
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInhibitor-conjugated harmonic nanoparticles targeting fibroblast activation protein†
Raphaël
De Matos
a,
Jérémy
Vuilleumier
a,
Christophe
Mas
b,
Samuel
Constant
bc,
Davide
Staedler
 d and
Sandrine
Gerber-Lemaire
*a
d and
Sandrine
Gerber-Lemaire
*a
aInstitute of Chemical Sciences and Engineering, Ecole Polytechnique Fédérale de Lausanne, Group for Functionalized Biomaterials, EPFL SB ISIC SCI-SB-SG, Station 6, CH-1015 Lausanne, Switzerland. E-mail: Sandrine.Gerber@epfl.ch
bOncotheis, 18 chemin des Aulx, CH-1228 Plan-les-Ouates, Geneva, Switzerland
cEpithelix, 18 chemin des Aulx, CH-1228 Plan-les-Ouates, Geneva, Switzerland
dDepartment of Pharmacology and Toxicology, Faculty of Biology and Medicine, University of Lausanne, Lausanne, CH-1011, Switzerland
First published on 4th October 2019
Abstract
The recent progress in the engineering of nanosized inorganic materials presenting tailored physical properties and reactive surface for post-functionalization has opened promising avenues for the use of nanoparticles (NPs) in diagnosis and therapeutic intervention. Surface decoration of metal oxide NPs with ligands modulating circulation time, cellular uptake, affinity and extravasation through active targeting led to efficient cancer specific bioimaging probes. The most relevant cancer biomarkers studied so far include surface and transmembrane cancer cell receptors. More recently, tumor microenvironments and more specifically the fibroblastic element of the tumor stroma have emerged as a valuable target for diagnosis and treatment of several types of cancers. In this study, a low molecular weight ligand targeting fibroblast activation protein α (FAP), which is specifically expressed by activated fibroblasts of the tumor stroma, was synthesized. This ligand demonstrated nanomolar inhibition of FAP with high selectivity with respect to prolyl oligopeptidase (PREP) and dipeptidyl peptidase (DPP) IV, as well as good biocompatibility toward a human lung tissue model. Bismuth ferrite (BFO) harmonic nanoparticles (HNPs) conjugated to this ligand showed target-specific association to FAP as demonstrated by reverse ELISA-type assay using Human Fibroblast Activation Protein alpha/FAP Alexa Fluor® 594-conjugated Antibody and multiphoton multispectral microscopy experiments. These functionalized HNPs may provide new nanocarriers to explore the role of FAP in tumorigenesis and to target the fibroblastic component of the tumor microenvironment.
Introduction
The ability to produce inorganic and organic nanoparticles (NPs) of tunable size and composition, combined with their high agent loading capacity and surface properties suitable for tailored chemical modifications, have generated high expectations that nanomaterials could provide unprecedented diagnosis and therapeutic tools.1 In particular, inorganic nanostructures appear as highly promising candidates for cancer diagnosis and therapy due to their intrinsic optical and physical properties, as well as their capacity to be transformed into multifunctional platforms for the co-delivery of multiple payloads.2 In addition, the opportunity to activate modalities (e.g. photothermal and photodynamic effects) can enhance and complement the chemotherapeutic effects.3 Major achievements in the field were reported with silica-based nanocarriers,4–7 magnetic NPs,8,9 gold nanocomposites10–15 and quantum dots.16 While conventional chemotherapies are generally poorly selective toward specific tissues, cells and intracellular compartments, inorganic NPs benefit from specific uptake by macrophages of the reticuloendothelial system or from the enhanced permeability and retention (EPR) effect.17 Substantial advances in the design of NP-based delivery systems was achieved by the introduction of targeting ligands at the surface of the nanocarriers to promote specific interaction with cancer cell biomarkers such as prostate specific membrane antigen, folate receptor, vascular endothelial growth factor receptors, αvβ3 integrin and epithelial growth factor receptors.18,19 However, addressing malignant tumor cells only remains ineffective for several types of cancer. Evidence has accumulated that the microenvironment of a given tumor plays a crucial role in its progression, metastasis and response to chemotherapeutic intervention. In particular, the fibroblastic element of the tumor stroma, which is predominantly characterized by the abundance of activated fibroblasts often designated as cancer-associated fibroblasts (CAFs), appears as a promising target for both cancer diagnosis and treatment,20,21 including the development of targeted nanotheranostic systems.22,23 The tumor-stroma symbiotic cross-talk was even proposed as a new cancer hallmark by Sverdlov in 2018.24 A specific characteristic of CAFs is the expression of fibroblast activation protein α (FAP), a transmembrane protease which is not found in normal, healthy adult tissues, except in granulation tissue of healing wounds.25–27 However, the mechanisms underlying the physiological and pathological functions of FAP are not fully elucidated.28 The development of NP-based diagnostic and delivery systems targeting the proteolytic activity of FAP could contribute both to a better understanding of the role of FAP activity in modulating tumor behavior and to develop new anticancer treatments acting against the cancer-stroma symbiotic interactions.29,30Recently, harmonic nanoparticles (HNPs), which are based on non-centrosymmetric metal oxide nanocrystals, have emerged as promising nanomaterials for bioimaging applications31 due to several favorable properties,32–34 including their efficient nonlinear response to excitation from the UV to mid-IR,35 their long-term photostability under pulsed laser irradiation and their spectrally narrow emission signal.36 Upon screening of different types of HNPs, bismuth ferrite (BiFeO3, BFO) HNPs demonstrated very high second harmonic efficiency37,38 as well as excellent in vitro cell compatibility after surface modification with poly(ethylene glycol) (PEG) derived coatings.39 These properties were exploited in various bioimaging applications, including stem cell tracking in tissue depth40 exploiting multi-harmonic detection41 and in vitro lung cancer cells imaging.42 Other applications of BFO HNPs include contrast enhancement in magnetic resonance imaging and computed tomography,43 and synergistic photothermal–photodynamic combination for solid tumor ablation.44
Herein, we present the design and production of BFO-based imaging HNPs targeting the fibroblastic elements of cancer tumor stroma by covalent conjugation at the surface of PEG-coated BFO NPs of a low molecular weight ligand containing a highly selective inhibitor of FAP. First, we synthesized a selective low nanomolar inhibitor of FAP containing cyanopyrrolidine and quinolinoyl subunits, as suggested by the SAR study reported by Jansen et al.45 Further derivatization of this inhibitor with a 4-dibenzocyclooctyne (DIBO) moiety provided a targeting ligand for conjugation at the surface of coated BFO HNPs through copper-free azide–alkyne [3 + 2] cycloaddition (click reaction). Then, the resulting functionalized NPs were assessed for their association with human recombinant (hr) FAP, in the presence of a fluorescently labelled anti-FAP antibody, by co-localization of the second harmonic signal of the HNPs with the fluorescent signal from the antibody. This work enlarges the panel of multifunctional inorganic nanoparticles for cancer imaging by targeting the fibroblastic components of the tumor stroma rather than malignant tumor cell biomarkers.
Results and discussion
Preparation of FAP inhibitor and targeting ligand for conjugation to BFO HNPs
Among the family of human prolyl-specific proteases, FAP presents the unique property to display both endo- and exoproteolytic activities, while other closely related proteases are exclusively exopeptidases, such as amino-dipeptidyl peptidase (DPP) IV, or exclusively endopeptidases such as prolyl oligopeptidase (PREP).25 FAP, which is specifically associated to CAFs and stromal cells appears as a promising target for anti-cancer therapy. Due to its adhesive properties, DPP IV might also favor cancer progression. However, there is no evidence for the efficacy of inhibiting DPP IV in tumor growth reduction.46,47 The abnormal activity of PREP was linked to neurological disorders in mammals. This enzyme was thus identified as a potential target in neurodegenerative disorders such as amnesia, Alzheimer's disease and depression.48,49 In view of the similar hydrolytic mechanisms and kinetics characteristics of these three prolylpeptidases, the design of selective inhibitors is key to the development of therapeutic approaches based on their inhibition, to avoid undesired off-target activities. Importantly, N-acyl-Gly-Pro derived motifs were identified as key components to discriminate between FAP and the other prolyl peptidases DPP IV, VII, VIII, and IX.50 However, until the demonstration by Jansen et al. that a (4-quinolinoyl)glycyl-2-cyanopyrrolidine motif could avoid cross-inhibition of both DPPs and PREP,45 most of reported FAP inhibitors were not entirely specific.51,52 We investigated the functionalization of the quinoline ring's 6-position of the (4-quinolinoyl)glycyl-2-cyanopyrrolidine core to introduce a suitable spacer between the subunit targeting FAP and the imaging BFO NPs. Based on previous work,42 covalent conjugation to the surface of PEG-coated BFO NPs should be performed in (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) EtOH:H2O medium to ensure efficient copper-free azide–alkyne [3 + 2] cycloaddition. Therefore, a PEG3 linker was selected to modulate the polarity of the targeting ligand and a terminal acrylamide was envisaged for cross-coupling to the 6-position of the quinoline ring (Scheme 1). Preparation of the 6-bromoquinoline core started from 5-bromoisatin which was submitted to a Pfitzinger reaction53 in the presence of sodium pyruvate to produce the dicarboxylic intermediate 2 in 97% yield. Microwave assisted selective decarboxylation,54 followed by amide formation delivered 6-bromoquinoline-4-caboxamide (3) in high yield. Condensation with cyano-proline derivative 4 (synthesis described in ESI S-3†) afforded intermediate 5 which constitutes the central unit of the ligand targeting FAP. Introduction of the polar spacer was achieved by Pd-catalyzed Heck reaction55 in the presence of the acrylamide derivative 6 (synthesis described in ESI S-5†). Activation of the terminal alcohol as 4-nitrophenyl carbonate allowed condensation with the DIBO derivative 8 (synthesis described in ESI S-8†) under mild conditions to generate DIBO-PEG3-FAPi (68% yield, 2 steps), ready to be conjugated to PEG-coated BFO NPs. To evaluate the FAP targeting ability of this ligand, a similar pathway was applied on compound 9 to introduce a biotin label in place of the strained cyclooctyne moiety, affording Biotin-PEG3-FAPi (synthesis described in ESI S-11†). Introduction of a biotin residue resulted in improved solubility of the targeting ligand and allowed evaluation of its inhibitory activity in absence of the sensitive cyclooctyne subunit.
:1) EtOH:H2O medium to ensure efficient copper-free azide–alkyne [3 + 2] cycloaddition. Therefore, a PEG3 linker was selected to modulate the polarity of the targeting ligand and a terminal acrylamide was envisaged for cross-coupling to the 6-position of the quinoline ring (Scheme 1). Preparation of the 6-bromoquinoline core started from 5-bromoisatin which was submitted to a Pfitzinger reaction53 in the presence of sodium pyruvate to produce the dicarboxylic intermediate 2 in 97% yield. Microwave assisted selective decarboxylation,54 followed by amide formation delivered 6-bromoquinoline-4-caboxamide (3) in high yield. Condensation with cyano-proline derivative 4 (synthesis described in ESI S-3†) afforded intermediate 5 which constitutes the central unit of the ligand targeting FAP. Introduction of the polar spacer was achieved by Pd-catalyzed Heck reaction55 in the presence of the acrylamide derivative 6 (synthesis described in ESI S-5†). Activation of the terminal alcohol as 4-nitrophenyl carbonate allowed condensation with the DIBO derivative 8 (synthesis described in ESI S-8†) under mild conditions to generate DIBO-PEG3-FAPi (68% yield, 2 steps), ready to be conjugated to PEG-coated BFO NPs. To evaluate the FAP targeting ability of this ligand, a similar pathway was applied on compound 9 to introduce a biotin label in place of the strained cyclooctyne moiety, affording Biotin-PEG3-FAPi (synthesis described in ESI S-11†). Introduction of a biotin residue resulted in improved solubility of the targeting ligand and allowed evaluation of its inhibitory activity in absence of the sensitive cyclooctyne subunit.
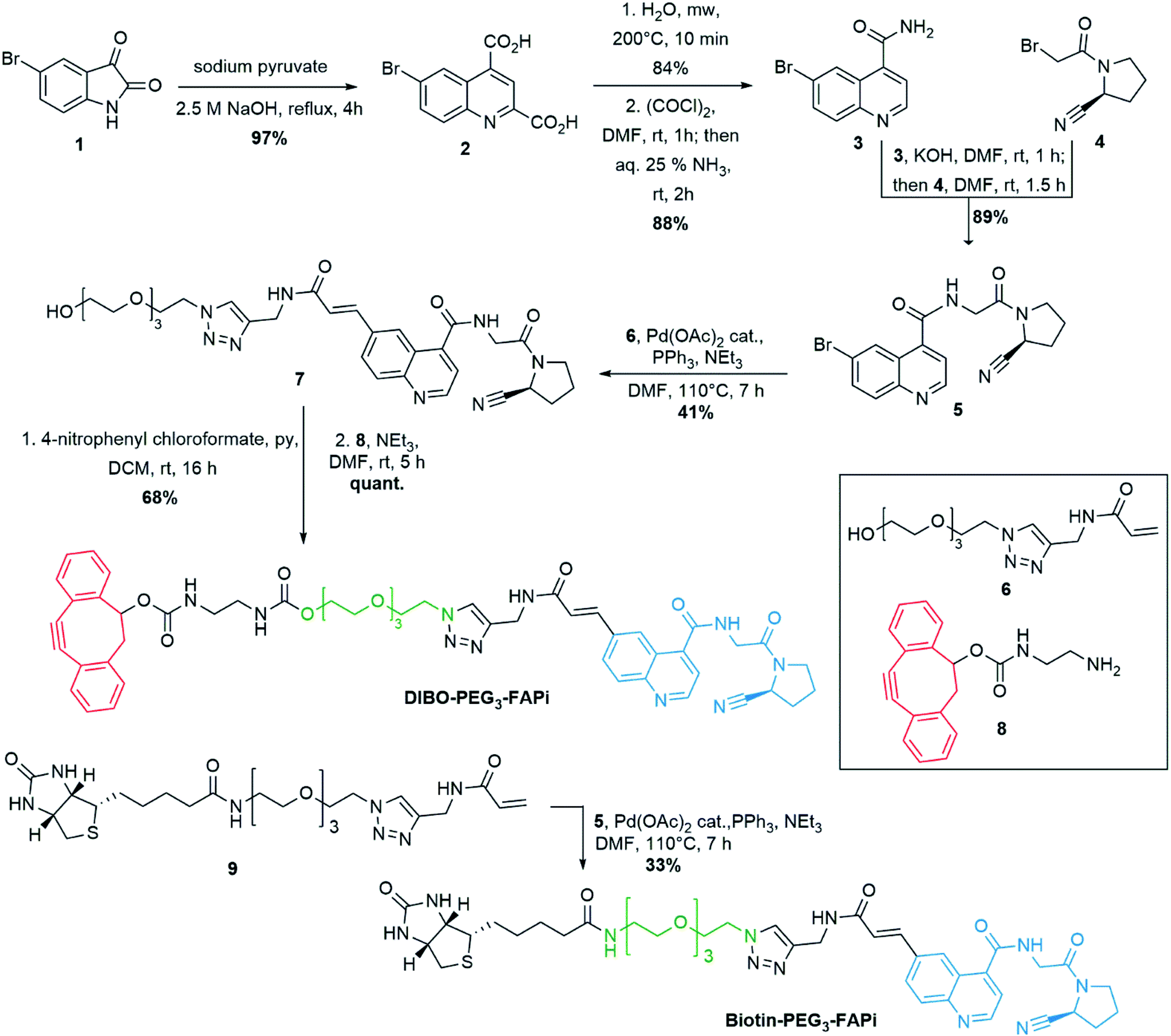 | ||
| Scheme 1 Synthesis of DIBO-PEG3-FAPi, targeting ligand for conjugation to the surface of imaging NPs. Synthesis of Biotin-PEG3-FAPi for evaluation of FAP targeting ability. | ||
Evaluation of the inhibitory activity on FAP, DPP IV and PREP
In order to assess the ability of the designed ligand to selectively target FAP, compounds 5 and Biotin-PEG3-FAPi were assessed for their inhibitory activity on human recombinant (hr) FAP, DPP IV and PREP (Table 1). Compound 5 showed potent inhibition of FAP and excellent selectivity with respect to PREP and DPP IV, which remained unaffected (selectivity index (SI) higher than 3300). Remarkably, addition of a PEG3 spacer significantly increased the potency against FAP as Biotin-PEG3-FAPi reached IC50 and Ki values of 9.2 and 3.8 nM, respectively. Importantly, a sharp selectivity with respect to PREP and DPP IV was observed (SIFAP/DPP IV and SIFAP/PREP higher than 10000), which confers to Biotin-PEG3-FAPi a highly favorable affinity/selectivity profile for the specific targeting of FAP.
| Compound | hrPREPa | hrDPP IVa | hrFAPa | |
|---|---|---|---|---|
| IC50 (μM) | IC50 (μM) | IC50 (μM) | K i (nM) | |
| a Enzymes were incubated with increasing concentrations (5, 10, 20, 50 and 100 nM) of compounds 5 and Biotin-PEG3-FAPi, for 30 min at 37 °C. Then, the residual enzymatic activity was measured after exposure to 50 μM of the appropriate substrates (Z-Gly-Pro-AMC for hrFAP and hrPREP, H-Gly-Pro-AMC for hrDPP IV). The half maximal inhibitory concentrations (IC50) were graphically determined and the inhibition constants (Ki) were calculated.57 | ||||
| 5 | >100 | >100 | 0.03 ± 0.005 | 12 ± 1.8 |
| Biotin-PEG3-FAPi | >100 | >100 | 0.0092 ± 0.0006 | 3.8 ± 0.23 |
In order to verify the compatibility of the targeting ligand in physiological environment, the effect of Biotin-PEG3-FAPi was evaluated on a model of human functional lung epithelium (MucilAir™-HF) based on the co-culture of bronchial cells and lung fibroblasts.56 MucilAir™-HF was submitted to repeated doses of Biotin-PEG3-FAPi, at 10 or 100 μM concentration, over 16 days. The cytotoxicity was measured by dosing Lactate Dehydrogenase (LDH) released in the culture supernatant at regular time points (Fig. 1A). At 10 μM concentration (3 basal applications per week), the targeting ligand did not affect cell viability in the tissue model. Cytotoxic effect started to be noticeable at higher doses (100 μM) from day 8. Similarly, the integrity and functionality of the epithelium, analyzed by trans-epithelial electrical resistance (TEER) and cilia beat frequency (CBF) measurements (Fig. 1B and C), were maintained upon repeated applications of Biotin-PEG3-FAPi, at 10 μM. Damages to the epithelium were only observed at high doses (100 μM) as TEER was affected and cilia beat was lost after day 8. These data suggest the good compatibility of the designed ligand in a concentration range which is relevant for labelling and imaging applications.
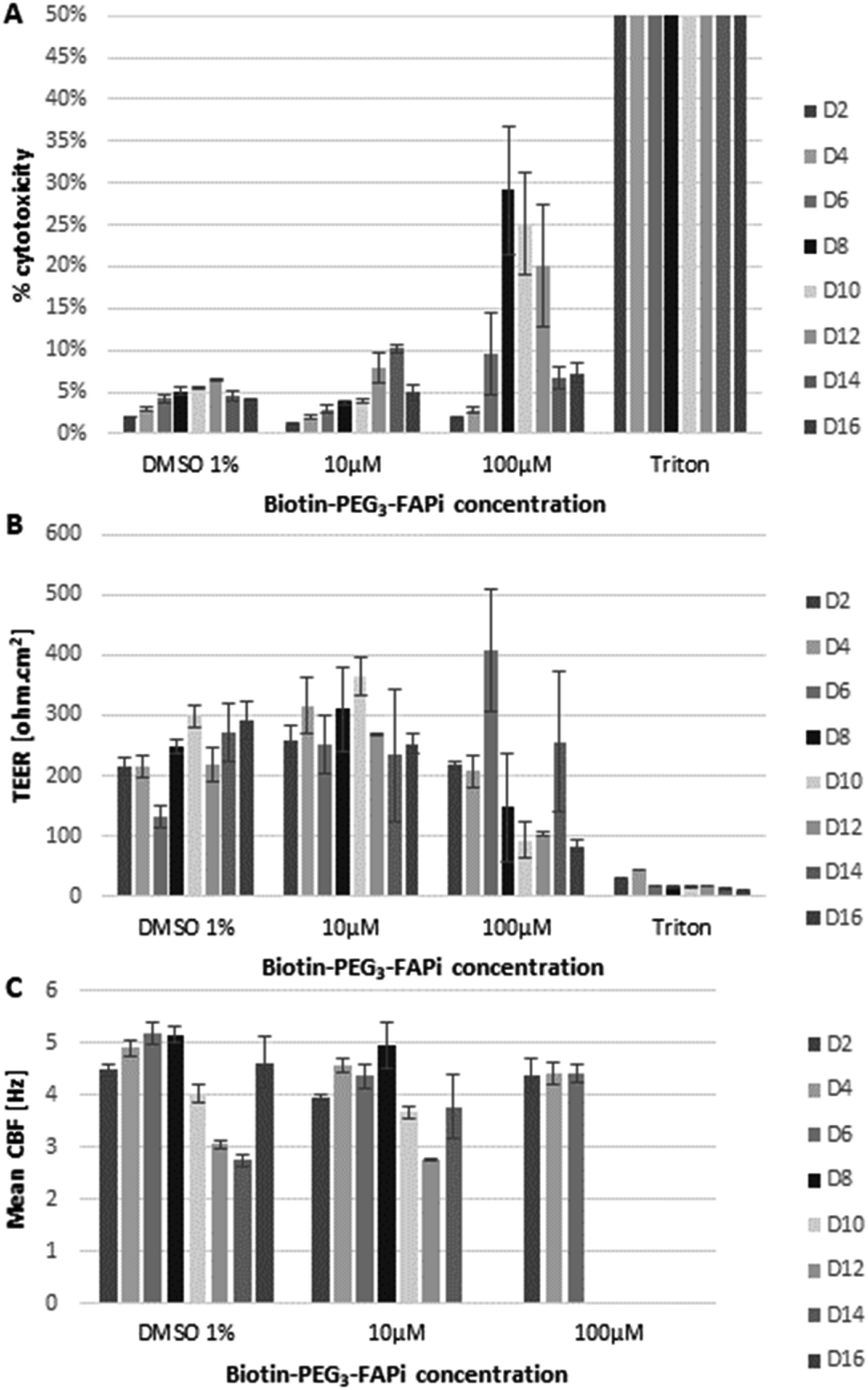 | ||
| Fig. 1 Evaluation of the cytocompatibility of the targeting ligand on the human lung tissue model MucilAir™-HF. Tissue was exposed to repeated doses (10 μM or 100 μM) of Biotin-PEG3-FAPi (day 0, 2, 4, 7, 9, 11 and 14) and measurements were made on different days (day 2, 4, 6, 8, 10, 12, 14 and 16). (A) Cytotoxicity was evaluated by dosing LDH in the culture supernatant. (B) Trans-epithelial electrical resistance (TEER) measurement. (C) Cilia beat frequency (CBF) measurement. Triton X-100 was used as positive control; DMSO (1%) was used as negative control. | ||
Conjugation of BFO NPs with targeting ligand
As previously established,39 BFO NPs were coated with a 1:1 mixture of linear PEG derivatives presenting end amino and azido groups (for detailed protocol, see ESI S-29†). Resulting PEGylated NPs were ultrasonicated in the presence of DIBO-PEG3-FAPi (16 h, 40 °C) allowing covalent conjugation of the targeting ligand through a 1,2,3-triazole moiety, under mild conditions (Fig. 2A). Separation from unreacted ligand was operated by cycles of emulsification in EtOAc followed by centrifugation, to afford BFO-PEG-FAPi NPs which were characterized for their size and surface charge. Their mean hydrodynamic diameter, measured by dynamic light scattering, was 50 ± 15 nm (Fig. 2B) and their zeta potential averaged −17.7 ± 1.6 mV. After coating and post-conjugation with the FAP targeting ligand, the average size of the functionalization layer was about 8 nm as measured on STEM images (ESI, Fig. S1†). The FT-IR spectrum of BFO-PEG-FAPi featured the apparition of bands at 1650 and 1375 cm−1, corresponding to alkene C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching and triazole C–N stretching (ESI, Fig. S2†). These data support the conjugation of DIBO-PEG3-FAPi to the surface of BFO-PEG NPs through a click reaction. According to the principles of nanoparticles design for biomedical applications presented by Blanco et al.,58 the size range and surface charge of BFO-PEG-FAPi NPs are expected to provide long-lasting systems in physiological environment.
C stretching and triazole C–N stretching (ESI, Fig. S2†). These data support the conjugation of DIBO-PEG3-FAPi to the surface of BFO-PEG NPs through a click reaction. According to the principles of nanoparticles design for biomedical applications presented by Blanco et al.,58 the size range and surface charge of BFO-PEG-FAPi NPs are expected to provide long-lasting systems in physiological environment.
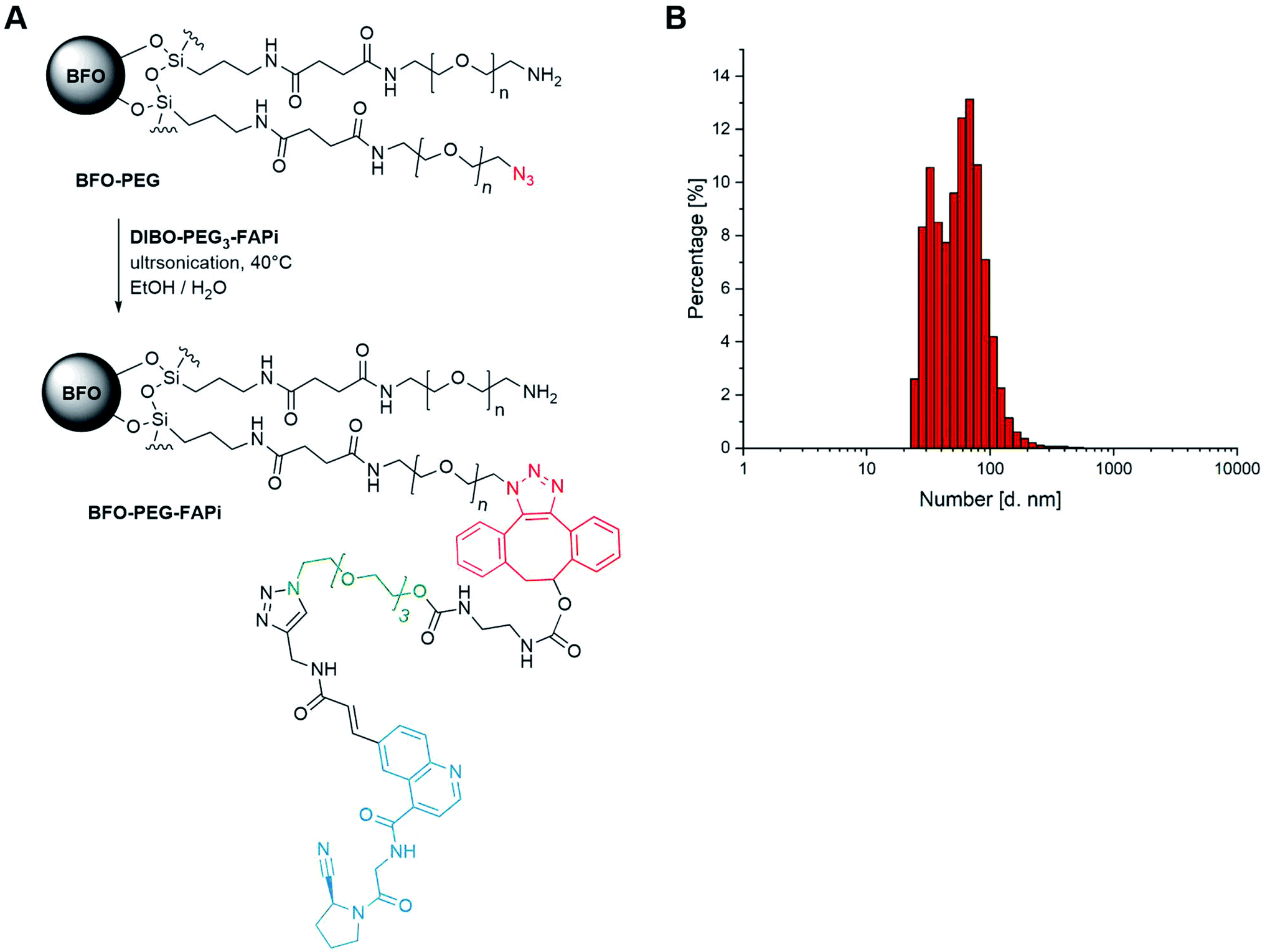 | ||
| Fig. 2 (A) Synthesis scheme of functionalized BFO-PEG-FAPi NPs; (B) dynamic light scattering analyses of functionalized nanoparticles. | ||
Association of BFO-PEG-FAPi NPs with FAP
The association of BFO-PEG-FAPi NPs with hrFAP, in comparison with BFO-PEG NPs was evaluated after incubation for 2 h at 37 °C of the functionalized or coated NPs with hrFAP, in the presence of an excess of Human Fibroblast Activation Protein alpha/FAP Alexa Fluor® 594-conjugated Antibody. The samples were centrifuged to remove the NPs from the supernatant, which was further analyzed on a fluorescence spectrophotometer (λex/λem = 590/645 nm) (Fig. 3). The fluorescence intensity of the solutions containing the anti-FAP antibody with hrFAP at the same concentrations than the ones used for the incubation experiments, was taken as reference. The incubation of BFO-PEG-FAPi NPs with hrFAP resulted in a significant decrease of the fluorescence intensity compared with the controls and to BFO-PEG NPs, indicating that the functionalized nanoparticles associated with FAP via a target-specific interaction.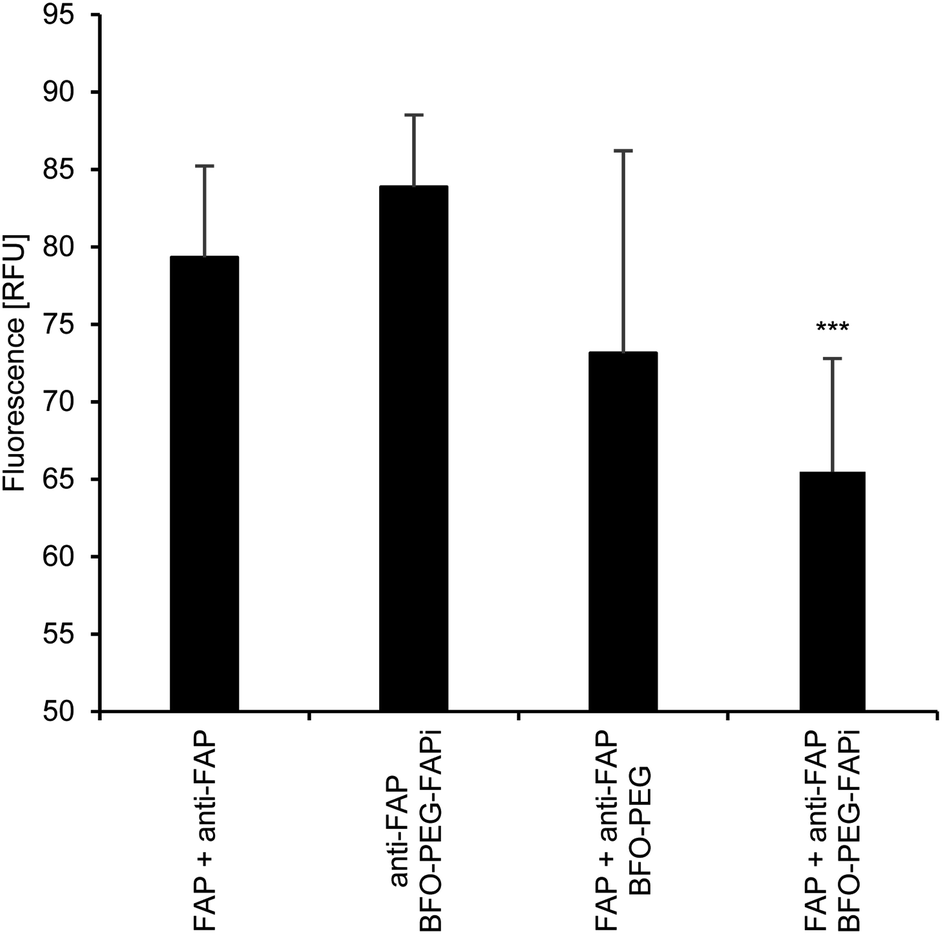 | ||
| Fig. 3 Association of BFO-PEG-FAPi NPs to FAP through target-specific interactions. HrFAP (FAP, 100 ng) was incubated for 2 h at 37 °C with BFO-PEG-FAPi NPs (100 μg mL−1) or BFO-PEG NPs (100 μg mL−1) in the presence of Human Fibroblast Activation Protein alpha/FAP Alexa Fluor® 594-conjugated Antibody (anti-FAP) (dilution 1:50). The samples were centrifuged (5 min, 10000 rpm) and the supernatants were transferred into a flat bottom 96-well plate. Fluorescence was measured in a multi-well plate spectrophotometer (λex/λem = 590/645 nm). HrFAP incubated with anti-FAP and BFO-PEG-FAPi NPs incubated with anti-FAP were used as controls. Results were analyzed using a Student's t-test. Fluorescence intensity of BFO-PEG-FAPi NPs incubated with FAP and anti-FAP was significantly lower than BFO-PEG NPs incubated with FAP and anti-FAP and the fluorescence intensity of the controls (all comparisons ***p < 0.001). | ||
The association of BFO-PEG-FAPi NPs to FAP was further analyzed by multiphoton microscopy. After incubation of hrFAP with BFO-PEG NPs or BFO-PEG-FAPi NPs for 2 hours at 37 °C, in the presence of the labeled anti-FAP antibody, the resulting conjugates were imaged by detection of the second harmonic (SH) signal from the NPs and of the fluorescent emission from the antibody, upon laser pulsed excitation at 840 nm (ESI, Fig. S3†). The ratio of conjugates emitting in both SHG (420 nm) and fluorescence (580–650 nm) channels to all structures emitting in the SHG channel was calculated in 4 different regions of each sample and averaged (Fig. 4). The number of conjugates generating both harmonic and fluorescence signals was significantly higher in the presence of BFO-PEG-FAPi NPs than in the presence of BFO-PEG NPs, thus confirming the target-specific association of functionalized BFO HNPs with FAP.
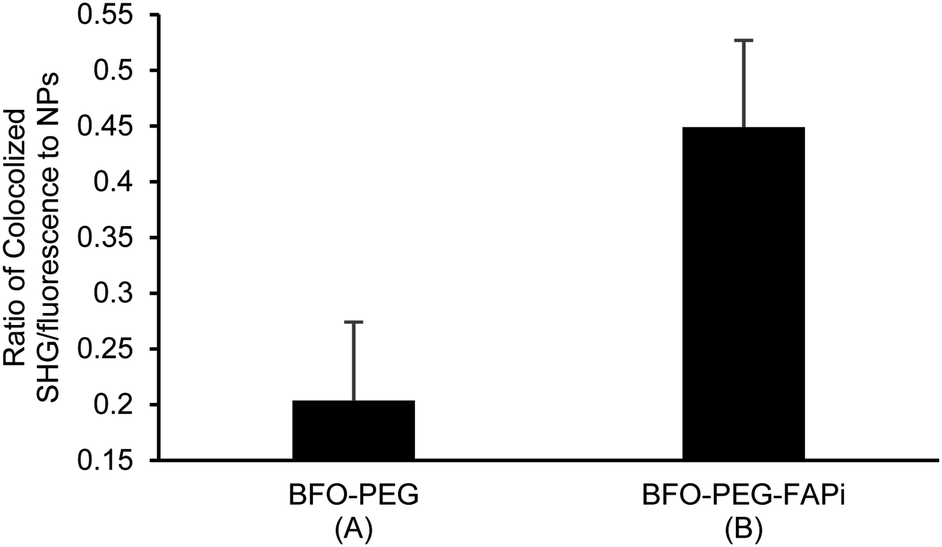 | ||
| Fig. 4 Association of BFO-PEG NPs and BFO-PEG-FAPi NPs to hrFAP quantified by multiphoton multispectral microscopy. HrFAP (100 ng) was incubated for 2 h at 37 °C with BFO-PEG NPs (100 μg mL−1) (A) or BFO-PEG-FAPi NPs (100 μg mL−1) (B) in the presence of Human Fibroblast Activation Protein alpha/FAP Alexa Fluor® 594-conjugated Antibody (dilution 1:5). SH and fluorescence signals were epi-collected upon focused excitation at 840 nm. The ratio of the area of conjugates emitting in both SH and fluorescence channels to the area of SH emitting structures was averaged from four distinct regions of each sample (635 × 635 μm). Error bars represent standard deviation from n = 4 calculations. | ||
While many of the proteases involved in cancer development and progression are expressed by both healthy and malignant cells, FAP holds the unique property to be mostly present at remodeling stroma in tumors and healing wounds.59 This remarkable feature makes FAP a highly promising target for both diagnosis and therapeutic intervention. While several inhibitors of FAP based on a pyrrolidine-2-boronic acid scaffold were evaluated as potential anti-cancer drugs, safety concerns were raised during clinical trials due to the cross inhibition of other related enzymes.60–62 Having previously disclosed the efficiency of PEGylated BFO NPs for selective imaging and cell tracking in tissue samples,40,42 we focused on the design and synthesis of a targeting ligand which would exhibit selective and high affinity for FAP while holding a suitable spacer to be conjugated at the surface of BFO-PEG NPs. Functionalization of a (4-quinolinoyl)glycyl-2-cyanopyrrolidine core by cross-coupling on the 6-position of the quinoline ring allowed the introduction of a small PEG spacer to modulate the polarity of the ligand for further post-conjugation reaction in EtOH/H2O medium. Conjugation of this inhibitory scaffold to N-(2-hydroxypropyl)methacrylamide copolymer was previously used to vizualize and isolate FAP from cell lysates.63 Interestingly, derivatization with a PEG spacer delivered Biotin-PEG3-FAPi which displayed effective binding to FAP (Ki = 3.8 ± 0.20 nM), high specific discrimination between FAP and its closely related proteases DPP IV and PREP (selectivity indexes above 10000), and good biocompatibility with the human 3D tissue model MucilAir™-HF.56 Combination of a N-acyl-Gly-Pro fragment with a (4-quinolinoyl)glycyl-2-cyanopyrrolidine moiety and a polar PEG spacer resulted in sharp specificity toward FAP. In agreement with our previous report,42 the use of a DIBO moiety allowed spontaneous azide to alkyne [3 + 2]-cycloaddition at the surface of PEG-BFO NPs, under mild conditions, for covalent coupling of the targeting ligand at the surface of the imaging NPs. While coated NPs did not show any significant interaction with FAP, functionalized BFO-PEG-FAPi NPs displayed efficient and stable association with FAP, assessed both by multiphoton microscopy and reverse ELISA-type assay using Human Fibroblast Activation Protein alpha/FAP Alexa Fluor® 594-conjugated Antibody. These results suggest the formation of a stable complex between BFO-PEG-FAPi NPs and FAP despite the potential reversibility of the covalent bond formation between the cyanopyrrolidine moiety and FAP active site serine residue.51 The possibility to exploit the simultaneous acquisition of second- and third-harmonic signals of BFO NPs for selective imaging in complex tissue samples41 combined with the stable association of BFO-PEG-FAPi NPs with FAP make these functionalized NPs promising nanodevices for cancer diagnosis applications.
Conclusions
Based on a (4-quinolinoyl)glycyl-2-cyanopyrrolidine core, we designed and synthesized a high affinity and selective targeting ligand for FAP, which was covalently conjugated at the surface of BFO-PEG NPs. The resulting BFO-PEG-FAPi nanoconjugates demonstrated stable association with FAP, thus providing promising labelling and imaging tools to study and target the fibroblastic element of the tumor stroma rather than malignant tumoral cells.Experimental section
Materials and methods
Reagent-grade solvents (Fluka, Riedel-de-Haën) and chemicals (Aldrich, Acros, Fluka, Sigma, Maybridge, TCI Chemicals, Apollo and Fluorochem) were used without further purification. BFO NPs (BF0011214) were obtained from TIBIO (Comano, Switzerland). All reactions were performed in flame-dried glassware under an inert atmosphere of nitrogen. All products were dried under vacuum (10−2 bar) before analytical characterization. Reactions were monitored by thin layer chromatography (TLC) on pre-coated aluminum plates SiO2 60 F254 (Merck, Darmstadt, Germany). The compounds were visualized by 254 nm light or stained with solutions of KMnO4, Pancaldi reagent [(NH4)6MoO4, Ce(SO4)2, H2SO4, H2O], ninhydrin or iodine vapors. Purifications were performed by flash chromatography (FC) on silica gel (Merck No. 9385 silica gel 60, 240–400 mesh, particle size 40–63 μm). NMR spectra were recorded on Bruker Avance III-400, Bruker Avance-400 or Bruker DRX-400 spectrometers (Bruker, Billerica, MA, USA) at room temperature (rt), unless otherwise stated. 1H frequency is at 400.13 MHz, 13C frequency is at 100.62 MHz. Chemical shifts are reported downfield from tetramethylsilane. 1H signals are reported in ppm with the internal chloroform signal at 7.26 ppm or the internal DMSO signal at 2.50 ppm or the internal CH3CN signal at 1.94 ppm, as internal references. 13C-NMR signals are reported in ppm with the internal chloroform signal at 77.16 ppm or the internal DMSO signal at 39.5 ppm or the internal CH3CN signal at 118.26 ppm, as internal references. The resonance multiplicity is described as s (singulet), d (doublet), t (triplet), q (quartet), quin (quintet), m (multiplet). Broad signals are indicated as br. Coupling constants (J) are given in hertz (Hz). IR spectra were recorded on a Jasco FT/IR-4100 spectrometer outfitted with a PIKE technology MIRacle™ ATR accessory as neat films compressed onto a Zinc Selenide window. The spectra are reported in cm−1 (w = weak, m = medium, s = strong, sh = shoulder). The qualitative accurate masses were measured by ESI-TOF using the Xevo G2-S QTOF (Waters) and nanoESI-FT-MS using the Elite™ Hybrid Ion Trap-Orbitrap (ThermoFisher) mass spectrometer (more details in ESI S-30†). For all new compounds, the following data are provided: accurate mass, 1H and 13C NMR spectra (with peak assignments), IR data. The purity of target compounds 5 and Biotin-PEG3-FAPi was tested on Waters Acquity UPLC® (gradient 98:2 to 2:98·H2O containing 0.5% formic acid:ACN containing 0.45% formic acid in 4 minutes, flow rate 0.8 mL min−1) with column Acquity UPLC® BEH C18 1.7 μm. The purity of all new compounds is higher than 95%. Measurements of the dynamic light scattering and zeta potential were obtained using a Malvern NanoZ instrument (Malvern Instruments, Malvern, UK). Centrifugations were performed on HERAEUS Biofuge 13 centrifugator. Scanning transmission electron microscopy (STEM) was performed at the Interdisciplinary Centre for Electron Microscopy (CIME, EPFL, Lausanne, Switzerland) on a FEI Titan Themis 60-300 microscope.
Synthesis protocols
Designation of the compounds refers to the chemical structures presented in Scheme 1.:1) to afford 5 as a brown foam (1.76 mmol, 682 mg, 89%). Analytical UPLC: Rt = 1.58 min. 1H NMR (400 MHz, chloroform-d) δ 2.19–2.43 (4H, m), 3.51–3.59 (1H, m), 3.69–3.76 (1H, m), 4.26 (1H, dd, J = 3.6, 17.8 Hz), 4.41 (1H, ddd, J = 2.8, 4.9, 17.8 Hz), 4.79 (1H, d, J = 5.2 Hz), 7.18 (1H, s), 7.54 (1H, d, J = 4.3 Hz), 7.83 (1H, dd, J = 2.2, 9.0 Hz), 8.00 (1H, d, J = 9.0 Hz), 8.49 (1H, d, J = 2.2 Hz) 8.95 (1H, dd, J = 2.3, 4.4 Hz). 13C NMR (101 MHz, chloroform-d) δ 25.2, 30.1, 42.6, 45.8, 46.9, 117.9, 119.6, 122.4, 125.7, 127.8, 131.7, 133.8, 139.8, 147.5, 150.3, 166.9, 167.0. HRMS m/z [M + H]+ calcd for C17H16BrN4O2+ 387.0451; found 387.0450. IR (νmax, cm−1): 3297 w, 3058 w, 2954 w, 2168 w, 2156 w, 2022 w, 2880 w, 1655 s, 1586 w, 1537 w, 1493 w, 1430 m, 1407 m, 1305 m, 1345 w, 1263 m, 1194 w, 1157 w, 1043 w, 978 w, 913 w, 867 w, 846 w, 830 w, 783 w, 730 m, 723 m, 702 w. 1H-NMR, 13C-NMR spectra and signal assignments, UPLC trace (ESI S-21†).
:1 then 10:1) to afford 7 as a white oil (0.29 mmol, 185 mg, 41%). 1H NMR (400 MHz, chloroform-d) δ 2.17–2.38 (4H, m), 3.48–3.62 (11H, m), 3.64–3.72 (2H, m), 3.70–3.76 (1H, m), 3.83 (t, J = 5.0 Hz), 4.26 (1H, dd, J = 4.8, 17.3 Hz), 4.37 (1H, dd, J = 5.4, 17.3 Hz), 4.50 (2H, t, J = 5.0 Hz), 4.55 (2H, dd, J = 3.7, 5.6 Hz), 4.79–4.84 (1H, m), 6.69 (1H, d, J = 15.7 Hz), 7.37 (1H, t, J = 5.6 Hz), 7.42 (1H, d, J = 4.3 Hz), 7.59 (1H, d, J = 15.6 Hz), 7.68 (1H, dd, J = 1.8, 8.9 Hz), 7.72 (1H, t, J = 5.2 Hz), 7.82 (1H, s), 7.94 (1H, d, J = 8.8 Hz), 8.40 (1H, d, J = 1.5 Hz), 8.79 (1H, d, J = 4.3 Hz). 13C NMR (101 MHz, chloroform-d) δ 25.3, 29.9, 35.1, 42.5, 45.9, 47.0, 50.4, 61.6, 69.5, 70.3, 70.5, 70.60, 70.62, 72.6, 118.4, 119.6, 123.0, 123.8, 124.4, 125.1, 129.1, 130.2, 134.3, 139.8, 141.6, 144.7, 148.8, 150.4, 165.9, 167.46, 167.51. HRMS m/z [M + Na]+ calcd for C31H38N8NaO7+ 657.2756; found 657.2762. IR (νmax, cm−1): 3307 w, 2929 w, 2873 w, 3057 w, 1711 w, 1658 m, 1577 w, 1535 w, 1428 w, 1410 w, 1366 w, 1310 w, 1266 w, 1210 w, 1196 w, 1120 m, 1103 m, 1062 m, 1036 w, 983 w, 934 w, 918 w, 862 w, 832 w, 732 s, 702 m, 665 w. 1H-NMR, 13C-NMR spectra and signal assignments (ESI S-23†).
:1) to afford intermediate (S,E)-2-(2-(2-(2-(4-((3-(4-((2-(2-cyanopyrrolidin-1-yl)-2-oxoethyl)carbamoyl)quinolin-6-yl)acrylamido)methyl)-1H-1,2,3-triazol-1-yl)ethoxy)ethoxy)ethoxy)ethyl (4-nitrophenyl) carbonate as a white solid (0.011 mmol, 8.7 mg, 68%). To a solution of this intermediate (1 equiv., 0.063 mmol, 50 mg) in anhydrous DMF (2 mL), were added NEt3 (3 equiv., 0.19 mmol, 26 μL) and DIBO derivative 8 (1.1 equiv., 0.069 mmol, 21 mg). The solution was stirred for 5 h at rt. The solvent was evaporated in vacuo and the crude product was purified by FCC (DCM/MeOH 15:1) to afford DIBO-PEG3-FAPi as a white oil (0.063 mmol, 61 mg, quant.). 1H NMR (400 MHz, acetonitrile-d3) δ 2.05–2.24 (4H, m), 2.75 (1H, dd, J = 4.0, 15.0 Hz), 3.07–3.18 (5H, m), 3.39–3.53 (11H, m), 3.62–3.69 (1H, m), 3.79 (2H, t, J = 5.1 Hz), 4.03 (2H, t, J = 4.7 Hz), 4.20 (2H, dd, J = 2.1, 5.7 Hz), 4.44 (2H, t, J = 5.0 Hz), 4.51 (2H, d, J = 5.8 Hz), 4.68–4.75 (1H, m), 5.28 (1H, s), 5.92 (1H, s), 6.26 (1H, s), 6.77 (1H, d, J = 15.7 Hz), 7.19 (1H, t, J = 5.8 Hz), 7.24–7.39 (7H, m), 7.44–7.55 (3H, m), 7.63 (1H, d, J = 15.7 Hz), 7.76 (1H, s), 7.86 (1H, dd, J = 1.9, 8.9 Hz), 8.00 (1H, d, J = 8.8 Hz), 8.54 (1H, d, J = 1.9 Hz), 8.86 (1H, d, J = 4.3 Hz). 13C NMR (101 MHz, acetonitrile-d3) δ 25.9, 30.6, 35.8, 41.6, 41.8, 43.0, 46.7, 47.0, 47.7, 50.9, 64.8, 70.0, 70.1, 71.0, 71.1, 71.2, 77.1, 110.7, 113.5, 120.1, 120.6, 121.8, 124.15, 124.20, 124.3, 125.0, 125.5, 126.78, 126.84, 127.1, 128.1, 128.2, 129.27, 129.34, 131.1, 131.2, 135.0, 140.1, 143.2, 145.8, 149.8, 151.8, 152.4, 153.3, 156.8, 166.3, 168.2, 168.5. Two Cq are not resolved. HRMS m/z [M + Na]+ calcd for C51H54N10NaO10+ 989.3917; found 989.3920. IR (νmax, cm−1): 3319 w, 3061 w, 2922 w, 2878 w, 2159 w, 1716 m, 1657 m, 1521 m, 1443 w, 1431 w, 1288 w, 1260 m, 1121 m, 1105 m, 1053 w, 1037 w, 981 w, 916 w, 860 w, 832 w, 764 m, 733 s, 699 m, 1970 w, 1840 w. 1H-NMR, 13C-NMR spectra and signal assignments (ESI S-26†).
Functionalization of BFO-PEG NPs
To a suspension of BFO-PEG NPs (4 mg) in EtOH (4 mL) was added distilled water (4 mL). A solution of DIBO-PEG3-FAPi (17.4 μmol, 17 mg) in DMF (200 μL) was added and the suspension was ultra-sonicated for 16 h at 40 °C. The mixture was then divided into eppendorfs and centrifuged (10 min, 13000 rpm). The supernatant was discarded and the NPs were resuspended in EtOAc (1 mL) and centrifuged (10 min, 13000 rpm). The procedure was repeated 3 times. BFO-PEG-FAPi NPs were stored in EtOH at a concentration of 1 mg mL−1. A sample (10 μL) was diluted with distilled water (1 mL) and ultra-sonicated for 30 min, before being analyzed with a Malvern NanoZ instrument to determine mean hydrodynamic diameter by dynamic light scattering and zeta potential.
Inhibition of hrFAP, hrDPP IV and hrPREP
The human recombinant enzymes were purchased from commercial sources: hrDPP IV and hrPREP (Enzo Life Sciences, Lausen, Switzerland), hrFAP (R&D systems, Abingdon, UK). The enzymatic activities were measured in flat bottom 96-well plates (Costar) containing in each well 0.01 μg of the enzymes and 50 μM of the appropriate substrates: Z-Gly-Pro-AMC for hrFAPα and hrPOP; H-Gly-Pro-AMC for hrDPP IV (both substrates from Bachem, Vionnaz, Switzerland) diluted in their respective assay buffers (50 mM Tris, 1 M NaCl, 1 mg mL−1 BSA, pH 7.5, for hrFAP; 50 mM Tris, 1 mg mL−1 BSA, pH 7.5, for hrPREP; 25 mM Tris and 1 mg mL−1 BSA, pH 8.0 for hrDPP IV). The enzyme solutions were incubated for 30 min at 37 °C with increasing concentrations (5, 10, 20, 50 and 100 nM) of compounds 5 or Biotin-PEG3-FAPi. The residual enzymatic activity was determined by measuring fluorescence increase for 60 min at 37 °C in a fluorescence multi-well plate reader (λex/λem = 360/460 nm, Synergy HT). Experiments were conducted in triplicate wells and repeated twice. The half maximal inhibitory concentrations (IC50) were graphically determined and the inhibition constants (Ki) were calculated.49Evaluation of cytocompatibility on MucilAir™-HF tissue samples
Airway cells were obtained from patients undergoing surgical polypectomy. All experimental procedures were explained in full, and all subjects provided informed consent. The study was conducted according to the declaration of Helsinki on biomedical research (Hong Kong amendment, 1989), and received approval from local ethics commission (Commission cantonale d'éthique de la recherche scientifique de Genève [CCER]). Airway epithelia co-cultured with fibroblasts were isolated from a mixture of human airway cells from 14 different donors (MucilAir™-Pool-HF) and maintained at the air–liquid interface (ALI) in MucilAir™ culture medium (EP04MM), ready-to-use, chemically defined, serum-free (Epithelix Sàrl, Geneva, Switzerland), in 24-well plates with 6.5 mm Transwell® inserts (cat #3470, Corning Incorporated, Tewksbury, USA). Biotin-PEG3-FAPi was first solubilized in DMSO and diluted to reach targeted concentrations (i.e. 10 μM and 100 μM) in MucilAir™ culture medium (EP04MM) with a fixed 1% DMSO for each dilution.Evaluation of the association of BFO-PEG-FAPi NPs with FAP
The protocol refers to the data presented in Fig. 3. Human Fibroblast Activation Protein alpha/FAP Alexa Fluor® 594-conjugated Antibody was purchased from R&D systems (Abingdon, UK). Incubations were carried out in hrFAP buffer (50 mM Tris, 1 M NaCl, 1 mg mL−1 BSA, pH 7.5) in 1.5 mL Eppendorf® tubes at 37 °C. The following combinations were evaluated in a final volume of 200 μL: (1) anti-FAP antibody (1:50 dilution), hrFAP (100 ng) in buffer (control); (2) anti-FAP antibody (1:50 dilution), BFO-PEG-FAPi NPs (100 μg mL−1) in buffer (control); (3) anti-FAP antibody (1:50 dilution), hrFAP (100 ng), BFO-PEG NPs (100 μg mL−1) in buffer; (4) anti-FAP antibody (1:50 dilution), hrFAP (100 ng), BFO-PEG-FAPi NPs (100 μg mL−1) in buffer. The Eppendorf® tubes containing the mixtures were incubated under gentle shaking for 2 h. The mixtures were centrifuged (5 min, 10000 rpm). The supernatants were collected and transferred to a flat bottom 96-well plate. The intensity of the fluorescent emission of the labelled anti-FAP antibody was measured on a fluorescence multi-well plate reader (λex/λem = 590/645 nm, Synergy HT). Experiments were conducted in five replicates. Results were analyzed using a student's t-test (all comparisons ***p < 0.001).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Drs Pascal Miéville and Aurélien Bornet for their support with NMR experiments, Dr Laure Menin for her support with MS experiments, as well as Dr Thomas LaGrange for STEM experiments. The authors are thankful to MER Dr Luigi Bonacina for granting access to the multiphoton microscopy platform of the Department of Applied Physics, University of Geneva. The authors acknowledge financial support from the France-Switzerland Interreg Program (Interreg fédéral, Vaud state, Geneva state).Notes and references
- K. T. Nguyen and Y. Zhao, Acc. Chem. Res., 2015, 48, 3016 CrossRef CAS.
- G. Chen, I. Roy, C. Yang and P. N. Prasad, Chem. Rev., 2016, 116, 2826 CrossRef CAS PubMed.
- L. Zhang, Y. Li and J. C. Yu, J. Mater. Chem. B, 2014, 2, 452 RSC.
- C.-F. Wang, M. P. Sarparanta, E. M. Mäkilä, M. L. K. Hyvönen, P. M. Laakkonen, J. J. Salonen, J. T. Hirvonen, A. J. Airaksinen and H. A. Santos, Biomaterials, 2015, 48, 108 CrossRef CAS.
- C. Argyo, V. Weiss, C. Bräuchle and T. Bein, Chem. Mater., 2014, 26, 435 CrossRef CAS.
- W. Q. Lim, S. Z. F. Phua, H. V. Xu, S. Sreejith and Y. Zhao, Nanoscale, 2016, 8, 12510 RSC.
- P. Kumar, P. Tambe, K. M. Paknikar and V. Gajbhiye, J. Controlled Release, 2018, 287, 35 CrossRef CAS.
- A. Semkina, M. Abakumov, N. Grinenko, A. Abakumov, A. Skorikov, E. Mironova, G. Davydova, A. G. Majouga, N. Nukolova, A. Kabanov and V. Chekhonin, Colloids Surf., B, 2015, 136, 1073 CrossRef CAS.
- J. Chomouka, J. Drbohlavova, D. Huska, V. Adam, R. Kizek and J. Hubalek, Pharmacol. Res., 2010, 62, 144 CrossRef.
- A. Ghoshal, U. Goswani, A. K. Sahoo, A. Chattopadhyay and S. S. Ghosh, ACS Biomater. Sci. Eng., 2015, 1, 1256 CrossRef CAS.
- J. Zhu, L. Zheng, S. Wen, Y. Tang, M. Shen, G. Zhang and X. Shi, Biomaterials, 2014, 35, 7635 CrossRef CAS PubMed.
- D. Dutta, A. K. Sahoo, A. Chattopadhyay and S. S. Ghosh, J. Mater. Chem. B, 2016, 4, 793 RSC.
- M. P. Melancon, M. Zhou and C. Li, Acc. Chem. Res., 2011, 44, 947 CrossRef CAS.
- A. A. Borran, A. Aghanejad, A. Farajollahi, J. Bara and Y. Omidi, Radiat. Phys. Chem., 2018, 152, 137 CrossRef CAS.
- W. Zhao, A. Li, A. Zhang, Y. Zheng and J. Liu, ChemMedChem, 2018, 13, 2134 CrossRef CAS.
- C. Ding, A. Zhu and Y. Tian, Acc. Chem. Res., 2014, 47, 20 CrossRef CAS.
- Y. Nakamura, A. Mochida, P. L. Choyke and H. Kobayashi, Bioconjugate Chem., 2016, 27, 2225 CrossRef CAS.
- J. E. Rosen, L. Chan, D.-B. Shieh and F. X. Gu, Nanomedicine, 2012, 8, 275 CrossRef CAS.
- R. Bazak, M. Houri, S. El Achy, S. Kamel and T. Refaat, J. Cancer Res. Clin. Oncol., 2016, 141, 769 CrossRef.
- J. Paulsson and P. Micke, Semin. Cancer Biol., 2014, 25, 61 CrossRef CAS.
- M. Li, M. Li, T. Yin, H. Shi, Y. Wen, B. Zhang, M. Chen, G. Xu, K. Ren and Y. Wei, Mol. Med. Rep., 2016, 13, 2476 CrossRef CAS.
- C. Fernandes, D. Suares and C. Mayur Yergeri, Front. Pharmacol., 2018, 9, 1230 CrossRef CAS PubMed.
- T. Cui, X. Li, Y. Shu, X. Huang, Y. Wang and W. Zhang, Int. J. Pharm., 2018, 552, 16 CrossRef CAS.
- E. Sverdlov, BioEssays, 2018, 40, 1800079 CrossRef.
- L. Juillerat-Jeanneret and S. Gerber-Lemaire, Mini-Rev. Med. Chem., 2009, 2, 215 CrossRef.
- D. T. Fearon, Cancer Immunol. Res., 2014, 2, 187 CrossRef CAS.
- F. Zi, J. He, D. He, Y. Li, L. Yang and Z. Cai, Mol. Med. Rep., 2015, 11, 3203 CrossRef CAS.
- P. Busek, R. Mateu, M. Zubal, L. Kotackova and A. Sedo, Front. Biosci., 2018, 23, 1933 CrossRef CAS.
- A. Loktev, T. Lindner, W. Mier, J. Debus, A. Altmann, D. Jäger, F. Giesel, C. Kratochwil, P. Barthe, C. Roumestand and U. A. Haberkorn, J. Nucl. Med., 2018, 59, 1423 CrossRef CAS.
- K. W. Jackson, V. J. Christiansen, V. R. Yadav, R. Silasi-Mansat, F. Lupu, V. Awasthi, R. R. Zhang and P. A. McKee, Neoplasia, 2015, 17, 43 CrossRef CAS PubMed.
- L. Bonacina, Mol. Pharmaceutics, 2013, 7, 205 Search PubMed.
- W. P. Dempsey, S. E. Fraser and P. Pantazis, BioEssays, 2012, 34, 351 CrossRef CAS.
- Y. Pu and D. Psaltis, Appl. Opt., 2013, 52, 567 CrossRef CAS.
- L. Bonacina, Y. Mugnier, F. Courvoisier, R. Le Dantec, J. Extermann, Y. Lambert, V. Boutou, C. Galez and J.-P. Wolf, Appl. Phys. B, 2007, 87, 399 CrossRef CAS.
- J. Extermann, L. Bonacina, E. Cuna, C. Kasparian, Y. Mugnier, T. Feurer and J.-P. Wolf, Opt. Express, 2009, 17, 15342 CrossRef CAS PubMed.
- L. L. Xuan, C. Zhou, A. Slablab, D. Chauvat, C. Tard, S. Perruchas, T. Gacoin, P. Villela and J. F. Roch, Small, 2008, 4, 1332 CrossRef.
- D. Staedler, T. Magouroux, R. Hadli, C. Joulaud, J. Extermann, S. Schwung, S. Passemard, C. Kasparian, G. Clarke, M. Gerrmann, R. Le Dantec, Y. Mugnier, D. Rytz, D. Ciepielewski, C. Galez, S. Gerber-Lemaire, L. Juillerat-Jeanneret, L. Bonacina and J.-P. Wolf, ACS Nano, 2012, 6, 2542 CrossRef CAS.
- S. Schwung, A. Rogov, G. Clarke, C. Joulaud, T. Magouroux, D. Staedler, S. Passemard, T. Jüstel, L. Badie, C. Galez, J.-P. Wolf, Y. Volkov, A. Prina-Mello, S. Gerber-Lemaire, D. Rytz, Y. Mugnier, L. Bonacina and R. Le Dantec, J. Appl. Phys., 2014, 116, 114306 CrossRef.
- D. Staedler, S. Passemard, T. Magouroux, A. Rogov, C. M. Maguire, B. M. Mohamed, S. Schwung, D. Rytz, T. Juestel, S. Hwu, Y. Mugnier, R. Le Dantec, Y. Volkov, S. Gerber-Lemaire, A. Prina-Mello, L. Bonacina and J.-P. Wolf, Nanomedicine, 2015, 11, 815 CrossRef CAS.
- L. Dubreil, I. Leroux, M. Ledevin, C. Schleder, L. Lagalice, C. Lovo, R. Fleurisson, S. Passemard, V. Kilin, S. Gerber-Lemaire, A. M. Colle, L. Bonacina and K. Rouger, ACS Nano, 2017, 11, 6672 CrossRef CAS.
- A. Rogov, M. Irondelle, F. Ramos-Gomes, J. Bode, D. Staedler, S. Passemard, S. Courvoisier, Y. Yamamoto, F. Waharte, D. Ciepielewski, P. Rideau, S. Gerber-Lemaire, F. Alves, J. Salamero, L. Bonacina and J.-P. Wolf, ACS Photonics, 2015, 2, 1416 CrossRef CAS.
- S. Passemard, D. Staedler, G. Sonego, T. Magouroux, G. S. Schneiter, L. Juillerat-Jeanneret, L. Bonacina and S. Gerber-Lemaire, J. Nanopart. Res., 2015, 17, 414 CrossRef.
- R. Azimeh, X. Wensheng, L. Zhao, S. Wang, Y. Liu, Z. Wu, J. Wang and F. Si-She, J. Biomed. Nanotechnol., 2018, 14, 1159 CrossRef.
- C. Yang, Y. Chen, W. Guo, Y. Gao, C. Song, Q. Zhang, N. Zheng, X. Han and C. Guo, Adv. Funct. Mater., 2018, 28, 1706827 CrossRef.
- K. Jansen, L. Heirbaut, J. D. Cheng, J. Joossens, O. Ryabtsova, P. Cos, L. Maes, A.-M. Lambeir, I. De Meester, K. Augustyns and P. Van der Veken, ACS Med. Chem. Lett., 2013, 4, 491 CrossRef CAS.
- F. Kikkawa, H. Kajiyama, K. Shibata, K. Ino, S. Nomura and S. Mitztani, Biochim. Biophys. Acta, Proteins Proteomics, 2005, 1751, 45 CrossRef CAS.
- H. C. Cheng, M. Abdel-Ghany, S. Zhang and B. U. Pauli, Clin. Exp. Metastasis, 1999, 17, 609 CrossRef CAS.
- J. A. García-Horsman, P. T. Männistö and J. I. Venäläinen, Neuropeptides, 2007, 41, 1 CrossRef.
- J. Lawandi, S. Gerber-Lemaire, L. Juillerat-Jeanneret and N. Moitessier, J. Med. Chem., 2010, 53, 3423 CrossRef CAS.
- C. Y. Edosada, C. Quan, C. Wiesmann, T. Tran, D. Sutherlin, M. Reynolds, J. M. Eliott, H. Raab, W. Fairbrother and B. B. Wolf, J. Biol. Chem., 2006, 281, 7437 CrossRef CAS PubMed.
- S. E. Poplawski, J. H. Lai, Y. Li, Z. Jin, Y. Liu, W. Wu, Y. Wu, Y. Zhou, J. L. Sudmeier, D. G. Sanford and W. W. Bachovchin, J. Med. Chem., 2013, 56, 3467 CrossRef CAS PubMed.
- T.-Y. Tsai, T.-K. Yeh, X. Chen, T. Hsu, Y.-C. Jao, C.-H. Huang, J.-S. Song, Y.-C. Huang, C. H. Chien, J. H. Chiu, S.-C. Yen, H. K. Tang, Y.-S. Chao and W.-T. Jiaang, J. Med. Chem., 2010, 53, 6572 CrossRef CAS.
- J. N. Sangshetti, A. S. Zambare, I. Gonjari and D. B. Shinde, Mini-Rev. Org. Chem., 2014, 11, 225 CrossRef CAS.
- H. Zhu, R. F. Yang, L. H. Yun and J. Li, Chin. Chem. Lett., 2010, 21, 35 CrossRef CAS.
- I. P. Beletskaya and A. V. Cheprakov, Chem. Rev., 2000, 100, 3009 CrossRef CAS.
- K. Balogh Sivars, U. Sivars, E. Hornberg, H. Zhang, L. Brändén, R. Bonfante, S. Huang, S. Constant, I. Robinson, C. J. Betts and P. M. Âberg, Toxicol. Sci., 2018, 162, 301 CrossRef.
- R. Z. Cer, U. Mudunuri, R. Stephens and F. J. Lebeda, Nucleic Acids Res., 2009, 37(suppl. 2), W441 CrossRef CAS.
- E. Blanco, H. Shen and M. Ferrari, Nat. Biotechnol., 2015, 33, 941 CrossRef CAS.
- R. T. Aimes, A. Zijlstra, J. D. Hooper, S. M. Ogbourne, M. L. Sit, S. Fuchs, D. C. Gotley, J. P. Quigley and T. M. Antalis, Thromb. Haemostasis, 2003, 89, 561 CrossRef CAS.
- J. A. McIntyre and J. Castañer, Drugs Future, 2004, 29, 882 CrossRef CAS.
- J. Nemunaitis, S. J. Vukelja, D. Richards, C. Cunningham, N. Senzer, J. Nugent, H. Duncan, B. Jones, E. Haltom and M. J. Uprichard, Cancer Invest., 2006, 24, 553 CrossRef CAS.
- K. Narra, S. R. Mullins, H. O. Lee, R. Stzremkowski-Brun, K. Magalong, V. J. Christansen, P. A. McKee, B. Egleston, S. J. Cohen, L. M. Weiner, N. J. Meropol and J. D. Cheng, Cancer Biol. Ther., 2007, 7, 1691 CrossRef.
- P. Dvořáková, P. Bušek, T. Knedlik, J. Schimer, T. Etrych, L. Kostka, L. Stollinová Šromová, V. Šubr, P. Šácha, A. Šedo and J. Konvalinka, J. Med. Chem., 2017, 60, 8385–8393 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis protocols for the preparation of compounds 4, 6, 8 and Biotin-PEG3-FAPi; analytical data (1H and 13C NMR spectra for new synthesized compounds); analytical UPLC traces for compounds 5 and Biotin-PEG3-FAPi; procedure for the coating of BFO NPs with heterobifunctional PEG derivatives; STEM and FT-IR characterization of BFO-PEG-FAPi; qualitative analysis by ESI-HRMS; multiphoton multispectral microscopy. See DOI: 10.1039/c9ra05299b |
| This journal is © The Royal Society of Chemistry 2019 |