
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStepwise engineering of Saccharomyces cerevisiae to produce (+)-valencene and its related sesquiterpenes†
Xiaodan
Ouyang
 a,
Yaping
Cha
a,
Wen
Li
a,
Chaoyi
Zhu
a,
Muzi
Zhu
b,
Shuang
Li
*a,
Min
Zhuo
a,
Shaobin
Huang
c and
Jianjun
Li
b
a,
Yaping
Cha
a,
Wen
Li
a,
Chaoyi
Zhu
a,
Muzi
Zhu
b,
Shuang
Li
*a,
Min
Zhuo
a,
Shaobin
Huang
c and
Jianjun
Li
b
aSchool of Biology and Biological Engineering, South China University of Technology, Higher Education Mega Center, Guangzhou 510006, China. E-mail: shuangli@scut.edu.cn; Fax: +86 20 3938 0601; Tel: +86 20 3938 0601
bState Key Laboratory of Applied Microbiology Southern China, Guangdong Provincial Key Laboratory of Microbial Culture Collection and Application, Guangdong Institute of Microbiology, Guangzhou 510070, China
cSchool of Environment and Energy, South China University of Technology, Higher Education Mega Center, Guangzhou 510006, China
First published on 24th September 2019
Abstract
(+)-Valencene and (+)-nootkatone are high value-added sesquiterpenoids found in grapefruit. The synthesis of (+)-nootkatone by chemical oxidation from (+)-valencene cannot meet the increasing demand in natural aromatics markets. Development of a viable bioprocess using microorganisms is attractive. According to the yields of β-nootkatol and (+)-nootkatone by strains harboring different expression cassettes in the resting cell assay, premnaspirodiene oxygenase from Hyoscyamus muticus (HPO), cytochrome P450 reductase from Arabidopsis thaliana (AtCPR) and alcohol dehydrogenase (ADH1) from Saccharomyces cerevisiae were finally selected and overexpressed in CEN·PK2-1Ca, yielding β-nootkatol and (+)-nootkatone with 170.5 and 45.6 mg L−1 ethyl acetate, respectively. A combinational engineering strategy including promoter change, regulator ROX1 knockout, squalene pathway inhibition, and tHMGR overexpression was performed to achieve de novo (+)-valencene production. Subsequent culture investigations found that galactose as the induced carbon source and a lower temperature (25 °C) were beneficial to target accumulation. Also, replacing the inducible promoters (GAL1) of HPO and AtCPR with constitutive promoters (HXT7 and CYC1) dramatically increased the β-nootkatol accumulation from 108.2 to 327.8 mg L−1 ethyl acetate in resting-cell experiments using (+)-valencene as a substrate. Finally, the total terpenoid titer of the engineered strain of PK2-25 using glucose as a carbon source was improved to 157.8 mg L−1 cell culture, which was 56 times the initial value. We present a new candidate for production of (+)-valencene and its related sesquiterpenoids with attraction for industry.
1. Introduction
Sesquiterpenes are a class of abundant natural products derived from the 15-carbon linear precursor, farnesyl pyrophosphate (FPP). (+)-Valencene is an important sesquiterpene that is an aroma ingredient in orange peel oil. Its odor characteristics include fresh citrus notes with sweet, orange and woody balsamic profiles. (+)-Nootkatone is an oxidized product of (+)-valencene. It is the main contributor to the aroma component of grapefruit and citrus-derived odorants with relatively low perception threshold.1,2 Applications of these compounds include food and beverages, fragrances, personal care and home products. It is on the authorized flavor list with a 0.5 mg kg−1 concentration level for food in the European Regulations by the European Food Safety Authority.3 (+)-Nootkatone is reported to have several pharmacological properties, such as antioxidant, antiseptic and antiallergic activities, and is also an effective repellent against such various insects as mosquitoes, bed bugs, termites and ticks.4 Recently, (+)-nootkatone was considered as protective agent against air pollution-induced respiratory effects.5Employing conventional extraction processes from natural sources, it takes 2.5 million kilograms of oranges to obtain a single kilogram of (+)-valencene. The trace amount of (+)-valencene and (+)-nootkatone in natural plants leads to high price of these fragrance compounds.6 The high cost, limited supply and inconsistent product quality have restricted their applications. Several chemical synthetic strategies have been developed via the Robison annulation, intramolecular Sakurai reaction, and Diels–Alder/Aldol tandem reaction, etc.7 However, the mentioned above generally provide racemic (±)-nootkatone. One-pot synthesis of (+)-nootkatone through singlet oxygenation of (+)-valencene has been realized with carcinogenic tert-butyl hydroperoxide in combination with Co(OAc)2, Cu(OAc)2 or V(OAc)2 catalysts.7 The explosive, corrosive, and toxic heavy metal chemocatalyst requirements of the chemical approaches call for greener and more effective methods for (+)-valencene and (+)-nootkatone preparation. Previously, (+)-valencene extracted from Valencia orange was converted into (+)-nootkatone by biotransformation using Aspergillus, Rhodococcus, Chlorella, Mucor species Botryodiplodia theobromae 1368, Yarrowia lipolytica 2.2ab, and Phanerochaete chrysosporium.1,8 These reported biotransformation approaches suffered frequently from long reaction time, relatively low yields and troublesome purification procedures.9
Recent rapid developments in metabolic engineering by rewiring cellular metabolism provide an attractive alternative approach for producing plant terpenoids.6,10,11Saccharomyces cerevisiae has been widely used in terpenoid biosynthesis because it is generally regarded as safe (GRAS),10 industrially robust and able to functionally express eukaryotic cytochrome P450 enzymes.12 There are many discussions about terpenoid production through metabolic engineering of S. cerevisiae at hierarchical levels of genetic manipulation tools, biological parts, pathways, organelles and systems.10,12–15 Although significant progresses have been achieved in the past decades, there are still a lot of efforts to be made.
In S. cerevisiae, the mevalonate (MVA) pathway is naturally tightly regulated at transcriptional and post-transcriptional levels, limiting the overall metabolic flux for sesquiterpene production. To increase the target sesquiterpene accumulation, genetic modifications that block the competition branch flux could be considered. The squalene synthase (ERG9) is a branch-point enzyme in the mevalonate (MVA) pathway.16–18 As sterols are essential for yeast cells, so it is not feasible to thoroughly knock out squalene synthase (ERG9). Down-regulating the expression of ERG9 by weakening its promoter is an alternative way. In addition, the ROX1 is a transcription factor that inhibits expression of hypoxia-induced genes in the presence of oxygen and also represses target genes during osmotic stress.19 Previous study showed that transcriptional regulation of ROX1 mediates the transcriptional regulation of ERG genes. Especially, ERG9 could be activated by ROX1 at the transcription level.20
In this study, we constructed and optimized the (+)-nootkatone biosynthesis gene cluster in S. cerevisiae to develop a new yeast-based platform for the biosynthesis of (+)-valencene and its related sesquiterpenes, combining the disruption of ROX1 gene, the repression of ERG9 through 45-bp deletion in its promoter, and the overexpression of a truncated version of HMG-CoA reductase 1 (tHMGR) (Scheme 1). Moreover, the carbon sources in the induction medium, the culture conditions and promoters of HPO and AtCPR were investigated to intensify the valencene and its related sesquiterpenes productivities.
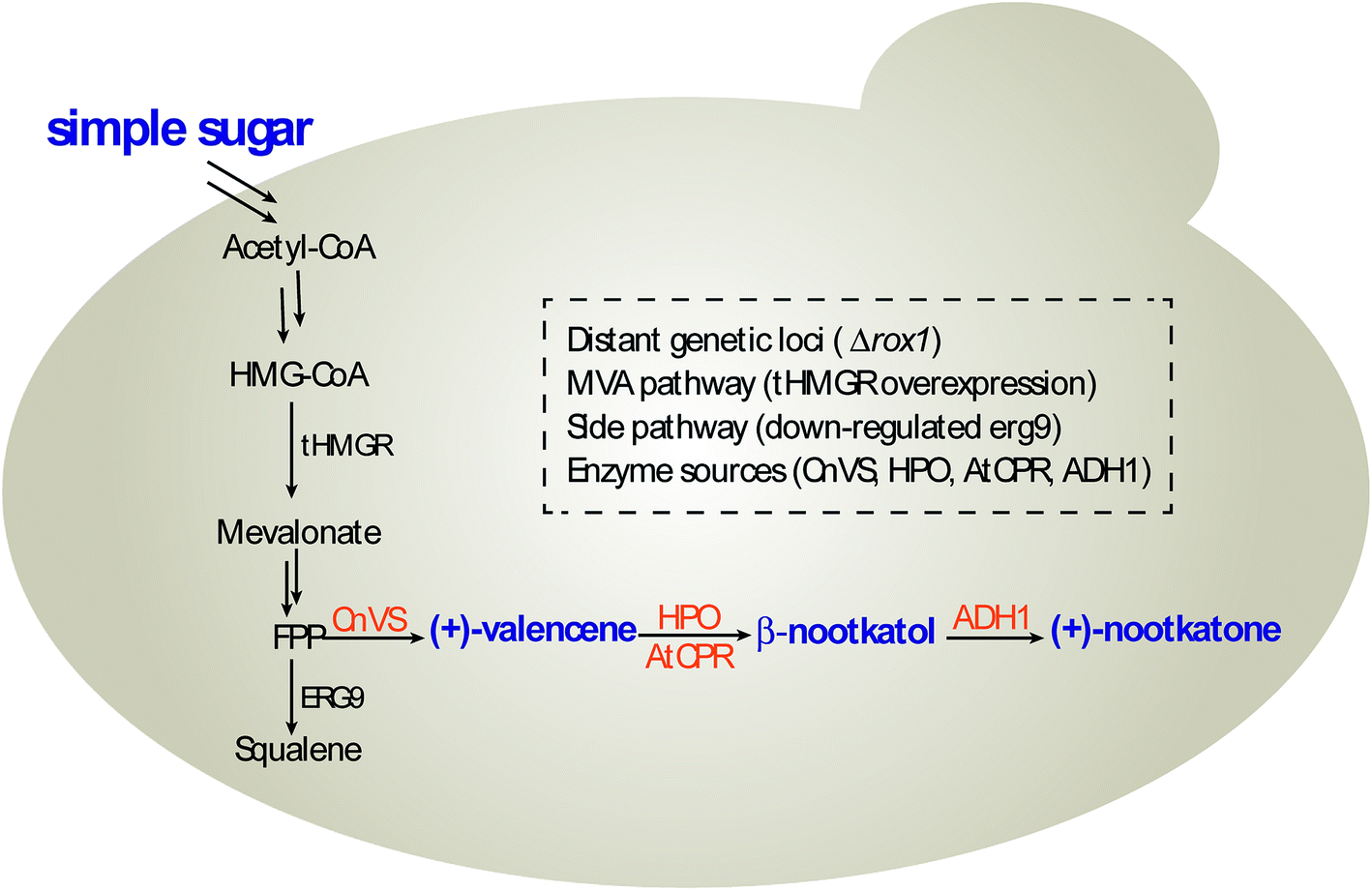 | ||
| Scheme 1 (+)-Nootkatone biosynthesis pathway in S. cerevisiae. | ||
2. Material and methods
2.1 General
DNA polymerase of PrimeSTAR HS was purchased from TaKaRa (Dalian, China). KOD FX DNA polymerase was from Toyobo Co. Ltd. (Osaka, Japan). Restriction enzymes and T4 DNA ligase were bought from Fermentas of Thermo Fisher Scientific (Pittsburgh, PA, USA). DNA preparations kits for plasmid extraction and DNA purification were purchased from Tiangen (Beijing, China). (+)-Valencene, β-nootkatol, (+)-nootkatone and isolongifolene were provided by Tokyo Chemical Industry Co. Ltd. (TCI, Tokyo, Japan). For all other procedures, the highest purity grade chemicals were used.2.2 Strains, media and genetic materials
E. coli DH5α was used for gene cloning and plasmid maintenance. All recombinant E. coli cells were cultured in Luria-Bertani (LB) medium with appropriate antibiotics (100 μg mL−1 ampicillin). All recombinant S. cerevisiae strains in this study were constructed from BY4741, BJ5464 or CEN·PK2-1Ca. Complete medium of yeast extract peptone dextrose (YPD, containing 1% (w/v) yeast extract, 2% (w/v) peptone and 2% (w/v) D-glucose) was used for yeast cultivation. Engineered yeast strains were grown in minimal synthetic defined (SD) medium (Clontech, Mountain View, USA), in which leucine and/or uracil were omitted for auxotrophic selection.21,22 For the expression of galactose-inducible genes, SG, SGR and SGG medium were used, in which the SD medium was changed as the medium containing 2% (w/v) galactose instead of glucose, or 2% (w/v) galactose supplemented with 0.7% (w/v) raffinose, or 2% (w/v) galactose combined with 10% (w/v) glycerol, respectively.All heterologous genes used in this study were synthesized by Sangon Biotech (Shanghai, China) with codon optimization for expression in S. cerevisiae. The truncated version of HMG1 (tHMGR), alcohol dehydrogenase isozymes (ADH1 and ADH3) were amplified from CEN·PK2-1Ca genomic DNA. Genes used in this study are listed in Table S1.† Plasmids and strains used in this work are listed in Tables S2 and S3,† respectively.
2.3 Cloning and integration of pathway in S. cerevisiae
The assembly of synthetic operon in YEp352 was performed according to the BioBrick principles23 described by Zelcbuch et al.24 To further simplify the cloning process, a seamless, recombination-based cloning strategy was applied for the synthetic operon construction in YEplac181 using the ClonExpress II One Step Cloning Kit (Vazyme, Nanjing, China). The knockout of ROX1 and knockdown of ERG9 were achieved via CRISPR/Cas9 technologies.25 For the CRISPR genome editing, yeast cells were pre-transformed with p414-TEF1p-Cas9-CYC1t plasmid (Addgene). All the PCR products were analyzed on 1% agarose gels and positive colonies were grown overnight for plasmid extraction using the Tianprep Rapid Mini Plasmid Kit (Tiangen, Beijing China) according to the manufacture's instructions. All clones were verified through the expression cassette sequencing (Sangon, China). The constructed plasmids containing the whole expression operon were chemically transformed into the yeast by S. cerevisiae EasyComp™ Transformation Kit (Invitrogen, CA, United States) and aliquots were plated on the corresponding auxotrophic minimal media. Table S4† lists the primers used to construct these plasmids and strains.2.4 Resting cell assays for β-nootkatol and (+)-nootkatone production
Recombinant yeast strains transformed freshly or from −80 °C freezer were cultivated on SD plates at 30 °C for two days. The recombinant single yeast colonies were inoculated into 15 mL tubes containing 5 mL of SD medium supplemented with appropriate amino acids for auxotrophic selection. Cultivation of strains (OD600 ≈ 1 to 2) was scaled up to 50 mL of total volume in 250 mL baffled culture flasks to a starter OD600 of 0.1. When cell density (OD600) reached about 3.0 after incubation for several hours, cells were recovered by centrifugation at 1060 × g for 10 min and resuspended in 50 mL of SG minimal medium. After 8 h induction, 150 OD600 units of cell pellets were harvested and transferred to sterilized 20 mL tubes containing 1 mL of 50 mM potassium phosphate buffer (KPi, pH 7.4). The resting cell assays were started by the addition of 20 μL of (+)-valencene stock solutions (100 mM (+)-valencene and 1% Triton X-100 (v/v) dissolved in DMSO). Tubes containing cell suspensions were incubated for 18 h. All cells were cultured at 30 °C, 200 rpm. Samples were taken and extracted into GC vials for further analysis.2.5 Bi-phasic batch cultivation in shake flasks
Pre-cultures of the respective strains were grown for 24–30 h at 30 °C, 200 rpm in 5 mL SD medium. The seed cultures were inoculated to an OD600 of 0.05 and grown at 30 °C and 200 rpm in 50 mL baffled shake flasks containing 10 mL SD medium. At an OD600 of about 0.8–1.0, cells were harvested by centrifugation at 1060 × g for 10 min and resuspended in 10 mL of SG, SGR or SGG medium combined with 2 mL of n-dodecane.26 The biocatalysis was performed at 25 or 30 °C under shaking at 200 rpm for different hours. 500 μL of immiscible n-dodecane layer was taken out at 24, 48, 72 and 96 h and added with 500 μL of ethyl acetate for GC analysis.2.6 Analysis of (+)-valencene, β-nootkatol and (+)-nootkatone
For resting cell assays, the (+)-valencene, β-nootkatol and (+)-nootkatone were extracted using equal volume of ethyl acetate. The mixture was vortexed at room temperature for 30 min and centrifuged.27 500 μL of the top organic layer was transferred into a new tube containing 500 μL of ethyl acetate. For bi-phasic fermentation, 500 μL of n-dodecane layer was taken out and mixed with equal volume of ethyl acetate. Samples were filtered into GC vials through 0.22 μm sterile filter. The concentrations of (+)-valencene, β-nootkatol and (+)-nootkatone were expressed as microgram per liter (mg L−1 ethyl acetate or mg L−1 cell culture).Analysis of the sesquiterpenes were determined by gas chromatography (GC) using a Hewlett-Packard 7890 Gas Chromatography (Agilent) equipped with a flame ionization detector (FID). An HP-5 column (crosslinked 5% Ph-Me siloxane; 30 m × 0.32 mm × 0.25 μm) was used with hydrogen as carrier gas. The temperatures of injector and detector were set as 250 °C and 320 °C, respectively. During analysis, 1 μL of sample was injected with a split ratio of 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 at a rate of 0.4 mL min−1 in constant flow mode (49 cm s−1 linear velocity). The initial oven temperature was set to 100 °C for 10 min with a ramp rate for separation as 10 °C per min, and maintained at 200 °C for 8 min.16 Analyses were carried out in triplicate, and concentrations were determined using 50 μM isolongifolene (Sigma Aldrich, St. Louis, MO) as an internal standard.
:1 at a rate of 0.4 mL min−1 in constant flow mode (49 cm s−1 linear velocity). The initial oven temperature was set to 100 °C for 10 min with a ramp rate for separation as 10 °C per min, and maintained at 200 °C for 8 min.16 Analyses were carried out in triplicate, and concentrations were determined using 50 μM isolongifolene (Sigma Aldrich, St. Louis, MO) as an internal standard.
Analysis by GC/MS using a GC/MS-HP7890 (Agilent) equipped with a 5975C series mass selective detector (MSD). Same analysis methods were used as GC-FID detection. MS data were recorded at 70 eV (EI), m/z (rel. intensity in %) as TIC, total ion current. The data were collected in full scan mode (m/z 50–650). Compounds in the samples were identified by the comparison of retention time and mass GC/MS spectra to the commercially available chemical standards and mass spectra data of the NIST Standard Reference Database.
3. Results and discussion
3.1 (+)-Valencene hydroxylation using resting cells
Firstly, two reported cytochrome P450s (HPO from Hyoscyamus muticus28 and CnVO from Callitropsis nootkatensis29) were investigated for (+)-valencene hydroxylation. The coding genes were separately introduced into the YEplac181 under the control of galactose inducible promoter of PGAL1. The cytochrome P450 reductase (CPR) from Arabidopsis thaliana was cloned into the vector of YEp352 containing a GAL1 promoter, generating p352-CPR. To this end, biohydroxylation of (+)-valencene in S. cerevisiae CEN·PK2-1Ca expressing different cytochrome P450s in combination with CPR were performed. 150 OD600 units of recombinant resting cells were collected and resuspended in 1 mL 50 mM KPi buffer containing 20 μL of (+)-valencene stock solution. After 18 h at 30 °C 200 rpm, the products were extracted using ethyl acetate and detected by GC-FID (Fig. 1A). The GC-MS analysis confirmed the β-nootkatol and (+)-nootkatone by comparison of the mass fragmentation pattern to an authentic standard chemicals (Fig. 1B). No β-nootkatol was detected in the extract of the PK2-C strain containing the empty plasmids. PK2-01 containing HPO/AtCPR expression cassettes catalyzed (+)-valencene hydroxylation to β-nootkatol and (+)-nootkatone, detected at 48.4 ± 4.5 and 8.9 ± 1.6 mg L−1 ethyl acetate corresponding to a 13.02% (mol mol−1) conversion of substrate to product (Fig. 2A). The whole-cell biotransformation of (+)-valencene in PK2-01 was not consistent with the previous reported results that HPO failed to synthesize (+)-nootkatone in S. cerevisiae, although the engineered strain could catalyze the hydroxylation of (+)-valencene to β-nootkatol.28,30 For the CnVO, formations of β-nootkatol and (+)-nootkatone was reduced to about 10–15% of those catalyzed by HPO (Fig. 2A). In this study, effects of double mutation combinations of V482I/A484I in HPO were also investigated. Results showed that no higher titers of β-nootkatol were obtained, indicating that in vitro enzyme-kinetic data is often not verifiable in practice.28,31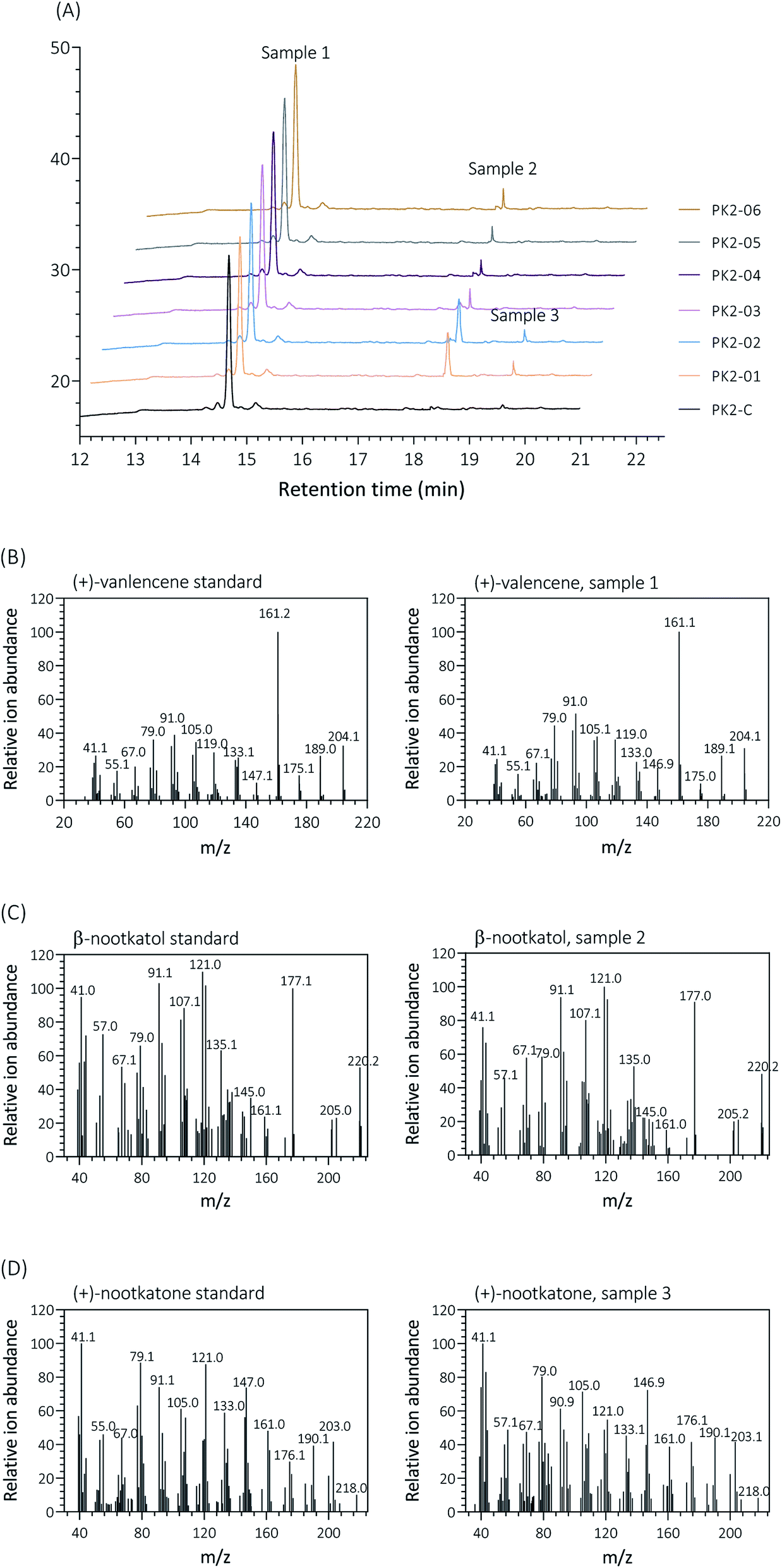 | ||
| Fig. 1 Analysis and identification of the hydroxylation of (+)-valencene (sample 1) to β-nootkanol (sample 2) and (+)-nootkatone (sample 3) by different S. cerevisiae strains expressing HPO, CnVO or their mutants, respectively. (A) GC-FID chromatogram patterns of the products obtained by 150 OD600 yeast cells incubated with 2 mM (+)-valencene at 30 °C for 18 h. Retention times of (+)-valencene, β-nootkanol and (+)-nootkatone were 14.639 min, 18.349 min and 19.540 min, respectively. Mass spectra of the products are compared with mass patterns of authentic standards (+)-valencene (B), β-nootkatol (C) and (+)-nootkatone (D) standard. | ||
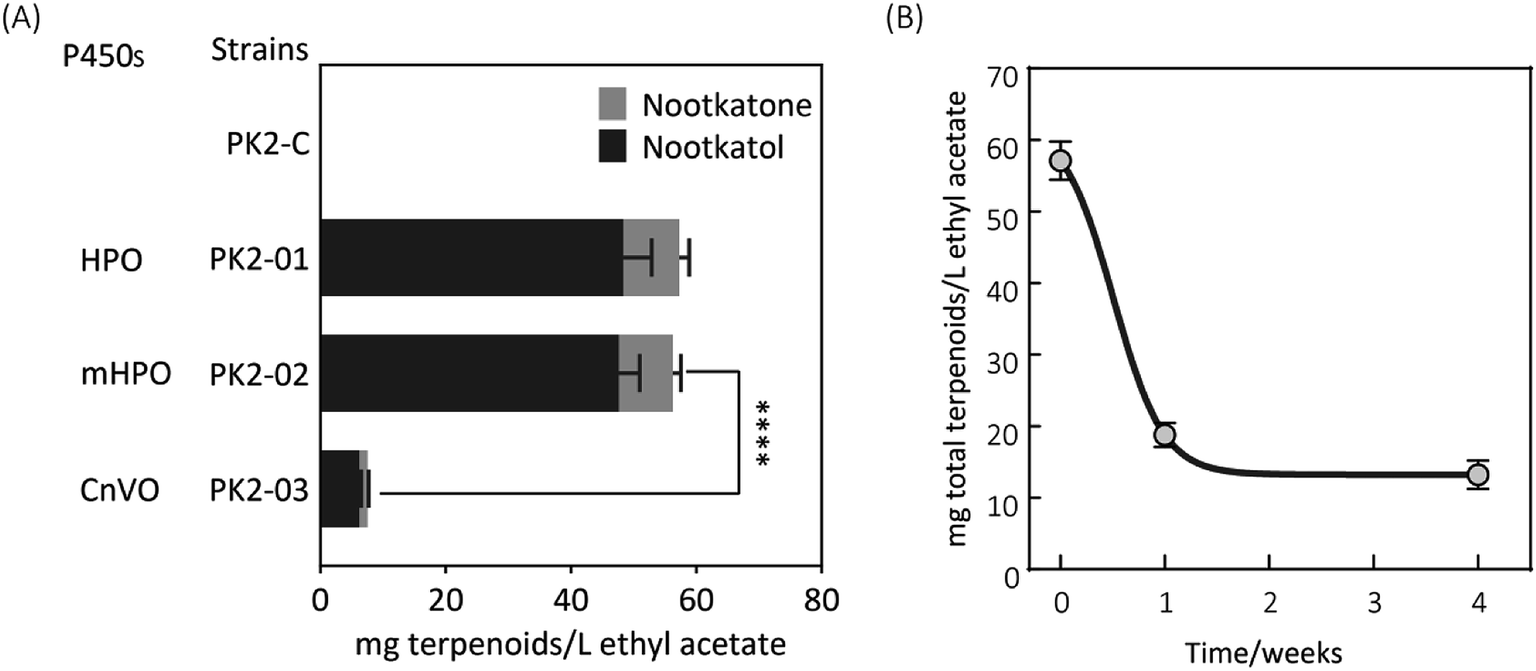 | ||
| Fig. 2 (A) Resting cell assays of (+)-valencene hydroxylation to β-nootkanol and (+)-nootkatone using different cytochrome P450s under the control of galactose inducible promoter of PGAL1 in YEplac181 in S. cerevisiae. Amino acid exchange of V482I/A484I (mHPO) in HPO was achieved through site-directed mutagenesis. No β-nootkanol or (+)-nootkatone was detected in strain PK2-C. The asterisk (****) indicates statistically significant differences in terpenoids formation (p < 0.0001, student's t-test). (B) Changes of (+)-valencene oxidation capabilities in yeast PK2-01 when cells incubated in glycerol stocks at during one month of storage. Recombinant yeast strains transformed freshly or from −80 °C freezer were cultivated on SD plates at 30 °C for two days. Then the recombinant single yeast colonies were inoculated into tubes containing 5 mL of SD medium supplemented with appropriate amino acids for auxotrophic selection. Cultivation of strains (OD600 ≈ 1 to 2) was scaled up to 50 mL of total volume in 250 mL baffled culture flasks to a starter OD600 of 0.1, which was prepared for subsequent resting cell assays. The total terpenoids was calculated as the sum of β-nootkanol and (+)-nootkatone in mg L−1 ethyl acetate. All experiments were performed in triplicates. | ||
When the recombinant PK2-01 was stored in −80 °C ultra-low temperature freezer for only one week, the total terpenoids production by the recovered strain decreased dramatically about 65%. Further kept at the same conditions, almost 80% of (+)-valencene hydroxylation activity was lost (Fig. 2B). In order to conquer the instability, the HPO and AtCPR were both introduced into S. cerevisiae CEN·PK2-1 Ca under the control of galactose inducible promoter of PGAL1 using the plasmids of YEplac181 or YEp352, denoted as PK2-30 and PK2-31, respectively (Fig. 3A). Surprisingly, great improvements of β-nootkatol and (+)-nootkatone production were obtained as 108.2 ± 4.5 and 19.1 ± 2.1 mg L−1 ethyl acetate, respectively, when the two HPO and AtCPR expression cassettes were both located on YEp352. While using YEplac181 as construction backbone, no similar results were observed.
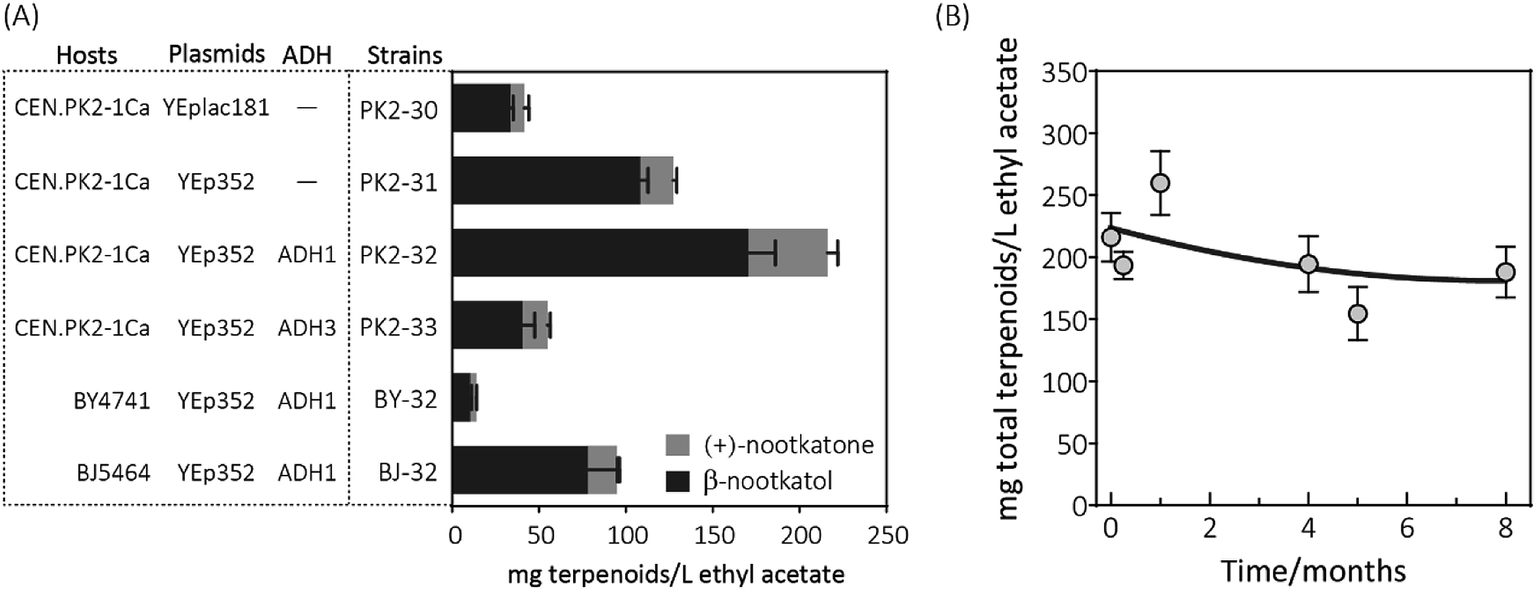 | ||
| Fig. 3 (A) Resting cell assays of (+)-valencene hydroxylation to β-nootkanol and (+)-nootkatone using different constructs in S. cerevisiae. Hosts (CEN·PK2-1Ca, BY4741 and BJ5464), constructed based on backbone (YEplac181 and YEp352) and alcohol dehydrogenase isozymes (ADH1 and ADH3) were compared. (B) Changes of (+)-valencene oxidation capabilities in yeast PK2-32 when cells incubated over a period of 8 months. The total terpenoids was calculated as the sum of β-nootkanol and (+)-nootkatone in mg L−1 ethyl acetate. Data is representative of triplicate determinations and error bars indicate standard deviations. | ||
Considering the fact that 19.1 mg L−1 ethyl acetate of (+)-nootkatone was detected in PK2-31, we speculated that the intrinsic alcohol dehydrogenase (ADH) activity accomplishes this conversion from β-nootkatol. Two different ADH genes (ADH1 and ADH3) were selected for overexpression under the constitutive promoter of PTDH3. When co-expressed with ADH1, (+)-nootkatone production was dramatically increased by 2.4-fold in the newly created strain PK2-32 as compared to PK2-31. Surprisingly, great improvement of β-nootkatol production was also observed (170.5 ± 15.5 mg L−1 ethyl acetate, Fig. 3A). The total oxidation rate of (+)-valencene to terpenoids by PK2-32 reached as 49.1% (mol mol−1), which was higher than that by Y. lipolytica 2-2ab previously reported.8 However, strain PK2-33 containing ADH3 showed decreases for both β-nootkatol and (+)-nootkatone production compared to PK2-32. ADH1 is generally regarded as primarily responsible for the NAD+ regeneration from NADH through the acetaldehyde reduction,32–34 which is beneficial for the β-nootkatol oxidation to (+)-nootkatone.
The capabilities of whole-cell (+)-valencene oxidation in three frequently employed industrial S. cerevisiae strains were further evaluated. The host strain CEN·PK2-1 Ca harboring p352-A1HC (PK2-32) produced the highest amounts of β-nootkatol and (+)-nootkatone, followed by the strain of BJ-32 (BJ5464 containing p352-A1HC). The lowest β-nootkatol and (+)-nootkatone production were monitored in S288c derived strain of BY-32 (10.3 ± 0.8 and 3.7 ± 0.2 mg L−1 ethyl acetate, respectively). The premnaspirodiene oxygenase (encoded by HPO), according to Marjohn (2016),35 is anchored in the endoplasmic reticulum (ER) membrane. The deletion of PEP4 and PRB1 in the yeast strain BJ5464 disordered the vacuolar degradation, which is important for heterologous ER resident protein expression.36 This might be the main reason that less than half of amounts of β-nootkatol and (+)-nootkatone (45.8 and 36%, respectively) were detected in BJ-32 than did in PK2-32. Whole genome sequencing of S. cerevisiae revealed that CEN·PK strains possessed significant higher abilities in galactose uptake and metabolism, and the great higher expression of HMG-CoA reductase (encoded by HMG1) compared to S288c.18
The storage stability of PK2-32 was also determined by incubating at −80 °C freezer. After 8 months incubation, almost 85% of (+)-valencene hydroxylation activities were still retained (Fig. 3B).
3.2 In-situ production of (+)-valencene from FPP
To synthesize (+)-valencene from FPP in S. cerevisiae strains, the open-reading frame of CnVS from Callitropsis nootkatensis was cloned under the control of TDH3 promoter, yielding p181-V1 and transformed into CEN·PK2-1Ca. After 2 days fermentation, 2.6 ± 0.2 mg L−1 cell culture of (+)-valencene was detected in 10 mL cultures overlaid with 20% (v/v) n-dodecane in conical flasks (Fig. 4). No (+)-valencene was detected from a CEN·PK2-1Ca culture containing an empty plasmid (data not shown). It was reported that the CnVS expressed under the control of inducible promoter PGAL1 in WAT11 only led to 1.36 mg L−1 (+)-valencene.37 The WAT11 strain was engineered with NADPH-cytochrome P450 reductase gene but not optimized for sesquiterpene production.38 As previously studied, the ergosterol biosynthetic pathway enrichment was detected in the popular production host of S. cerevisiae CEN·PK strains,18 which might supply higher FPP concentration in vivo and showed stronger capability in (+)-valencene biosynthesis.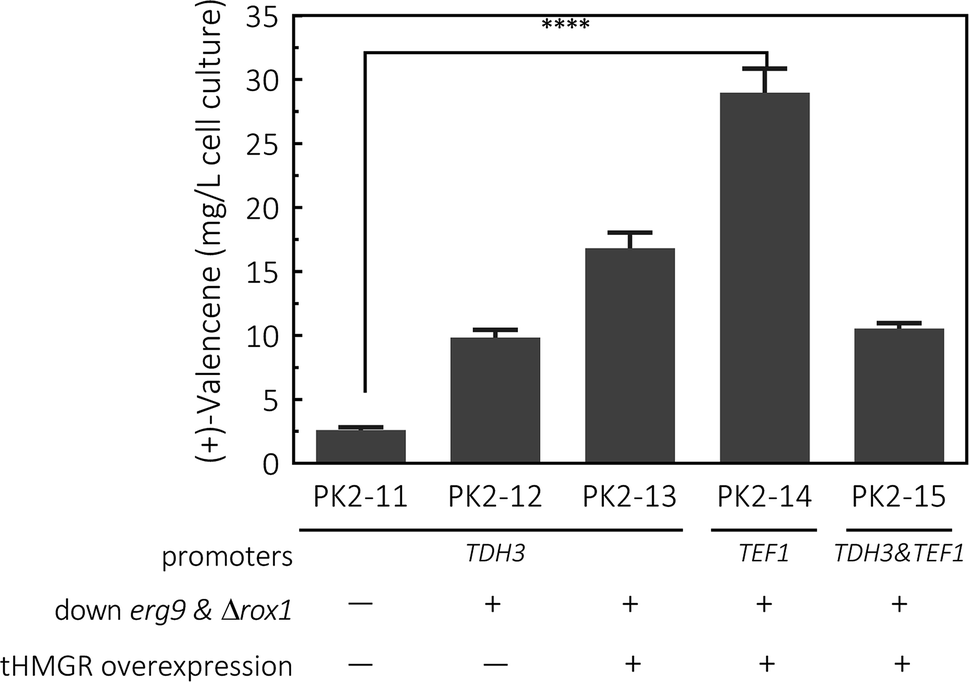 | ||
| Fig. 4 De novo production of (+)-valencene in different S. cerevisiae strains cultured in 50 mL flask cultures containing 10 mL containing 20% n-dodecane. The valencene synthase (CnVS) from C. nootkatensis was cloned under different promoters of TDH3 and TEF1. The platform was based on CEN·PK2-1Ca combining the suppression of erg9, knockout of the negative regulator rox1 and overexpression of truncated form of HMG1 gene (tHMGR). Error bars represent standard deviation from a triplicate analysis. The asterisk (****) indicates statistically significant differences in terpenoids formation (p < 0.0001, student's t-test). | ||
To enhance yields, down-regulation of ERG9 together with knockout of negative transcriptional regulator of ROX1 was applied to increase metabolic flux toward (+)-valencene synthesis, generating PK2-12. This strategy resulted in a 3.8-fold improvement in the (+)-valencene content compared to PK2-11. Then we overexpressed the truncated HMG1 gene (tHMGR), which is a major rate limiting enzyme in the MVA pathway,22,39–41 under the control of the TDH3 promoter and integrated the entire overexpression cassette into p181-V1. As expected, there was a further improvement in (+)-valencene production, reaching titer of 16.8 ± 1.2 mg L−1 yeast culture (PK2-13 in Fig. 4). Considering the toxic effects of (+)-valencene to S. cerevisiae,42 we placed the (+)-valencene synthase gene under a weaker endogenous promoter of TEF1 (PTEF1), yielding the recombinant yeast of PK2-14. Also, the expression vector p181-V1HV2 containing tHMGR and two CnVS (PTDH3 and PTEF1 controlled, respectively) expression cassettes, was also constructed and transformed into yeast to generate PK2-15. The (+)-valencene production was surprisingly enhanced to 28.9 ± 1.9 mg L−1 cell culture in PK2-14. PTDH3 and PTEF1 are the common-used constitutive promoters in the yeast S. cerevisiae. There is a general view that the promoter strength of PTDH3 is stronger than that of PTEF1 under different growth phases.43,44 However, the high-level expression of enzymes in some certain pathway might result in detrimental effects on cell growth.45 Actually, the final cell density of OD600 for PK2-14 was dramatically increased to 5.23 from 3.77 for PK2-13, indicating the product potential toxicity inhibited the cell growth and (+)-valencene synthesis.42 This might possibly explain the less productivity of cell factories of PK2-13. The significance decrease of 63.6% in (+)-valencene levels was observed when two CnVS expression cassettes existed in one cell (PK2-15) compared to PK2-14, which further demonstrated that the up-regulation of CnVS expression was not beneficial to the (+)-valencene accumulation. These results provide insights in promoter selection for practical application in sesquiterpenoids cell factory construction.
3.3 De novo production of (+)-valencene related terpenoids through culture condition optimization
Having enhanced the (+)-valencene accumulation through stepwise metabolic engineering approaches, the combination of (+)-valencene production from FPP and (+)-valencene oxidation to β-nootkatol and (+)-nootkatone was carried out. Co-expression of CnVS with HPO, AtCPR and ADH1 in the metabolic altered yeast CEN·PK2-1Ca mutant created a new yeast strain designated as PK2-24.Yeast can grow on a variety of carbon sources including fermentable and nonfermentable substances. Raffinose is a trisaccharide composed of galactose, glucose and fructose. Usually raffinose is added to minimal medium containing galactose for protein expression induction.27 As an organic compound, glycerol assimilation can occur in the presence of galactose in yeast. Earlier work indicated that the glycerol or raffinose addition might be favor to noscapine biosynthesis.6,46 We first explored the effects of galactose, raffinose and glycerol as carbon sources on the production of (+)-valencene related terpenoids from PK2-24. In 50 mL bi-phasic shake flask cultures, supplementation of raffinose or glycerol resulted in more efficient synthesis of (+)-valencene and led to statistically significant improvements with about 1.3-fold increase of (+)-valencene titers (Fig. 5A). However, an in-significant decrease in the β-nootkatol productivities was monitored in the case of raffinose or glycerol addition.
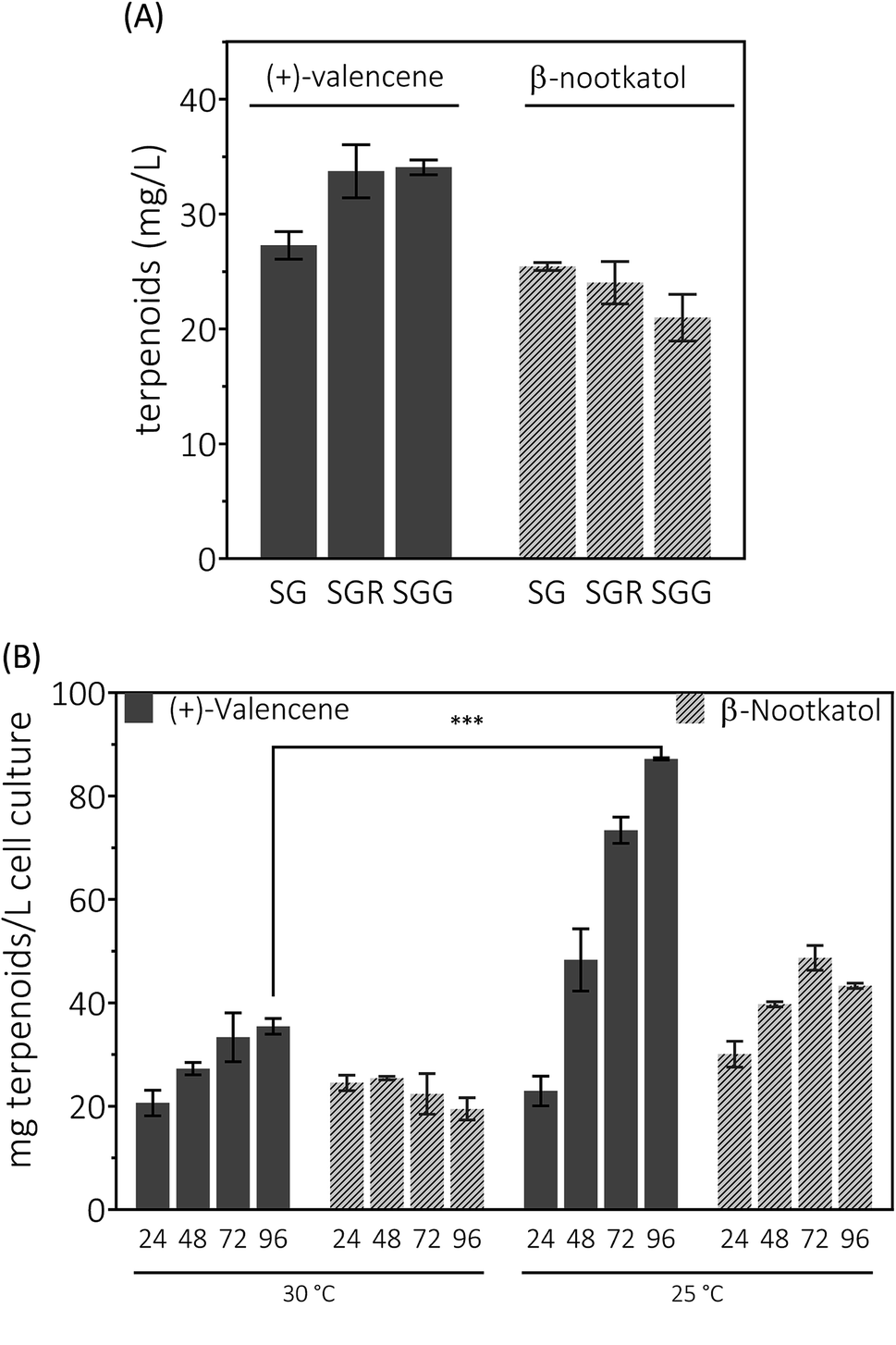 | ||
| Fig. 5 De novo production of (+)-valencene and β-nootkatol using PK2-24 in 50 mL bi-phasic conical flask cultures. One μL aliquots of n-dodecane phase for each sample were analyzed by GC-FID. (A) Carbon source in induction medium effects on the products accumulation. Yeasts were cultured in 20 g L−1 glucose at 30 °C for 14–16 h for cell growth. Cells were collected by centrifugation and transferred into fresh induction medium containing galactose to induce HPO and AtCPR expression, SG (2% galactose), SGG (2% galactose and 10% glycerol) and SGR (2% galactose and 0.7 raffinose), respectively. (B) Time and temperature responses of product accumulation. 24, 48, 72 and 96 represent the cultivation time in hours. Mean values and standard deviations of biological triplicates are shown. The asterisk (***) indicates statistically significant differences in (+)-valencene formation (p < 0.001, student's t-test). | ||
We continued to investigate the roles of temperature and incubation time played in (+)-valencene accumulation in SG medium (Fig. 5B). As previous culturing conditions, recombinant cells were cultured at 30 °C. The (+)-valencene levels were found to increase up to about 1.7-fold over 96 h fermentation time. The β-nootkatol titers in the n-dodecane layer remained generally constant at about 22–24 mg L−1 cell culture. However, when PK2-24 cells shifted to 25 °C, tremendous improvements in (+)-valencene levels was observed as culture time elongation (from 22.9 ± 2.8 mg L−1 at 24 h to 87.2 ± 0.2 mg L−1 at 96 h). A relatively moderate increase in β-nootkatol titers was observed in 0–72 h range, followed by a slight decrease in the final 24 h cultivation. Lower incubation temperature and longer cultivation time resulted in an increase of the final (+)-valencene and β-nootkatol from 20.6 ± 2.5 and 24.5 ± 1.5 up to 87.2 ± 0.2 and 43.3 ± 0.5 mg L−1 cell culture, respectively.
Generally speaking, the (+)-valencene contents in n-dodecane layer were improved dramatically, and the β-nootkatol concentration increased by a moderate amount through culture condition optimization. At the end of fermentation, even decreased β-nootkatol levels were monitored under many culture conditions. The results indicated the activity of HPO and AtCPR was not sufficient to transform the in situ produced (+)-valencene into β-nootkatol. As many cytochrome P450 enzymes, the instability is a major hurdle for the (+)-valencene hydroxylation for industrial processes. Wriessnegger et al.30 reported that reduced level of HPO and CPR enzymes were observed with extended cultivation times. Efforts must be made to identify or design proteins with improved stability.47 The more disappointing part is that no (+)-nootkatone was detected in the strain PK2-24. The reasons for this might be the accumulation of (+)-nootkatone in yeast endomembranes.42
3.4 Improve the level of β-nootkatol by promoter optimization
Expression of HPO and CPR from the GAL1 promoter was subjected to galactose induction and glucose inhibition. Considering the facts that (1) the preferred carbon sources for yeast are glucose and fructose, (2) the inducer galactose has a relatively high cost, and (3) glucose–galactose transitions would cause major metabolic changes. In order to develop a glucose-based production system, we changed the promoter of HPO and AtCPR from GAL1 to HXT7, encoding a hexose transporter. Great improvements of β-nootkatol and (+)-nootkatone production were observed as 244.3 ± 19.3 and 48.4 ± 0.9 mg L−1 in PK2-34 (Fig. 6A). In consideration of the possible negative effects on genes expression by using the same promoters and terminators for two genes in one plasmid, AtCPR was put under another commonly used promoter (CYC1) in S. cerevisiae, yielding the plasmid p352-HC3. As expectations, the titers of β-nootkatol and (+)-nootkatone were improved to 327.9 ± 10.7 mg L−1 and 53.7 ± 5.1 mg L−1 in PK2-35 (Table 1). The total oxidation efficiency of (+)-valencene to terpenoids by PK2-35 reached as 87.0% (mol mol−1), which was increased by 3.1-fold compared to PK2-31. This suggested that expressing genes with different promoters was better than that with the same promoters.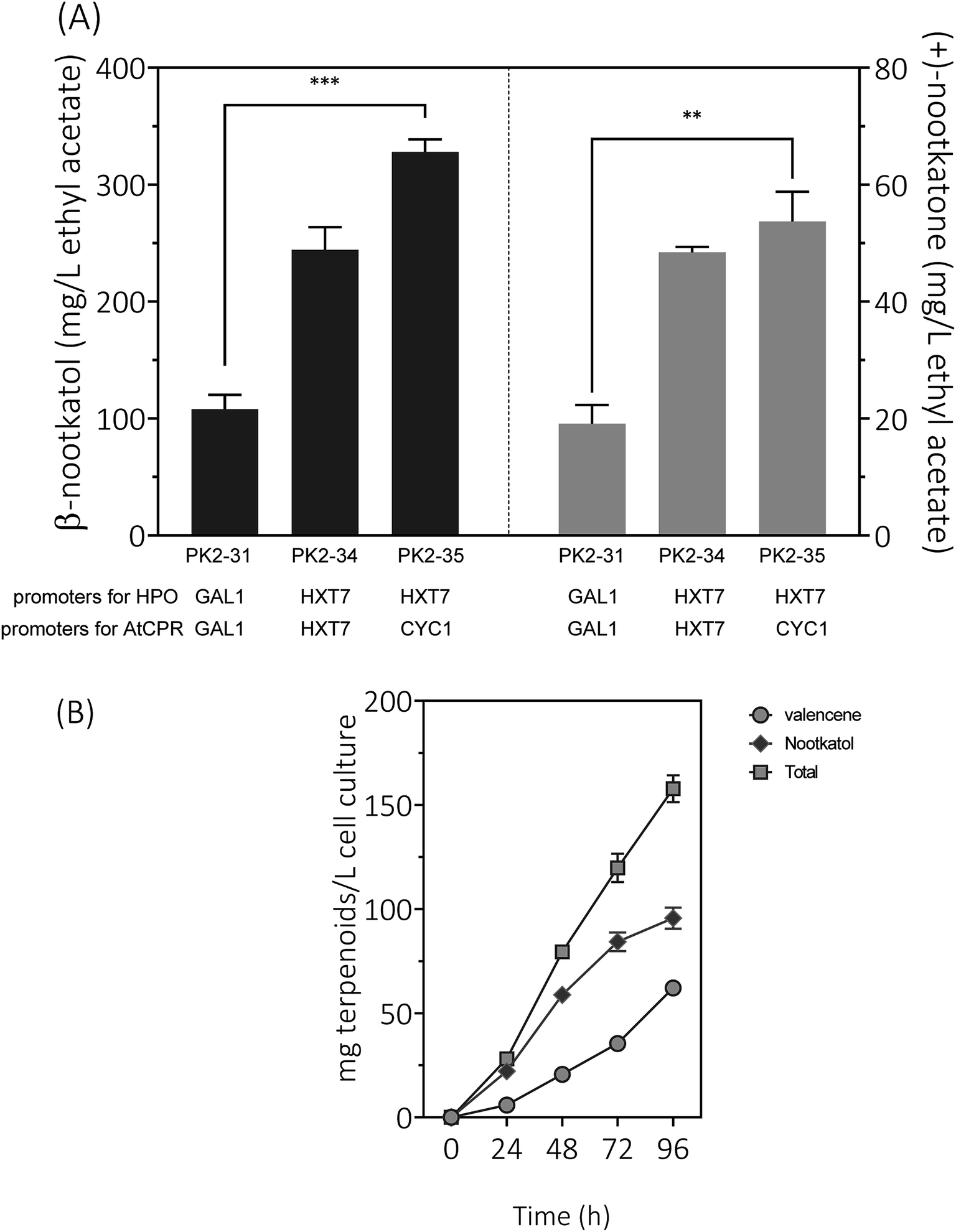 | ||
| Fig. 6 (A) Resting cell assays of (+)-valencene hydroxylation to β-nootkanol and (+)-nootkatone using different constructs in S. cerevisiae CEN·PK2-1Ca. Promoters of HPO and AtCPR were noted at the bottom of the diagram. (B) Time responses of product accumulation. 24, 48, 72 and 96 represent the cultivation time in hours with PK2-25 harboring p181-V2H and p352-A1HC3. Mean values and standard deviations of biological triplicates are shown. The asterisk (***) indicates statistically significant differences in β-nootkanol formation (p < 0.001, student's t-test). The asterisk (**) indicates statistically significant differences in (+)-nootkatone formation (p < 0.05, student's t-test). | ||
| Host | (+)-Valencene addition (mM) | β-Nootkatol (mg L−1) | (+)-Nootkatone (mg L−1) | β-Nootkatol plus (+)-nootkatone (mg L−1) | Conditions | Reference |
|---|---|---|---|---|---|---|
| a Calculated from figures in the literature. | ||||||
| S. cerevisiae WAT11 | 0.4 | 5 | 3 | 8 | 24 h at 28 °C | 42 |
| 1 | — | — | 6a | |||
| 2 | — | — | 4.5a | |||
| P. pastoris | 2 | 0 | 40a | 40 | 300 OD600 per mL cells, 12 h at 28 °C | 30 |
| S. cerevisiae W303 | 4 | 560 | 25a | 585 | 300 OD600 per mL cells, 16 h at 28 °C | 27 |
| S. cerevisiae CEN·PK2-1Ca | 2 | 327.8 | 53.7 | 381.5 | 150 OD600 per mL cells, 18 h at 30 °C | PK2-35 this study |
Inspired by the prominent effects in resting cell assays, we tried to produce (+)-valencene and its related terpenoids by the recombinant yeast PK2-25 containing p181-V2H and p352-A1HC3 through using simple carbon source glucose, which was much cheaper than galactose. As previous optimized culture conditions, the strain PK2-25 was cultured at 25 °C in SD medium. As expected, the yield of β-nootkatol was increased to 95.7 ± 5.1 mg L−1 over 96 h, which was nearly 2-fold as compared to PK2-24 (Fig. 6B). At the same time, the production of (+)-valencene was 62.1 ± 1.4 mg L−1. It was obvious that β-nootkatol accumulated rapidly during 0–72 h (1.17 mg L−1 h−1) and increased slowly in last 24 h (0.47 mg L−1 h−1). Conversely, the accumulation of (+)-valencene was slow during 0–72 h (0.49 mg L−1 h−1) and fast in last 24 h (1.11 mg L−1 h−1). This indicated that the expression level of HPO and AtCPR were improved by using the constitutive promoters, not the inducible promoters adopted in PK2-24. The total amount of terpenoids was up to 157.8 ± 6.4 mg L−1 cell culture, which was 29.6 mg per L per OD600. It was much higher than that of P. pastoris (0.86 mg per L per OD600) (Table 2).
| Host | Overexpressed enzymes | (+)-Valencene (mg L−1) | β-Nootkatol (mg L−1) | (+)-Nootkatone (mg L−1) | Total terpenoids (mg L−1) | Terpenoids (mg per L per OD600) | Reference |
|---|---|---|---|---|---|---|---|
| a Different sources of valencene oxidase were overexpressed in corresponding hosts. b Different sources of cytochrome P450 reductase (CPR) were overexpressed in corresponding hosts. c Calculated from figures in the literature. | |||||||
| S. cerevisiae WAT11 | VSa | 1.36 | — | — | 1.36 | — | 48 |
| VSa, P450 & CPRb | 0.15 | 0.92 | 0.04 | 1.11 | — | ||
| R. sphaeroides | VSa | 57.5 | — | — | 57.5 | — | 37 |
| VSa, MVA operon | 352 | — | — | 352 | — | ||
| S. cerevisiae W303 | VSa, P450 & CPRb, tHMG1, ICE2 | 1c | 30 | — | 31 | — | 27 |
| S. cerevisiae WAT11 | VSa, P450 | 1.305 | 0.116 | 0.144 | 1.565 | — | 29 |
| P. pastoris | VSa, P450/CPRb, RAD52 | 9c | 94 | 3c | 106 | — | 49 |
| P. pastoris | VSa | 51 | — | — | 51 | — | 30 |
| VSa, P450/CPRb | 2 | 8 | 0.3 | 10.3 | — | ||
| VSa, P450/CPRb, ADH1, tHMG1 | 7 | 18 | 17 | 42 | 0.86c | ||
| Y. lipolytica | VSa, P450/CPRb tHMG1, ERG20 | 22.8 | — | 0.98 | 23.78 | — | 50 |
| S. cerevisiae CEN·PK2-1Ca | VSa, P450/CPRb, ADH1, tHMG1, ΔROX1, down-regulated ERG9 | 62 | 96 | — | 158 | 29.64 | PK2-25 this study |
4. Conclusions
A (+)-valencene related sesquiterpenoids biosynthesis pathway in S. cerevisiae was successfully constructed and optimized. Finally we were able to increase the synthesis of β-nootkatol by 91-fold (from 3.6 to 327.9 mg L−1) and (+)-nootkatone by 62-fold (from 0.87 to 53.7 mg L−1) through resting cell transformation. In vivo, the level of total terpenoids was improved by 56-fold through combining the suppression of ERG9, knockout of the negative regulator ROX1 and overexpression of truncated form of HMG1 gene (tHMGR) etc. The results demonstrated the important influences of key enzymes, expression cassettes, hosts and culture temperature on sesquiterpenes production in S. cerevisiae, which could also be adapted for the bioproduction of other terpenoids and chemicals.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was financially supported by the Natural Science Foundation of China (Grant 21878104 and 31800063, U1701243), the Science and Technology Planning Project of Guangdong Province, China (Grant 2017A010105019) and the Project on the Integration of Industry, Education, and Research of Guangzhou, China (Grant 201704020183 and 201903010086).References
- M. Furusawa, T. Hashimoto, Y. Noma and Y. Asakawa, Chem. Pharm. Bull., 2006, 53, 1513–1514 CrossRef PubMed.
- H. G. Haring, F. Rijkens, H. Boelens and A. Van der Gen, J. Agric. Food Chem., 1972, 20, 1018–1021 CrossRef CAS.
- FEEDAP, EFSA J., 2016, 14, 4475 Search PubMed.
- B. C. R. Zhu, G. Henderson, A. M. Sauer, Y. Yu, W. Crowe and R. A. Laine, J. Chem. Ecol., 2003, 29, 2695–2701 CrossRef CAS.
- A. Nemmar, S. Al-Salam, S. Beegam, P. Yuvaraju, N. Hamadi and B. H. Ali, Nutrients, 2018, 10, 263 CrossRef.
- S. L. Yanran Li, K. Thodey, I. Trenchard, A. Cravens and C. D. Smolke, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 3922–3931 CrossRef PubMed.
- B. Hong, R. Lebeuf, S. Delbaere, P. Alsters and V. Nardello-Rataj, Catalysts, 2016, 6, 184–195 CrossRef.
- D. M. Palmerín-Carreño, O. M. Rutiaga-Quiñones, J. R. Verde Calvo, A. Prado-Barragán and S. Huerta-Ochoa, LWT–Food Sci. Technol., 2015, 64, 788–793 CrossRef.
- T.-D. Nguyen, G. MacNevin and D.-K. Ro, De novo synthesis of high-value plant sesquiterpenoids in yeast, in Methods in enzymology, ed. D. A. Hopwood, Academic Press, 2012, vol. 517, pp. 261–278 Search PubMed.
- J. Lian, S. Mishra and H. Zhao, Metab. Eng., 2018, 50, 85–108 CrossRef CAS.
- C. M. Humphreys and N. P. Minton, Curr. Opin. Biotechnol., 2018, 50, 174–181 CrossRef CAS.
- A. Krivoruchko and J. Nielsen, Curr. Opin. Biotechnol., 2015, 35, 7–15 CrossRef CAS.
- Y. Zhang, J. Nielsen and Z. Liu, FEMS Yeast Res., 2017, 17, fox080 CrossRef.
- S. Ostergaard, L. Olsson and J. Nielsen, Microbiol. Mol. Biol. Rev., 2000, 64, 34–50 CrossRef CAS.
- Q. Yan and S. S. Fong, Curr. Opin. Biotechnol., 2018, 53, 254–263 CrossRef CAS PubMed.
- E. M. Paradise, J. Kirby, R. Chan and J. D. Keasling, Biotechnol. Bioeng., 2008, 100, 371–378 CrossRef CAS.
- J. Hong, S. H. Park, S. Kim, S. W. Kim and J. S. Hahn, Appl. Microbiol. Biotechnol., 2019, 103, 211–223 CrossRef CAS PubMed.
- J. M. Otero, W. Vongsangnak, M. A. Asadollahi, R. Olivares-Hernandes, J. Maury, L. Farinelli, L. Barlocher, M. Osterås, M. Schalk, A. Clark and J. Nielsen, BMC Genomics, 2010, 11, 723 CrossRef.
- A. Matthew, R. B. Kennedy and M. Bard, Biochim. Biophys. Acta, 1999, 1445, 110–122 Search PubMed.
- K. W. N. Henry, J. T. Edlind and D. Thomas, Eukaryotic Cell, 2002, 1, 1041–1044 CrossRef CAS.
- D. C. Amberg, D. J. Burke, D. Burke and J. N. Strathern, Methods in yeast genetics: a cold spring harbor laboratory course manual, Cold Spring Harbor Laboratory Press, 2005 Search PubMed.
- W. Xie, L. Ye, X. Lv, H. Xu and H. Yu, Metab. Eng., 2015, 28, 8–18 CrossRef CAS.
- R. P. Shetty, D. Endy and T. F. Knight Jr, J. Biol. Eng., 2008, 2, 5 CrossRef.
- L. Zelcbuch, N. Antonovsky, A. Bar-Even, A. Levin-Karp, U. Barenholz, M. Dayagi, W. Liebermeister, A. Flamholz, E. Noor, S. Amram, A. Brandis, T. Bareia, I. Yofe, H. Jubran and R. Milo, Nucleic Acids Res., 2013, 41, e98 CrossRef CAS.
- J. DiCarlo, J. Norville, P. Mali, X. Rios, J. Aach and G. Church, Nucleic Acids Res., 2013, 41, 4336–4343 CrossRef CAS.
- M. Girhard, K. Machida, M. Itoh, R. Schmid, A. Arisawa and V. Urlacher, Microb. Cell Fact., 2009, 8, 36–48 CrossRef.
- A. Emmerstorfer, M. Wimmer-Teubenbacher, T. Wriessnegger, E. Leitner, M. Muller, I. Kaluzna, M. Schurmann, D. Mink, G. Zellnig, H. Schwab and H. Pichler, Biotechnol. J., 2015, 10, 623–635 CrossRef CAS.
- S. Takahashi, Y.-S. Yeo, Y. Zhao, P. E. O'Maille, B. T. Greenhagen, J. P. Noel, R. M. Coates and J. Chappell, J. Biol. Chem., 2007, 282, 31744–31754 CrossRef CAS PubMed.
- K. Cankar, A. van Houwelingen, M. Goedbloed, R. Renirie, R. M. de Jong, H. Bouwmeester, D. Bosch, T. Sonke and J. Beekwilder, FEBS Lett., 2014, 588, 1001–1007 CrossRef CAS.
- T. Wriessnegger, P. Augustin, M. Engleder, E. Leitner, M. Müller, I. Kaluzna, M. Schürmann, D. Mink, G. Zellnig, H. Schwab and H. Pichler, Metab. Eng., 2014, 24, 18–29 CrossRef CAS.
- K. van Eunen and B. M. Bakker, Perspectives in Science, 2014, 1, 126–130 CrossRef.
- O. De Smidt, J. du Preez and J. Albertyn, FEMS Yeast Res., 2008, 8, 967–978 CrossRef CAS.
- O. De Smidt, J. du Preez and J. Albertyn, FEMS Yeast Res., 2012, 12, 33–47 CrossRef CAS.
- B. M. Bakker, C. Bro, P. Kötter, M. A. Luttik, J. P. van Dijken and J. T. Pronk, J. Bacteriol., 2000, 182, 4730–4737 CrossRef CAS.
- C. N. Marjohn, S. Jae-Young, N. Franz Marielle, K. Me-Sun, J. Yu Jin, K. Kwon-Kyoo, N. Illsup and C. Yong-Gu, J. Plant Biotechnol., 2016, 43, 422–431 CrossRef.
- C. L. Young, D. L. Raden and A. S. Robinson, Traffic, 2013, 14, 365–381 CrossRef CAS.
- J. Beekwilder, A. van Houwelingen, K. Cankar, A. D. van Dijk, R. M. de Jong, G. Stoopen, H. Bouwmeester, J. Achkar, T. Sonke and D. Bosch, Plant Biotechnol. J., 2014, 12, 174–182 CrossRef CAS PubMed.
- P. Urban, C. Mignotte, M. Kazmaier, F. Delorme and D. Pompon, J. Biol. Chem., 1997, 272, 19176–19186 CrossRef CAS.
- M. A. Asadollahi, J. Maury, M. Schalk, A. Clark and J. Nielsen, Biotechnol. Bioeng., 2010, 106, 86–96 CAS.
- M. Z. Ding, H. F. Yan, L. F. Li, F. Zhai, L. Q. Shang, Z. Yin and Y. J. Yuan, PLoS One, 2014, 9, e109348 CrossRef.
- Q. Li, Z. Sun, J. Li and Y. Zhang, FEMS Microbiol. Lett., 2013, 345, 94–101 CrossRef CAS.
- C. Gavira, R. Hofer, A. Lesot, F. Lambert, J. Zucca and D. Werck-Reichhart, Metab. Eng., 2013, 18, 25–35 CrossRef CAS.
- L. Xiong, Y. Zeng, R.-Q. Tang, H. S. Alper, F.-W. Bai and X.-Q. Zhao, Microb. Cell Fact., 2018, 17, 58 CrossRef PubMed.
- S. Partow, V. Siewers, S. Bjørn, J. Nielsen and J. Maury, Yeast, 2010, 27, 955–964 CrossRef CAS PubMed.
- T. C. Williams, L. K. Nielsen and C. E. Vickers, ACS Synth. Biol., 2013, 2, 136–149 CrossRef CAS.
- N. Guaragnella, M. Zdralevic, P. Lattanzio, D. Marzulli, T. Pracheil, Z. Liu, S. Passarella, E. Marra and S. Giannattasio, Biochimica et Biophysica Acta, 2013, 1833, 2765–2774 CrossRef CAS PubMed.
- R. Nelson David, Philos. Trans. R. Soc., B, 2013, 368, 201204–201230 Search PubMed.
- K. Cankar, A. v. Houwelingen, D. Bosch, T. Sonke, H. Bouwmeester and J. Beekwilder, FEBS Lett., 2011, 585, 178–182 CrossRef CAS.
- T. Wriessnegger, S. Moser, A. Emmerstorfer-Augustin, E. Leitner, M. Muller, I. Kaluzna, M. Schurmann, D. Mink and H. Pichler, Fungal Genet. Biol., 2016, 89, 114–125 CrossRef CAS.
- X. Guo, J. Sun, D. Li and W. Lu, Biochem. Eng. J., 2018, 137, 125–131 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra05558d |
| This journal is © The Royal Society of Chemistry 2019 |