
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAsymmetric retro-[1,4]-Brook rearrangement of 3-silyl allyloxysilanes via chirality transfer from silicon to carbon†
Ya Wua,
Hua Chenb,
Wenyu Yangb,
Yu Fanb,
Lu Gao*b,
Zhishan Su c,
Changwei Huc and
Zhenlei Song*b
c,
Changwei Huc and
Zhenlei Song*b
aDepartment of Biological and Chemical Engineering, Chongqing University of Education, Chongqing 400067, China
bSichuan Engineering Laboratory for Plant-Sourced Drug, Research Center for Drug Industrial Technology, Key Laboratory of Drug-Targeting and Drug Delivery System of the Education Ministry, West China School of Pharmacy, Sichuan University, Chengdu, 610041, China. E-mail: lugao@scu.edu.cn; zhenleisong@scu.edu.cn
cKey Laboratory of Green Chemistry & Technology, Ministry of Education, College of Chemistry, Sichuan University, Chengdu, 610064, China
First published on 21st August 2019
Abstract
An asymmetric retro-[1,4]-Brook rearrangement of 3-silyl allyloxysilanes has been developed via Si-to-C chirality transfer. Mechanistic studies reveal that the silyl group migrates with retention of configuration. The stereochemical outcome of the newly formed stereogenic carbon center, which has remained a longstanding question, is also clarified, suggesting a diastereoselective Si to C chirality transfer without loss of enantiomeric excess.
Introduction
Intramolecular O-to-C silyl migration, now called retro-Brook (or West) rearrangement, was first reported by Speier and later systematically studied by West.1 The retro-Brook rearrangement occurs only under special circumstances2 and so has been less investigated than Brook rearrangement.3 But, it comprises a powerful synthetic tool because diverse organosilanes could be constructed from more accessible silyl ethers by a rapid and regio- and stereoselective manner. A covalent Si–O bond is cleaved and a Si–C bond is formed via silyl migration. Thus, the stereochemical courses at the migrating silicon center and the stereochemical control at the forming carbon center comprise two important stereochemical issues. Tomooka and co-workers4 reported the first example of practically useful level of retro-[1,4]-Brook rearrangement of allyloxysilane by use of HMPA as a co-solvent. In this work, they also showed, for the first time, that the silyl migration proceeded with retention of configuration at the silicon center (Scheme 1). In contrast, more efforts have been directed toward diastereoselective formation of the Si–C bond to generate synthetically useful chiral organosilanes. Nearly all previous studies have used stereogenic C1,2b,d,e C2 (ref. 2h) or C3 (ref. 2a and j) centers in substrate I to control diastereoselective formation of the Si–C bond in III. When the migrating silicon is stereogenic,5 it might be used as a stereochemical controller by Si to C chirality transfer, which was redefined by Oestreich.6 Achieving this in practice is quite challenging. There are only two examples we know come from a preliminary study by Tomooka and co-workers.4 The 3-Me allyloxysilane with SiOMePht-Bu as the migrating silyl group afforded a dr of 83![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :17, while the corresponding 3-SiMe3 allyloxysilane only migrated with a dr of 66:34. In both cases, the stereochemistry of the formed stereogenic carbon center were not determined.
:17, while the corresponding 3-SiMe3 allyloxysilane only migrated with a dr of 66:34. In both cases, the stereochemistry of the formed stereogenic carbon center were not determined.
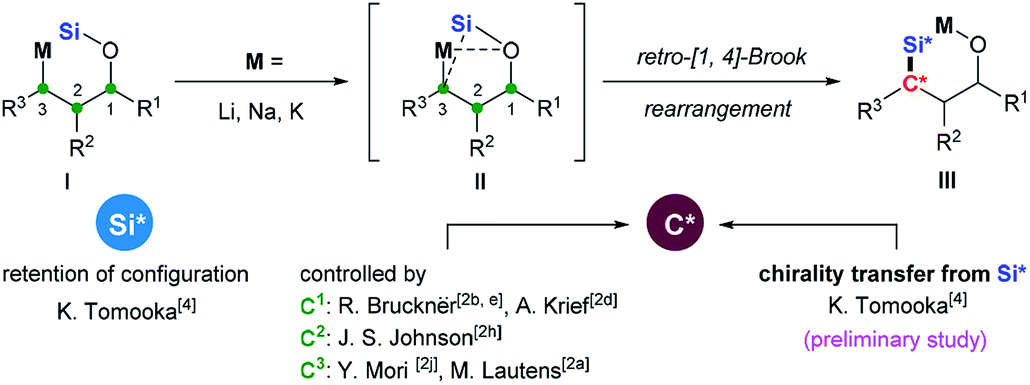 | ||
| Scheme 1 Diastereoselective retro-Brook rearrangement. | ||
Oestreich rationalized the difficulties in achieving high diastereoselectivity during Si-to-C chirality transfer as follows.6 The relatively long Si–C bond disfavors formation of a compact transition state II, which weakens diastereoselectivity. At the same time, all three substituents on the migrating silyl group can affect the stereochemical course, requiring the careful selection of three substituents that together allow efficient stereochemical control. Despite these difficulties, Oestreich described an intermolecular Pd- catalyzed asymmetric hydrosilylation using chiral silane (Scheme 2a),7 and Leighton demonstrated an intramolecular Hosomi–Sakuraiallylation involving a chiral allylsilane intermediate (Scheme 2b).8 In both of these cases, either cyclic silanes or acyclic silane with three distinct, sterically demanding substituents were used to achieve the high stereochemical control.
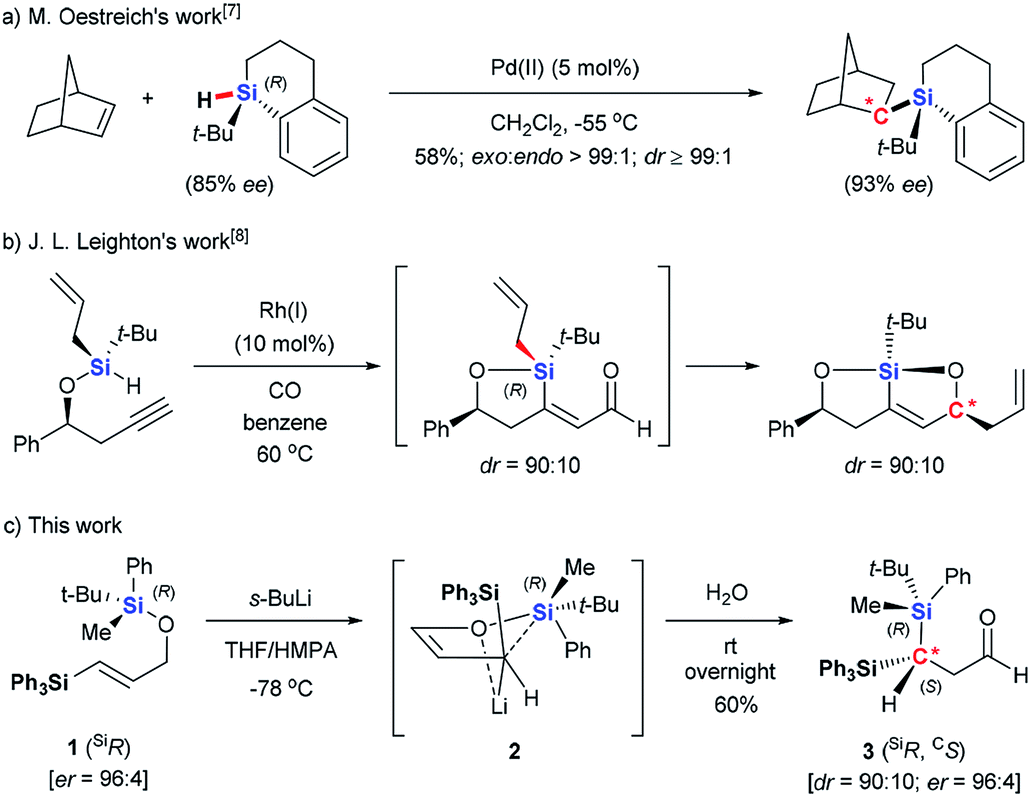 | ||
| Scheme 2 Chirality Transfer from Silicon to Carbon. | ||
Here we report an asymmetric retro-[1,4]-Brook rearrangement of 3-silyl allyloxysilanes 1 via an efficient Si-to-C chirality transfer (Scheme 2c). The combination of SiMePht-Bu as the migrating silyl group and SiPh3 as the terminal silyl group proved most effective, giving geminal bis(silyl) aldehyde 3 and enol derivatives 4 in good yield with high diastereoselectivity. The overall stereochemical outcome of the migrating silicon center and the newly formed carbon center were clarified by detailed mechanistic studies.
Results and discussion
This project arose from our interest in developing chiral geminal bis(silanes) reagents and synthons.9 These species contain two different silyl groups, making the carbon to which they are attached a stereogenic center. In previous work, we achieved asymmetric C–C or C–H bonds formation via 3,3-sigma tropic rearrangement of optically pure 3,3-bis(silyl) allylic alcohols, allowing asymmetric synthesis of crotyl geminal bis(silanes).10 We were curious whether asymmetric C–Si bond formation could be another efficient strategy to construct chiral geminal bis(silanes). Our s-BuLi-promoted retro-[1,4]-Brook rearrangement of 3-silyl allyloxysilanes11 appeared to be a suitable model to test this possibility. The reaction tolerates a wide range of migrating and non-migrating silyl groups, making it practical for identifying the best pair of silyl groups.We initially fixed t-BuPhMeSi as the migrating silyl group (Table 1). Entries 1–7 showed an obvious steric bias for the non-migrating silicons (Si1) at the 3-position of 1. When Si1 was an SiMe3 group, geminal bis(silyl) aldehydes 3a were generated as a nearly 1:1 mixture of two diastereomers (entry 1). Even when one methyl was replaced with a phenyl group, dr did not improve for the corresponding products 3b (entry 2). These results imply that the small methyl group does not permit good diastereochemical control. Diastereoselectivity improved progressively when steric demand at Si1 increased from SiMe3 to SiEt3, Si(n-Pr)3, Si(i-Pr)3 and finally SiPh3 (entries 3–6). The largest SiPh3 group imposed the strongest stereochemical control, providing 3f at the highest dr of 90:10 (entry 6). Interestingly, an inverse steric bias for Si1 was observed when the migrating silicon was switched from t-BuPhMeSi to 1-NpPhMeSi. The largest SiPh3 group afforded a dr of only 65:35, while the smallest SiMe3 provided the best dr of 86:14 (entries 7–9). We also tested the silicon combination in which the t-BuPhMeSi functioned as a chiral auxiliary, while SiPh3 migrated (entry 10). The reaction gave the aldehyde 3f with a dr of 74:26 lower than that obtained in entry 5.
|
||||||
|---|---|---|---|---|---|---|
| Entry | Sub. | Si1 | Si2 | Prod. | Yieldb | drc |
| a Reaction conditions: 1 (0.15 mmol), s-BuLi (0.60 mmol), HMPA (0.6 mmol), 0.5 mL of THF, at −78 °C for 0.5 h, then H2O at rt overnight.b Isolated yields.c Ratios were determined from 1H NMR analysis of crude product. | ||||||
| 1 | 1a | Me3Si | t-BuPhMeSi | 3a | 62% | 56:44 |
| 2 | 1b | Me2PhSi | t-BuPhMeSi | 3b | 65% | 47:53 |
| 3 | 1c | Et3Si | t-BuPhMeSi | 3c | 53% | 67:33 |
| 4 | 1d | (n-Pr)3Si | t-BuPhMeSi | 3d | 50% | 65:35 |
| 5 | 1e | (i-Pr)3Si | t-BuPhMeSi | 3e | 45% | 83:17 |
| 6 | 1f | Ph3Si | t-BuPhMeSi | 3f | 60% | 90:10 |
| 7 | 1g | Ph3Si | 1-NpPhMeSi | 3g | 65% | 65:35 |
| 8 | 1h | Et3Si | 1-NpPhMeSi | 3h | 73% | 83:17 |
| 9 | 1i | Me3Si | 1-NpPhMeSi | 3i | 63% | 86:14 |
| 10 | 1j | t-BuPhMeSi | Ph3Si | 3f | 55% | 74:26 |
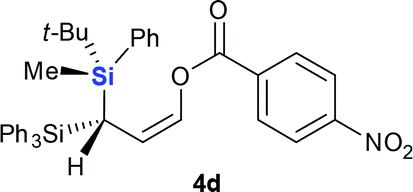
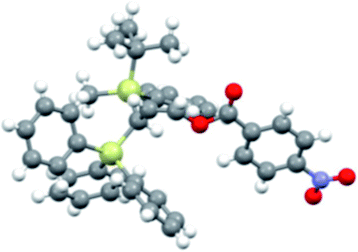
Next we examined the scope of electrophiles for quenching the lithium enolate intermediate generated from 1f. The reaction tolerated triethylsilyl chloride (entry 1), various acyl chlorides (entries 2–9) and chlorocarbonates (entries 10 and 11) to provide 3,3-bis(silyl) enol derivatives 4 in good yields with high diastereoselectivity (Table 2). The enol double bond formed exclusively with Z-selectivity. The relative stereochemistry of the products was unambiguously established based on X-ray diffraction analysis of 4d crystals.12 Methyl iodide was also a suitable electrophile, but less reactive than acyl chloride, giving 4l in 40% yield with O-alkylation selectivity (entry 12).
|
|||
|---|---|---|---|
| Entry | Electrophiles | Product | Yieldb |
| a Reaction conditions: 1f (0.15 mmol), s-BuLi (0.60 mmol), HMPA (0.6 mmol), 0.5 mL of THF, at −78 °C for 0.5 h, then electrophile at rt for 2 h.b Isolated yields.c Ratios were determined from crude 1H NMR analysis of product.d 10.0 equiv. of MeI. | |||
| 1 | Et3SiCl | 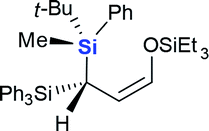 |
4a (60%) |
| 2 | 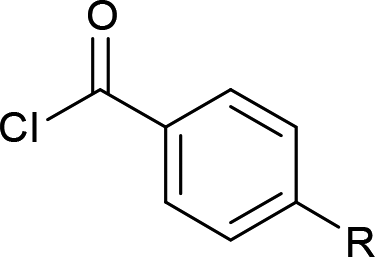 |
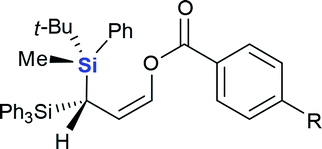 |
4b (R = H, 65%) |
| 3 | 4c (R = Br, 65%) | ||
| 4 | 4d (R = NO2, 60%) | ||
| 5 | 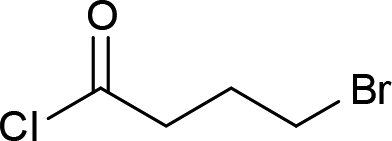 |
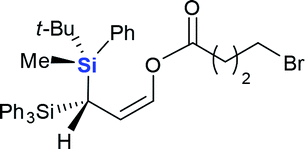 |
4e (67%) |
| 6 | 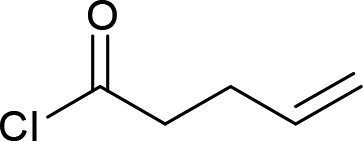 |
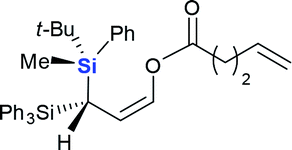 |
4f (50%) |
| 7 | 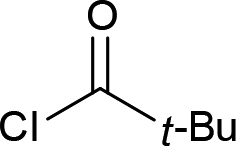 |
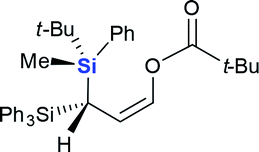 |
4g (66%) |
| 8 | 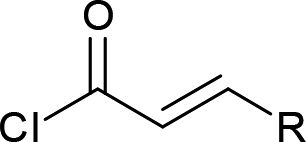 |
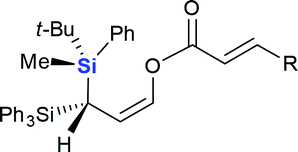 |
4h (R = Me, 50%) |
| 9 | 4i (R = Ph, 55%) | ||
| 10 | 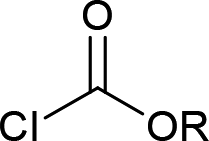 |
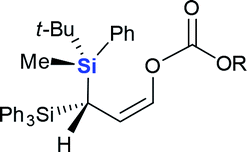 |
4j (R = Me, 70%) |
| 11 | 4k (R = Ph, 70%) | ||
| 12 | Meld | 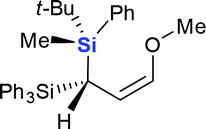 |
4l (40%) |
 |
 |
||
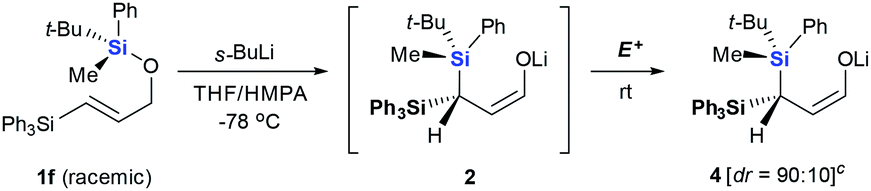
The silicon can migrate with either retention or inversion of configuration. Thus, the relative stereochemistry of 3f may not reflect the stereochemical course of the migrating silicon, or how it controls the stereochemical outcome of the resulting stereogenic carbon center. In particular, if the enantiomerically defined silyl group racemizes during migration, the carbon center can be constructed diastereoselectively, but not enantioselectively. The observation by Tomooka and co-workers that silicon migrates with retention of configuration in their simple allyloxy system4 does not necessarily apply to our case, since the non-migrating silicon may affect the stereochemical course. To gain a definitive answer to this question, we used enantiomerically defined 10 as a stereochemical probe (Scheme 3).13 Following the procedure developed by Oestreich,14 a 1:1 mixture of 5 was separated by several cycles of silica gel chromatography, affording 6 in diastereomerically pure form. Reduction of 6 with DIBAL-H provided hydrosilane 7 in 77% yield. Subsequent chlorination of 7 with CCl4 delivered chlorosilane 8, which directly reacted with the potassium salt of 9, giving 10 in 90% yield. The high er of 10 (96:4) suggests that transformation from 6 to 10 proceeds in a stereospecific manner at the silicon center, and should follow the known sequence of retention-retention-inversion.15 Thus, the absolute configuration of the silicon in 10 was assigned as R. Under the optimal retro-[1,4]-Brook rearrangement conditions, 10 was converted into aldehyde 11. The major isomer showed an er of 96:4, indicating that the silicon migrated stereospecifically. X-ray diffraction analysis of 1116 unambiguously confirmed the R-configuration of the silicon, indicating that migration proceeds with retention of configuration as in Tomooka's case. The X-ray diffraction analysis of 11 also established the S-configuration of the new stereogenic carbon center. The result revealed that the migration proceeded by a diastereoselective Si to C chirality transfer without loss of enantiomeric excess.
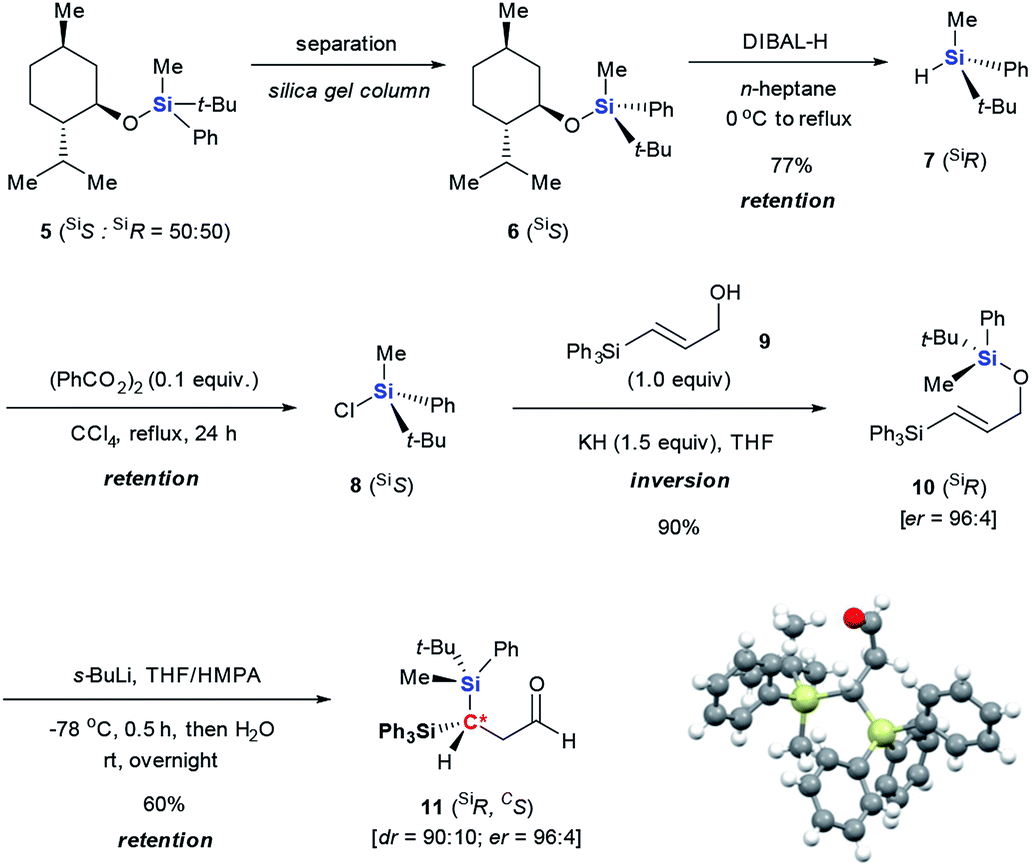 | ||
| Scheme 3 Preparation of enantiomerically defined 10 and its retro-[1,4]-Brook rearrangement to form 11. | ||
A plausible mechanism to explain our results is proposed in Scheme 4, based on the model we proposed for the racemic version of the reaction.11 The α-deprotonation of 10 gives the corresponding allylic anion, which adopts the endo-orientation assisted by Li–O coordination.17 The O-to-C silyl migration takes place irreversibly via two possible pentacoordinated silicate transition states or intermediates, TS-1 and TS-2.18 In this way, the configuration of the silicon center is retained without racemization. While TS-2 suffers a severely steric repulsion between the Ph group on Si1 and one of the Ph groups on Si2, the interaction between the Me group on Si1 and the Ph group on Si2 appears being tolerable in the case of TS-1. These considerations are supported by the preliminary results from density functional theory calculations, which showed TS-1 to be more stable than TS-2 by 6.2 kJ mol−1. Our model also explains the observed steric bias for substituents on Si1. Substituents smaller than the Ph group might not be large enough to create an appreciable difference between the non-bonded interaction with the Me group in TS-1 and with the Ph group in TS-2. As a result, 3 forms with poor diastereoselectivity (Table 1, entries 1–4).
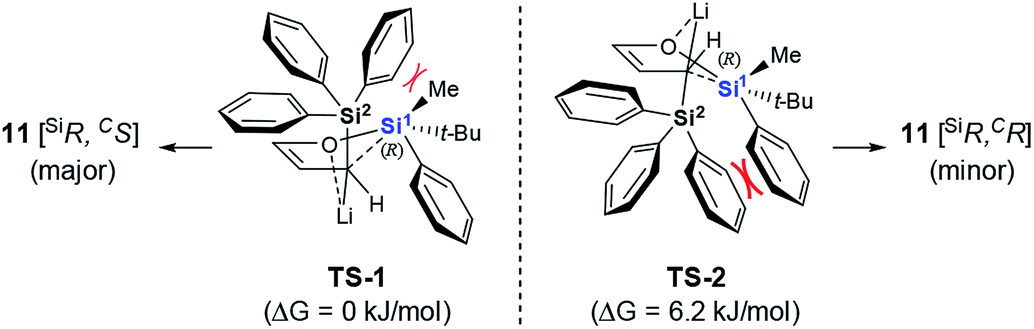 | ||
| Scheme 4 Plausible reaction mechanism. | ||
Conclusions
In summary, Si-to-C chirality transfer has been used as an efficient strategy to achieve asymmetric retro-[1,4]-Brook rearrangement of 3-silyl allyloxysilanes. The SiMePht-Bu and SiPh3 groups, in which SiMePht-Bu migrates, function as the best combination to give geminal bis(silyl) aldehyde and enol derivatives with high diastereoselectivity. The silyl group migrates with retention of configuration. Enantioselective generation of the stereogenic carbon center suggests that Si-to-C chirality transfer is a promising method to construct optically pure chiral organosilanes. Further applications of this strategy are being explored in our group.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for financial support from the NSFC (21622202, 21502125).Notes and references
- (a) J. L. Speier, J. Am. Chem. Soc., 1952, 74, 1003 CrossRef CAS; (b) R. West, R. Lowe, H. F. Stewart and A. Wright, J. Am. Chem. Soc., 1971, 93, 282 CrossRef CAS.
- (a) M. Lautens, P. H. M. Delanghe, J. B. Goh and C. H. Zhang, J. Org. Chem., 1992, 57, 3270 CrossRef CAS; (b) C. Gibson, T. Buck, M. Noltemeyer and R. Brückner, Tetrahedron Lett., 1997, 38, 2933 CrossRef CAS; (c) T. N. Mitchell, M. Schtüze and F. Gießelmann, Synlett, 1997, 183 CrossRef CAS; (d) J. Bousbaa, F. Ooms and A. Krief, Tetrahedron Lett., 1997, 38, 762 CrossRef; (e) C. Gibson, T. Buck, M. Walker and R. Brückner, Synlett, 1998, 201 CrossRef CAS; (f) T. N. Mitchell and M. Schütze, Tetrahedron, 1999, 55, 1285 CrossRef CAS; (g) B. M. Comanita, S. Woo and A. G. Fallis, Tetrahedron Lett., 1999, 40, 5283 CrossRef CAS; (h) M. R. Nahm, X. L. Hu, J. R. Potnick, C. M. Yates, P. S. White and J. S. Johnson, Angew. Chem., Int. Ed., 2005, 44, 2377 CrossRef CAS PubMed; (i) E. N. Onyeozili and R. E. Maleczka, Tetrahedron Lett., 2006, 47, 6565 CrossRef CAS; (j) Y. Mori, Y. Futamura and K. Horisaki, Angew. Chem., Int. Ed., 2008, 47, 1091 CrossRef CAS PubMed; (k) J. A. Brekan, D. Chernyak, K. L. White and K. A. Scheidt, Chem. Sci., 2012, 3, 1205 RSC; (l) Y. P. He, H. T. Hu, X. G. Xie and X. G. She, Tetrahedron, 2013, 69, 559 CrossRef CAS; (m) Y. P. He, B. Ma, J. Yang, X. G. Xie and X. G. She, Tetrahedron, 2013, 69, 5545 CrossRef CAS; (n) V. Bariak, A. Malastová, A. Almássy and R. Šebesta, Chem.–Eur. J., 2015, 21, 13445 CrossRef CAS PubMed; (o) Y. J. Kwon, Y. K. Jeon, H. B. Sim, I. Y. Oh, I. Shin and W. S. Kim, Org. Lett., 2017, 19, 6224 CrossRef CAS PubMed; (p) L. Biancalana, S. Zacchini, N. Ferri, M. G. Lupo, G. Pampaloni and F. Marchetti, Dalton Trans., 2017, 46, 16589 RSC.
- For reviews, see: (a) A. G. Brook, Acc. Chem. Res., 1974, 7, 77 CrossRef CAS; (b) P. C. Bulman-Page, S. S. Klair and S. Rosenthal, Chem. Soc. Rev., 1990, 19, 147 RSC; (c) P. Jankowski, P. Raubo and J. Wicha, Synlett, 1994, 985 CrossRef CAS; (d) M. A. Brook, Silicon in Organic, Organometallic, and Polymer Chemistry, John Wiley and Sons, New York, 2000 Search PubMed; (e) W. H. Moser, Tetrahedron, 2001, 57, 2065 CrossRef CAS; (f) M. Kira and T. Iwamoto, The Chemistry of Organic Silicon Compounds, Z. Rappoport and Y. Apeloig, ed. John Wiley and Sons, New York, 2001, pp. 853–948 Search PubMed; (g) E. Schaumann and A. Kirschning, Synlett, 2007, 177 CrossRef CAS; (h) A. B. Smith and C. M. Adams, Acc. Chem. Res., 2004, 37, 365 CrossRef CAS PubMed; (i) H. J. Zhang, D. L. Priebbenow and C. Bolm, Chem. Soc. Rev., 2013, 42, 8540 RSC; (j) M. Sasaki and K. Takeda, Molecular Rearrangements in Organic Synthesis, ed. C. M. Rojas, John Wiley and Sons: New York, 2015, pp. 151–183 Search PubMed; (k) G. Eppe, D. Didier and I. Marek, Chem. Rev., 2015, 115, 9175 CrossRef CAS PubMed.
- A. Nakazaki, T. Nakai and K. Tomooka, Angew. Chem., Int. Ed., 2006, 45, 2235 CrossRef CAS PubMed.
- For reviews of enatioselective generation of stereogenic silicon center, see: (a) M. Oestreich, Synlett, 2007, 1629 CrossRef CAS; (b) L. W. Xu, L. Li, G. Q. Lai and J. X. Jiang, Chem. Soc. Rev., 2011, 40, 1777 RSC; (c) L. W. Xu, Angew. Chem., Int. Ed., 2012, 51, 12932 CrossRef CAS PubMed; (d) Y. Wu, L. Gao and Z. L. Song, Chem. Bull., 2015, 78, 676 CAS; (e) R. Shintani, Asian J. Org. Chem., 2015, 4, 510 CrossRef CAS; (f) J. O. Bauer and C. Strohmann, Eur. J. Inorg. Chem., 2016, 2868 CrossRef CAS; (g) Y. M. Cui, Y. Lin and L. W. Xu, Coord. Chem. Rev., 2017, 330, 37 CrossRef CAS. For selected progresses, see: (h) K. Igawa, J. Takada, T. Shimono and K. Tomooka, J. Am. Chem. Soc., 2008, 130, 16132 CrossRef CAS PubMed; (i) Y. Yasutomi, H. Suematsu and T. Katsuki, J. Am. Chem. Soc., 2010, 132, 4510 CrossRef CAS PubMed; (j) R. Shintani, H. Otomo, K. Ota and T. Hayashi, J. Am. Chem. Soc., 2012, 134, 7305 CrossRef CAS PubMed; (k) J. O. Bauer and C. Strohmann, Angew. Chem., Int. Ed., 2014, 53, 720 CrossRef CAS PubMed; (l) R. Shintani, C. Takagi, T. Ito, M. Naito and K. Nozaki, Angew. Chem., Int. Ed., 2015, 54, 1616 CrossRef CAS PubMed; (m) R. Shintani, R. Takano and K. Nozaki, Chem. Sci., 2016, 7, 1205 RSC; (n) L. Chen, J. B. Huang, Z. Xu, Z. J. Zheng, K. F. Yang, Y. M. Cui, J. Cao and L. W. Xu, RSC Adv., 2016, 6, 67113 RSC; (o) K. Igawa, D. Yoshihiro, Y. Abe and K. Tomooka, Angew. Chem., Int. Ed., 2016, 55, 5814 CrossRef CAS PubMed; (p) Q. W. Zhang, K. An, L. C. Liu, Q. Zhang, H. F. Guo and W. He, Angew. Chem., Int. Ed., 2017, 56, 1125 CrossRef CAS PubMed; (q) X. F. Bai, J. F. Zou, M. Y. Chen, Z. Xu, L. Li, Y. M. Cui, Z. J. Zheng and L. W. Xu, Chem.–Asian J., 2017, 12, 1730 CrossRef CAS PubMed; (r) Y. Sato, C. Takagi, R. Shintani and K. Nozaki, Angew. Chem., Int. Ed., 2017, 56, 9211 CrossRef CAS PubMed; (s) G. Zhan, H. L. Teng, Y. Luo, S. J. Lou, M. Nishiura and Z. M. Hou, Angew. Chem., Int. Ed., 2018, 57, 12342 CrossRef CAS PubMed; (t) H. Chen, Y. Chen, X. X. Tang, S. F. Liu, R. P. Wang, T. B. Hu, L. Gao and Z. L. Song, Angew. Chem., Int. Ed., 2019, 58, 4695 CrossRef CAS PubMed.
- M. Oestreich, Chem.–Eur. J., 2006, 12, 30 CrossRef CAS PubMed.
- (a) M. Oestreich and S. Rendler, Angew. Chem., Int. Ed., 2005, 44, 1661 CrossRef CAS PubMed; (b) S. Rendler and M. Oestreich, Beilstein J. Org. Chem., 2007, 3, 9 Search PubMed.
- D. R. Schmidt, S. J. O'Malley and J. L. Leighton, J. Am. Chem. Soc., 2003, 125, 1190 CrossRef CAS PubMed.
- (a) Q. Luo, C. Wang, Y. X. Li, K. B. Ouyang, L. Gu, M. Uchiyama and Z. F. Xi, Chem. Sci., 2011, 2, 2271 RSC; (b) H. Li, L. T. Liu, Z. T. Wang, F. Zhao, S. G. Zhang, W. X. Zhang and Z. F. Xi, Chem.–Eur. J., 2011, 17, 7399 CrossRef CAS PubMed; (c) K. Groll, S. M. Manolikakes, X. M. du Jourdin, M. Jaric, A. Bredihhin, K. Karaghiosoff, T. Carell and P. Knochel, Angew. Chem., Int. Ed., 2013, 52, 6776 CrossRef CAS PubMed; (d) H. Y. Cui, J. Y. Zhang and C. M. Cui, Organometallics, 2013, 32, 1 CrossRef CAS; (e) S. T. Ding, L. J. Song, L. W. Chung, X. H. Zhang, J. W. Sun and Y. D. Wu, J. Am. Chem. Soc., 2013, 135, 13835 CrossRef CAS PubMed; (f) V. Werner, T. Klatt, M. Fujii, J. Markiewicz, Y. Apeloig and P. Knochel, Chem.–Eur. J., 2014, 20, 8338 CrossRef CAS PubMed; (g) R. Shintani, H. Kurata and K. Nozaki, Chem. Commun., 2015, 51, 11378 RSC; (h) Z. X. Liu, H. C. Tan, T. R. Fu, Y. Xia, D. Qiu, Y. Zhang and J. B. Wang, J. Am. Chem. Soc., 2015, 137, 12800 CrossRef CAS PubMed; (i) Z. J. Liu, X. L. Lin, N. Yang, Z. S. Su, C. W. Hu, P. H. Xiao, Y. Y. He and Z. L. Song, J. Am. Chem. Soc., 2016, 138, 1877 CrossRef CAS PubMed; (j) T. Kosai, S. Ishida and T. Iwamoto, Angew. Chem., Int. Ed., 2016, 55, 15554 CrossRef CAS PubMed; (k) M. Das, A. Manvar, M. Jacolot, M. Blangetti, R. C. Jones and D. F. O'Shea, Chem.–Eur. J., 2015, 21, 8737 CrossRef CAS PubMed; (l) M. Das and D. F. O'Shea, Org. Lett., 2016, 18, 336 CrossRef CAS PubMed; (m) Y. Mizuhata, S. Fujimori, T. Sasamori and N. Tokitoh, Angew. Chem., Int. Ed., 2017, 56, 4588 CrossRef CAS PubMed; (n) H. Kinoshita, A. Ueda, H. Fukumoto and K. Miura, Org. Lett., 2017, 19, 882 CrossRef CAS PubMed; (o) Y. B. Zhang, Q. Y. Guo, X. W. Sun, J. Lu, Y. J. Cao, Q. Pu, Z. W. Chu, L. Gao and Z. L. Song, Angew. Chem., Int. Ed., 2018, 57, 942 CrossRef CAS PubMed; (p) H. Hazrati and M. Oestreich, Org. Lett., 2018, 20, 5367 CrossRef CAS PubMed; (q) S. Xu, R. Chen, Z. H. Fu, Y. P. Gao and J. B. Wang, J. Org. Chem., 2018, 83, 6186 CrossRef CAS PubMed; (r) M. H. Yang, J. Lian, W. Sun, T. Z. Qiao and S. F. Zhu, J. Am. Chem. Soc., 2019, 141, 4579 CrossRef PubMed; (s) J. Guo, H. L. Wang, S. P. Xing, X. Hong and Z. Lu, Chem, 2019, 5, 881 CrossRef CAS.
- (a) Z. W. Chu, K. Wang, L. Gao and Z. L. Song, Chem. Commun., 2017, 53, 3078 RSC; (b) Y. Chu, Q. Pu, Z. X. Tang, L. Gao and Z. L. Song, Tetrahedron, 2017, 73, 3707 CrossRef CAS.
- Z. L. Song, Z. Lei, L. Gao, X. Wu and L. J. Li, Org. Lett., 2010, 12, 5298 CrossRef CAS PubMed.
- CCDC 1901967 [4d] contains the supplementary crystallographic data for this paper.†.
- (a) S. Rendler, M. Oestreich, C. P. Butts and G. C. Lloyd-Jones, J. Am. Chem. Soc., 2007, 129, 502 CrossRef CAS PubMed; (b) S. Rendler and M. Oestreich, Angew. Chem., Int. Ed., 2008, 47, 5997 CrossRef CAS PubMed; (c) K. Igawa, D. Yoshihiro, N. Ichikawa, N. Kokan and K. Tomooka, Angew. Chem., Int. Ed., 2012, 51, 12745 CrossRef CAS PubMed; (d) T. T. Metsänen, P. Hrobárik and M. Oestreich, J. Am. Chem. Soc., 2014, 136, 6912 CrossRef PubMed; (e) T. Fallon and M. Oestreich, Angew. Chem., Int. Ed., 2015, 54, 12488 CrossRef CAS PubMed.
- V. T. Trepohl, R. Fröhlich and M. Oestreich, Tetrahedron, 2009, 65, 6510 CrossRef CAS.
- Reduction of (R)-10 with DIBAL-H giving (S)-7 in 81% yield. This result supports that the configuration of the silicon center in 10 should be R. See ESI† for details.
- CCDC 1901968 [11] contains the supplementary crystallographic data for this paper.†.
- (a) W. C. Still and T. L. Macdonald, J. Am. Chem. Soc., 1974, 96, 5561 CrossRef CAS; (b) M. Schlosser and S. Strunk, Tetrahedron, 1989, 45, 2649 CrossRef CAS; (c) A. R. Katritzky, M. Piffl, H. Lang and E. Anders, Chem. Rev., 1999, 99, 665 CrossRef CAS PubMed.
- (a) A. Wright and R. West, J. Am. Chem. Soc., 1974, 96, 3227 CrossRef CAS; (b) T. Kawashima, K. Naganuma and R. Okazaki, Organometallics, 1998, 17, 367 CrossRef CAS; (c) K. Naganuma, T. Kawashima and R. Okazaki, Chem. Lett., 1999, 1139 CrossRef CAS; (d) T. Kawashima, J. Organomet. Chem., 2000, 611, 256 CrossRef CAS; (e) T. Kawashima, Bull. Chem. Soc. Jpn., 2003, 76, 471 CrossRef CAS; (f) E. P. A. Couzijn, M. Schakel, F. J. J. de Kanter, A. W. Ehlers, M. Lutz, A. L. Spek and K. Lammertsma, Angew. Chem., Int. Ed., 2004, 43, 3440 CrossRef CAS PubMed; (g) Q. Liu, Y. Chen, X. Zhang, K. N. Houk, Y. Liang and A. B. Smith, J. Am. Chem. Soc., 2017, 139, 8710 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterisation data for new compounds. CCDC 1901967 and 1901968. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra05482k |
| This journal is © The Royal Society of Chemistry 2019 |