
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign of highly porous Fe3O4@reduced graphene oxide via a facile PMAA-induced assembly†
Huan Wanga,
Madumali Kalubowilageb,
Stefan H. Bossmann b and
Placidus B. Amama*a
b and
Placidus B. Amama*a
aTim Taylor Department of Chemical Engineering, Kansas State University, Manhattan, KS 66506, USA. E-mail: pamama@ksu.edu
bDepartment of Chemistry, Kansas State University, Manhattan, KS 66506, USA
First published on 4th September 2019
Abstract
Advances in the synthesis and processing of graphene-based materials have presented the opportunity to design novel lithium-ion battery (LIB) anode materials that can meet the power requirements of next-generation power devices. In this work, a poly(methacrylic acid) (PMAA)-induced self-assembly process was used to design super-mesoporous Fe3O4 and reduced-graphene-oxide (Fe3O4@RGO) anode materials. We demonstrate the relationship between the media pH and Fe3O4@RGO nanostructure, in terms of dispersion state of PMAA-stabilized Fe3O4@GO sheets at different surrounding pH values, and porosity of the resulted Fe3O4@RGO anode. The anode shows a high surface area of 338.8 m2 g−1 with a large amount of 10–40 nm mesopores, which facilitates the kinetics of Li-ions and electrons, and improves electrode durability. As a result, Fe3O4@RGO delivers high specific-charge capacities of 740 mA h g−1 to 200 mA h g−1 at various current densities of 0.5 A g−1 to 10 A g−1, and an excellent capacity-retention capability even after long-term charge–discharge cycles. The PMAA-induced assembly method addresses the issue of poor dispersion of Fe3O4-coated graphene materials—which is a major impediment in the synthesis process—and provides a facile synthetic pathway for depositing Fe3O4 and other metal oxide nanoparticles on highly porous RGO.
Introduction
Smart or stimuli-responsive polymers are suitable to construct a morphology-controllable network due to their conformation and structural change in response to applied external stimuli such as pH, temperature, light, and electrical or magnetic fields.1–3 Slight external triggers can induce significant changes in the structure and properties of smart polymers including the change of degree of cross-linking, expansion, or shrinkage of polymer volume, or the detachment of polymer functional groups. Poly(methacrylic acid) (PMAA), a common pH-responsive polymer, has already attracted intense attention due to its interesting dependence of charge density on the solution.4–6 PMAA contains a large number of ionizable carboxyl groups, which makes it a type of water-soluble weak polyelectrolyte with a pKa value of 4.9.7 The dissociation degree of carboxyl groups is largely influenced by the environmental pH; and PMAA chains can undergo reversible transitions between tightly coiled conformations at low pH and highly extended conformations at high pH.8Due to its super electrochemical properties and structural moldability, graphene has emerged as an excellent candidate for replacing commercial graphite anodes in lithium-ion batteries (LIBs).9 Large scalable preparation of graphene-based electrodes utilize graphene oxide (GO) as a host combining other high capacity metal/metal oxide active materials. Porous 3D graphene architecture10,11 has been widely developed to accommodate the restacking issue of GO sheets during common fabrication processes. Porous graphene serves as efficient conductive channels and buffer skeleton network, which can facilitate charge transfer and protect other active nanoparticles during the cycling process. Previous zeta-potential analyses12,13 indicate that GO sheets are negatively charged in aqueous media over a wide pH range of 1 to 11. The charge density of GO varies with pH, enabling GO sheets to also be considered as a pH-responsive material. Recently, some pH-sensitive GO-based materials have been prepared by grafting PMAA polymer via covalent or noncovalent interactions.14,15 In general, prior studies mainly focused on tuning pH-responsive properties of polymer chains for application in biomedical engineering,2,3 while applications in energy storage, especially LIBs, are yet to be reported.
Fe3O4 has a high theoretical capacity of 924 mA h g−1 and is widely used in combination with nanocarbons for designing LIB anodes.16–18 Fe3O4 nanoparticles of controlled size distribution can be deposited onto GO sheets (Fe3O4@GO) via in situ aqueous reaction, resulting in coordination bonds between Fe atoms and COOH groups on a GO surface.16 However, the concentration of Fe3O4@GO aqueous suspension is usually very low (<0.6 mg ml−1) due to the reduced number of repelling functional groups after depositing Fe3O4 onto a GO surface. The use of PMAA as a stabilizer can result in a higher concentration of Fe3O4@GO suspension in water. More importantly, it is possible to control the dispersion state of PMAA-stabilized Fe3O4@GO sheets in water by changing the pH, due to the high pH response of PMAA; we emphasize this simple strategy can be employed in the design of high-performance electrode materials for LIB application. Previous studies of developing a desired Fe3O4@graphene electrode mainly focused on seeking novel synthesis reactions of a Fe3O4@graphene nanocomposite,19–22 controlling the size of Fe3O4 nanoparticles,23–25 and optimizing structural design of the composites mostly by sacrificial template methods.18,26,27 However, self-assembly of Fe3O4@graphene has received little attention, especially using a pH-responsive polymer as the assembly agent. As mentioned above, in spite of the immense potential of pH-responsive polymers in LIB application, current publications on the subject are limited.1,2,4,8 As a common pH-responsive polymer, PMAA is worthwhile to investigate in terms of designing the desired Fe3O4@GO structure with high porosity and well-dispersed Fe3O4 for high-performance anodes. Furthermore, the underlying mechanism of self-assembly behavior of Fe3O4@GO sheets using PMAA is poorly understood and will benefit from further examination.
Herein, we report the rational design of highly porous Fe3O4@RGO anodes via a facile PMAA-induced self-assembly method that involves adjusting the pH of the aqueous media. Synthesis steps for porous Fe3O4@RGO anodes are illustrated in Fig. 1. Fe3O4@GO sheets form a network and PMAA acts as a stabilizer, a cross-linking agent, and a driving force for structural change. The Fe3O4@GO–PMAA sheets demonstrate phase transfer behavior at different pH since PMAA is a weak polyanion, exhibiting a negative and neutral charge at high and low pH, respectively.4–6 The study also evaluates the effectiveness of PMAA in dispersing Fe3O4@GO in water and probes the pH-responsive behavior of the stabilized suspensions. Various techniques (rheology measurement, UV-Vis, electron microscopy, and electrophoretic light scattering) were used to characterize the dispersion of Fe3O4@GO.16,20,21,28,29 The results indicate that PMAA is influential in controlling the dispersion state of Fe3O4@GO in water, and the resulting porosity of Fe3O4@RGO after drying and annealing. Furthermore, lower weight ratio of PMAA to Fe3O4@GO shows different degrees of stabilization of Fe3O4@GO in comparison to the same sample with the same pH solution, but with a higher weight ratio. Based on the results, a stabilizing mechanism is proposed to explain the pH-responsive self-assembly of Fe3O4@GO sheets in water. This work provides fresh insights into the dispersion and stabilization of Fe3O4@GO in water using weak polyelectrolytes and a novel synthetic pathway for controlled fabrication of super-mesoporous Fe3O4@RGO composites. The PMAA-induced self-assembly method addresses the issue of poor dispersion of Fe3O4-coated graphene materials—which is a major challenge during synthesis of graphene-based nanocomposites—and provides a new and facile synthetic pathway for producing highly porous RGO framework that serve as a support for well-dispersed Fe3O4 or other metal oxide nanoparticles.
 | ||
| Fig. 1 Schematic illustration of self-assembly of crumpled Fe3O4/RGO anodes via adjusting pH of dispersion. | ||
Results and discussion
pH-Response of Fe3O4@GO–PMAA in aqueous dispersion
Fig. 2a shows the molecular structures of citric acid and PMAA. The COOH groups on both molecules can form coordinative bonds with Fe3O4, and thus can be used as stabilizers for Fe3O4@GO dispersion. However, citric acid is a much smaller molecule than the long-chain PMAA polymer, which makes citric acid a type of non pH-sensitive electrolyte. The FTIR spectrum (Fig. S1†) shows strong characteristic wide absorption peaks around 3000 cm−1 that are associated with COOH groups on the PMAA chains. The conformational changes the PMAA chain undergoes as a function of pH is schematically shown in Fig. 2b, which are consistent with previous research publications.5,8,30 PMAA is a type of weak polyacid with isoelectric point at pH ∼ 2.30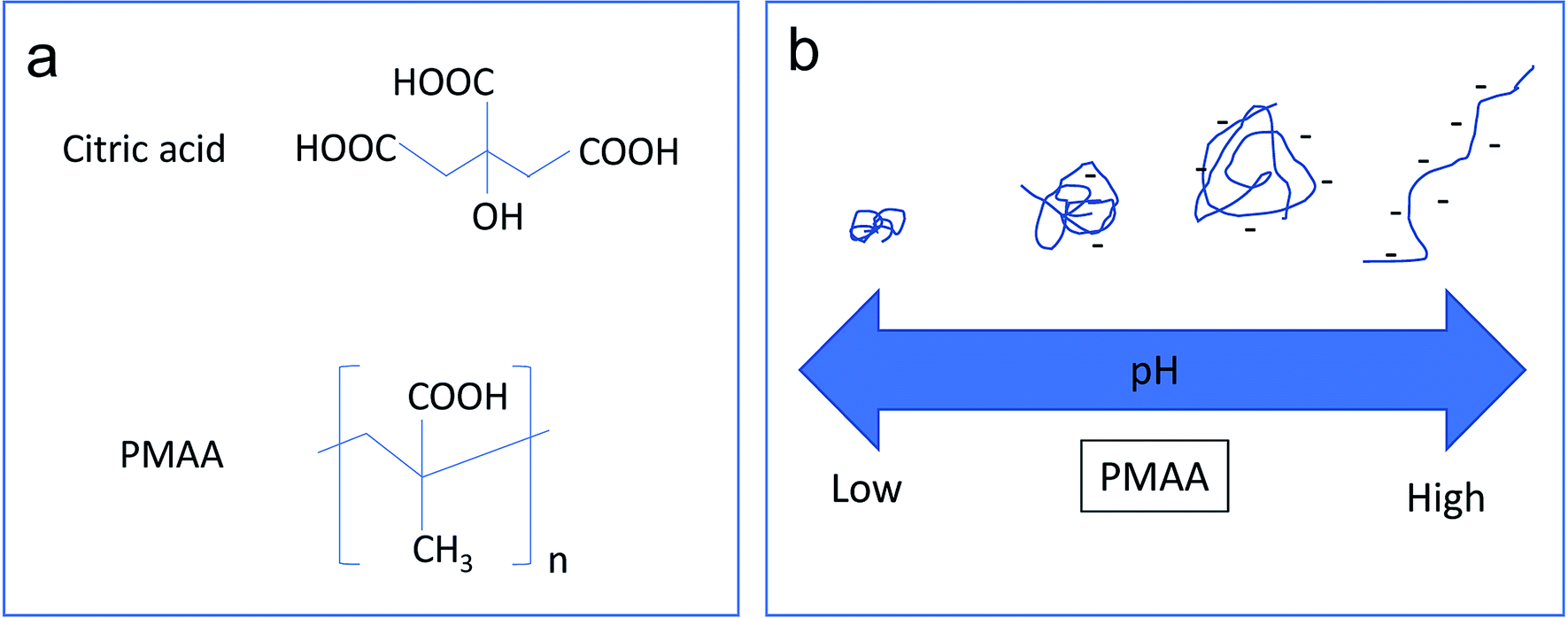 | ||
| Fig. 2 (a) Structure of citric acid and PMAA. (b) Schematic illustration of the effect of pH on the chain conformations of PMAA. | ||
Therefore, PMAA can form a transparent and homogeneous water solution (1.2 mg ml−1 and 4.8 mg ml−1) at any pH higher than 1.0 due to ionization of COOH associated with negative zeta potential values.30 At high pH (>5), the PMAA chains are in an ionized state and behave as a randomly coiled or extended configuration. The high negative-charge density along the polymer backbone causes the polymer chain to repel itself, and thus further stabilize the attached Fe3O4@GO sheets. At low pH (<4), PMAA exhibits significant intramolecular hydrogen bonding, resulting in a more crumpled or globular-like conformation. A previous study5 confirmed that sudden conformational change of PMAA occurs from a collapsed conformation to a random coil in the range of pH 4–6.
PMAA can generally stabilize Fe3O4@GO sheets in water media at pH between 5 and 9, as shown by the pictures of PMAA aqueous solutions and Fe3O4@GO–PMAA suspensions in Fig. 3; a summary of their dispersion states is presented in Table 1. Two PMAA aqueous solutions with 1.2 mg ml−1 and 4.8 mg ml−1 were used for this study. The black Fe3O4@GO suspensions were stable even after storing for a long time (>1 month). However, at pH <4 or >9, the Fe3O4@GO sheets are highly aggregated. Note at specific pH (pH 5, 7, and 9), there are differences in the degree of dispersion for Fe3O4@GO stabilized by 1.2 mg ml−1 PMAA and 4.8 mg ml−1 PMAA. The stability of Fe3O4@GO dispersion with 4.8 mg ml−1 PMAA decreases from “highly dispersed” to “moderately dispersed”, and then to “slightly dispersed” with increasing pH from 5 to 7 and then to 9, respectively. In particular, at pH 9, some particles in Fe3O4@GO dispersion with 4.8 mg ml−1 PMAA can be observed at the bottom of the container after one month of storage; whereas for Fe3O4@GO dispersion with 1.2 mg ml−1 PMAA, the best stability was achieved at pH 7, and dispersions at pH 5 and 9 exhibited moderate stabilities.
 | ||
| Fig. 3 Pictures of PMAA aqueous solution and Fe3O4@GO aqueous suspensions stabilized with 1.2 mg ml−1 and 4.8 mg ml−1 PMAA at different pH levels. | ||
| pH | PMAA dispersion | pH | Fe3O4@GO dispersion (1.2 mg ml−1 PMAA) | Fe3O4@GO dispersion (4.8 mg ml−1 PMAA) |
|---|---|---|---|---|
| 1 | Precipitated | 1–3 | Precipitated | Precipitated |
| 2–3 | Mostly crumbled | 4 | Slightly dispersed | Slightly dispersed |
| 4–6 | Crumpled to coiled | 5 | Moderately dispersed | Highly dispersed |
| 7 | Mostly coiled | 7 | Highly dispersed | Moderately dispersed |
| 9–11 | Mostly extended | 9 | Moderately dispersed | Slightly dispersed |
| 11 | Precipitated | Precipitated |
To rationalize the observed difference in the stability of the dispersions at pH 5, 7, and 9, zeta potential, UV-Vis spectroscopic and viscosity measurements were carried out. Zeta potential represents the equilibrium electric potential at the shear plane (or slip plane) of particles in a liquid.31,32 Typically, the more positive or negative zeta potentials correspond to higher stability of dispersed particles. Table 2 shows zeta potentials for Fe3O4@GO–PMAA aqueous suspensions with 1.2 mg ml−1 and 4.8 mg ml−1 PMAA at pH 5, 7, and 9. The zeta potential exhibits an increasing trend with pH from 5 to 9, regardless of the concentration of PMAA; this is mainly attributed to the increasing amount of COOH groups ionized from PMAA as the pH increases. Besides, at the same pH, 4.8 mg ml−1 PMAA-stabilized Fe3O4@GO suspensions show higher zeta potentials than those with 1.2 mg ml−1 PMAA. Interestingly, even though the most negative zeta potential occurs at pH 9 for the Fe3O4@GO–PMAA suspensions at various concentrations, the highest stabilities were observed at much lower pH (pH 5 for 4.8 mg ml−1 PMAA-stabilized Fe3O4@GO and pH 7 for 1.2 mg ml−1 PMAA-stabilized Fe3O4@GO). This ‘paradox’ has been explained by UV-Vis spectroscopic and particle-size data presented in the following discussion.
UV-Vis spectroscopic analysis was used to determine the optimum pH for stabilizing Fe3O4@GO (Fig. 4a). The stability of 2 mg ml−1 Fe3O4@GO with 1.2 mg ml−1 PMAA and 4.8 mg ml−1 PMAA dispersions were systematically studied at different pH. After diluting and resting the aforementioned dispersions for four days, the upper stable dispersions were tested in order to minimize the scattering influence of agglomerated particles on increasing UV absorbance. It has been reported that suspensions containing more dispersed particles have higher UV absorbance compared to suspensions containing agglomerated ones.8,33 An amount of 1.2 mg ml−1 PMAA-stabilized Fe3O4@GO at pH 7 shows the highest UV-Vis absorption, while for 4.8 mg ml−1 PMAA-stabilized Fe3O4@GO, the highest absorption occurs at a pH of 5. Since pure PMAA (1.2 or 4.8 mg ml−1) shows almost zero UV-Vis absorbance, the mentioned difference in the UV-Vis absorption may be due to the presence of more COO− in 4.8 mg ml−1 PMAA-stabilized Fe3O4@GO sheets than in 1.2 mg ml−1 PMAA-stabilized Fe3O4@GO, which has been verified by the zeta potential data in Table 2. So, for 4.8 mg ml−1 PMAA-stabilized Fe3O4@GO, at a relatively lower pH of 5, there is enough negative-charge density (−36.5 mV of zeta potential) along the GO sheet for repulsing other neighboring GO sheets, even though PMAA are still in coiled conformation at that acidic pH. In comparison, for 1.2 mg ml−1 PMAA-stabilized Fe3O4@GO, enough charge density can be achieved at pH 7 (−26.9 mV of zeta potential) instead of at pH 5 (−21.4 mV of zeta potential). As pH increases from 5 to 9, more carboxylate groups emerge, resulting in a fully extended configuration of PMAA chains formed eventually. Note that it is highly possible that one extended PMAA chain attaches to more than one surrounding GO sheet via bonding with Fe3O4; as a result, the GO sheets can be pulled much closer, resulting in restacking. Ditsch et al.34 reported a similar phenomenon for the poly acrylic acid (PAA)-stabilized Fe3O4, demonstrating that one PAA chain can link to multiple Fe3O4 particles and then form ‘bridges’ between Fe3O4 particles. For 4.8 mg ml−1 PMAA-stabilized Fe3O4@GO, UV-Vis absorption decreases as pH increases from 5 to 9; this result supports the ‘bridging’-induced restacking hypothesis at high pH range. Interestingly, the UV-Vis absorption profiles for 1.2 mg ml−1 PMAA stabilized Fe3O4@GO are somewhat different at different pH. We conclude from the results that enough charge density of PMAA cannot be achieved at pH 5, but can be at pH 7, and thus the highest absorption occurs at pH 7; whereas at pH 9, the restacking of GO sheets also occurs, albeit insignificantly, compared to that of 4.8 mg ml−1 PMAA-stabilized Fe3O4@GO.
 | ||
| Fig. 4 (a) UV-Vis spectra of Fe3O4@GO–PMAA aqueous suspensions with 1.2 mg ml−1 and 4.8 mg ml−1 PMAA at different pH levels. (b) Effective particle diameter of PMAA-stabilized Fe3O4@GO aqueous suspensions with 1.2 mg ml−1 and 4.8 mg ml−1 PMAA at different pH levels. | ||
Size of Fe3O4@GO–PMAA in Fig. 4b supports the ‘bridging’ phenomenon observed in the UV-Vis data. DLS technology was used here to observe mean effective particle diameter (z-average diameter) and standard size deviations of Fe3O4@GO–PMAA. The smallest effective particle diameter was achieved at pH 7 for the 1.2 mg ml−1 PMAA-stabilized Fe3O4@GO, which suggests the optimum dispersion state under these conditions, while the particle size increases at pH 5 due to the lower charge density on Fe3O4@GO sheets. At pH 9, even though the charge density reaches a high value (−51.1 mV of zeta potential), the stability of the suspension decreases because of the ‘bridge’ between neighboring Fe3O4@GO sheets. In comparison, for the 4.8 mg ml−1 PMAA-stabilized Fe3O4@GO, the effective particle diameter keeps increasing from pH 5 to 9. The most severe ‘bridging’ occurs at pH 9, which results in a slightly dispersed suspension, as observed in Fig. 3. TEM images (Fig. S2†) also support that at pH 9, 4.8 mg ml−1 PMAA-stabilized Fe3O4@GO sheets suffer severe agglomeration; unlike an extended sheet-like structure, flower-shaped patterns of Fe3O4@GO sheets were induced by the bridging effect at high PMAA concentration. Given that at pH 9, particles in 4.8 mg ml−1 PMAA-stabilized Fe3O4@GO experience slight aggregation during the DLS (dynamic light scattering) testing,29 we performed testing for shorter duration (<10 seconds) to determine the correct effective particle diameter for this sample. Therefore, based on the UV-Vis and particle-size results, we conclude that in general, Fe3O4@GO suspensions show better stability at pH 5 or 7 than at pH 9, and the point where ‘bridging’ occurs is related to the concentration of PMAA (or the weight ratio of PMAA to Fe3O4@GO).
Suspensions with well-dispersed GO sheets are known to show higher viscosities compared to those that have agglomerated sheets.35 Furthermore, it was shown that below the critical concentration of 90 mg ml−1, PMAA–water solution does not show shear thinning or thickening behavior with an increase of shear rate.36 The observed shear-thinning behavior in Fe3O4@GO–PMMA suspensions suggests the presence of highly dispersed Fe3O4@GO sheets. In an effort to investigate this pH-dependent behavior, viscosity measurements were obtained for 1.2 mg ml−1 and 4.8 mg ml−1 PMAA-stabilized Fe3O4@GO aqueous suspensions (Fig. 5) at pH 5 and 7. The concentration of PMAA polymer denominates the viscosity of suspension due to the sticky nature of polymer. In general, 4.8 mg ml−1 PMAA-stabilized Fe3O4@GO suspensions show higher viscosity compared to those of 1.2 mg ml−1 PMAA-stabilized Fe3O4@GO suspensions. The similar viscosities at pH 5 and 7 for 4.8 mg ml−1 PMAA-stabilized Fe3O4@GO also reveal the bigger influence of PMAA concentration on viscosity, although the dispersion state at pH 5 is better than pH 7, based on UV-Vis absorption results. After comparing all viscosity profiles shown in Fig. 5, we conclude again that 1.2 mg ml−1 PMAA-stabilized Fe3O4@GO at pH 7 and 4.8 mg ml−1 PMAA-stabilized Fe3O4@GO at pH 5 are the two conditions for the highest dispersion due to their high viscosities.
 | ||
| Fig. 5 Viscosity as a function of shear rate for Fe3O4@GO–PMAA aqueous suspensions with 1.2 mg ml−1 and 4.8 mg ml−1 PMAA at different pH levels. | ||
Chemical and structural characterization of Fe3O4@RGO anode
As shown in Fig. 1, Fe3O4@RGO anode materials are produced after annealing the Fe3O4@GO–PMAA sheets at 600 °C for 3 h. GO sheets can be thermally reduced to RGO; meanwhile, PMAA can be removed with only 7 percent residual amorphous carbon (TGA, Fig. S3†). The porous structure of Fe3O4@RGO was designed by adjusting the pH of 1.2 mg ml−1 PMAA-stabilized Fe3O4@GO suspension from 9 to 4. Fe3O4@GO sheets were first stabilized by PMAA at pH 9, and under this condition, PMAA chains are completely extended and behave like connecting ‘bridges’ by linking neighboring Fe3O4@GO sheets. Therefore, neighboring GO sheets are constrained by PMAA chains instead of randomly dispersing in water. The distance between GO sheets decreased significantly when compared to that of citric acid-stabilized GO sheets. After decreasing pH from 9 to 4, the PMAA chains shrink to a highly coiled structure and the attached Fe3O4@GO sheets are forced to crumple at the same time.The as-synthesized Fe3O4@RGO anode material was analyzed by XRD first (Fig. 6a). Diffraction peaks at 2θ = 28.9°, 34.9°, 42.4°, 57.8°, and 62.0° are assigned to the (220), (311), (400), (511) and (440) planes of face-centered cubic Fe3O4 (JCPDS No. 63-3107), respectively. Remarkably, the diffraction peak (002) associated with RGO is identified at 20–30°, indicating the crumpled and partially stacked states of graphene layers after adjusting pH from 9 to 4. Raman spectrum of the Fe3O4@RGO (Fig. 6b) shows a typical peak of magnetite ∼680 cm−1 and characteristic peaks of the D- and G-bands from graphene at around 1338 and 1600 cm−1.
 | ||
| Fig. 6 (a) XRD pattern of Fe3O4@RGO. (b) Raman spectrum of Fe3O4@RGO. | ||
SEM was used to support the proposed self-assembly mechanism of Fe3O4@GO sheets induced by PMAA, as well as showing the surface morphology of the resulting Fe3O4@RGO anode after annealing treatment. Fig. 7a reveals a rough surface for PMMA-induced Fe3O4@RGO materials. For comparison, the citric acid-stabilized Fe3O4@RGO was analyzed under similar conditions and a clear contrast was observed, revealing a relatively smoother surface (Fig. 7b). Panels (c) and (d) of Fig. 7 clearly exhibit the porosity difference between PMAA-induced and citric acid-stabilized Fe3O4@RGO anodes; the former shows more mesopores (∼10–40 nm) than the latter. Furthermore, for PMAA-induced Fe3O4@RGO, there are fewer agglomerated Fe3O4 nanoparticles compared to those in the citric acid-stabilized Fe3O4@RGO material. Additional SEM images of PMAA-induced Fe3O4@RGO materials are presented in the Fig. S4.† The morphology of Fe3O4 particles on RGO sheets was characterized by high-resolution TEM image (Fig. S5†); the average size of Fe3O4 is ∼10 nm with narrow size distribution.
 | ||
| Fig. 7 (a and b) SEM images of PMAA-induced porous Fe3O4@RGO. (c and d) Citric acid-assisted stacked Fe3O4@RGO. (e) Nitrogen adsorption–desorption isotherms and pore-size distribution of PMAA-induced porous Fe3O4@RGO (inset figure). (f) TGA curve of Fe3O4@RGO acquired with a temperature ramp of 5 °C min−1 in air. | ||
To further investigate the porous structure and surface area of PMAA-induced Fe3O4@RGO material, N2 adsorption–desorption isotherms were obtained, as shown in Fig. 7e. The adsorption–desorption profile is close to type IV with an evident hysteresis loop in the 0.4–1.0 range of relative pressure, indicating the mesoporous structure of the Fe3O4@RGO composites. In addition, the inset in Fig. 7e shows the pore-size distribution (obtained from the Barrett–Joyner–Halenda model) for PMAA-induced Fe3O4@RGO, in which the pore diameters are predominantly below ∼10 nm. Therefore, it can be concluded that PMAA-induced Fe3O4@RGO possesses a hierarchical porous structure composed of micropores and mesopores. Furthermore, the specific surface area of the composite is ∼338.8 m2 g−1, which is clearly higher than many state-of-the-art Fe3O4@RGO anodes reported in the literature (Table S1†).12,28,37–39 To conclude, our porous structure of Fe3O4@RGO material not only offers a buffer room for huge volume expansion of Fe3O4 during the charge/discharge cycles, but also facilitates the transport of lithium-ions and electrolyte molecules, which makes active sites accessible and eventually results in enhanced anode performance in LIBs.
Electrochemical performance of Fe3O4@RGO anode
From the TGA data (Fig. 7f), the composition of Fe3O4 is ∼60 wt%. Based on this weight ratio, the Fe3O4@RGO anode shows a theoretical capacity of ∼703 mA h g−1 (924 mA h g−1 × 60 percent + 372 mA h g−1 × 40 percent).16 Fig. 8a shows the first four and the 200th charge–discharge curves of PMAA-induced Fe3O4@RGO half-cell at a current density of 2 A g−1, with a voltage window of 0.002–3.0 V. The first discharge capacity is about 1470 mA h g−1, while the second capacity decreases to 770 mA h g−1. The irreversible capacity loss is related to the formation of the solid-electrolyte interphase (SEI) film.40,41 After 200 cycles, the capacity stabilizes at 480 mA h g−1. For comparison, the citric acid-stabilized Fe3O4@RGO anode is also cycled under the same conditions (Fig. 8b). It is obvious the specific capacity of citric acid-stabilized Fe3O4@RGO decreases faster than that of PMAA-induced Fe3O4@RGO. The capacity gradually fades to 200 mA h g−1 during 200 discharge/charge cycles. Fig. S6† shows 1–100 cycling performance of PMAA-induced Fe3O4@RGO and citric acid-stabilized Fe3O4@RGO anode at a low current rate (0.5 A g−1), with the former showing superior performance. At this relatively lower cycling rate, the fading of capacities becomes slower for both samples compared to those at higher current (2 A g−1). | ||
| Fig. 8 (a) Selected discharge–charge profiles (1st, 2nd, 3rd, 4th, and 200th cycles) of a PMAA-induced Fe3O4/RGO anode, and (b) a citric acid-stabilized Fe3O4@RGO anode at a current density of 2 A g−1 in the potential window of 3.0–0.002 V (vs. Li/Li+); (c) cycling performance of a PMAA-induced Fe3O4/RGO anode; and (d) a citric acid-stabilized Fe3O4@RGO anode at different cycling rates of 0.5 A g−1, 1 A g−1, 2 A g−1, 5 A g−1, and 10 A g−1 with a cutoff voltage window of 3.0–0.002 V. | ||
The rate capability of PMAA-induced Fe3O4@RGO was studied at different current rates, from 0.5 to 10 A g−1 (Fig. 8c). Fig. 8c shows the decrease in the capacity with increasing C rates. A specific charge capacity of 740 mA h g−1 is obtained at 0.5 A g−1, and even at a high current density of 10 A g−1, it still delivers 200 mA h g−1. In contrast, the capacity of citric acid-stabilized Fe3O4@RGO fades dramatically to 70 mA h g−1 at 10 A g−1 (Fig. 8d). In Fig. 8c, a high capacity of 780 mA h g−1 can be measured when the current rate reduces back from 10 A g−1 to 0.5 A g−1, indicating a high stability as well as excellent reversibility. As shown in Fig. 9, the Li storage capacity of our Fe3O4@RGO is comparable to Fe3O4-based nanocarbon anodes with a higher weight percentage (69–89 wt%) of Fe3O4.12,28,40–43 Fe3O4 shows a high theoretical capacity (924 mA h g−1), therefore energy density of anodes is largely dependent on weight ratio of Fe3O4 to graphene or other nanocarbon component. However, excessive Fe3O4 in the anode often leaves it unprotected by nanocarbon and inevitably exposed to the electrolyte, thus inducing volume expansion and capacity fading during the discharging/charging process. In this study, our anode composite only contains 60 wt% of Fe3O4, which enables the high energy density to be preserved while mitigating its structural pulverization. The improved stability and rate performance of the PMAA-induced Fe3O4@RGO is attributed to its hierarchical porous structure that facilitates fast Li+ diffusion and an effective electron transport during the charge–discharge cycles.
 | ||
| Fig. 9 Comparison of anode performance of Fe3O4@RGO with state-of-the-art Fe3O4-nanocarbons (containing different weight percentages of Fe3O4). | ||
Conclusions
To summarize, our results elucidate the relationship between the media pH and nanomaterial structure, in terms of dispersion state of Fe3O4@GO and porosity of Fe3O4@RGO. By adjusting media pH, configuration and charge density of PMAA chains change, which largely determines the stability of Fe3O4@GO in water. The concentration ratio of PMAA and Fe3O4@GO plays another important role in the stability of Fe3O4@GO due to the ‘bridging’ phenomenon that dominates at different pH. The PMAA-induced self-assembly method is a facile method for designing high-performance Fe3O4@RGO anode materials. The presence of large amounts of mesopores improve the porosity of Fe3O4@RGO materials and provide buffer space for Fe3O4 particles, as well as accommodate the huge volume expansion during long-term discharge/charge cycles. The resulting Fe3O4@RGO anode with only 60 wt% Fe3O4 can still deliver high capacities of 740 mA h g−1 to 200 mA h g−1 at various current densities of 0.5 A g−1 to 10 A g−1, which are comparable to other Fe3O4-based anodes with a higher weight ratio of Fe3O4. The PMAA-induced self-assembly method addresses the issue of poor dispersion of iron oxide-coated graphene materials and provides a highly porous graphene framework for depositing other higher-capacity active nanoparticles.Experimental
Synthesis of PMAA
We added 150 ml of ethanol, 67 g of methacrylic acid (MAA), and 0.7 g of 2, 2′-azobis(2-methylpropionamidine) dihydrochloride (AIBA) to a 500 ml flask equipped with a thermocouple for monitoring the temperature and a mechanical stirring device. The reaction mixture was purged with argon at ambient temperature for 30 min under stirring. The temperature was then raised to 65 °C and the mixture was kept at 65 °C for 6 h. The reaction mixture was then cooled to 25 °C, and the sample was dried and stored for further analysis. The solid content (or percentage of PMAA) was ∼35 wt% in the solvent.Preparation of Fe3O4@GO–PMAA aqueous solution
The method for decoration of Fe3O4 nanoparticles on GO sheets is described in detail in our previous work.16 We added 259.6 mg FeCl2·4H2O and 708.4 mg FeCl3·4H2O to GO dispersion (50 ml of 1 mg ml−1) at 80 °C under N2 atmosphere. For a lower weight ratio of PMAA to Fe3O4@GO composite, we mixed 12 mg of PMAA with 20 mg Fe3O4@GO in 10 ml water, and sonicated it at room temperature for 1 h to form coordinate bonds between Fe3O4 and PMAA; while for the sample with a higher ratio, we added 48 mg of PMAA in the dispersion. Ammonia and HCl solutions in water were used to adjust pH between 1 and 9 for the Fe3O4@GO–PMAA dispersed in water. In comparison, an Fe3O4@GO–citric acid dispersion was also prepared following the steps in our previous work.16Self-assembly of porous Fe3O4@RGO anodes
The as-prepared Fe3O4@GO–PMAA dispersion (∼2 ml) was drop-cast on the Cu current collector (9 μm thick) and dried at 40 °C in a vacuum oven. Thereafter, the dried Fe3O4/GO was annealed at 600 °C for 3 h in an argon atmosphere to reduce GO to RGO.29 Synthesis steps for porous Fe3O4@RGO anodes are illustrated in Fig. 1. PMAA is able to stabilize Fe3O4@GO in water and facilitate the formation of a concentrated Fe3O4@GO dispersion (∼2 mg ml−1). Thereafter, by changing the pH of the solution from basic to acidic, crumpled Fe3O4@GO sheets were formed. An Fe3O4@RGO anode stabilized via citric acid treatment was also prepared for comparison.Materials characterization
XRD patterns were collected on a Rigaku Miniflex II X-ray diffractometer with Cu Kα radiation (λ = 1.5418 Å) at 40 kV and 40 mA. The morphological characterization of the samples was conducted using a field-emission scanning electron microscope (SEM, Hitachi S5200) and a transmission electron microscope (TEM, FEI Talos). For TEM imaging, a small amount of the Fe3O4/RGO sample was dispersed in isopropanol via ultrasonication; a drop of the homogeneous suspension was deposited on a holey carbon TEM grid and examined at 120 and 200 kV. Thermogravimetric analysis (TGA) was performed on a Q500 TGA analyzer (TA Instruments) with a temperature ramp of 5 °C min−1 from room temperature (RT) to 900 °C under a stream of air flow for Fe3O4@RGO, and from RT to 600 °C under nitrogen flow for PMAA. UV-Vis measurements were performed on a UV-2600 spectrometer (Shimadzu) using plastic cuvettes. Suspensions were first diluted to a suitable value followed by four days of rest. The upper stable suspensions were finally used for UV-Vis measurements. Viscosity of the suspensions as a function of shear rate was measured using a Brookfield rheometer equipped with 40 mm parallel-plate geometry. The annealing treatment was performed in a chemical vapor deposition (CVD) system by heating it to 600 °C for 3 h in argon flow (250 sccm). Brunauer–Emmett–Teller (BET) specific surface areas were determined from N2 adsorption using a Quantachrome (Autosorb-1) instrument at liquid nitrogen temperature. Raman spectra were measured using a Horiba 550 Raman spectrometer with a laser wavelength of 532 nm. The zeta potentials and effective particle sizes of Fe3O4@GO–PMAA were measured on a Brookhaven ZetaPALS analyzer. For reporting of accurate effective particle sizes, mean z-average diameter along with the standard deviations were derived based on seven replicate measurements. The pH dependence of zeta potential for Fe3O4@GO–PMAA aqueous suspension at 23 °C was measured at different pH values.Electrochemical measurements
The half-cell assembly was carried out in an argon-filled glovebox with concentrations of moisture and oxygen below 5 ppm. The as-prepared Fe3O4@RGO nanocomposite on the Cu foil current collector was used as an anode for cells. The separator was a microporous polypropylene membrane and the counter electrode was Li foil. The electrolyte was prepared by dissolving 1 M of LiPF6 in an ethylene carbonate (EC)/dimethyl carbonate (DMC)/diethyl carbonate (DEC) mixture (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1, in wt%). The mass loading of anode materials was controlled around 4.6 mg cm−2 on the current collector. Galvanostatic cycling experiments of the cells were performed on a Maccor 4300 battery test system in the voltage range of 0.001–3.00 V versus Li+/Li at room temperature.
:1:1, in wt%). The mass loading of anode materials was controlled around 4.6 mg cm−2 on the current collector. Galvanostatic cycling experiments of the cells were performed on a Maccor 4300 battery test system in the voltage range of 0.001–3.00 V versus Li+/Li at room temperature.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to the National Science Foundation (Grant Number 1653527) and Kansas State University for their support of this work. Publication of this article was funded in part by the Kansas State University Open Access Publishing Fund. Dr Lauren Chlebanowski and Dr Prem Thapa are acknowledged for their assistance with TGA and TEM characterization, respectively.References
- L. Zhai, Chem. Soc. Rev., 2013, 42, 7148–7160 RSC.
- L. Qiu, D. Liu, Y. Wang, C. Cheng, K. Zhou, J. Ding, V. T. Truong and D. Li, Adv. Mater., 2014, 26, 3333–3337 CrossRef CAS PubMed.
- C. De las Heras Alarcón, S. Pennadam and C. Alexander, Chem. Soc. Rev., 2005, 34, 276–285 RSC.
- G. D. Mendenhall, Y. Geng and J. Hwang, J. Colloid Interface Sci., 1996, 184, 519–526 CrossRef CAS PubMed.
- X. Wang, X. Ye and G. Zhang, Soft Matter, 2015, 11, 5381–5388 RSC.
- J. Zhang and N. A. Peppas, Macromolecules, 2000, 33, 102–107 CrossRef CAS.
- M. R. de Moura, F. A. Aouada and L. H. Mattoso, J. Colloid Interface Sci., 2008, 321, 477–483 CrossRef CAS PubMed.
- K. C. Etika, M. A. Cox and J. C. Grunlan, Polymer, 2010, 51, 1761–1770 CrossRef CAS.
- K. Wu, H. Yang, L. Jia, Y. Pan, Y. Hao, K. Zhang, K. Du and G. Hu, Green Chem., 2019, 21, 1472–1483 RSC.
- K. Wu, K. Du and G. Hu, J. Mater. Chem. A, 2018, 6, 3444–3453 RSC.
- K. Wu, D. Liu and Y. Tang, Electrochim. Acta, 2018, 263, 515–523 CrossRef CAS.
- W. Wei, S. Yang, H. Zhou, I. Lieberwirth, X. Feng and K. Müllen, Adv. Mater., 2013, 25, 2909–2914 CrossRef CAS PubMed.
- H. Wei, W. Yang, Q. Xi and X. Chen, Mater. Lett., 2012, 82, 224–226 CrossRef CAS.
- X. Zhao, L. Yang, X. Li, X. Jia, L. Liu, J. Zeng, J. Guo and P. Liu, Bioconjug. Chem., 2015, 26, 128–136 CrossRef CAS PubMed.
- T. Gao, Q. Ye, X. Pei, Y. Xia and F. Zhou, J. Appl. Polym. Sci., 2013, 127, 3074–3083 CrossRef CAS.
- H. Wang, J. Xie, M. Follette, T. C. Back and P. B. Amama, RSC Adv., 2016, 6, 83117–83125 RSC.
- H. Wang, X. Li, M. Baker-Fales and P. B. Amama, Curr. Opin. Chem. Eng., 2016, 13, 124–132 CrossRef.
- C. Kibby, K. Jothimurugesan, T. Das, H. S. Lacheen, T. Rea and R. J. Saxton, Catal. Today, 2013, 215, 131–141 CrossRef CAS.
- L. Li, A. Kovalchuk, H. Fei, Z. Peng, Y. Li, N. D. Kim, C. Xiang, Y. Yang, G. Ruan and J. M. Tour, Adv. Energy Mater., 2015, 5, 1500171–1500176 CrossRef.
- L. Zhao, M. Gao, W. Yue, Y. Jiang, Y. Wang, Y. Ren and F. Hu, ACS Appl. Mater. Interfaces, 2015, 7, 9709–9715 CrossRef CAS PubMed.
- X. Fan, S. Li, H. Zhou and L. Lu, Electrochim. Acta, 2015, 180, 1041–1049 CrossRef CAS.
- M. Zhang and M. Jia, J. Alloys Compd., 2013, 551, 53–60 CrossRef CAS.
- M. Ren, M. Yang, W. Liu, M. Li, L. Su, C. Qiao, X. Wu and H. Ma, Electrochim. Acta, 2016, 194, 219–227 CrossRef CAS.
- Y. Chen, B. Song, L. Lu and J. Xue, Nanoscale, 2013, 5, 6797–6803 RSC.
- L. Li, P. Gao, S. Gai, F. He, Y. Chen, M. Zhang and P. Yang, Electrochim. Acta, 2016, 190, 566–573 CrossRef CAS.
- D. Chen, G. Ji, Y. Ma, J. Y. Lee and J. Lu, ACS Appl. Mater. Interfaces, 2011, 3, 3078–3083 CrossRef CAS PubMed.
- S. H. Choi and Y. C. Kang, Carbon, 2014, 79, 58–66 CrossRef CAS.
- J. Su, M. Cao, L. Ren and C. Hu, J. Phys. Chem. C, 2011, 115, 14469–14477 CrossRef CAS.
- H. Wang, J. Xie, H. Almkhelfe, V. Zane, R. Ebini, C. Sorensen and P. B. Amama, J. Mater. Chem. A, 2017, 5, 23228–23237 RSC.
- S. Yu and G. M. Chow, J. Mater. Chem., 2004, 14, 2781–2786 RSC.
- M. Zembala, Adv. Colloid Interface Sci., 2004, 112, 59–92 CrossRef CAS PubMed.
- P. K. Koutsoukos, P. G. Klepetsanis and N. Spanos, Zeta-Potential Calculation, CRC Press, 2016 Search PubMed.
- J. C. Grunlan, L. Liu and O. Regev, J. Colloid Interface Sci., 2008, 317, 346–349 CrossRef CAS PubMed.
- A. Ditsch, P. E. Laibinis, D. I. Wang and T. A. Hatton, Langmuir, 2005, 21, 6006–6018 CrossRef CAS PubMed.
- J. Xiao, Y. Tan, Y. Song and Q. Zheng, RSC Adv., 2016, 6, 41392–41403 RSC.
- C. Robin, C. Lorthioir, C. Amiel, A. Fall, G. Ovarlez and C. Le Cœur, Macromolecules, 2017, 50, 700–710 CrossRef CAS.
- N. A. Zubir, C. Yacou, J. Motuzas, X. Zhang and J. C. D. Da Costa, Sci. Rep., 2014, 4, 4594 CrossRef PubMed.
- X. Li, X. Huang, D. Liu, X. Wang, S. Song, L. Zhou and H. Zhang, J. Phys. Chem. C, 2011, 115, 21567–21573 CrossRef CAS.
- Z.-S. Wu, S. Yang, Y. Sun, K. Parvez, X. Feng and K. Müllen, J. Am. Chem. Soc., 2012, 134, 9082–9085 CrossRef CAS PubMed.
- P. Lian, X. Zhu, H. Xiang, Z. Li, W. Yang and H. Wang, Electrochim. Acta, 2010, 56, 834–840 CrossRef CAS.
- Z. Zhang, F. Wang, Q. An, W. Li and P. Wu, J. Mater. Chem. A, 2015, 3, 7036–7043 RSC.
- S. Hao, B. Zhang, Y. Wang, C. Li, J. Feng, S. Ball, M. Srinivasan, J. Wu and Y. Huang, Electrochim. Acta, 2018, 260, 965–973 CrossRef CAS.
- L. Jun, X. Xijun, H. Renzong, Y. Lichun and Z. Min, Adv. Energy Mater., 2016, 6, 1600256 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Table S1: Surface areas of Fe3O4@RGO; Fig. S1: FTIR spectrum of PMAA; Fig. S2: TEM images of Fe3O4@RGO; Fig. S3: TGA curve of PMAA; Fig. S4: SEM images of Fe3O4@RGO; Fig. S5: TEM image of Fe3O4@RGO; Fig. S6: Cycling performance of PMMA-induced and citric acid-stabilized Fe3O4@RGO. See DOI: 10.1039/c9ra04980k |
| This journal is © The Royal Society of Chemistry 2019 |