
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTriphala inhibits alpha-synuclein fibrillization and their interaction study by NMR provides insights into the self-association of the protein†
Mandar Bopardikar a,
Anusri Bhattacharyab,
Veera Mohana Rao Kakitab,
Kavitha Rachinenib,
Lalit C. Bordec,
Sinjan Choudharyb,
Sri Rama Koti Ainavarapu*a and
Ramakrishna V. Hosur*ab
a,
Anusri Bhattacharyab,
Veera Mohana Rao Kakitab,
Kavitha Rachinenib,
Lalit C. Bordec,
Sinjan Choudharyb,
Sri Rama Koti Ainavarapu*a and
Ramakrishna V. Hosur*ab
aDepartment of Chemical Sciences, Tata Institute of Fundamental Research, Homi Bhabha Road, Colaba, Mumbai 400005, India. E-mail: koti@tifr.res.in; rvhosur53@gmail.com
bUM-DAE Centre for Excellence in Basic Sciences, University of Mumbai, Kalina Campus, Santacruz, Mumbai 400098, India
cDepartment of Biological Sciences, Tata Institute of Fundamental Research, Homi Bhabha Road, Colaba, Mumbai 400005, India
First published on 10th September 2019
Abstract
The process of assembly and accumulation of the intrinsically disordered protein (IDP), alpha-synuclein (αSyn) into amyloid fibrils is a pathogenic process leading to several neurodegenerative disorders such as Parkinson's disease, multiple system atrophy and others. Although several molecules are known to inhibit αSyn fibrillization, the mechanism of inhibition is just beginning to emerge. Here, we report the inhibition of fibrillization of αSyn by Triphala, a herbal preparation in the traditional Indian medical system of Ayurveda. Triphala was found to be a rich source of polyphenols which are known to act as amyloid inhibitors. ThT fluorescence and TEM studies showed that Triphala inhibited the fibrillization of αSyn. However, it was observed that Triphala does not disaggregate preformed αSyn fibrils. Further, native-PAGE showed that Triphala reduces the propensity of αSyn to oligomerize during the lag phase of fibrillization. Our NMR results showed that certain stretches of residues in the N-terminal and NAC regions of αSyn play an anchor role in the self-association process of the protein, thereby providing mechanistic insights into the early events during αSyn fibrillization.
Introduction
Alpha-synuclein (αSyn), a 140 amino acid intrinsically disordered protein (IDP),1 is expressed in human neuronal cells.2 The accumulation of this protein in the form of fibrillar aggregates, which are the major constituents of Lewy bodies (LB), in the cytoplasm of dopaminergic neurons, is a characteristic of neurodegenerative disorders like Parkinson's disease (PD), Lewy body dementia (LBD), multiple system atrophy (MSA).3,4 The duplication or triplication of the gene encoding αSyn (SNCA) is linked to familial early onset PD.5There is no preventive therapy available for PD as yet. However, small organic molecules capable of inhibiting αSyn fibrillization represent one of the potential therapeutic strategies for PD. In the past few years, several studies have reported the inhibition of αSyn fibrillization by small organic molecules.6–32 Interestingly, most of these molecules are polyphenols. In this context, we utilized a herbal preparation frequently used in the traditional medicinal system of India, commonly known as Ayurveda. The past couple of decades have witnessed an increase in the research on various herbal formulations prescribed by Ayurveda.33–37 The scientific validation of the pharmaceutical properties of these Ayurvedic formulations is beginning to emerge.33–37 Triphala, an Ayurvedic formulation containing naturally occurring polyphenols, is known to possess a wide range of medicinal properties such as free radical scavenging, antioxidant, anti-inflammatory, antimutagenic, antistress, hypoglycaemic and radioprotective which have been recently reviewed.38 Some of these properties of Triphala are believed to be due to its polyphenolic constituents.39,40 Recently, aqueous extract of Triphala was found to inhibit cancer cell proliferation for HeLa, PANC-1 and MDA-MB-231 cell lines.41 However, a detailed chemical characterization of Triphala has not yet been reported. Therefore, in the current investigation, we performed a thorough chemical characterization of aqueous extract of Triphala. Indeed, Triphala is found to be abundant in polyphenolic content. Further, we analyzed Triphala for its ability to inhibit αSyn fibrillization as well as disaggregate preformed αSyn fibrils in vitro. We find that Triphala inhibits αSyn fibrillization, while it has no effect on preformed αSyn fibrils.
Materials and methods
Preparation of aqueous extract of Triphala
Triphala capsules were purchased from the Himalaya Drug Company, India. 20% (w/v) Triphala powder was suspended in Milli-Q water. This partially soluble sample was then heated to 65 °C for 30 min. The sample was then centrifuged at 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm and the soluble fraction was lyophilized with the help of a Labconco Free Zone 2.5 lyophilizer, USA. Required quantities of the lyophilized powder were weighed and dissolved in phosphate buffer saline (PBS) (10 mM Na2HPO4, 1.8 mM KH2PO4, 137 mM NaCl, 2.7 mM KCl and 0.01% (w/v) sodium azide) pH 7.4. The pH of the Triphala solution was adjusted to 7.4 before utilizing the sample for experiments.
000 rpm and the soluble fraction was lyophilized with the help of a Labconco Free Zone 2.5 lyophilizer, USA. Required quantities of the lyophilized powder were weighed and dissolved in phosphate buffer saline (PBS) (10 mM Na2HPO4, 1.8 mM KH2PO4, 137 mM NaCl, 2.7 mM KCl and 0.01% (w/v) sodium azide) pH 7.4. The pH of the Triphala solution was adjusted to 7.4 before utilizing the sample for experiments.
Protein overexpression and purification
αSyn gene contained in the vector pT7-7 baring ampicillin resistance was expressed in Escherichia coli BL21 (DE3) cells. 10 ml of LB containing 100 μg ml−1 of freshly prepared ampicillin was inoculated with the glycerol stock of over-expressed αSyn and grown overnight at 37 °C with shaking (180 rpm). 10 ml (1% inoculum) of the overnight culture was then transferred to 1 litre of LB or M9 culture containing 100 μg ml−1 ampicillin and allowed to grow till an OD600 of 0.8–1.0. Subsequently, the culture was induced with 1 mM IPTG for 6–10 h. Finally, the culture was centrifuged to obtain a cell pellet which was stored at −80 °C. αSyn was purified by a previously reported non-chromatographic method.42,43 Briefly, frozen cell pellets were resuspended in Tris buffer (50 mM Tris, 10 mM EDTA and 150 mM NaCl) pH 8 and sonicated for 10 min. The suspension was placed in a boiling water-bath for 20 min and then centrifuged for 10 min. The supernatant was collected into a fresh tube and streptomycin sulfate (136 μl of 10% solution/ml of supernatant) and glacial acetic acid (228 μl ml−1 of supernatant) were added followed by centrifugation for 10 min. Again, the supernatant was collected and then precipitated with saturated ammonium sulfate which was added as 1:1 (v/v) to supernatant. Precipitated protein was washed with ammonium sulfate solution (1:1 (v/v) saturated ammonium sulfate and water). The pellet was resuspended in 3 ml of 100 mM ammonium acetate and precipitated with addition of an equal volume of ethanol. Resuspension in ammonium acetate and precipitation in ethanol was repeated twice more. Finally, the pellet was resuspended in 100 mM ammonium acetate, followed by freezing in liquid nitrogen and lyophilization. Pure αSyn was obtained as confirmed by mass spectrometry (Fig. S1†).
αSyn amyloid fibril formation
The fibrillization reaction was commenced with 150 μM small oligomeric αSyn in 2 ml Protein LoBind Eppendorf tube in PBS pH 7.4. This protein sample either in the presence or absence of various concentrations of Triphala was subjected to mechanical agitation at 65 rpm and 37 °C. 5 μl aliquots were withdrawn from the fibrillating αSyn sample at various intervals of time. Each aliquot was uniformly mixed with 245 μl of 25 μM ThT solution in PBS pH 7.4 and fluorescence was immediately recorded with the help of an Agilent spectrofluorometer with excitation at 444 nm and emission from 455 to 500 nm. Both the excitation and emission slit widths were 5 nm for all the measurements. The fluorescence intensity at 485 nm was plotted against time.Transmission electron microscopy (TEM)
αSyn samples at the end of fibrillization (198 h) were diluted to 30 μM in distilled water. These were spotted onto a carbon-coated Formvar grid, incubated for 5 min, stained with a freshly prepared 1% (w/v) aqueous solution of uranyl acetate filtered through 0.22 μm syringe filter by incubating for 1 min and then finally washed with distilled water. The samples were then imaged on a Libra 120EFTEM electron microscope from Carl Zeiss (Germany).Circular dichroism (CD) spectroscopy
In order to monitor the changes in secondary structural features during αSyn fibrillization, far UV CD spectra were recorded at different time intervals. All the spectra were recorded on a Jasco J-1500 spectropolarimeter. The spectra were acquired in the wavelength range of 200–250 nm. Six scans were obtained and averaged to produce each spectrum. Each spectrum was baseline corrected by subtracting the buffer or Triphala signal.Nuclear magnetic resonance (NMR) spectroscopy
All the experiments were performed on a 800 MHz Bruker Avance NMR spectrometer. 1H–15N HSQC spectra were recorded for 150 μM 15N isotopically enriched αSyn in PBS pH 7.4 in the presence and absence of 0.75 mg ml−1 Triphala. The 2D data was collected with 256 complex data points along the indirect dimension. Both the samples were incubated overnight at 37 °C under fibrillating condition prior to HSQC experiment. The temperature for the experiments was 25 °C. Spectra were recorded in phase-sensitive mode and quadrature detection was performed with the help of States-TPPI method.44 Data was processed with the help of Topspin 3.5 (Bruker, USA) and analysed with CARA (Computer-Aided Resonance Assignment, Institute for Molecular Biology and Biophysics, Zurich, Switzerland). Chemical shift assignments were obtained by transfer from BMRB (accession number 16300) and other previous reports.45,46 Chemical shift perturbations (CSP) were calculated as (δH2 + 0.1(δN2))1/2,47 where δN and δH represent the difference in the nitrogen and proton chemical shifts of αSyn in the presence of Triphala with respect to that of free αSyn, respectively.Native-polyacrylamide gel electrophoresis (native-PAGE)
αSyn (100 μM) was incubated overnight with or without 0.75 mg ml−1 Triphala at 37 °C under agitation. After the incubation period, 12 μl aliquots were withdrawn from each of the samples, thoroughly mixed with 3 μl of staining dye (containing no SDS) and loaded on to an 11% polyacrylamide gel (prepared without the addition of SDS). Electrophoresis was carried out at 90 V. Various species which were resolved on the gel, were then visualized by Coomassie staining.Results and discussion
Triphala is rich in polyphenols
Extensive chemical characterization of aq. Triphala extract (referred to as Triphala from here onwards) was performed with the help of LC-MS. A detailed list of the chemical constituents that were detected is provided, including the polyphenols (Table S1†) and other constituents (Table S2†). Here it is worth noting that quercetin is one of the constituents of Triphala, which has been previously reported to inhibit αSyn fibrillization.7,11 However, in 0.5 mg ml−1 Triphala, the concentration of quercetin comes out to be ≈5 μM which is far less than what would be required for inhibiting the aggregation of 150 μM αSyn.7,11Triphala inhibits αSyn fibrillization
The process of fibrillization of αSyn was monitored in the presence of varying concentrations of Triphala with the help of thioflavin T (ThT) binding assay48–50 in a time-dependent manner. αSyn fibrillization when probed by ThT fluorescence shows a sigmoidal curve which is characterized by three different phases namely, nucleation, elongation and saturation phase.51 It was observed that ThT fluorescence systematically decreased with an increase in the concentration of Triphala (Fig. 1A). Particularly, in the saturation phase of the αSyn fibrillization process, the ThT fluorescence intensity decreased consistently with an increase in the concentration of Triphala (Fig. S2†). In order to study if the process of fibril formation initiated at a time point beyond the duration shown in (Fig. 1A), ThT fluorescence was collected up to 330 h (Fig. S3†). However, no increase in ThT fluorescence was observed for αSyn in the presence of 0.5 mg ml−1 Triphala (Fig. S3†). It appeared that 0.5 mg ml−1 Triphala was capable of completely inhibiting αSyn fibrillization (Fig. 1A). However, there is a possibility that Triphala might interfere with ThT fluorescence. Therefore, transmission electron microscopy (TEM) studies were performed. It was observed that 0.5 mg ml−1 Triphala almost completely prohibited the formation of αSyn fibrils for 150 μM of the protein (Fig. 1B and C). Since a TEM image shows only a small region on the grid, therefore different regions on each grid were imaged (Fig. S4†). This comparison clearly shows the inhibitory effect of 0.5 mg ml−1 Triphala on the fibrillization of 150 μM αSyn (Fig. S4†).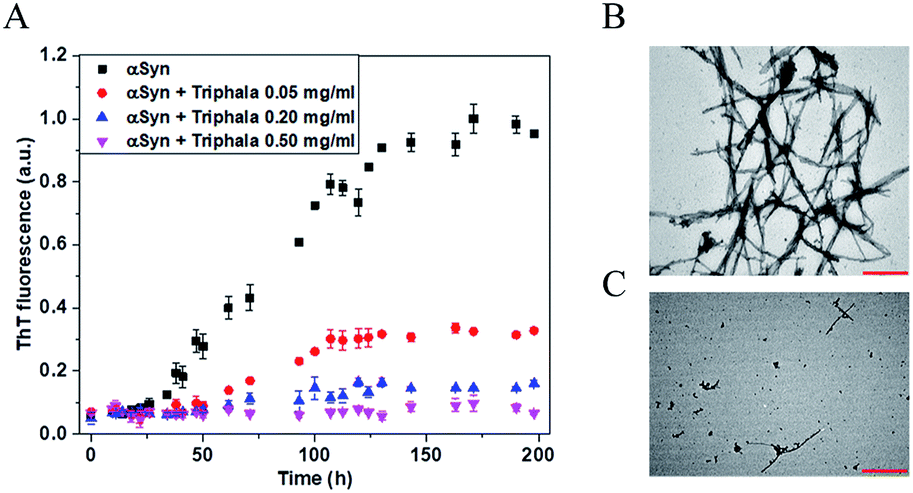 | ||
| Fig. 1 Effect of Triphala on the fibrillization of αSyn. (A) The time-course of the fibrillization of 150 μM αSyn in the presence of different concentrations of Triphala monitored by ThT fluorescence. ThT fluorescence saturates to different levels in a Triphala concentration-dependent manner. Each data point was an average of two independent measurements. The error bars are SD. (B and C) Morphological characterization of the effect of Triphala on αSyn. TEM images of αSyn at the end of fibrillization reaction in the absence (B) and presence (C) of 0.5 mg ml−1 Triphala. The scale bar is 500 nm in panels (B) and (C). | ||
Triphala prevents ‘random coil to β-sheet’ structural transition essential for αSyn fibrillization
In order to investigate the effect of Triphala on the secondary structure of αSyn, far UV circular dichroism (CD) spectra were recorded for αSyn under fibrillating conditions in a time-dependent manner, both in the presence and the absence of 0.75 mg ml−1 Triphala (Fig. 2). Initially, αSyn was observed to be largely random coil (≈58%) as calculated by BESTSEL deconvolution algorithm (Fig. 2A and Table S3†).52,53 However, minor anti-parallel β-sheet content (≈24%) was also observed (Table S3†). This observation of a major random coil along with a minor anti-parallel β-sheet conformation of αSyn is consistent with earlier reports.54,55 A clear structural transition from random coil to β-sheet was observed for αSyn after 37 hours of incubation under fibrillating condition (Fig. 2A). At this point in time, it was found that αSyn developed minor parallel β-sheet content which steadily increased with the passage of time (Table S3†). In this context, our results are in agreement with previous findings by Chakraborty et al.25 and Celej et al.,56 where it was reported that αSyn fibrillization is characterized by ‘random coil → anti-parallel β-sheet → parallel β-sheet’ secondary structural changes, however, differ from the results obtained by Ghosh et al.,57 where it was observed that αSyn, forms fibrils via an α-helix rich intermediate. On the other hand, in the presence of 0.75 mg ml−1 Triphala, αSyn did not show any structural change even up to 22 days of incubation and remained largely random coil (Fig. 2B and Table S3†). This suggests that the presence of Triphala causes stabilization of αSyn in its natively unfolded conformation.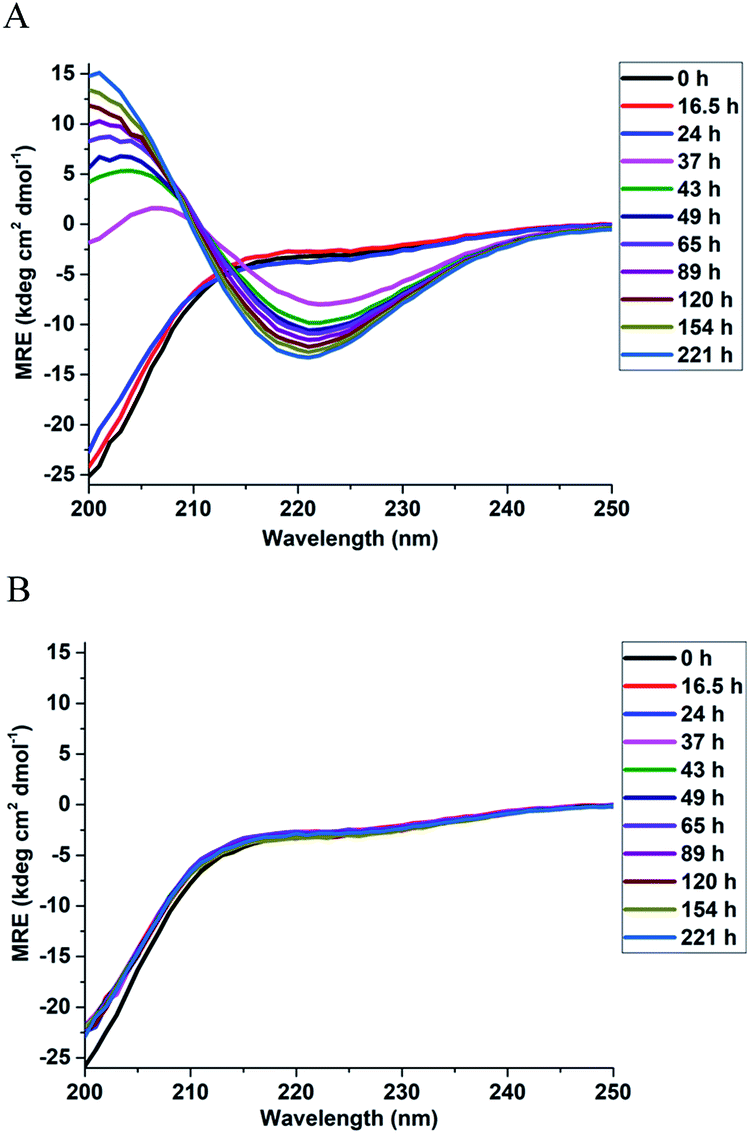 | ||
| Fig. 2 Effect of Triphala on the secondary structure of αSyn. The time evolution of the secondary structure of 150 μM αSyn during fibrillization in the absence (A) and presence (B) of 0.75 mg ml−1 Triphala studied using CD spectroscopy. | ||
Triphala influences the self-association of αSyn and reduces its oligomerization propensity
The observation that Triphala hinders the fibrillization of αSyn raises the question as to how and by what mechanism? In this respect, NMR and native-PAGE have provided valuable insight into the mechanism lying underneath. 1H–15N HSQC spectra (Fig. 3A) were recorded for αSyn in the presence and absence of 0.75 mg ml−1 of Triphala under fibrillating conditions in the lag phase (see Materials and methods). The individual spectra along with a spectrum recorded under identical conditions in the presence of 0.5 mg ml−1 Triphala (Fig. S5†) show a systematic change in the αSyn HSQC spectrum with increase in Triphala concentration. A comparative analysis of the spectra (shown in Fig. 3A) revealed interesting features: (i) absence of several peaks in the αSyn spectrum (in red) which appeared in the presence of Triphala (spectrum in blue). (ii) The residues for which peaks have vanished in the αSyn spectrum belong to the non-amyloid-β component (NAC) and N-terminal regions. A close inspection of these peaks reveals that they belong to sequential stretches of residues along the αSyn primary sequence: (a) T72, G73, T75 and A76; (b) T81, G84, G86 and S87; (c) T64, V66, G67, G68, A69; (d) A53, T54; (e) G41, K43, T44; (f) T92, G93; (g) G31, T33. (iii) It is to be noted that all these stretches contain primarily threonine and serine residues and their neighboring residues along the sequence. (iv) Chemical shift perturbation (CSP) measurements for the peaks which were present in both the spectra (Fig. 3B) showed insignificant peak shift due to the presence of Triphala. (v) The presence of Triphala caused an increase in the peak intensities for residues in the N-terminal and NAC regions (Fig. 3C). On the other hand, the intensities for C-terminal residues were found to be decreased in the presence of Triphala (Fig. 3C).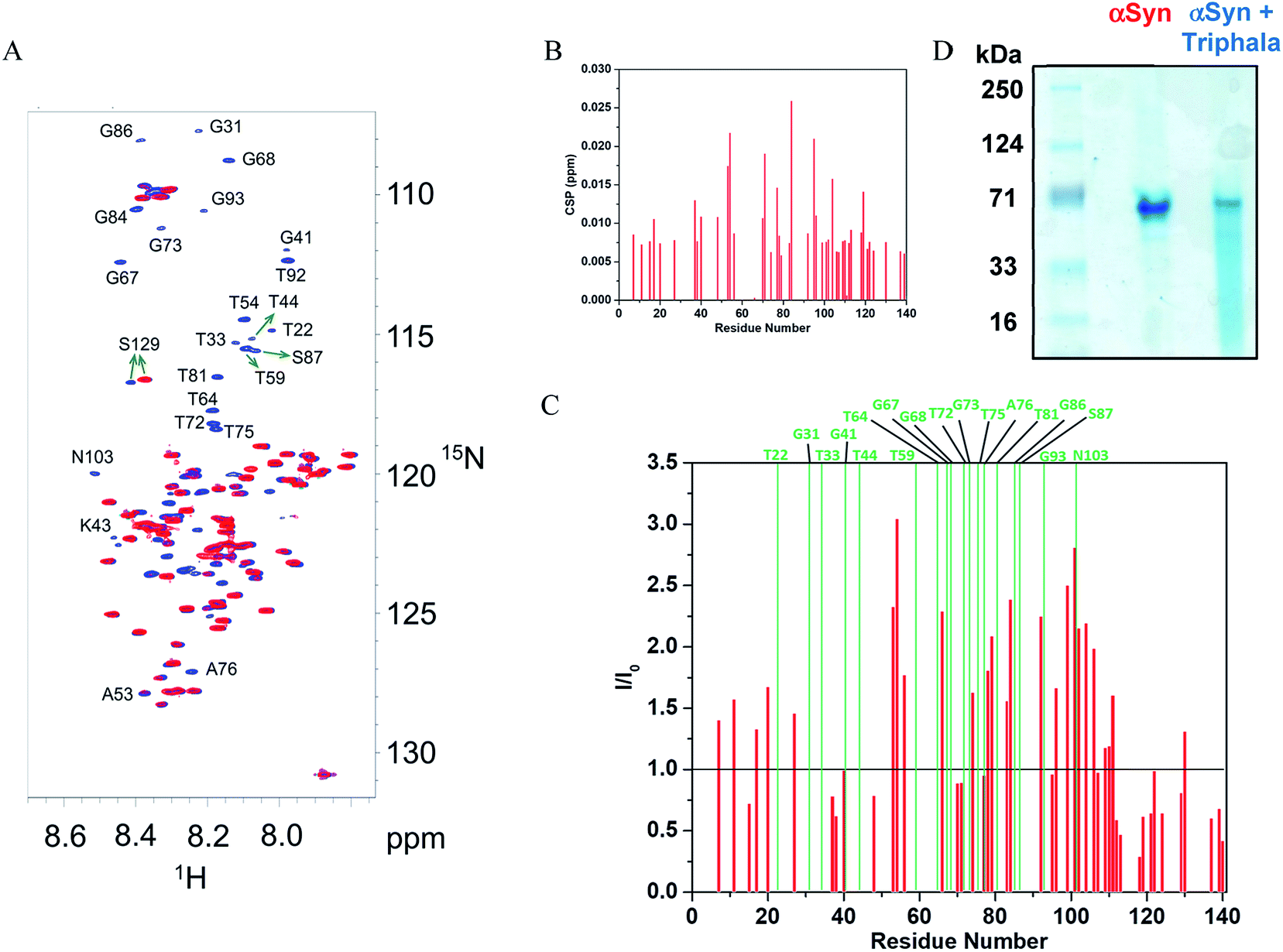 | ||
| Fig. 3 Mode of interaction of Triphala with αSyn and its effect on the early self-association of αSyn. (A) 1H–15N HSQC spectrum of αSyn in the presence (blue) and absence (red) of 0.75 mg ml−1 Triphala. The peaks were assigned by transfer of assignments from BMRB (accession number 16300) and other previous reports.45,46 (B) Chemical shift perturbation (CSP) for 150 μM αSyn observed in 1H–15N HSQC spectra in the presence of 0.75 mg ml−1 Triphala relative to free αSyn. (C) Ratios of the peak heights of 150 μM αSyn observed in 1H–15N HSQC spectra in the presence of 0.75 mg ml−1 Triphala relative to peak heights in αSyn 1H–15N HSQC spectrum in the absence of Triphala are shown as red bars. The green bars indicate residues which were absent in 1H–15N HSQC spectrum of αSyn. (D) Native-PAGE gel showing the oligomerization status of αSyn in both the absence as well as the presence of 0.75 mg ml−1 Triphala. | ||
It may be mentioned here that the absence of peaks cannot be attributed to chemical exchange between the backbone amide protons and water (solvent) for the following reasons: a backbone amide 1H resonance will be broadened due to exchange with water if kex ≈ |ΩNH − ΩH2O|,58 where kex = rate of exchange, ΩNH = amide 1H frequency and ΩH2O = water 1H frequency. For αSyn, ΩNH varies within the range of 8.7 to 7.7 (ppm) (Fig. 3A). Accordingly, average ΩNH turns out to be 8.2 (ppm). ΩH2O is equal to 4.7 (ppm). The difference turns out to be 3.5 (ppm), which for a basic 1H larmor frequency of 800 (MHz), equals 2800 (s−1). Now kex values for the residues in αSyn have been explicitly measured by Bhattacharyya et al.59 under similar conditions, and they turn out to be extremely slow (<8 s−1). Hence, it is clear that kex ≪ |ΩNH − ΩH2O|, and hence exchange with solvent cannot account for vanishing of peaks. Now it is known that αSyn exists in oligomeric form during the lag phase of fibrillization process.56,57,60–74 So then a question arises that is the disappearance of peaks for stretches of residues in the free αSyn HSQC spectrum (Fig. 3A) due to the oligomerization of the protein? If the answer to this question turns out to be yes, then the fact that these peaks appear in the presence of Triphala would mean Triphala is reducing the capability of the protein to oligomerize. In order to address the above question, native-PAGE was performed for αSyn samples in both the absence as well as presence of Triphala. Untreated αSyn was observed to migrate at ≈70 kDa (Fig. 3D). Monomeric αSyn has been previously shown to migrate at ≈16 kDa in native-PAGE experiments.67,75 This suggests that major αSyn population is an oligomer, possibly tetramer, under our experimental conditions. Whereas, in the presence of Triphala, a large smear was observed in the lower molecular mass region starting from the main oligomer band (≈70 kDa) right up to the expected monomer mass of ≈16 kDa (Fig. 3D). In order to check for the possibility of protein degradation due to Triphala, mass spectrum of αSyn was obtained both with and without Triphala (Fig. S6†). The spectra appear to be very similar which indicates that Triphala does not cause degradation of αSyn (Fig. S6†). The presence of a continuous smear rather than discrete bands (Fig. 3D) could be due to (i) the existence of equilibrium between the oligomers of different sizes and monomer (ii) the binding of a varying number of Triphala constituents to αSyn or (iii) the prevalence of both (i) and (ii). The native gel (Fig. 3D) clearly shows that treatment with Triphala causes a breakdown of oligomeric αSyn into smaller oligomers and monomer. These observations suggest that the disappearance of peaks in HSQC spectrum for untreated αSyn (Fig. 3A) is due to a major oligomer population, where the peaks come from the locally flexible residues. Whereas, the peaks for the stretches of residues in the N-terminal and NAC regions (mentioned above) which participate in the formation of the monomer–monomer contacts that are responsible for the integrity of the oligomer vanish from the spectrum (Fig. 3A). A schematic for αSyn oligomer is shown (Fig. 4). The red cylinders represent the following stretches of residues: (i) G31, T33 (ii) G41, K43, T44 (iii) A53, T54 (iv) T64, V66, G67, G68, A69 (v) T72, G73, T75, A76 (vi) T81, G84, G86, S87 (vii) T92, G93 (Fig. 4). It is these regions which are probably responsible for the formation of monomer–monomer contacts leading to the formation of oligomers (Fig. 4). The various regions along the primary sequence of αSyn are shown (Fig. S7A†) and the above stretches of residues (represented as red cylinders in Fig. 4) are highlighted (Fig. S7B†). It can be seen that these residues belong to the N-terminal and NAC regions. Triphala probably interferes in the formation of these interactions, as evident from the appearance of the peaks for these stretches of residues in the HSQC spectrum (Fig. 3A). The low CSP values suggest that the reduction in the ability of αSyn to oligomerize is brought about by the Triphala constituents by mode of weak interactions (Fig. 3B). On the other hand, the ratio of HSQC peak intensities (Fig. 3C) indicate that some of the Triphala constituents probably also interact with C-terminal residues causing an inhomogeneous broadening as is evident from the decrease in the peak intensities due to the presence of Triphala (Fig. 3C). This is particularly clear from the splitting of S129 into two peaks (Fig. 3C) in the presence of Triphala which is likely to be due to the slow exchange between bound and unbound state.58 For the other C-terminal residues, the peak splitting could be small compared to the spectral resolution and hence, causes an inhomogeneous peak broadening instead of showing up as two separate peaks.
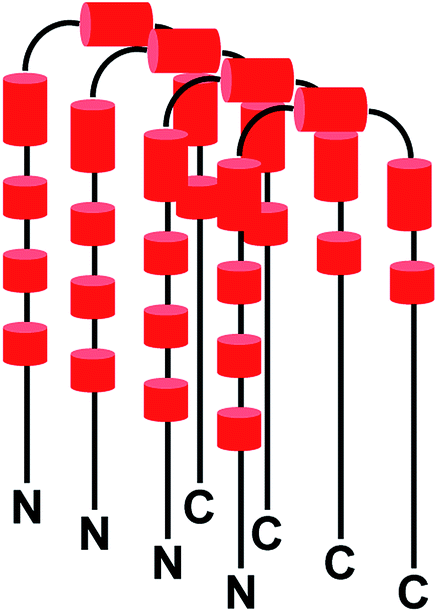 | ||
| Fig. 4 Schematic for αSyn oligomer. The black solid lines depict flexible regions within the oligomer. The red cylinders represent different regions responsible for the formation of monomer–monomer contacts within the oligomer as observed in HSQC experiments. ‘N’ and ‘C’ represent the N- and C-terminals respectively. | ||
Triphala does not disaggregate αSyn amyloid fibrils
It has been shown earlier that Triphala breaks down lysozyme fibrils into smaller and thinner fibrils.76 Therefore, in order to check the stability of αSyn fibrils in the presence of Triphala, preformed fibriller αSyn was incubated with 0.5 mg ml−1 Triphala. A spontaneous decrease in ThT fluorescence was observed upon the introduction of 0.5 mg ml−1 Triphala to prefibrillized αSyn (Fig. S8†). This suggested that Triphala may be capable of destabilizing αSyn fibrils. In order to confirm the above possibility, TEM images were recorded. Prefibrillized αSyn after treatment with 0.5 mg ml−1 Triphala did not show any significant change in the morphology, shape or size of the fibrils even after incubation for a period of 5 and half days (Fig. S9†). These results showed that 0.5 mg ml−1 Triphala is not capable of disaggregating αSyn fibrils. In view of these results, the decrease in ThT fluorescence upon addition of Triphala (Fig. S8†) could be due to (i) competitive binding of ThT and Triphala with αSyn fibrils or (ii) inner filter effect (Triphala might absorb in the region 400–450 nm, where ThT also absorbs). However, it was observed that Triphala does not absorb in this region (Fig. S10†). Therefore, the decrease in ThT fluorescence upon addition of Triphala (Fig. S8†) may be attributed to competitive binding of ThT and Triphala with αSyn fibrils.In order to check if a higher concentration of Triphala is required to cause disaggregation of αSyn fibrils, the fibrils were treated with 2.5 mg ml−1 Triphala. The effect of 2.5 mg ml−1 Triphala on αSyn fibrils was monitored in a time-dependent manner by far-UV CD spectroscopy. Fibriller αSyn showed a global minimum at 220 nm which is characteristic of β-sheet secondary structure as expected for amyloid fibrils (Fig. S11†). However, upon addition of Triphala, no change whatsoever was observed in the β-sheet character of fibrillar αSyn even after 5 and half days of incubation time (Fig. S11†). This suggested that even 2.5 mg ml−1 Triphala may not be capable of destabilizing αSyn fibrils. In order to see if this higher concentration of Triphala caused any minor changes in the morphology of αSyn fibrils without leading to any detectable changes in their secondary structure, TEM images were taken. It was observed that the rod-like filamentous shape of the fibrils remained unchanged (Fig. S12†). The width of the fibrils was found to be the same (≈13 nm) in the presence and absence of Triphala (Fig. S12†). These results indicate that even a high concentration i.e. 2.5 mg ml−1 of Triphala does not disaggregate preformed αSyn fibrils. In order to understand why Triphala is incapable of disaggregating preformed αSyn fibrils, it is important to know the residues of αSyn which interact with Triphala. As discussed above, certain stretches of residues of αSyn are responsible for oligomerization (Fig. S7B†) and Triphala probably interacts with these regions and thus arrests oligomerization of αSyn. Now, in the fibriller state of αSyn, the residues which form the fibril core are known77 and are highlighted along the primary sequence of αSyn (Fig. S7C†). A comparison of the residues (Fig. S7B and C†) suggests that the ones which interact with Triphala in the native state of αSyn are not available in the fibriller state of the protein since they are buried in the fibril core. Probably it is the inaccessibility of these residues, in the fibriller state, which is responsible for Triphala being incapable of disaggregating αSyn fibrils.
Conclusions
In summary, aqueous extract of Triphala, a natural herbal preparation has been shown to inhibit fibrillization of αSyn. While various biophysical tools have been used to study the course of αSyn fibrillization, NMR analysis of the effect of Triphala on the process have yielded insights into the nature of the residues involved in the self-association of αSyn leading to the formation of oligomers, which is one of the early events preceding the formation of mature amyloid fibrils. We demonstrate that stretches of residues which lie in the N-terminal and the NAC regions of the protein chain play important role in the early association process, and the Triphala components, rich in polyphenols, interfere with this association. It is likely that different polyphenol components of Triphala act in a synergistic manner to bring about the arrest of αSyn self-association. Our results clearly state that Triphala has a wealth of molecules which are amyloid inhibitors. Future studies will determine the particular constituents which are responsible for bringing about the inhibition of αSyn fibrillization.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are thankful to Tata Institute of Fundamental Research (TIFR), Department of Atomic Energy (DAE) for financial support. R. V. H. is a DAE sponsored Raja Ramanna Fellow. αSyn plasmid was a kind gift from Vinod Subramaniam, Vrije Universiteit Amsterdam. Mass Spectrometry analysis was performed at the DBT-BIRAC supported Center for Applications of Mass Spectrometry (CAMS) at Venture Center BioIncubator at CSIR-NCL, Pune, India. The authors thank Francois-Xavier Theillet and Christian Griesinger for fruitful discussions. The authors acknowledge Shreyada Save, UM-DAE CEBS, Mumbai 400098, India, for her help in ThT fluorescence experiments. The authors are thankful to Mamata Kombrabail, Department of Chemical Sciences, TIFR, Mumbai 400005, India, for her help in CD and fluorescence experiments. The authors would like to thank Shrabasti Bhattacharya and Vaibhav Shukla for their inputs on CD experiments and CD data analysis. The authors are also grateful to Geetanjali Dhotre for helping with MALDI experiments and mass calibration.References
- P. H. Weinreb, W. Zhen, A. W. Poon, K. A. Conway and P. T. Lansbury Jr, Biochemistry, 1996, 35, 13709–13715 CrossRef CAS PubMed.
- D. F. Clayton and J. M. George, Trends Neurosci., 1998, 21, 249–254 CrossRef CAS.
- J. Q. Trojanowski, M. Goedert, T. Iwatsubo and V. M. Lee, Cell Death Differ., 1998, 5, 832–837 CrossRef CAS PubMed.
- M. Hashimoto and E. Masliah, Brain Pathol., 1999, 9, 707–720 CrossRef CAS.
- A. B. Singleton, M. Farrer, J. Johnson, A. Singleton, S. Hague, J. Kachergus, M. Hulihan, T. Peuralinna, A. Dutra, R. Nussbaum, S. Lincoln, A. Crawley, M. Hanson, D. Maraganore, C. Adler, M. R. Cookson, M. Muenter, M. Baptista, D. Miller, J. Blancato, J. Hardy and K. Gwinn-Hardy, Science, 2003, 302, 841 CrossRef CAS PubMed.
- K. A. Conway, J. C. Rochet, R. M. Bieganski and P. T. Lansbury Jr, Science, 2001, 294, 1346–1349 CrossRef CAS PubMed.
- M. Zhu, S. Han and A. L. Fink, Biochim. Biophys. Acta, 2013, 1830, 2872–2881 CrossRef CAS PubMed.
- M. Caruana, T. Högen, J. Levin, A. Hillmer, A. Giese and N. Vassallo, FEBS Lett., 2011, 585, 1113–1120 CrossRef CAS.
- J. Li, M. Zhu, S. Rajamani, V. N. Uversky and A. L. Fink, Chem. Biol., 2004, 11, 1513–1521 CrossRef CAS PubMed.
- B. Ahmad and L. J. Lapidus, J. Biol. Chem., 2012, 287, 9193–9199 CrossRef CAS.
- X. Meng, L. A. Munishkina, A. L. Fink and V. N. Uversky, Parkinson's Dis., 2010, 1–16, DOI:10.4061/2010/650794.
- X. Meng, L. A. Munishkina, A. L. Fink and V. N. Uversky, Biochemistry, 2009, 48, 8206–8224 CrossRef CAS PubMed.
- D. E. Ehrnhoefer, J. Bieschke, A. Boeddrich, M. Herbst, L. Masino, R. Lurz, S. Engemann, A. Pastore and E. E. Wanker, Nat. Struct. Mol. Biol., 2008, 15, 558–566 CrossRef CAS PubMed.
- P. K. Singh, V. Kotia, D. Ghosh, G. M. Mohite, A. Kumar and S. K. Maji, ACS Chem. Neurosci., 2013, 4, 393–407 CrossRef CAS.
- G. R. Lamberto, A. Binolfi, M. L. Orcellet, C. W. Bertoncini, M. Zweckstetter, C. Griesinger and C. O. Fernandez, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 21057–21062 CrossRef CAS PubMed.
- L. Breydo, J. W. Wu and V. N. Uversky, Biochim. Biophys. Acta, 2012, 1822, 261–285 CrossRef CAS PubMed.
- M. Perni, C. Galvagnion, A. Maltsev, G. Meisl, M. B. D. Müller, P. K. Challa, J. B. Kirkegaard, P. Flagmeier, S. I. A. Cohen, R. Cascella, S. W. Chen, R. Limbocker, P. Sormanni, G. T. Heller, F. A. Aprile, N. Cremades, C. Cecchi, F. Chiti, E. A. A. Nollen, T. P. J. Knowles, M. Vendruscolo, A. Bax, M. Zasloff and C. M. Dobson, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E1009–E1017 CrossRef CAS PubMed.
- N. N. Jha, R. Kumar, R. Panigrahi, A. Navalkar, D. Ghosh, S. Sahay, M. Mondal, A. Kumar and S. K. Maji, ACS Chem. Neurosci., 2017, 8, 2722–2733 CrossRef CAS PubMed.
- T. Ibrahim and J. McLaurin, Biochem. Biophys. Res. Commun., 2016, 469, 529–534 CrossRef CAS PubMed.
- A. A. Deeg, A. M. Reiner, F. Schmidt, F. Schueder, S. Ryazanov, V. C. Ruf, K. Giller, S. Becker, A. Leonov, C. Griesinger, A. Giese and W. Zinth, Biochim. Biophys. Acta, 2015, 1850, 1884–1890 CrossRef CAS PubMed.
- K. Ji, Y. Zhao, T. Yu, Z. Wang, H. Gong, X. Yang, Y. Liu and K. Huang, Food Funct., 2016, 7, 409–416 RSC.
- N. Ahsan, S. Mishra, M. K. Jain, A. Surolia and S. Gupta, Sci. Rep., 2015, 5, 9862 CrossRef CAS.
- N. N. Jha, D. Ghosh, S. Das, A. Anoop, R. S. Jacob, P. K. Singh, N. Ayyagari, I. N. N. Namboothiri and S. K. Maji, Sci. Rep., 2016, 6, 28511 CrossRef CAS PubMed.
- N. Ahsan, I. A. Siddique, S. Gupta and A. Surolia, Eur. J. Med. Chem., 2018, 143, 1174–1184 CrossRef CAS PubMed.
- R. Chakraborty, S. Sahoo, N. Halder, H. Rath and K. Chattopadhyay, ACS Chem. Neurosci., 2019, 10, 573–587 CrossRef CAS PubMed.
- J. Pujols, S. Peña-Díaz, D. F. Lázaro, F. Peccati, F. Pinheiro, D. González, A. Carija, S. Navarro, M. Conde-Giménez, J. García, S. Guardiola, E. Giralt, X. Salvatella, J. Sancho, M. Sodupe, T. F. Outeiro, E. Dalfó and S. Ventura, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 10481–10486 CrossRef CAS PubMed.
- D. Yedlapudi, G. S. Joshi, D. Luo, S. V. Todi and A. K. Dutta, Sci. Rep., 2016, 6, 38510 CrossRef CAS PubMed.
- S. Ghosh, A. Kundu and K. Chattopadhyay, Sci. Rep., 2018, 8, 5481 CrossRef PubMed.
- N. Joshi, S. Basak, S. Kundu, G. De, A. Mukhopadhyay and K. Chattopadhyay, Langmuir, 2015, 31, 1469–1478 CrossRef CAS PubMed.
- D. Ghosh, S. Mehra, S. Sahay, P. K. Singh and S. K. Maji, Int. J. Biol. Macromol., 2017, 100, 37–54 CrossRef CAS PubMed.
- H. Nedaei, A. A. Saboury, A. A. Meratan, L. Karami, L. Sawyer, B. Kaboudin, N. Jooyan and A. Ghasemi, J. Mol. Liq., 2018, 266, 291–298 CrossRef CAS.
- D. L. Melnikova, V. D. Skirda and I. V. Nesmelova, J. Phys. Chem. B, 2017, 121, 2980–2988 CrossRef CAS PubMed.
- T. Kuboyama, C. Tohda and K. Komatsu, Biol. Pharm. Bull., 2014, 37(6), 892–897 CrossRef CAS PubMed.
- S. Sivasankar, R. Lavanya, P. Brindha and N. Angayarkanni, PLoS One, 2015, 10(3), e0120512 CrossRef PubMed.
- S. R. Varma, T. O. Sivaprakasam, A. Mishra, L. M. S. Kumar, N. S. Prakash, S. Prabhu and S. Ramakrishnan, PLoS One, 2016, 11(1), e0145921 CrossRef PubMed.
- K. Lu, D. Chakroborty, C. Sarkar, T. Lu, Z. Xie, Z. Liu and S. Basu, PLoS One, 2012, 7(8), e43934 CrossRef CAS PubMed.
- N. Uabundit, J. Wattanathorn, S. Mucimapura and K. Ingkaninan, J. Ethnopharmacol., 2010, 127, 26–31 CrossRef PubMed.
- M. S. Baliga, S. Meera, B. Mathai, M. P. Rai, V. Pawar and P. L. Palatty, Chin. J. Integr. Med., 2012, 18, 946–954 CrossRef PubMed.
- D. P. Singh, R. Govindarajan and A. K. Rawat, Phytochem. Anal., 2008, 19, 164–168 CrossRef CAS PubMed.
- V. Pawar, P. Lahorkar and D. B. Ananthanarayana, Indian J. Pharm. Sci., 2009, 71, 382–386 CrossRef CAS PubMed.
- S. Cheriyamundath, T. Mahaddalkar, S. N. Save, S. Choudhary, R. V. Hosur and M. Lopus, Biomed. Pharmacother., 2018, 98, 76–81 CrossRef CAS PubMed.
- R. Jakes, M. G. Spillantini and M. Goedert, FEBS Lett., 1994, 345, 27–32 CrossRef CAS PubMed.
- M. J. Volles and P. T. Lansbury Jr, J. Mol. Biol., 2007, 366, 1510–1522 CrossRef CAS PubMed.
- D. Marion, M. Ikura, R. Tschudin and A. Bax, J. Magn. Reson., 1989, 85, 393–399 CAS.
- J. G. Reddy and R. V. Hosur, J. Biomol. NMR, 2014, 59, 199–210 CrossRef CAS PubMed.
- C. Bodner, A. S. Maltsev, C. Dobson and A. Bax, Biochemistry, 2010, 49, 862–871 CrossRef CAS PubMed.
- J. Cavanagh, W. J. Fairbrother, A. G. I. Palmer, M. Rance and N. J. Skelton, Protein NMR Spectroscopy: Principles and Practice, Elsevier Inc., 2nd edn, 2007 Search PubMed.
- M. Biancalana and S. Koide, Biochim. Biophys. Acta, 2010, 1804(7), 1405–1412 CrossRef CAS PubMed.
- M. R. H. Krebs, E. H. C. Bromley and A. M. Donald, J. Struct. Biol., 2005, 149, 30–37 CrossRef CAS PubMed.
- C. Xue, T. Y. Lin, D. Chang and Z. Guo, R. Soc. Open Sci., 2017, 4, 160696 CrossRef PubMed.
- M. F. Bishop and F. A. Ferrone, Biophys. J., 1984, 46, 631–644 CrossRef CAS PubMed.
- A. Micsonai, F. Wien, L. Kernya, Y. H. Lee, Y. Goto, M. Refregiers and J. Kardos, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E3095–E3103 CrossRef CAS PubMed.
- A. Micsonai, F. Wien, E. Bulyaki, J. Kun, E. Moussong, Y. H. Lee, Y. Goto, M. Refregiers and J. Kardos, Nucleic Acids Res., 2018, 46, W315–W322 CrossRef CAS PubMed.
- M. Sandal, F. Valle, I. Tessari, S. Mammi, E. Bergantino, F. Musiani, M. Brucale, L. Bubacco and B. Samori, PLoS Biol., 2008, 6(1), e6 CrossRef PubMed.
- H. Yu, W. Han, W. Ma and K. Schulten, J. Chem. Phys., 2015, 143, 243142 CrossRef PubMed.
- M. S. Celej, R. Sarroukh, E. Goormaghtigh, G. D. Fidelio, J. M. Ruysschaert and V. Raussens, Biochem. J., 2012, 443, 719–726 CrossRef CAS PubMed.
- D. Ghosh, P. K. Singh, S. Sahay, N. N. Jha, R. S. Jacob, S. Sen, A. Kumar, R. Riek and S. K. Maji, Sci. Rep., 2015, 5, 9228 CrossRef CAS PubMed.
- M. P. Williamson, Prog. Nucl. Magn. Reson. Spectrosc., 2013, 73, 1–16 CrossRef CAS PubMed.
- D. Bhattacharyya, R. Kumar, S. Mehra, A. Ghosh, S. K. Maji and A. Bhunia, Chem. Commun., 2018, 54, 3605–3608 RSC.
- L. Giehm, D. I. Svergun, D. E. Otzen and B. Vestergaard, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 3246–3251 CrossRef CAS PubMed.
- N. Zijlstra, C. Blum, I. M. J. Segers-Nolten, M. M. A. E. Claessens and V. Subramaniam, Angew. Chem., Int. Ed., 2012, 51, 8821–8824 CrossRef CAS PubMed.
- W. Paslawski, S. Mysling, K. Thomsen, T. J. D. Jørgensen and D. E. Otzen, Angew. Chem., Int. Ed., 2014, 53, 7560–7563 CrossRef CAS PubMed.
- G. M. Mohite, R. Kumar, R. Panigrahi, A. Navalkar, N. Singh, D. Datta, S. Mehra, S. Ray, L. G. Gadhe, S. Das, N. Singh, D. Chatterjee, A. Kumar and S. K. Maji, Biochemistry, 2018, 57, 5183–5187 CrossRef CAS PubMed.
- J. I. Gallea and M. S. Celej, J. Biol. Chem., 2014, 289, 26733–26742 CrossRef CAS PubMed.
- N. Lorenzen, S. B. Nielsen, A. K. Buell, J. D. Kaspersen, P. Arosio, B. S. Vad, W. Paslawski, G. Christiansen, Z. Valnickova-Hansen, M. Andreasen, J. J. Enghild, J. S. Pedersen, C. M. Dobson, T. P. J. Knowles and D. E. Otzen, J. Am. Chem. Soc., 2014, 136, 3859–3868 CrossRef CAS PubMed.
- N. Cremades, S. I. A. Cohen, E. Deas, A. Y. Abramov, A. Y. Chen, A. Orte, M. Sandal, R. W. Clarke, P. Dunne, F. A. Aprile, C. W. Bertoncini, N. W. Wood, T. P. J. Knowles, C. M. Dobson and D. Klenerman, Cell, 2012, 149, 1048–1059 CrossRef CAS PubMed.
- S. W. Chen, S. Drakulic, E. Deas, M. Ouberai, F. A. Aprile, R. Arranz, S. Ness, C. Roodveldt, T. Guilliams, E. J. De-Genst, D. Klenerman, N. W. Wood, T. P. J. Knowles, C. Alfonso, G. Rivas, A. Y. Abramov, J. M. Valpuesta, C. M. Dobson and N. Cremades, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E1994–E2003 CrossRef CAS PubMed.
- G. Fusco, S. W. Chen, P. T. F. Williamson, R. Cascella, M. Perni, J. A. Jarvis, C. Cecchi, M. Vendruscolo, F. Chiti, N. Cremades, L. Ying, C. M. Dobson and A. D. Simone, Science, 2017, 358, 1440–1443 CrossRef CAS PubMed.
- J. A. Varela, M. Rodrigues, S. De, P. Flagmeier, S. Gandhi, C. M. Dobson, D. Klenerman and S. F. Lee, Angew. Chem., Int. Ed., 2018, 57, 4886–4890 CrossRef CAS PubMed.
- H. Kumar, J. Singh, P. Kumari and J. B. Udgaonkar, J. Biol. Chem., 2017, 292(41), 16891–16903 CrossRef CAS PubMed.
- B. D. van Rooijen, M. M. Claessens and V. Subramaniam, Biochim. Biophys. Acta, 2009, 1788(6), 1271–1278 CrossRef CAS PubMed.
- B. D. van Rooijen, K. A. van Leijenhorst-Groener, M. M. Claessens and V. Subramaniam, J. Mol. Biol., 2009, 394, 826–833 CrossRef CAS PubMed.
- S. Mysling, C. Betzer, P. H. Jensen and T. J. D. Jorgensen, Biochemistry, 2013, 52, 9097–9103 CrossRef CAS PubMed.
- H. Y. Kim, M. K. Cho, A. Kumar, E. Maier, C. Siebenhaar, S. Becker, C. O. Fernandez, H. A. Lashuel, R. Benz, A. Lange and M. Zweckstetter, J. Am. Chem. Soc., 2009, 131, 17482–17489 CrossRef CAS PubMed.
- A.-C. Hoffmann, G. Minakaki, S. Menges, R. Salvi, S. Savitskiy, A. Kazman, H. V. Miranda, D. Mielenz, J. Klucken, J. Winkler and W. Xiang, Sci. Rep., 2019, 9, 544 CrossRef PubMed.
- S. N. Save and S. Choudhary, RSC Adv., 2017, 7, 20460 RSC.
- M. Vilar, H. T. Chou, T. Luhrs, S. K. Maji, D. R. Loher, R. Verel, G. Manning, H. Stahlberg and R. Riek, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 8637–8642 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra05551g |
| This journal is © The Royal Society of Chemistry 2019 |