef.
- F. Lu and L. Y. Foo, Food Chem., 2001, 75, 197–202 CrossRef.
- S. N. Nivitabishekam, M. Asad and V. S. Prasad, Chem.-Biol. Interact., 2009, 177, 247–253 CrossRef CAS.
- S. S. Cheng, C. G. Huang, Y. J. Chen, J. J. Yu, W. J. Chen and S. T. Chang, Bioresour. Technol., 2009, 100, 452e6 Search PubMed.
- Y. H. Choi, G. Y. Kim and H. H. Lee, Drug Des., Dev. Ther., 2014, 8, 1941e53 Search PubMed.
- E. J. Yang, E. Y. Yim, G. Song, G. O. Kim and C. G. Hyun, Interdiscip. Toxicol., 2009, 2, 245e9 Search PubMed.
- Y. J. Ko, G. Ahn, Y. M. Ham, S. M. Song, E. Y. Ko, S. H. Cho, W. J. Yoon and K. N. Kim, EXCLI J., 2017, 29(16), 1103–1113 Search PubMed.
- C. Y. Shen, T. T. Zhang, W. L. Zhang and J. G. Jiang, Food Funct., 2016, 7, 4451–4459 RSC.
- J. J. Haddad, N. E. Saade and B. Safieh-Garabedian, Cell. Signalling, 2003, 15, 255e67 CrossRef.
- B. J. Pettus, J. Bielawski, A. M. Porcelli, D. L. Reames, K. R. Johnson, J. Morrow, C. E. Chalfant, L. M. Obeid and Y. A. Hannun, FASEB J., 2003, 11, 1411e21 Search PubMed.
- A. Ito, Y. Itoh, Y. Sasaguri, M. Morimatsu and Y. Mori, Arthritis Rheum., 1992, 35, 1197e201 CrossRef.
- B. Raychaudhuri, C. J. Fisher, C. F. Farver, A. Malur, J. Drazba, M. S. Kavuru and M. J. Thomassen, Cytokines, 2000, 12, 1348e55 Search PubMed.
- L. Das and M. Vinayak, PLoS One, 2015, 10(4), e0124000 CrossRef.
| This journal is © The Royal Society of Chemistry 2019 |