
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCombining autoclave and LCWM reactor studies to shed light on the kinetics of glucose oxidation catalyzed by doped molybdenum-based heteropoly acids†
Dorothea Voßa,
Sebastian Ponceb,
Stefanie Wesingera,
Bastian J. M. Etzoldb and
Jakob Albert *a
*a
aLehrstuhl für Chemische Reaktionstechnik, Friedrich-Alexander-Universität Erlangen-Nürnberg, Egerlandstraße 3, 91058 Erlangen, Germany. E-mail: jakob.albert@fau.de
bLehrstuhl für Technische Chemie, Technische Universität Darmstadt, Alarich-Weiss-Straße 8, 64287 Darmstadt, Germany
First published on 17th September 2019
Abstract
In this work we combined kinetic studies for aqueous-phase glucose oxidation in a high-pressure autoclave setup with catalyst reoxidation studies in a liquid-core waveguide membrane reactor. Hereby, we investigated the influence of Nb- and Ta-doping on Mo-based Keggin-polyoxometalates for both reaction steps independently. Most importantly, we could demonstrate a significant increase of glucose oxidation kinetics by Ta- and especially Nb-doping by factors of 1.1 and 1.5 compared to the classical HPA-Mo. Moreover, activation energies for the substrate oxidation step could be significantly reduced from around 80 kJ mol−1 for the classical HPA-Mo to 61 kJ mol−1 for the Ta- and 55 kJ mol−1 for the Nb-doped species, respectively. Regarding catalyst reoxidation kinetics, the doping did not show significant differences between the different catalysts.
Introduction
Multiphase chemical reactions, especially gaseous–liquid reactions, are often applied to make valuable chemicals from biomass. One intensively studied approach is the selective partial oxidation of carbohydrates to carboxylic acids, especially formic acid1,2 and acetic acid.3,4 Oxygen is usually activated by metal catalysts. Hereby, mostly vanadium-containing catalysts like polyoxometalates (POMs),5–7 or water-soluble vanadium salts are used.8,9 In particular POMs are highly promising catalysts for these kinds of reaction due to their strong Brønsted acidity combined with a high redox activity and high stability in aqueous solution.10,11 Within the redox-cycle, shown schematically in Fig. 1, POM catalysts allow the use of molecular oxygen as oxidant. Mostly used in homogeneously catalyzed reactions are POMs from the so-called Keggin-type [XM12O40]n−.12–14 Nyman summarized that studies on polyoxometalates containing W, Mo and V are abundant as these structures could be easily obtained through simple acidification while investigations on the POM chemistry of Nb and Ta are poor.15 However, especially polyoxoniobates have received certain attention in the past two decades. Several groups have shown that Nb can also be introduced into Keggin-type structures, i.e. vanadoniobate clusters like {VNb12O40(VO)2},16 [PM2Nb12O40]9−![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 17 or [SiNb12O40]12−.18 Recently, also a few Ta-containing POMs could be synthesized as well.19 Nevertheless, up to now the influence of Nb or Ta on the catalytic properties of Keggin-POMs for biomass oxidation to formic acid is not studied.
17 or [SiNb12O40]12−.18 Recently, also a few Ta-containing POMs could be synthesized as well.19 Nevertheless, up to now the influence of Nb or Ta on the catalytic properties of Keggin-POMs for biomass oxidation to formic acid is not studied.
 | ||
| Fig. 1 Schematic illustration of the catalyst redox cycle. | ||
Traditionally, kinetic studies of such gaseous–liquid reactions are carried out in stirred tank reactors.20 E.g. for the two vanadium-substituted POMs H5PV2Mo10O40 (HPA-2) and H8PV5Mo7O40 (HPA-5) the oxidation kinetics of monosaccharides like glucose, fructose, sorbitol, and gluconic acid as well as disaccharides like cellobiose and sucrose, up to water-insoluble biomass like beech and spruce wood were determined.21 Moreover, Yokoyama et al. performed kinetic studies on the oxidation of non-phenolic lignin subunits by tungsten-based Keggin-POMs in water.22 Besides, these autoclave studies, which allow to operate close to a later on technical system, employing micro-scale reactors for kinetic studies can show significant advantages.23,24 These reactors enable studies with minimal sample consumption under precise control of reaction parameters in combination with enhanced mass- and heat-transfer, either to suppress transfer limitations or to enable kinetic transient response studies.25 Recently, some of us introduced a new optofluidic microreactor which combines for gas/liquid reactions very fast and continuous gas/liquid mass transfer with in situ spectroscopy and the general advantages of microreactors.26 These so called liquid core waveguide membrane (LCWM) microreactor was used to study independently the kinetics of the reduction and reoxidation step (see Fig. 1) of four different Keggin-type POM-catalysts with varying vanadium substitution degree.27
In this work we prepared two Nb- and Ta-doped Keggin-type POMs as also a non-substituted one (HPA-Mo) as reference. Combining autoclave and LCWM-reactor studies we deduce the influence of the Nb and Ta doping on the oxidation as also reoxidation kinetics and resulting product distribution with glucose as a model substrate.
Experimental details
Chemicals
All chemicals were obtained commercially and used as received without further purification. The Keggin-type polyoxometalate catalyst H3PMo12O40 (HPA-Mo) was synthesized according to the literature.6,28 The tantalum and niobium doped HPA-Mo species (HPA-Ta and HPA-Nb) were synthesized according to a modified synthesis procedure by Nyman.29 Oxygen, (4.5 GA 201, Linde AG) was used as the oxidizing agent. Demineralized water was used as a solvent. D(+)-glucose (99.9%, Merck KGaA) was used as a substrate.Experimental setup for the high pressure autoclave experiments
The oxidation reactions were carried out in a 600 mL autoclave made of Hastelloy C276 and equipped with a gas entrainment impeller. A flat Teflon sealing located at the reactor head was used. All pipes, valves and fittings were made of stainless steel 1.4571. The reactor was equipped with a cooling coil made of Hastelloy C276 and a heating jacket to control the reactor temperature. The oxygen was fed into the system by a mass flow controller to keep up the required pressure. A pressure control valve was installed to prevent higher pressures than 65 bar. Additionally, the reactor was connected to a rupture disk with a burst pressure maximum of 100 bar.For the batch experiments, the autoclave was filled with 0.25 mol L−1 glucose, 0.01 mol L−1 catalyst and 100 g water as the solvent. The system was purged twice with 20 bar oxygen pressure to remove the residual air out of the reactor. In the following, the reactor was pre-pressurized with about 40 bar oxygen pressure, the stirrer was set to 300 rpm and the heating was switched on. When the desired temperature was reached, the oxygen pressure was increased to 60 bar and the stirring speed was set to 1000 rpm in order to start the gas entrainment. This moment was set as starting time of the experiment.
The kinetic experiments were performed as described above. As soon as the desired temperature and pressure were reached, a liquid sample was taken for the determination of the initial substrate and product concentrations. Then, the stirrer speed was set to 1000 rpm in order to start the reaction. Liquid samples were taken each 15 minutes during the first hour, each 30 minutes during the second hour, as well as after three and four hours, respectively. During sampling, the gas entrainment was stopped by reduction of the stirrer speed to 300 rpm. After the total reaction time of four hours, a gas sample was taken in order to analyze the gas composition and to close the carbon mass balance.
Liquid core waveguide membrane microreactor setup
Fig. 2 shows a scheme of the reactor apparatus. For a detailed explanation on the set-up and working principle please refer to ref. 26. Briefly, the samples are pumped through the inlet fluidic cell, which also connects the light source (DH-2000, Ocean Optics, Tungsten-Halogen Lamps) through a standard optical fiber. At the outlet, a second fluidic cell delivers the liquid stream to the waste, and transmitted light to the spectrometer (Spectro 320 (D) R5, Instrument Systems) through a second optical fiber. Before and after the inlet and outlet fluidic cells, respectively, two valves control the liquid-phase pressure. Additionally, between the fluidic cells, the Teflon tube (AF-2400, Biogeneral, 1.0 mm i.d., 1.6 mm o.d) is enclosed in a steel tube, in which gas flows (2 L h−1) at constant pressure. Finally, a heating wire surrounds the outer tube, enabling temperature control during the experiments (up to 150 °C). | ||
| Fig. 2 Schematic illustration of the LCWM setup used in the experiments. FCi, o: fluidic cells in and out, P: pressure indicator, T: temperature indicator, BPR: backpressure regulator, v: two-way valve. | ||
For the reduction experiments, a constant temperature was set on the LCWM reactor (130 °C). The steel outer tube was flushed with nitrogen and the pressure was set to 2 bar. Subsequently, the system was filled with double-distilled water and a background spectrum was taken. The aqueous solutions with catalyst (40 μmol L−1) and D-glucose substrate (0.75 mmol L−1) were pumped into the reactor, and the liquid inlet and outlet valves were closed after a constant liquid pressure of ≈ 3 bar was reached. Finally, optical absorption spectra between 400–900 nm were recorded.
Before performing reoxidation experiments, catalysts were pre-reduced (HPA-nred) by glucose in a magnetically stirred two-neck glass reactor (25 mL) equipped with a reflux condenser and temperature control. The reactions took at least 1 hour, with constant vigorous stirring (700 rpm) in a nitrogen-pre-saturated solution at 100 °C. The initial concentration ratio between glucose and the catalysts was 50:1.
For the reoxidation experiments itself, a constant temperature was set on the LCWM reactor (80–150 °C). The steel outer tube was set to 2 bar of nitrogen gas. Subsequently, the system was filled with double-distilled water and a background spectrum was taken. Aqueous solutions of HPA-nred catalysts (2–20 μmol L−1) were pumped into the reactor, and the inlet and outlet valves were closed to keep a constant liquid pressure of ≈6 bar. Continuous absorption spectra measurements between 500 to 800 nm were started. After two minutes, rapid exchange of the gas phase was carried out by discharging N2 into the fume, flushing with 5 bar of synthetic air, while adsorption spectra were continuously recorded.
Kinetic description
For the kinetic description we applied the following power law (eqn (1)) and Arrhenius law (eqn (2)).Observable reaction rate:
| robs = kobscglucosea | (1) |
| kobs = k1ccatalyst,oxb |
Arrhenius equation:
 | (2) |
For determination of the effective substrate oxidation order and rate constant as well as effective activation energy we used a linearization of the logarithmic forms (eqn (3) and (4)).
Logarithmic power law:
|
lnrobs = lnkobs + alnc0,glucose
| (3) |
Logarithmic Arrhenius equation:
 | (4) |
For reoxidation experiments developed within the LCWM setup, initial reaction rates were obtained from the initial slope of the concentration versus time curve (eqn (5)). Due to the fast gaseous/liquid mass transfer, the oxygen concentration in the liquid-phase was assumed to be constant. Activation energy values were calculated by applying eqn (4) for the reoxidation reaction rate constant (kox) values and temperatures.
 | (5) |
Analytics
Characterization of the used HPA-catalysts has been carried out using different analytical techniques. Elemental analysis has been done using a PerkinElmer Plasma 400 ICP-OES device. The determination of hydration water content was done by performing thermogravimetric analysis (TGA) on a Setsys 1750 CS Evolution. FTIR spectra were recorded in a range between 2000 and 400 cm−1 on a Jasco FT/IR-4100 spectrometer equipped with a PIKE GladiATR ATR-accessory with a resolution of 4 cm−1 at ambient conditions. 31P-NMR spectra were recorded on a JEOL ECX-400 MHz at room temperature without resolution enhancement in D2O-solution showing the same chemical shifts according to the literature.6,28 Powder X-ray diffraction was measured using a STADIP from Stoe & Cie GmbH. All XRD measurements were carried out on a flat sample holder in transmission geometry (4–60°/2θ, λ = 1.54060 Å, Ge[111]-monochromator) using a Mythen1K (Dectris, Baden, Switzerland) detector. Moreover, we measured Raman-spectroscopy for all applied catalysts with Bruker Senterra 1 using a NDYAG laser at a wavelength of 532 nm. Electrochemical measurements (cyclic voltammetry and square wave voltammetry) were performed in a three-electrode system connected to a PARSTAT MC Multichannel potentiostat with a platinum working electrode, a platinum coil counter electrode, and an Ag/AgCl reference electrode. All experiments were carried out at room temperature in a 0.5 M Na2SO4 aqueous electrolyte solution. The sample solutions were purged with nitrogen for 3 min then maintained for 1 min for stabilization.Analysis of reaction mixtures has been carried out using liquid and gaseous analysis. Liquid products were analyzed by means of HPLC measurements using a High performance liquid chromatography from Jasco equipped with a 300 mm × 8 mm SH1011 Shodex column. As eluent a 5 mmol aqueous sulfuric acid solution was applied. Gaseous product analysis was performed using a Varian GC 450 equipped with a 2 m × 0.75 mm ID ShinCarbon ST column.
Determination of yields
The yields of the liquid and gaseous products formic acid (FA), acetic acid (AA), the liquid intermediates glycolaldehyde (GA) and 5-hydroxymethylfurfural (HMF), CO2 and CO were calculated by eqn (6).
 | (6) |
The total carbon yield (TOC) was calculated by eqn (7) and served as a representative for the substrate conversion as other products than FA, AA, GA, HMF, CO2 and CO were not detected using the described analytical methods.
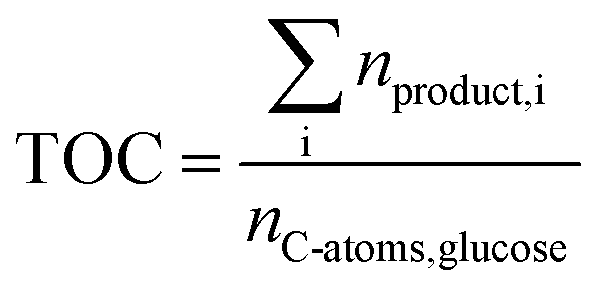 | (7) |
Results and discussion
Catalyst characterization
The synthesized Keggin-type polyoxometalate catalysts HPA-Mo, HPA-Ta and HPA-Nb were characterized by different analytical methods. Elementary analysis using ICP-OES resulted in loadings of 0.23 wt% Nb for HPA-Nb and 0.47 wt% Ta for HPA-Ta. Moreover, the elemental composition of the HPA-Mo was determined to P/Mo = 1/11.94. Thermogravimetric analysis resulted in hydration water contents of 8 molecules for HPA-Mo, 9 molecules for HPA-Nb and 14 molecules for HPA-Ta, respectively. Additionally, FTIR, 31P-NMR, Raman and XRD measurements were performed to confirm the Keggin species (see characterization of polyoxometalate catalysts in the ESI, Fig. S1–S6†).Furthermore, for comparison, the UV/Vis spectra of the fully reduced, partially reduced and fully oxidized forms of the catalysts were obtained within the LCWM microreactor (see Fig. S7†). For all catalysts, the spectra show a quasi-isosbestic point with the continuous disappearance of a band (<500 nm) related to Mo peroxo species, and the appearance of a broad band (>600 nm) typical for the known ‘heteropoly blues’. Nevertheless, as compared in Fig. S8,† HPA-Ta and HPA-Nb partially-reduced spectra present a slightly deviation from the HPA-Mo behavior. It is evident a broadening of the heteropoly blue band, reducing visibility of the presence of Mo(V) transition systems, and the homo-intervalence-charge-transfer-peak bands of Mo–O–Mo groups (approx. at 750 and 860 nm, respectively).12 To get further evidence of this deviation, both catalysts were electrochemically characterized (see Fig. S9 and S10†). Square wave voltammograms of both catalysts show the appearance of a defined peak at −0.4 V in comparison to HPA-Mo. An additional peak at +0.033 V is only observed for HPA-Ta. Metal doping therefore also affects the measured electrochemical first reduction potential of the catalysts, which decreases in the order HPA-Mo > HPA-Nb > HPA-Ta (−0.433, −0.443, −0.447 V, respectively). Similar behavior has been previously observed in V-substituted HPA-Mo complexes27 where a decrease in the redox potential was also correlated to an increase in the oxidation rate constants.
Influence of Nb- and Ta-doping on product distribution at high glucose conversion
Motivated by a former work using different Keggin-type POMs as catalyst,27 we performed long-term batch experiments using glucose as a substrate at 80 °C in order to check the influence of Nb- and Ta-doping on the product selectivity (see Table S1 in the ESI†). However, we could not detect remarkable product yields for all three catalysts. Consequently, we increased the reaction temperature to 160 °C (Table 1). Hereby, we could detect high glucose conversion resulting in total carbon yields between 86 and 96%. As expected, formic acid (FA) was produced as the major liquid product with yields of around 30% followed by acetic acid (AA) with yields of around 7%. Moreover, glycol aldehyde (GA) (approx. 3%) could be detected in all samples. Regarding the gas phase products, CO2 was the major product with yields between 43% (HPA-Nb) and 51% (HPA-Ta). Additionally, 4–5% of CO were formed in each experiment.| Entry | Catalyst | FA-yield [%] | AA-yield [%] | GA-yield [%] | CO2-yield [%] | CO-yield [%] | Total carbon yield [%] |
|---|---|---|---|---|---|---|---|
| 1 | HPA-Mo | 28.2 | 6.7 | 3.2 | 46.9 | 4.0 | 89.0 |
| 2 | HPA-Ta | 30.0 | 7.0 | 2.3 | 51.3 | 5.1 | 95.7 |
| 3 | HPA-Nb | 28.5 | 7.3 | 3.0 | 42.6 | 5.0 | 86.4 |
This product distribution is well in line with literature proposed reaction pathways.7–9 GA is the intermediate product of the oxidation of glucose to formic acid and CO2, while AA is most likely formed by thermal induced conversion of glucose to HMF and levulinic acid (LA) (see Fig. 3). However, the last two intermediates could not be detected maybe due to too low concentrations near the detection limit of our used HPLC. CO could be formed due to the high temperatures by thermal decomposition of the intermediates or FA.
 | ||
| Fig. 3 Reaction scheme of glucose conversion to formic acid and acetic acid using Keggin-type POMs.7–9 | ||
Fig. 3 shows the reaction scheme for the acid catalyzed (upper pathway) as well as the HPA-induced (lower pathway) of glucose oxidation by molecular oxygen in aqueous media.
Influence of Nb- and Ta-doping on kinetics of the substrate oxidation step
The selectivity and product distribution are mainly influenced by the substrate oxidation step (r1 in Fig. 1) as also non-catalytic side reactions. Therefore, the influence of the Nb- and Ta-doping on the product distribution was studied together with the kinetics of the substrate oxidation step and within a high-pressure autoclave experiment allowing for product analysis. In these experiments the influence of the catalyst reoxidation step (r2 in Fig. 1), was suppressed by applying oxygen pressures of 60 bar, where the catalyst reoxidation was shown to be much faster than the substrate oxidation.21To obtain further insight into the kinetics of the oxidation step r1 samples were taken from the autoclave during the first 4 h. For the determination of the effective activation energies, the experiments were performed at different reaction temperatures (120 °C, 140 °C and 160 °C). The liquid product distribution during the experiments is exemplarily shown for the catalyst HPA-Nb in Fig. 4.
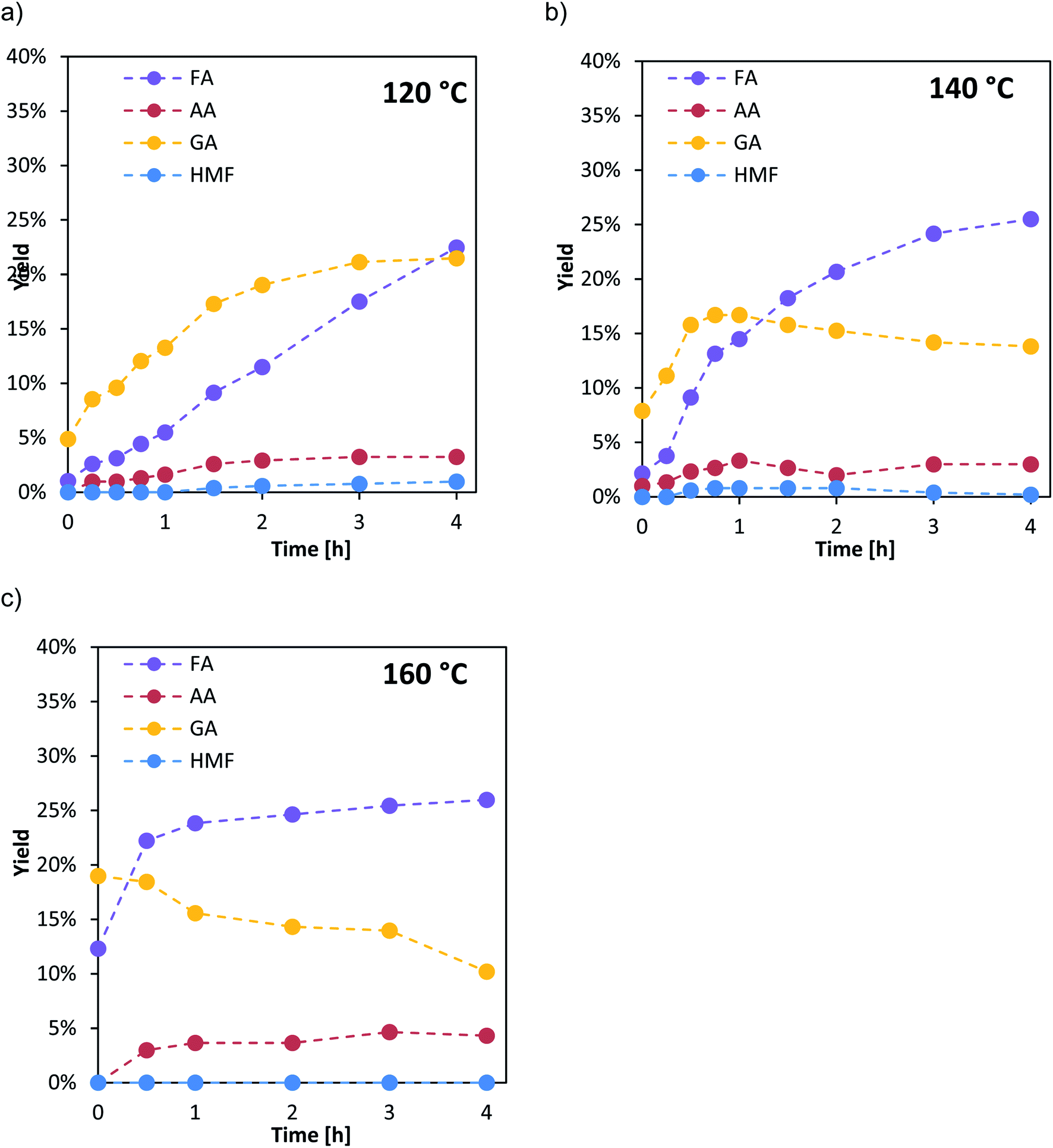 | ||
| Fig. 4 HPA-Nb catalyst: time depended yield distribution of the liquid products for different reaction temperatures: (a) 120 °C, (b) 140 °C, (c) 160 °C: reaction conditions: 60 bar O2, 1000 rpm, 1 mmol HPA-Nb, 100 g water, 0.25 mol L−1 glucose, 4 h fed-batch, and high-pressure autoclave. | ||
The temperature variation shows that at low temperature (120 °C, top left), the HPA catalyzed reaction pathway (see Fig. 3) is dominant. Hereby, the intermediate GA is predominantly formed to a maximum yield of 20% after 4 h reaction time. With increasing reaction time, formic acid yield also increases and becomes dominant after 4 h. The thermal induced reaction pathway leading to AA with HMF as an intermediate plays only a minor role at this temperature.
At 140 °C (Fig. 4, top right), still the HPA catalyzed reaction pathway is dominant but GA is formed to its maximum yield of 18% already within the first hour. Afterwards, its amount slightly decreases within the next 3 h. FA yield drastically increases from the beginning and is already the major species after 1 h reaction time. Again, the thermal induced reaction pathway with HMF as an intermediate on the way to AA plays only a minor role with yields below 5%.
At the highest applied temperature of 160 °C, the maximum amount of GA is already reached after pre-heating at reaction time zero and then consequently converted to FA. The amount of FA is sharply increasing within the first hour and then increases slowly for the next three hours. Here, significantly higher AA yields (ca. 5%) could also be detected over the whole reaction time whereby HMF yield is still close to the detection limit.
The other two catalysts HPA-Ta and HPA-Mo show the same trends with the HPA-catalyzed reaction pathway being the dominant reaction pathway at all applied temperatures (see ESI, Fig. S11 and S12†).
The activation energies for the three different catalysts (Fig. 5a–c) were determined from the temperature variation.
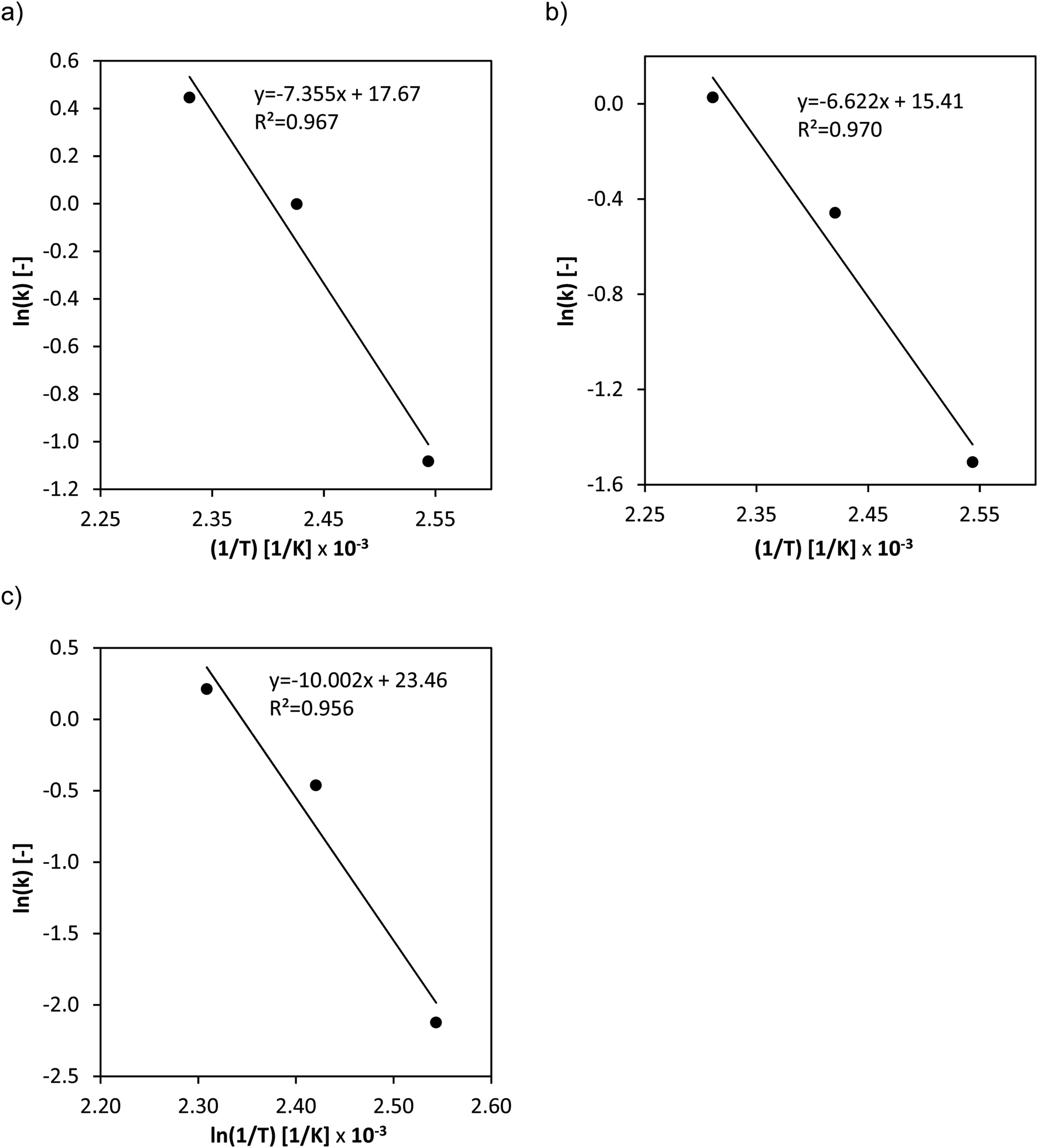 | ||
| Fig. 5 Arrhenius plots of the temperature variation for the substrate oxidation step for (a) HPA-Nb, (b) HPA-Ta and (c) HPA-Mo. Reaction conditions: 120–160 °C, 60 bar O2, 1000 rpm, 1 mmol HPA, 100 g water, 0.25 mol L−1 glucose, 4 h fed-batch, and high-pressure autoclave. | ||
Moreover, the reaction orders of the catalysts were determined from logarithmic plots of the substrate oxidation reaction rate vs. natural logarithm of the initial concentration of glucose. The normalized concentration–time profiles (Fig. S13–S15†) as well as the logarithmic plots of reaction rate vs. initial glucose concentration (Fig. S16 and 17) are shown in the ESI.† For HPA-Mo, analogous experiments have been performed showing an effective reaction order of 1.12 with an effective reaction rate constant of 1.10 mol L−1 h−1.27 The kinetic parameters obtained are summarized in Table 2.
| Catalyst | Effective reaction order nobs [—] | Effective rate constant kobs [mol L−1 h−1] | Effective activation energy [kJ mol−1] |
|---|---|---|---|
| HPA-Mo27 | 1.12 | 1.10 | 83.2 |
| HPA-Ta | 1.20 | 1.16 | 55.1 |
| HPA-Nb | 1.40 | 1.44 | 61.1 |
The determined effective activation energies show significant differences between the two doped catalysts and the conventional HPA-Mo catalyst. The Nb- respectively Ta-doping has a positive effect on the activation barrier which could significantly be reduced from 83 kJ mol−1 for HPA-Mo to around 60 kJ mol−1 for the two doped species. Moreover, the doping seems to have a positive effect on the substrate oxidation kinetics as it increases the effective rate constants.
Influence of Nb- and Ta-doping on the catalyst reoxidation kinetics
While the overall selectivity is mainly influenced by the substrate oxidation step, the catalyst re-oxidation step (r2 in Fig. 1) is at envisaged low oxygen pressures rate limiting and thus critical for the overall reaction rate. As reported by us for vanadium substituted HPA catalysts the UV/Vis signal of the HPAs can be employed to deduce within the LCWM reactor the concentration change from reduced to oxidized HPA catalyst.27 To obtain the influence of the Nb and Ta substitution on the reoxidation kinetics a transient response experiment was carried out within the LCWM reactor, where for fully reduced HPA catalysts the concentration change was determined after exchanging fast from an inert to an oxygen gas atmosphere.Fig. 6a shows exemplary the corresponding time dependent UV/Vis spectra for the reoxidation experiment for HPA-Nb catalyst at 120 °C. Employing the absorption at 750 nm the concentration change of the HPA-Nbred species is given in Fig. 6b for reoxidation experiments carried out from 80 to 140 °C. Similar results were obtained for the HPA-Tared and HPA-Mored catalyst and are given in the ESI (see Fig. S18†). Qualitatively the reoxidation of all three catalysts started to be observable at 100 °C. For a more quantitative comparison the initial reaction rates were deduced for all temperatures and activation energies derived (see eqn (4) and (5) and values in Table 3). Independent on the metal doping, the reoxidation reactions showed order of magnitude for the reoxidation rate. The activation energy of HPA-Ta and HPA-Mo is similar and approx. 20 kJ mol−1. A slight difference can be observed for HPA-Nb, with a slightly increased activation energy of 28 kJ mol−1. Thus, the Nb substitution can increase the reoxidation kinetics at high temperatures slightly. Nevertheless, the benchmarking reoxidation rates of vanadium substituted HPAs,27 which can be reoxidized already at 80 °C, cannot be achieved.
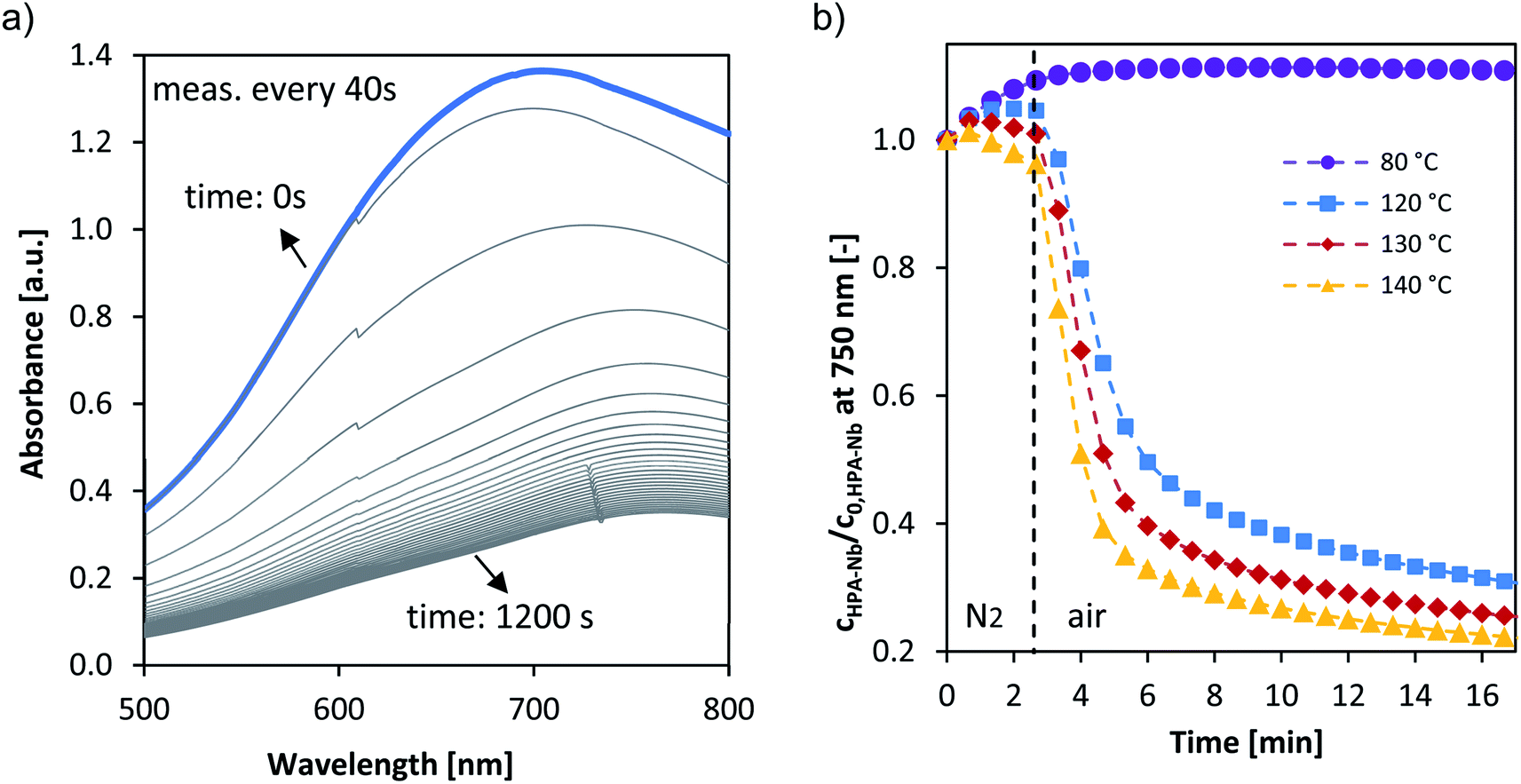 | ||
| Fig. 6 (a) Example absorption spectra of the reoxidation of pre-reduced HPA-Nb at 120 °C (the blue spectra represents t = 0 min). (b) Normalized concentration–time profiles at 750 nm of the reoxidation of pre-reduced HPA-Nb catalyst. Reaction conditions: 80–140 °C, 5 bar synthetic air saturation, 11 μM pre-reduced HPA-Nb initial concentration, LCWM reactor. | ||
| Entry | Catalyst | Reaction order at 120 °C [—] | kreoxidation, at 120 °C [min−1] | Effective activation energy [kJ mol−1] |
|---|---|---|---|---|
| 1 | HPA-Mo26 | 0.99 | 0.219 | 20.43 |
| 2 | HPA-Ta | 0.96 | 0.236 | 19.04 |
| 3 | HPA-Nb | 0.96 | 0.203 | 28.84 |
A variation of the catalyst concentration from 1.8 to 12 μM at 120 °C allowed to deduce the reaction orders regarding the catalyst concentration (Fig. 7a) and first-order rates resulted for all three HPAs (Table 3).
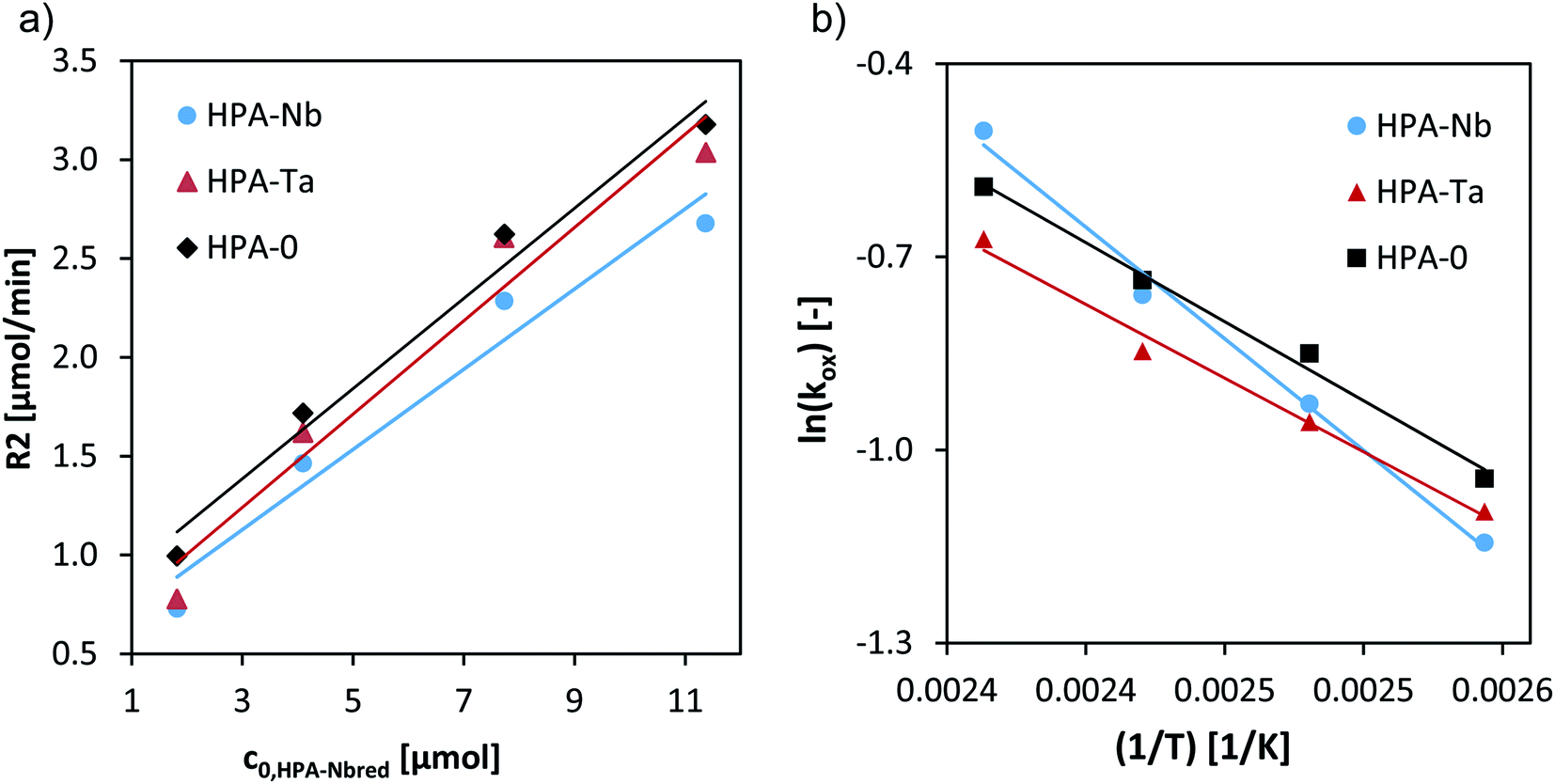 | ||
| Fig. 7 (a) Initial reaction rates vs. initial concentration of HPA-Nbred, HPA-Tared, and HPA-MOred catalysts at 120 °C and Arrhenius plot for the reoxidation of HPA-Nbred, HPA-Tared, HPA-MOred catalysts. Reaction conditions: 80–140 °C, 5 bar synthetic air saturation, 1.8–12 μM pre-reduced HPA-Nb, HPA-Ta, HPA-MO initial concentration, LCWM reactor (see eqn (4) and (5) and Arrhenius plot in (b)). | ||
Conclusion
Selective liquid-phase oxidation of biomass-derived glucose to formic and acetic acid in aqueous media could be demonstrated using Nb- and Ta-doped Mo-based Keggin-type polyoxometalates. Hereby, we could show that these vanadium-free catalysts require significant higher reaction temperatures of 120–160 °C to show full glucose conversion in a high-pressure autoclave setup. The main reaction pathway for the three used Mo-based POM catalysts leads to formic acid yields of around 30% at 160 °C whereby the intermediate glycol aldehyde is the dominant species between 120–140 °C with a maximum yield of 20%. Moreover, only small amounts of acetic acid and its intermediate HMF could be detected for the whole temperature range.Regarding the kinetics of the glucose oxidation step, the Ta- and especially the Nb-doping lead to a significant increase for the measured effective rate constants as well as the effective reaction orders compared to the non-doped HPA-Mo catalyst. Moreover, the effective activation energies could be significantly reduced from 80 kJ mol−1 for the HPA-Mo catalyst to 61 kJ mol−1 for the HPA-Nb and 55 kJ mol−1 for the HPA-Ta catalyst.
The influence of the Nb- and Ta-doping on the reoxidation kinetics was carried out within a LCWM reactor performing a transient response experiment, where for fully reduced HPA catalysts the concentration change was determined after exchanging fast from an inert to an oxygen gas atmosphere. The activation energy for the reoxidation of HPA-Ta and HPA-Mo was similar and approx. 20 kJ mol−1 whereby a slight increase could be observed for HPA-Nb with 28 kJ mol−1. Thus, the Nb substitution can increase the reoxidation kinetics at high temperatures slightly. Nevertheless, the benchmarking reoxidation rates of vanadium substituted HPAs, which could be reoxidized already at 80 °C, could not be achieved.
To sum up, we could show that Mo-based Keggin-type POMs could successfully be applied for selective glucose oxidation especially to glycol aldehyde at temperatures of around 120 °C. With increasing temperature, the consecutive reaction leading to formic acid was favored. The Ta- and especially Nb-doping significantly increases the selectivity-determining substrate oxidation kinetics and thus opens new interesting perspectives for liquid-phase oxidation reactions.
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
DV, SW and JA thank Julian Mehler supporting the catalyst synthesis.References
- J. Albert and P. Wasserscheid, Expanding the scope of biogenic substrates for the selective production of formic acid from water-insoluble and wet waste biomass, Green Chem., 2015, 17, 5164–5171, 10.1039/c5gc01474c.
- J. Albert, R. Wölfel, A. Bösmann and P. Wasserscheid, Selective oxidation of complex, water-insoluble biomass to formic acid using additives as reaction accelerators, Energy Environ. Sci., 2012, 5(7), 7956–7962, 10.1039/c2ee21428h.
- Z. Zhang and G. W. Huber, Catalytic oxidation of carbohydrates into organic acids and furan chemicals, Chem. Soc. Rev., 2018, 47, 1351–1390, 10.1039/c7cs00213k.
- J. Zhang, M. Sun, X. Liu and Y. Han, Catalytic oxidative conversion of cellulosic biomass to formic acid and acetic acid with exceptionally high yields, Catal. Today, 2014, 233, 77–82, DOI:10.1016/j.cattod.2013.12.010.
- T. Lu, M. G. Niu, Y. C. Hou, W. Z. Wu, S. H. Ren and F. Yang, Catalytic oxidation of cellulose to formic acid in H5PV2Mo10O40 + H2SO4 aqueous solution with molecular oxygen, Green Chem., 2016, 18, 4725–4732, 10.1039/c6gc01271j.
- J. Albert, D. Lüders, A. Bösmann, D. M. Guldi and P. Wasserscheid, Spectroscopic and electrochemical characterization of heteropoly acids for their optimized application in selective biomass oxidation to formic acid, Green Chem., 2014, 16, 226–237, 10.1039/c3gc41320a.
- J. Li, D. J. Ding, L. Deng, Q. X. Guo and Y. Fu, Catalytic Air Oxidation of Biomass-Derived Carbohydrates to Formic Acid, ChemSusChem, 2012, 5, 1313–1318, DOI:10.1002/cssc.201100466.
- M. Niu, Y. Hou, S. Ren, W. Wang, Q. Zheng and W. Wu, The relationship between oxidation and hydrolysis in the conversion of cellulose in NaVO3 – H2SO4 aqueous solution with O2, Green Chem., 2015, 17, 335–342, 10.1039/c4gc00970c.
- J. Albert, M. Mendt, M. Mozer and D. Voß, Explaining the role of vanadium in homogeneous glucose transformation reactions using NMR and EPR spectroscopy, Appl. Catal., A, 2019, 570, 262–270, DOI:10.1016/j.apcata.2018.10.030.
- C. L. Hill and C. M. Prosser-McCartha, Homogeneous catalysis by transition metal oxygen anion clusters, Coord. Chem. Rev., 1995, 143, 407–455, DOI:10.1016/0010-8545(95)01141-B.
- A. Proust, B. Matt, R. Villaneau, G. Guillemot, P. Gouzerh and G. Izzet, Functionalization and post-functionalization: a step towards polyoxometalate-based materials, Chem. Soc. Rev., 2012, 41, 7605–7622, 10.1039/c2cs35119f.
- I. V. Kozhevnikov, Catalysis by heteropoly acids and multicomponent polyoxometalates in liquid-phase reactions, Chem. Rev., 1998, 98(1), 171–198, DOI:10.1021/cr960400y.
- H. N. Miras, J. Yan, D. L. Long and L. Cronin, Engineering polyoxometalates with emergent properties, Chem. Soc. Rev., 2012, 41, 7403–7430, 10.1039/c2cs35190k.
- S. Omwoma, C. T. Gore, Y. Ji, C. Hu and Y.-F. Song, Environmentally benign polyoxometalate materials, Coord. Chem. Rev., 2015, 286, 17–29, DOI:10.1016/j.ccr.2014.11.013.
- M. Nyman, Polyoxoniobate chemistry in the 21st century, Dalton Trans., 2011, 40, 8049–8058, 10.1039/c1dt10435g.
- G. Guo, Y. Xu, J. Cao and C. Hu, Synthesis, structures and applications of electron-rich polyoxometalates, Chem. Commun., 2011, 47, 9411–9413, DOI:10.1038/s41570-018-0112.
- J.-H. Son and H. Casey, Reversible capping/uncapping of phosphorous-centered Keggin-type polyoxoniobate clusters, Chem. Commun., 2015, 51, 1436–1438, 10.1039/c4cc05689b.
- M. Nyman, F. Bonhomme, T. M. Alam, M. A. Rodriguez, B. R. Cherry, J. L. Krumhansl, T. M. Nenoff and A. M. Sattler, A general synthetic procedure for heteropolyniobates, Science, 2002, 297, 996–998, DOI:10.1126/science.1073979.
- P. A. Abramov, M. N. Sokolov and C. Vicent, Polyoxoniobates and Polyoxotantalates as Ligands—Revisited, Inorganics, 2015, 3, 160–177, DOI:10.3390/inorganics3020160.
- V. Hessel, P. Angeli, A. Gavriilidis and H. Löwe, Gas−Liquid and Gas−Liquid−Solid Microstructured Reactors: Contacting Principles and Applications, Ind. Eng. Chem. Res., 2005, 44, 9750–9769, DOI:10.1021/ie0503139.
- J. Reichert and J. Albert, Detailed Kinetic Investigations on the Selective Oxidation of Biomass to Formic Acid (OxFA Process) Using Model Substrates and Real Biomass, ACS Sustainable Chem. Eng., 2017, 5, 7383–7392, DOI:10.1021/acssuschemeng.7b01723.
- T. Yokoyama, H.-M. Chang, R. S. Reiner, R. H. Atalla, I. A. Weinstock and J. F. Kadla, Polyoxometalate oxidation of non-phenolic lignin subunits in water: effect of substrate structure on reaction kinetics, Holzforschung, 2004, 58(2), 116–121, DOI:10.1515/HF.2004.016.
- K. Jähnisch, V. Hessel, H. Löwe and M. Baerns, Chemistry in Microstructured Reactors, Angew. Chem., Int. Ed., 2004, 43(4), 406–446, DOI:10.1002/anie.200490000.
- T. Noel and V. Hessel, Membrane microreactors: gas–liquid reactions made easy, ChemSusChem, 2013, 6(3), 405–407, DOI:10.1002/cssc.201200913.
- P. Sobieszuk, J. Aubin and R. Pohorecki, Hydrodynamics and mass transfer in gas-liquid flows in microreactors, Chem. Eng. Technol., 2012, 35(8), 1346–1358, DOI:10.1002/ceat.201100643.
- S. Ponce, H. Christians, A. Drochner and B. J. M. Etzold, An Optical Microreactor Enabling In Situ Spectroscopy Combined with Fast Gas-Liquid Mass Transfer, Chem. Ing. Tech., 2018, 90, 1855–1863, DOI:10.1002/cite.201800061.
- S. Ponce, M. Trabold, A. Drochner, J. Albert and B. J. M. Etzold, Insights into the redox kinetics of vanadium substituted heteropoly through liquid core waveguide membrane microreactor studies, Chem. Eng. J., 2019, 369, 443–450, DOI:10.1016/j.cej.2019.03.103.
- V. F. Odyakov and E. G. Zhizhina, A novel method of the synthesis of molybdovanadophosphoric heteropoly acid solutions, React. Kinet. Catal. Lett., 2008, 95, 21–29, DOI:10.1007/s11144-008-5374-7.
- M. Nyman, F. Bonhomme, T. M. Alam, M. A. Rodriguez, B. R. Cherry, J. L. Krumhansl, T. M. Nenoff and A. M. Sattler, A General Synthetic Procedure for Heteropolyniobates, Science, 2002, 297, 996–998, DOI:10.1126/science.1073979.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra05544d |
| This journal is © The Royal Society of Chemistry 2019 |