
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceObservation of enhanced magnetic entropy change near room temperature in Sr-site deficient La0.67Sr0.33MnO3 manganite
B. Arunab,
V. R. Akshayab and
M. Vasundhara *ab
*ab
aMaterials Science and Technology Division, CSIR-National Institute for Interdisciplinary Science and Technology, Trivandrum, India. E-mail: mvas@niist.res.in; vasu.mutta@gmail.com; Tel: +91 4712515491
bAcademy of Scientific and Innovative Research (AcSIR), CSIR-Human Resource Development Centre, Ghaziabad, Uttar Pradesh, India
First published on 30th July 2019
Abstract
The effect of Sr-site deficiency on the structural, magnetic and magnetic entropy change of La0.67Sr0.33−yMnO3−δ (y = 0.18 and 0.27) compounds was investigated. The compounds were prepared by the conventional solid-state route and powder X-ray diffraction technique along with Rietveld refinement was carried out to confirm the structure and phase purity. Lattice parameters and unit cell volumes are found to increase with the increase in Sr-deficiency due to the electrostatic repulsion from the neighbouring oxygen ions. A mixed valence state of Mn2+/Mn3+/Mn4+ was confirmed using the X-ray photoelectron spectroscopy technique and it was observed that the change of state from Mn3+ + Mn3+ pairs to Mn2+ + Mn4+ pairs is different for both the studied compounds. A second order ferromagnetic–paramagnetic transition with an enhancement in magnetization in comparison to the pristine compound (La0.67Sr0.33MnO3) was observed due to multiple double exchange interactions. The La0.67Sr0.15□0.18MnO3−δ compound exhibits a magnetic entropy change (ΔSM) of 4.61 J kg−1 K−1 at 310 K, and the La0.67Sr0.06□0.27MnO3−δ compound exhibits a ΔSM of 4.11 J kg−1 K−1 at 276 K under a field of 50 kOe. In our previous work, we reported a large value of ΔSM but at higher temperatures, around 350 K. However, in the present case, we have achieved a near room temperature (310 K) MCE with a significant ΔSM value (4.61 J kg−1 K−1) which is larger than that reported for numerous perovskite manganites. Thus, the studied material could be a potential candidate for room temperature magnetic refrigeration applications.
Introduction
Magnetic refrigeration is a green and energy efficient technology based on the principle of the magnetocaloric effect (MCE). The recent trends in magnetic refrigeration confirm that it is a cost-effective and environmentally sound alternative to the conventional vapour compression refrigeration system.1–3 In comparison to the conventional technique, the magnetic refrigeration system has better energy efficiency, adaptability and does not release any greenhouse gases. In the entire process, the magnetocaloric material is subjected to repeated magnetization and demagnetization cycles, and as a result, the magnetic material heats up and cools down respectively due to the magneto-thermodynamic effect.4,5 However, the figure of merit of magnetocaloric materials is reported in terms of magnetic entropy change (ΔSM) and adiabatic temperature change (ΔTad).6,7 From the literature, it could be seen that intermetallic compounds (Gd5(Si2Ge2), La(Fe1−xSix), MnAs1−xSbx, Ni–Mn–X (X = Ga, In, Sn), MnFe(P1−xSix) etc.) and oxide compounds (mixed valence manganites, ErCo2, Ho2O3, etc.) are reported as potential candidates for magnetic refrigeration applications.8–18 Even though the intermetallic compounds show significant ΔSM values, they are detrimental to magnetic refrigeration applications due to their high cost, complicated synthesis routes, large thermal and field hysteresis etc. On the other hand, oxide materials especially perovskite manganites having an ABO3 structure are reported to be potential candidates for magnetic refrigeration applications due to their low cost, simple synthesis route, high chemical stability and low eddy current heating.19Doped manganites having a formula of 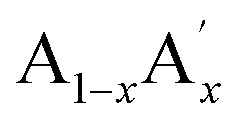 MnO3 are important candidates due to the correlation between charge, spin and orbital orders and their significant contribution towards the MCE. In doped manganites, rare Earth ions occupy the A-site, and alkaline rare Earth ions occupy the A′- site.20,21 The different oxidation state of rare Earth and alkaline rare Earth ions create a mixed valence state of manganese (Mn2+/Mn3+/Mn4+ etc.) and eventually determine the strength of the exchange interactions. Moreover, different rare Earth ions having the same oxidation state such as
MnO3 are important candidates due to the correlation between charge, spin and orbital orders and their significant contribution towards the MCE. In doped manganites, rare Earth ions occupy the A-site, and alkaline rare Earth ions occupy the A′- site.20,21 The different oxidation state of rare Earth and alkaline rare Earth ions create a mixed valence state of manganese (Mn2+/Mn3+/Mn4+ etc.) and eventually determine the strength of the exchange interactions. Moreover, different rare Earth ions having the same oxidation state such as 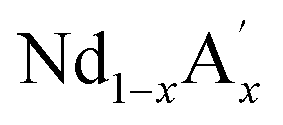 MnO3,
MnO3, 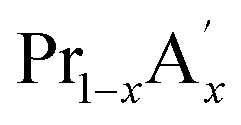 MnO3 and
MnO3 and 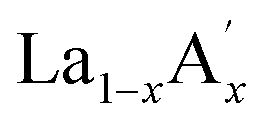 MnO3 also exhibit different exchange interactions as well as different magnetic and electrical transport properties due to the difference in the average A-site ionic radius.22–27 It is understood that, in doped manganites, the double exchange (DE) interaction via the Mn3+–O2−–Mn4+ ions is responsible for the ferromagnetic (FM) behaviour and the superexchange (SE) interaction through Mn3+–O2−–Mn3+ and Mn4+–O2−–Mn4+ ions are responsible for the antiferromagnetic (AFM) behaviour. This indicates that the strength of the competing exchange interactions and magnetic properties are eventually affected by different factors such as rare Earth substitution at the A-site, replacement of manganese by other transition elements, the introduction of deficiency at A and A′- sites, etc.28–37 Recently, many reports have demonstrated that the creation of deficiency at the rare Earth and alkaline rare Earth sites alter the Mn2+/Mn3+/Mn4+ ratio which severely affect the magnetic and magnetocaloric properties.37–49 The structure of manganites is imperfect and contain vacancy type point defects, called Schottky defects due to the cyclic temperature changes during synthesis and sintering process.50,51 The presence of cation (V(c)) and anion (V(a)) vacancies strongly influence the properties of the manganites and causing a change in the stability of the perovskite structure.52 The change in the valence state and coordination number of cations due to defect formation affects the magnetic phase transition temperature and magnetic ordering of the manganites. Moreover, a change in the bond length affects the packing of MnO6 octahedral and leads to a change in the structure and stability of the compounds. The interstitial point defects, called Frenkel defects is not formed in these compounds and only vacancy type point defects are exist due to elevated synthesis and sintering temperature conditions.50 The anion vacancies are formed during heating process due to the thermal dissociation of oxygen and that leads to the reduction of oxygen content to O3−δ.51 Moreover, the presence of vacancies at the oxygen site is compensated by a reduction in the average valence of manganese ions.52 On the other hand, the cation vacancies are formed during cooling process due to high mobility of cation sub lattices, and the formation of vacancies at the A-site leads to a decrease in the average valence of Mn-ions. The defectiveness and the stability in the perovskite structure can be controlled by changing the cooling conditions and the sintering temperature.51 Moreover, researchers have tried to tune the transition temperature (TC) and ΔSM by varying the vacancy content to a certain extent. Boujelben et al. reported the effect of Pr and Sr-deficiency in Pr0.7Sr0.3MnO3 compound and found that Pr-deficiency and Sr-deficiency have different effects on the magnetic properties.53 The effect of La deficiency in (La1−x)0.8Ca0.2MnO3 was reported by Phan et al. and found that the La-deficient compound exhibits a large ΔSM value than that of the pristine compound.54 Na-deficient La0.8Na0.05□0.15MnO3 and Sr-deficient La0.65Eu0.05Sr0.15MnO3 exhibit ΔSM values of 3.48 J kg−1 K−1 at 260 K and 4.96 J kg−1 K−1 at 280 K under a field change of 20 kOe.43,44 A tunable transition temperature with a ΔSM value of 3.42 J kg−1 K−1 (300 K) under 50 kOe field was reported by Skini et al. in K-deficient La0.8K0.1□0.1MnO3 compound.45
MnO3 also exhibit different exchange interactions as well as different magnetic and electrical transport properties due to the difference in the average A-site ionic radius.22–27 It is understood that, in doped manganites, the double exchange (DE) interaction via the Mn3+–O2−–Mn4+ ions is responsible for the ferromagnetic (FM) behaviour and the superexchange (SE) interaction through Mn3+–O2−–Mn3+ and Mn4+–O2−–Mn4+ ions are responsible for the antiferromagnetic (AFM) behaviour. This indicates that the strength of the competing exchange interactions and magnetic properties are eventually affected by different factors such as rare Earth substitution at the A-site, replacement of manganese by other transition elements, the introduction of deficiency at A and A′- sites, etc.28–37 Recently, many reports have demonstrated that the creation of deficiency at the rare Earth and alkaline rare Earth sites alter the Mn2+/Mn3+/Mn4+ ratio which severely affect the magnetic and magnetocaloric properties.37–49 The structure of manganites is imperfect and contain vacancy type point defects, called Schottky defects due to the cyclic temperature changes during synthesis and sintering process.50,51 The presence of cation (V(c)) and anion (V(a)) vacancies strongly influence the properties of the manganites and causing a change in the stability of the perovskite structure.52 The change in the valence state and coordination number of cations due to defect formation affects the magnetic phase transition temperature and magnetic ordering of the manganites. Moreover, a change in the bond length affects the packing of MnO6 octahedral and leads to a change in the structure and stability of the compounds. The interstitial point defects, called Frenkel defects is not formed in these compounds and only vacancy type point defects are exist due to elevated synthesis and sintering temperature conditions.50 The anion vacancies are formed during heating process due to the thermal dissociation of oxygen and that leads to the reduction of oxygen content to O3−δ.51 Moreover, the presence of vacancies at the oxygen site is compensated by a reduction in the average valence of manganese ions.52 On the other hand, the cation vacancies are formed during cooling process due to high mobility of cation sub lattices, and the formation of vacancies at the A-site leads to a decrease in the average valence of Mn-ions. The defectiveness and the stability in the perovskite structure can be controlled by changing the cooling conditions and the sintering temperature.51 Moreover, researchers have tried to tune the transition temperature (TC) and ΔSM by varying the vacancy content to a certain extent. Boujelben et al. reported the effect of Pr and Sr-deficiency in Pr0.7Sr0.3MnO3 compound and found that Pr-deficiency and Sr-deficiency have different effects on the magnetic properties.53 The effect of La deficiency in (La1−x)0.8Ca0.2MnO3 was reported by Phan et al. and found that the La-deficient compound exhibits a large ΔSM value than that of the pristine compound.54 Na-deficient La0.8Na0.05□0.15MnO3 and Sr-deficient La0.65Eu0.05Sr0.15MnO3 exhibit ΔSM values of 3.48 J kg−1 K−1 at 260 K and 4.96 J kg−1 K−1 at 280 K under a field change of 20 kOe.43,44 A tunable transition temperature with a ΔSM value of 3.42 J kg−1 K−1 (300 K) under 50 kOe field was reported by Skini et al. in K-deficient La0.8K0.1□0.1MnO3 compound.45
Recently, our group has reported the effect of La and Sr-deficiency on the magnetocaloric properties in La0.67Sr0.33MnO3 (LSMO) compound. La0.67−xSr0.33MnO3−δ (x = 0.09) and La0.67Sr0.33−yMnO3−δ (y = 0.09) termed as La-0.09 and Sr-0.09 compounds, were prepared by solid-state route and it was found that Sr-deficient compound has better magnetocaloric properties than that of the pristine as well as La-deficient compounds.37 Compound with a deficiency of y = 0.09 i.e., La0.67Sr0.24□0.09MnO3−δ displayed a ΔSM of 5.08 J kg−1 K−1 at 352 K and a adiabatic temperature change of 3.48 K for 50 kOe field. Thus the creation of Sr-deficiency in LSMO exhibits a promising MCE behaviour with an increase in magnetocaloric properties value and a decrease in TC towards room temperature from 365 K of the pristine compound. In the present study, as a continuing effort, we have further increased the Sr-deficiency content in La0.67Sr0.33−yMnO3−δ by a factor of y = 0.18 and 0.27 to further tune the TC towards room temperature. Thus we have synthesized La0.67Sr0.33−yMnO3−δ (y = 0.18 and 0.27) compounds using solid-state method and explored their structural, magnetic and magnetocaloric properties and compared the results with that of the pristine compound, reported in our previous study.37
Experimental
La0.67Sr0.15□0.18MnO3−δ and La0.67Sr0.06□0.27MnO3−δ are termed as Sr-0.18 and Sr-0.27 compounds and were prepared by conventional solid-state method using La2O3 (Alfa Aesar, 99%), SrCO3 (Sigma-Aldrich, 98%) and MnCO3 (Sigma-Aldrich, 99.9+ %) as starting materials. The raw materials were weighed according to the stoichiometry and mixed in an agate mortar using distilled water as solvent medium for 6 h. The homogenous mixture was dried and calcined at 1000 °C and 1200 °C for 12 h with intermediate grinding. The grounded powders were pelletized into cylindrical shape of 12 mm diameter, which was then sintered at 1350 °C for 12 h. The pellets were then powdered and then structural analysis was carried out by powder X-ray diffraction (XRD) technique using PANalytical X'Pert Pro diffractometer having Ni-filtered Cu Kα radiation. Rietveld refinement was done using GSAS-EXPGUI software to analyze the crystal structure and phase purity of the compounds. Iodometric titration was performed to check the oxygen stoichiometry. X-ray photoelectron spectroscopy (XPS) was done to study the surface elemental composition and oxidation states using a PHY 5000 Versa Probe II, ULVAC-PHI, Inc. Instrument having Al Kα as the X-ray source. Initially, a wide scan was done, and a pressure of 5 × 10−10 mbar was maintained in the XPS chamber during the measurement. The binding energy of the high resolution scans were corrected using C 1s = 284.8 eV as the reference. Magnetic properties were measured using the vibrating sample magnetometer attached to the physical property measurement system (Quantum Design Inc., USA).Results and discussion
From the Rietveld refinement of the powder XRD patterns, it is confirmed that both the studied compounds belong to rhombohedral structure with R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) c space group. The refined parameters obtained from the Rietveld analysis are shown in Table 1 and the refined XRD patterns are shown in Fig. 1. The experimental XRD patterns are in good agreement with the patterns calculated by the software, and shows better goodness of fit. In our previous study, it was confirmed that the pristine compound LSMO was phase pure, and no impurity peaks were detected.37 On the other hand, La-0.09 and Sr-0.09 compounds show an additional peak at 36.1° due to the presence of Mn3O4 as a secondary phase as reported in our previous work.37,48,49 However, in the present case, Sr-0.18 and Sr-0.27 compounds show some additional secondary peaks at 28.9°, 36.1°, 25.9° etc. due to the prominent presence of Mn3O4 secondary phases. Moreover, with increase in Sr-deficiency, the intensity of the secondary peaks also increases. It indicates that, with increase in Sr-deficiency content, the percentage of rhombohedral ferrimagnetic Mn3O4 secondary phase also increases.49 Even though the formation of Mn3O4 depends on the amount of deficiency, some reports show that the preparation conditions such as manual grinding sometimes does not ensure perfect homogeneity and produces Mn3O4 secondary phase.55 In La and Sr-deficient compounds, three oxidation states are possible for manganese ions (Mn2+/Mn3+/Mn4+).37,56,57 Out of these ions, Mn2+ ions (ionic radius of 1.13 Å) occupy the A-site along with La3+ (ionic radius of 1.36 Å) and Sr2+ (ionic radius of 1.44 Å).37,56 However, Mn3+ (0.645 Å) and Mn4+ (0.53 Å) occupy the B-site forming a MnO6 octahedral coordinated system. Recently, Hundley et al. suggested a charge disproportionation model in deficient manganites based on the stability of manganese pairs.58 According to the charge disproportionation model, in octahedrally coordinated systems, Mn2+–Mn4+ pair is the more stable than Mn3+–Mn3+ pair.58 Thus in deficient compounds, a change of state from Mn3+ + Mn3+ pair to Mn2+ + Mn4+ pair is possible because of the more stable nature of the later pair. Thus the presence of Mn2+ ions in Sr-0.18 and Sr-0.27 compounds can be attributed to the stable nature of Mn2+ + Mn4+ pairs as suggested by the charge disproportionation model and this indicates that a portion of the manganese ions form Mn3O4 secondary phase, and the other portion occupies the A-site as Mn2+ ions along with La3+ and Sr2+ ions. From Table 1, it can be seen that Sr-0.18 and Sr-0.27 compounds have greater unit cell volume (354.377 Å3 and 355.388 Å3 respectively) than that of the pristine (349.468 Å3) and Sr-0.09 (351.686 Å3) compounds.37 The increase in the unit cell volume of the Sr-deficient compounds cannot be explained on the basis of the ionic radius of Mn2+/Mn3+/Mn4+ ions or the amount of Mn3O4 impurity phase present in the compound. The increase in unit cell volume of Sr-site deficient compounds can be related to the decreasing concentration of oxygen due to the formation of anionic vacancies and the increase in radius of the deficient Sr-site.37,48,49,59 The presence of oxygen vacancies is compensated by a reduction in the average valence of manganese ions. This leads to the reduction of some Mn4+ to Mn3+and Mn3+ to Mn2+. Hence, due to defectiveness, the charge compensation is taking place and the average ionic radius and the unit cell volume increases. Moreover, the anion (V(a)) vacancies at the oxygen sites reduces bonding electrostatic force and eventually leads to the increase of unit cell volume.52 Due to the electrostatic repulsion from the neighbour oxygen ions, the radius of the deficient Sr-site (1.547 Å) is increased by 7% than the actual ionic radius of Sr-site (1.44 Å).37,49,60 This will eventually affect the unit cell volume of the Sr-deficient compounds and hence Sr-0.18 and Sr-0.27 compounds exhibit greater unit cell volume than that of the pristine and Sr-0.09 compounds. As the unit cell volume changes with Sr-deficiency, distortion occurs in the MnO6 octahedra due to the changes in the Mn– O bond lengths and the Mn–O–Mn bond angles. The Jahn–Teller active ion, Mn3+ distorts the MnO6 octahedra due to the strong Jahn–Teller effect, while Mn2+ and Mn4+ are Jahn–Teller inactive and do not tend to distort the octahedrally coordinated system. The iodometric titration method was performed to estimate the oxidation state of manganese and hence the stoichiometry of the oxygen ion.48,61 Powders of Sr-deficient compounds are weighed and dissolved in a mixture of 10 ml of 10 mass% KI aqueous solution and 2.5 ml of 2 M HCl. The liberated iodine is titrated against 0.1 N sodium thiosulphate standard volumetric solution using 1 mass% starch solution as an indicator. It is assumed that the oxidation state of manganese is compensated due to the creation of vacancy and the Sr-deficient compounds can be written as La0.673+Sr0.33−y2+Mn1−α3+Mnα4+O3−δ (y = 0.18 and 0.27). The value of α was determined from the titration and the amount of oxygen content was calculated for 5 titrations using the equation as
c space group. The refined parameters obtained from the Rietveld analysis are shown in Table 1 and the refined XRD patterns are shown in Fig. 1. The experimental XRD patterns are in good agreement with the patterns calculated by the software, and shows better goodness of fit. In our previous study, it was confirmed that the pristine compound LSMO was phase pure, and no impurity peaks were detected.37 On the other hand, La-0.09 and Sr-0.09 compounds show an additional peak at 36.1° due to the presence of Mn3O4 as a secondary phase as reported in our previous work.37,48,49 However, in the present case, Sr-0.18 and Sr-0.27 compounds show some additional secondary peaks at 28.9°, 36.1°, 25.9° etc. due to the prominent presence of Mn3O4 secondary phases. Moreover, with increase in Sr-deficiency, the intensity of the secondary peaks also increases. It indicates that, with increase in Sr-deficiency content, the percentage of rhombohedral ferrimagnetic Mn3O4 secondary phase also increases.49 Even though the formation of Mn3O4 depends on the amount of deficiency, some reports show that the preparation conditions such as manual grinding sometimes does not ensure perfect homogeneity and produces Mn3O4 secondary phase.55 In La and Sr-deficient compounds, three oxidation states are possible for manganese ions (Mn2+/Mn3+/Mn4+).37,56,57 Out of these ions, Mn2+ ions (ionic radius of 1.13 Å) occupy the A-site along with La3+ (ionic radius of 1.36 Å) and Sr2+ (ionic radius of 1.44 Å).37,56 However, Mn3+ (0.645 Å) and Mn4+ (0.53 Å) occupy the B-site forming a MnO6 octahedral coordinated system. Recently, Hundley et al. suggested a charge disproportionation model in deficient manganites based on the stability of manganese pairs.58 According to the charge disproportionation model, in octahedrally coordinated systems, Mn2+–Mn4+ pair is the more stable than Mn3+–Mn3+ pair.58 Thus in deficient compounds, a change of state from Mn3+ + Mn3+ pair to Mn2+ + Mn4+ pair is possible because of the more stable nature of the later pair. Thus the presence of Mn2+ ions in Sr-0.18 and Sr-0.27 compounds can be attributed to the stable nature of Mn2+ + Mn4+ pairs as suggested by the charge disproportionation model and this indicates that a portion of the manganese ions form Mn3O4 secondary phase, and the other portion occupies the A-site as Mn2+ ions along with La3+ and Sr2+ ions. From Table 1, it can be seen that Sr-0.18 and Sr-0.27 compounds have greater unit cell volume (354.377 Å3 and 355.388 Å3 respectively) than that of the pristine (349.468 Å3) and Sr-0.09 (351.686 Å3) compounds.37 The increase in the unit cell volume of the Sr-deficient compounds cannot be explained on the basis of the ionic radius of Mn2+/Mn3+/Mn4+ ions or the amount of Mn3O4 impurity phase present in the compound. The increase in unit cell volume of Sr-site deficient compounds can be related to the decreasing concentration of oxygen due to the formation of anionic vacancies and the increase in radius of the deficient Sr-site.37,48,49,59 The presence of oxygen vacancies is compensated by a reduction in the average valence of manganese ions. This leads to the reduction of some Mn4+ to Mn3+and Mn3+ to Mn2+. Hence, due to defectiveness, the charge compensation is taking place and the average ionic radius and the unit cell volume increases. Moreover, the anion (V(a)) vacancies at the oxygen sites reduces bonding electrostatic force and eventually leads to the increase of unit cell volume.52 Due to the electrostatic repulsion from the neighbour oxygen ions, the radius of the deficient Sr-site (1.547 Å) is increased by 7% than the actual ionic radius of Sr-site (1.44 Å).37,49,60 This will eventually affect the unit cell volume of the Sr-deficient compounds and hence Sr-0.18 and Sr-0.27 compounds exhibit greater unit cell volume than that of the pristine and Sr-0.09 compounds. As the unit cell volume changes with Sr-deficiency, distortion occurs in the MnO6 octahedra due to the changes in the Mn– O bond lengths and the Mn–O–Mn bond angles. The Jahn–Teller active ion, Mn3+ distorts the MnO6 octahedra due to the strong Jahn–Teller effect, while Mn2+ and Mn4+ are Jahn–Teller inactive and do not tend to distort the octahedrally coordinated system. The iodometric titration method was performed to estimate the oxidation state of manganese and hence the stoichiometry of the oxygen ion.48,61 Powders of Sr-deficient compounds are weighed and dissolved in a mixture of 10 ml of 10 mass% KI aqueous solution and 2.5 ml of 2 M HCl. The liberated iodine is titrated against 0.1 N sodium thiosulphate standard volumetric solution using 1 mass% starch solution as an indicator. It is assumed that the oxidation state of manganese is compensated due to the creation of vacancy and the Sr-deficient compounds can be written as La0.673+Sr0.33−y2+Mn1−α3+Mnα4+O3−δ (y = 0.18 and 0.27). The value of α was determined from the titration and the amount of oxygen content was calculated for 5 titrations using the equation as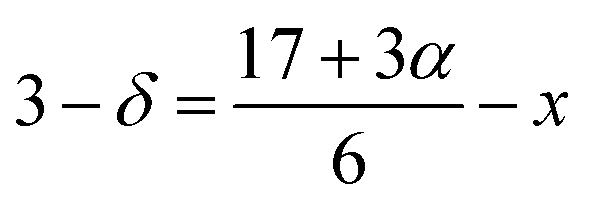
| Sr-0.18 | Sr-0.27 | |
|---|---|---|
| Cell parameters | ||
| a (Å) | 5.4309 (1) | 5.5379 (1) |
| b (Å) | 5.5309 (1) | 5.5379 (1) |
| c (Å) | 13.3767 (2) | 13.3805 (2) |
| V (Å3) | 354.377 (8) | 355.388 (7) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||
| Positional parameters | ||
| La/Sr x | 0 | 0 |
| La/Sr y | 0 | 0 |
| La/Sr z | 0.25 | 0.25 |
| Mn x | 0 | 0 |
| Mn y | 0 | 0 |
| Mn z | 0 | 0 |
| O x | 0.546 | 0.550 |
| O y | 0 | 0 |
| O z | 0.25 | 0.25 |
|
||
| Bond distance (Å) | ||
| 〈d Mn–O〉 | 1.963 (2) | 1.968 (2) |
|
||
| Bond angle (°) | ||
| Mn–O1–Mn | 165.1 (0) | 163.8 (0) |
|
||
| Agreement factors | ||
| RWP | 5.20 | 4.77 |
| RP | 3.85 | 3.59 |
| χ2 | 1.84 | 1.51 |
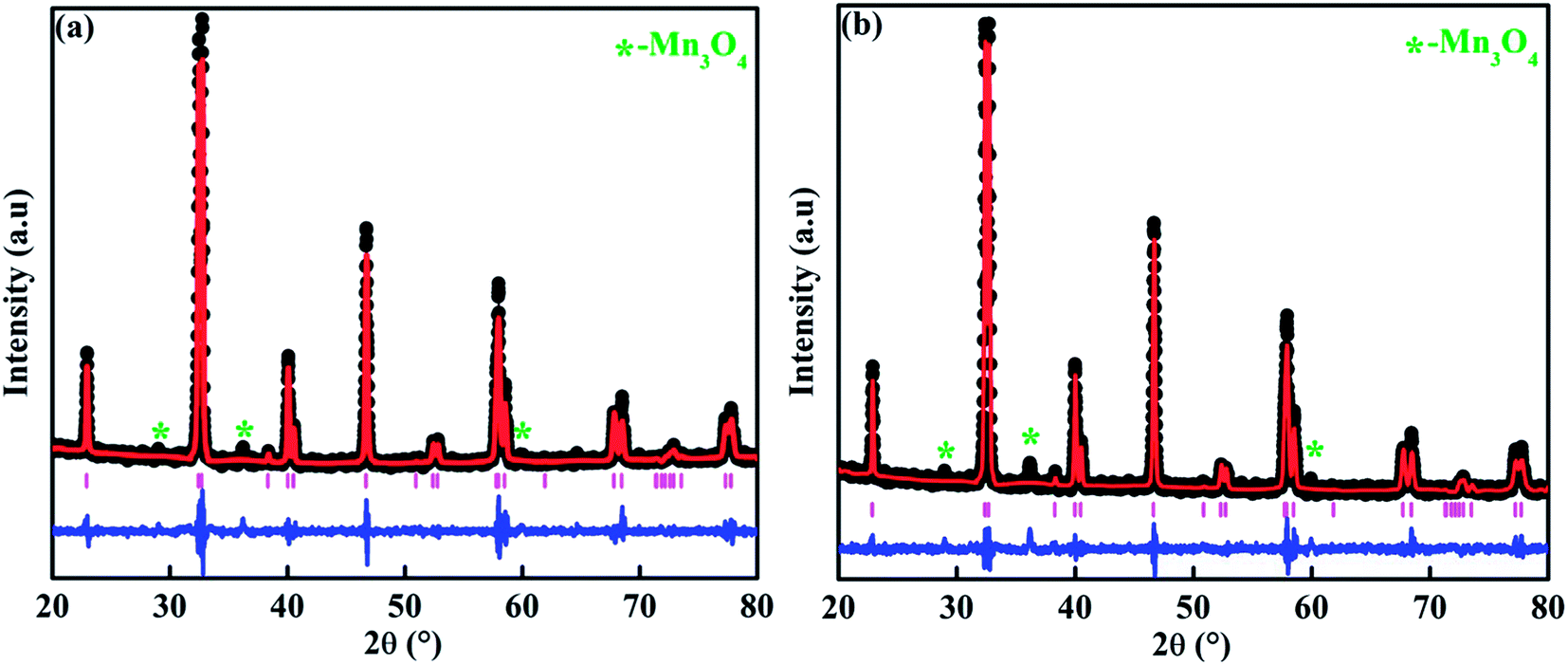 | ||
| Fig. 1 Rietveld refined XRD patterns of (a) La0.67Sr0.15□0.18MnO3−δ and (b) La0.67Sr0.06□0.27MnO3−δ compounds. Black colour indicates the experimental data. Red lines are theoretical fits to the XRD data; pink corresponds to the Bragg reflection, and blue lines correspond to the difference between the experimental and the calculated fits. | ||
The oxygen content decreases with increase in deficiency and it is found to be 2.74 and 2.69 for Sr-0.18 and Sr-0.27 compounds respectively with an accuracy of ± 0.03.
To determine the oxidation state of manganese and to identify the elemental composition on the surface, a detailed XPS analysis was performed. The Mn–2P3/2 spectra of Sr-0.18 and Sr-0.27 compounds were curve fitted using MultiPak spectrum: ESCA and are shown in Fig. 2(a) and (b) respectively. The fitting was done using a standard Shirley background, and the Mn–2P3/2 spectra was decomposed into three components for both Sr-0.18 and Sr-0.27 compounds. In Sr-0.18 compound, the three peak centers at 640.9, 642.4 and 643.4 eV corresponds to Mn2+, Mn3+ and Mn4+ oxidation states respectively.37,62–66 In Sr-0.27 compound, the peak centers of Mn2+, Mn3+ and Mn4+ are at 640.8, 642.3 and 643.2 eV respectively.37,62–66 It was confirmed in our previous report that in deficient compounds, three oxidation states (Mn2+/Mn3+/Mn4+) are possible for manganese while only two oxidation states (Mn3+ and Mn4+) exist in the pristine compound.37 The ratio of Mn2+/Mn3+/Mn4+ is calculated from the area of each component obtained from the peak fitting. For the pristine compound, the Mn3+/Mn4+ ratio is about 67.9:32.1, which is near to the expected ratio (67:33).37 However, in the present case, the ratio of Mn2+/Mn3+/Mn4+ is found to be 70.6/6.3/23.1 and 64.7/10.4/24.9 for Sr-0.18 and Sr-0.27 compounds respectively. Hence it is to be noted that the change of state from Mn3+ + Mn3+ pairs to Mn2+ + Mn4+ pairs under charge disproportionation phenomena are different for Sr-deficient compounds, and this has been confirmed from XPS analysis. Thus the ratio of Mn2+/Mn3+/Mn4+ ions alter with the Sr-deficiency content, which inturn severely affects the DE interactions and thereby the magnetic properties.
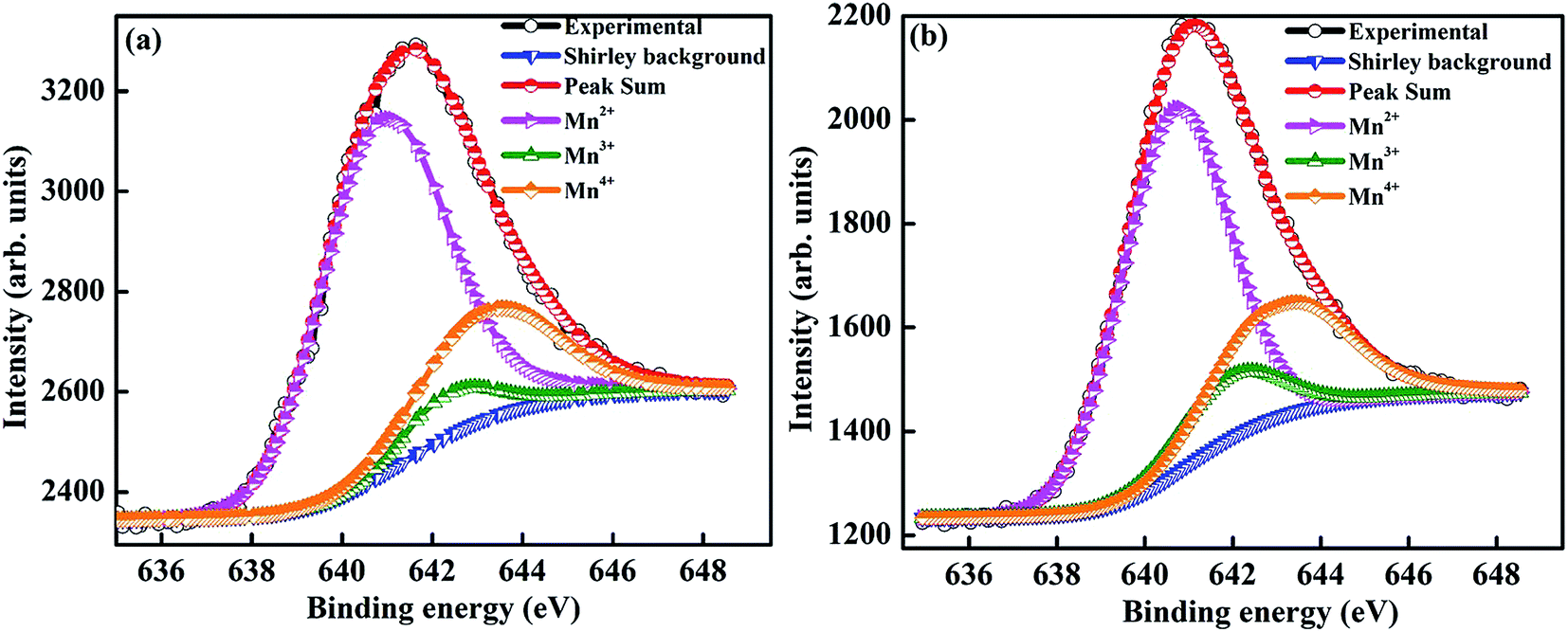 | ||
| Fig. 2 Mn–2P3/2 X-ray photo electron spectra of (a) La0.67Sr0.15□0.18MnO3−δ and (b) La0.67Sr0.06□0.27MnO3−δ compounds. | ||
Fig. 3 shows the magnetization as a function of temperature from 2–380 K for both the compounds. The measurement was carried out under 100 Oe field in zero-field-cooled (ZFC) and field cooled (FC) modes. Large thermo-magnetic irreversibility is witnessed in both the compounds due to the competing interaction between different magnetic states. Both the compounds show a hump like feature below 50 K, which was not seen in the magnetization curves of La-0.09 and Sr-0.09 compounds, reported in our earlier work.37 This low-temperature anomaly is due to the ordering of the ferrimagnetic Mn3O4 phase, which is very prominent with the increase in Sr-deficiency, this result corroborates with that obtained from the Rietveld refinement analysis.49 From our previous report, it can be seen that the magnetization of La-0.09 and Sr-0.09 compounds are greater than that of the pristine compound due to the increase in DE interaction.37 In the present case, the magnetization increases further for Sr-0.18 and then decreases for Sr-0.27, which means that Sr-0.18 compound shows maximum magnetization among all the Sr-deficient compounds. Thus the DE interaction and FM nature of Sr-0.18 and Sr-0.27 compounds do not merely destroy by Sr-deficiency. In the pristine compound, the exchange coupling is between Mn3+ ions having the electronic configuration of 3d4 (t3↑2g e↑g with S = 2) and Mn4+ ions having the electronic configuration of 3d3 (t3↑2g with S = 3/2). However, in deficient compounds, the exchange coupling is enhanced by the presence of Mn2+ ions due to its electronic configuration (t3↑2g e2↑g with S = 5/2) and spin only magnetic moment (5μB) compared to Mn3+ (4μB) and Mn4+ (3μB). In addition to the traditional DE mechanism, some additional charge hopping mechanisms take place in the deficient compounds due to the presence of Mn2+ ions.56 The Mn2+ ions at the A-site favour an additional hopping via Mn3+–O2−–Mn2+–O2−–Mn4+ path, called the multiple DE interactions and eventually the ferromagnetism and conductivity increases.37,56 Thus it can be understood that the high magnetization value of Sr-0.18 and Sr-0.27 compounds compared to that of the pristine compound is due to the multiple DE interaction through intervening Mn2+ ions at the A-site. It suggests that the coexistence of Mn2+/Mn3+/Mn4+ ions in Sr-0.18 and Sr-0.27 compounds can be corroborated with the result obtained from XPS analysis. However, the unconventional charge hopping mechanism completely depends on the ratio of Mn2+/Mn3+/Mn4+ ions, the Sr-0.18 compound shows maximum magnetization, and after that decreases for Sr-0.27 compound. It suggests that the existence of Mn2+ ions in deficient compounds is responsible for the increase in the DE interaction, which is in agreement with the coexistence of the mixed valence state of Mn2+/Mn3+/Mn4+ in deficient compounds as obtained from XPS analysis. Again, the paramagnetic (PM) to FM transition temperature was calculated from the derivative of M versus T graph (not shown), and found significant changes with the increase in Sr-content. It is to be noted that in our previous work, we have tuned the TC from 365 K (pristine) to 355 K for Sr-0.09 compound, however, in the present case, the same is further tuned down to 314 K and 277 K for Sr-0.18 and Sr-0.27 compounds respectively. The significant reduction in TC towards room temperature in both the compounds is due to the changes in the average ionic radius and the changes in the Goldschmidt tolerance factor.37,49 From the Rietveld refinement analysis, it is evident that the average A-site ionic radius and unit cell volume of both the compounds increases with increase in Sr-deficiency. As the tolerance factor of the compounds is directly proportional to the average A-site cationic radius, the decrease in TC with Sr-deficiency can be related to the increase in tolerance factor.37,49 The variation of inverse magnetic susceptibility χ−1 with temperature was analyzed and shown in Fig. 3. The magnetic susceptibility obey the Curie–Weiss law at T > TC, according to the relation
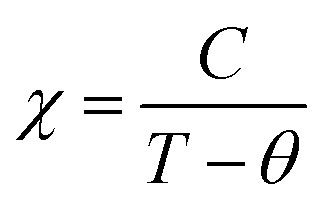
where, kB is the Boltzmann constant, μB is the Bohr magneton, Na is the Avogadro number and Mm is the molecular formula weight. The μmeaseff is found to be 5.83 μB 5.81 μB for Sr-0.18 and Sr-0.27 compounds. The theoretical effective magnetic moment μtheff of Sr-0.18 compound is calculated as follows
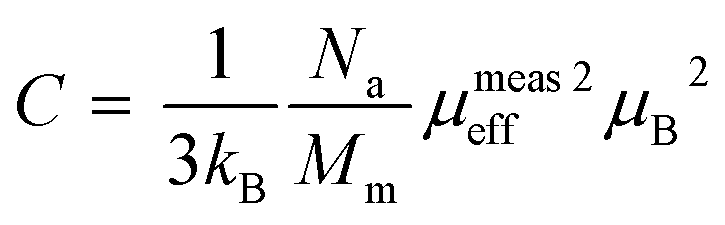
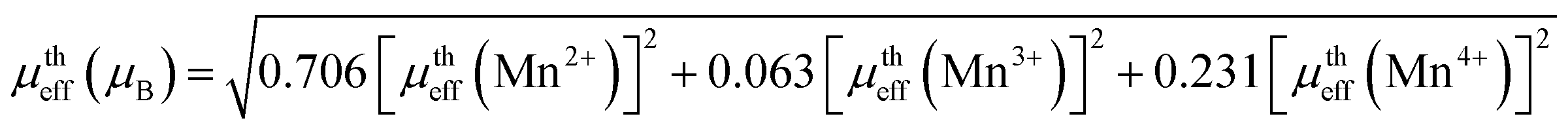
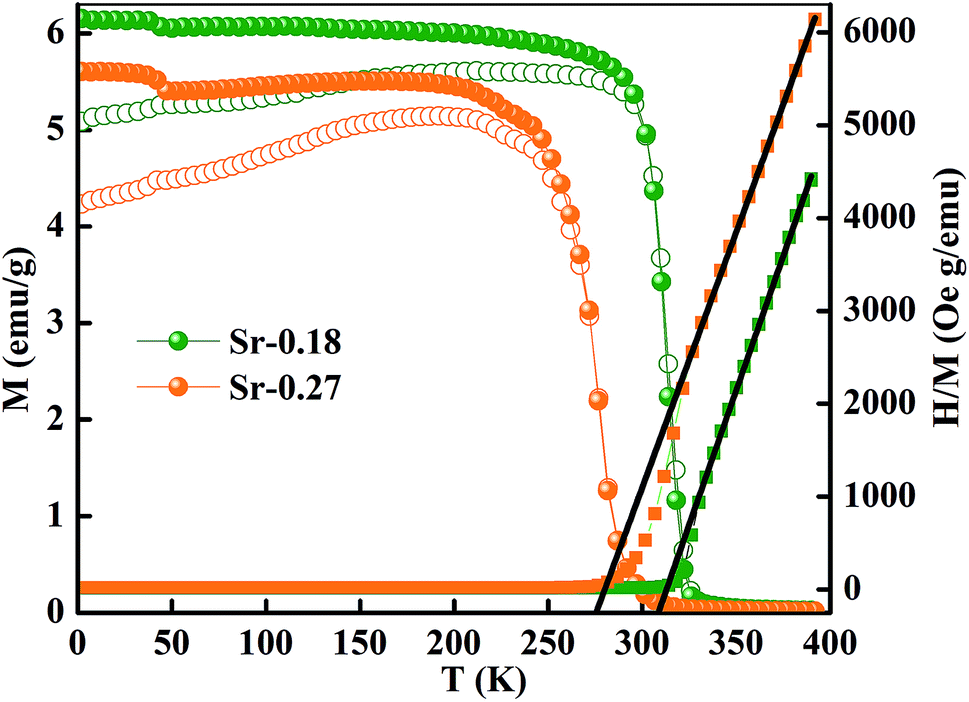 | ||
| Fig. 3 Temperature dependence of magnetization of La0.67Sr0.15□0.18MnO3−δ and La0.67Sr0.06□0.27MnO3−δ compounds under 100 Oe field. | ||
With μtheff(Mn2+) − 5.92μB, μtheff(Mn3+) − 4.90μB and μtheff(Mn4+) − 3.87μB and the ratio of Mn2+/Mn3+/Mn4+ is taken from the XPS analysis. The μtheff is found to be 5.45 μB 5.37 μB for Sr-0.18 and Sr-0.27 compounds.
Further, the isothermal magnetization measurement was carried out at 2 K and 300 K and are shown in Fig. 4(a) and (b) respectively. Both the compounds show a small coercive field with soft ferromagnetic nature. It can be seen that magnetization increases rapidly at low field and does not saturate even at a field of 90 kOe. The hysteresis loops at 2 K confirm the high FM nature of the compounds, and it is evident that the FM nature is not merely destroyed by the increase in Sr-deficiency. From the hysteresis loop at 300 K, it can be understood that Sr-0.18 compound shows more FM character than that of Sr-0.27 compound, which confirm the room temperature FM behaviour of the former. The competing exchange interaction between DE and SE due to the FM/AFM components vary with the Mn2+/Mn3+/Mn4+ ratio, which in turn severely affects the isothermal magnetization behaviour of the deficient compounds at 2 K and 300 K. Furthermore, the isothermal magnetization measurements around the respective TC was measured and are shown in Fig. 5(a) and (b). The magnetization was measured from 284 K to 354 K for Sr-0.18, and 242 K to 322 K for Sr-0.27 with an interval of 4 K. For T < TC, a FM nature with a non-saturating behaviour at higher fields is seen for both the compounds. However, for T > TC, a PM behaviour with a reduction in magnetization was observed. To further understand the nature of the magnetic transition, M2 versus H/M called Arrott plots were plotted and are shown in Fig. 5(c) and (d). The regular Arrott plot between M2 and H/M indicates that the transition governing the mean field model and the curve that pass through the origin should represent the TC. Moreover, based on the Banerjee's criterion, a positive slope in the Arrott plot confirm that both the compounds undergo second order transition.67
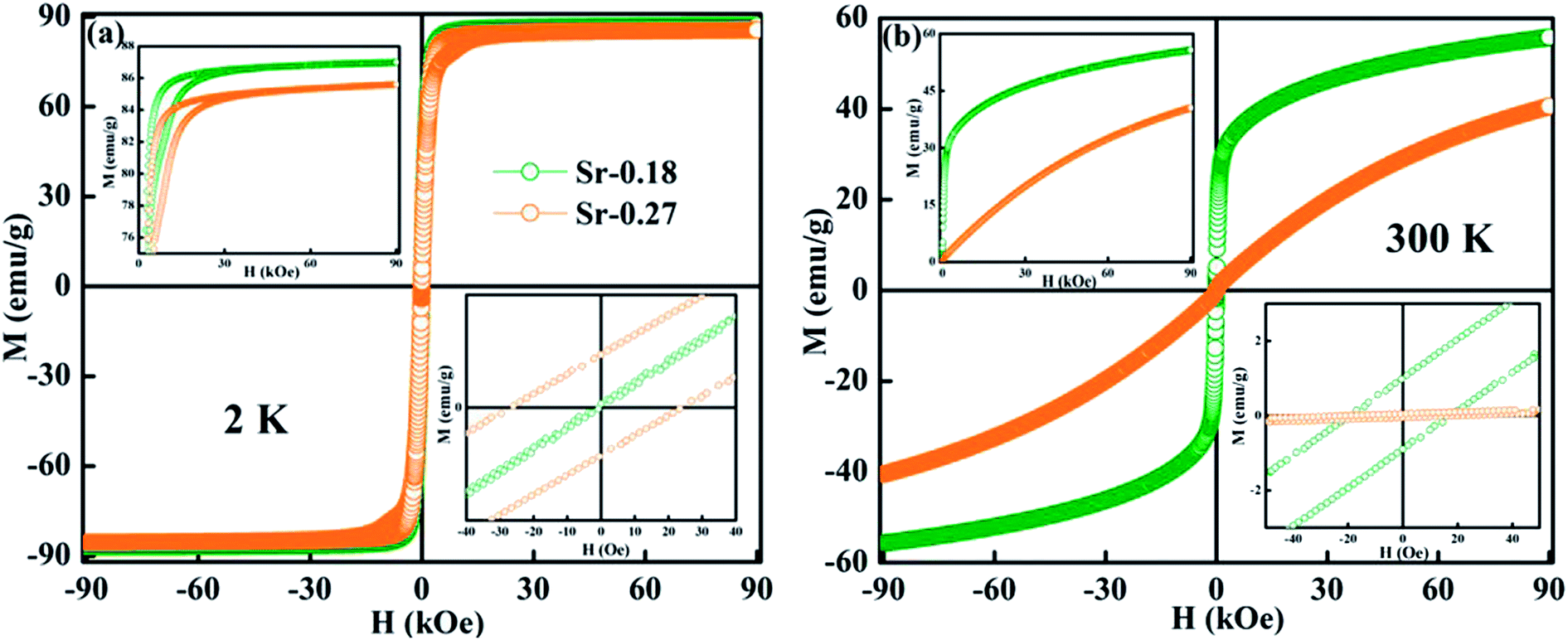 | ||
| Fig. 4 M–H loops at 2 K and 300 K of (a) La0.67Sr0.15□0.18MnO3−δ and (b) La0.67Sr0.06□0.27MnO3−δ compounds. The inset shows the enlarged view of the coercive field. | ||
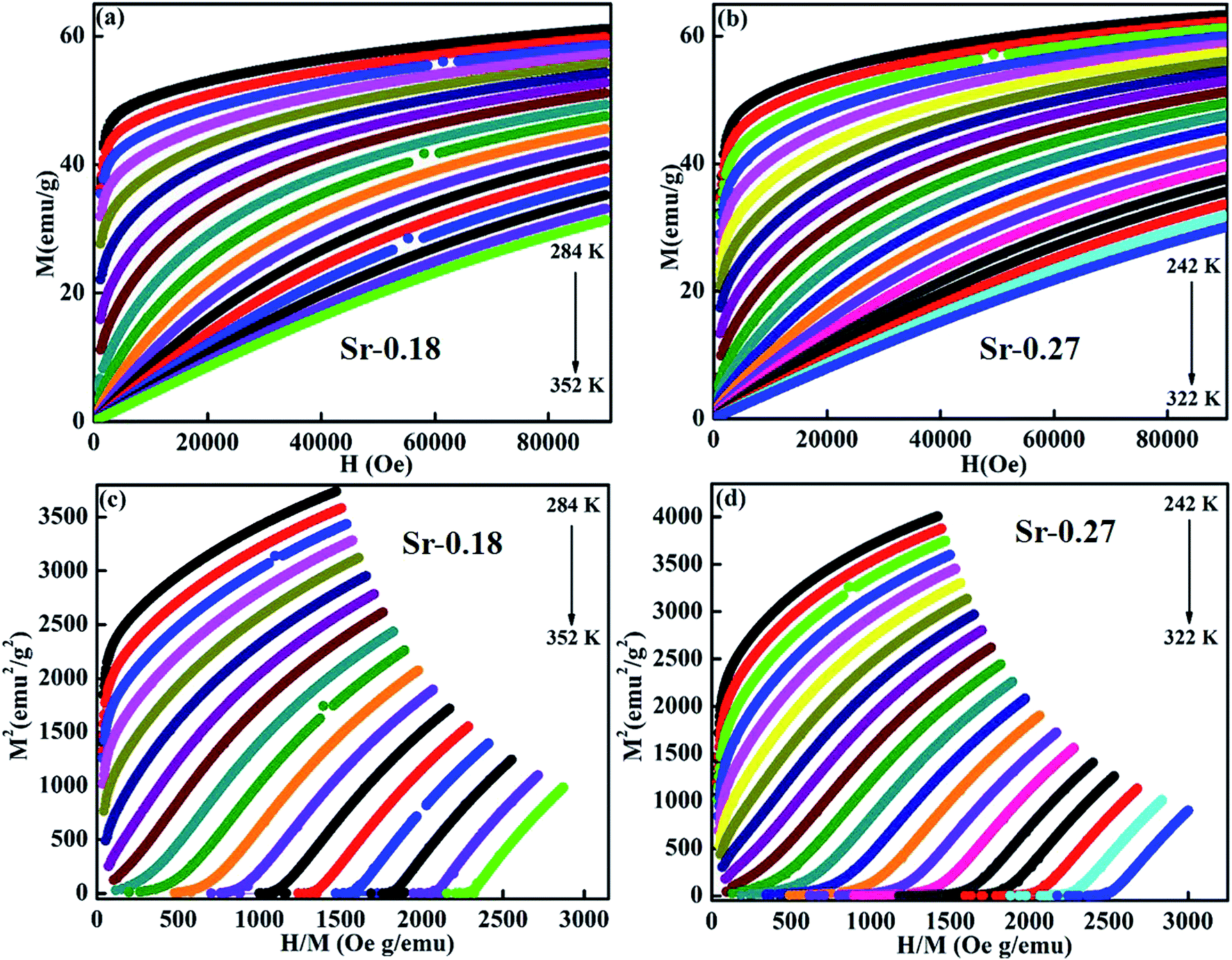 | ||
| Fig. 5 Isothermal field dependence magnetization of (a) La0.67Sr0.15□0.18MnO3−δ and (b) La0.67Sr0.06□0.27MnO3−δ and Arrott plot of (c) La0.67Sr0.15□0.18MnO3−δ and (d) La0.67Sr0.06□0.27MnO3−δ compounds. | ||
The magnetic entropy change is calculated from the isothermal magnetization curve based on the Maxwell relation as:68
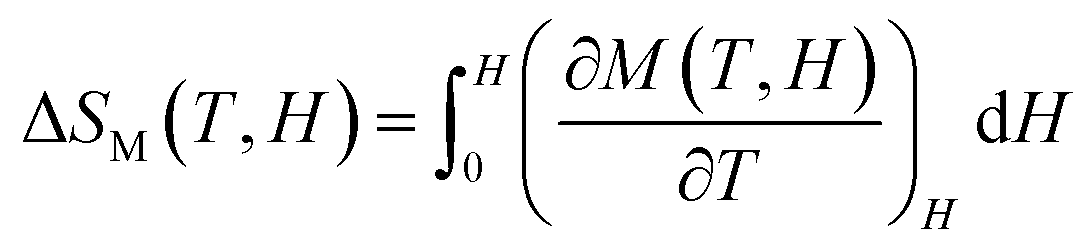
The ΔSM as a function of temperature for 10–50 kOe field is shown in Fig. 6. Both the compounds show a maximum ΔSM around TC, and a reduction is seen on either side of the curve. For a field of 10 kOe, the Sr-0.18 compound exhibit a maximum ΔSM value of 1.40 J kg−1 K−1 at 306 K, and for Sr-0.27 compound, the ΔSM reaches a maximum value of 1.17 J kg−1 K−1 at 272 K. It can be seen that with increase in the magnetic field, the maximum ΔSM value is slightly shifts to the high temperature region for both the compounds. For a field of 50 kOe, the Sr-0.18 compound exhibit a ΔSM value of 4.61 J kg−1 K−1 at 310 K, and for Sr-0.27 compound, the ΔSM reaches a maximum value of 4.11 J kg−1 K−1 at 276 K. In our previous work, we reported a ΔSM value of 4.78 J kg−1 K−1 at 364 K for the pristine compound and 5.08 J kg−1 K−1 at 352 K for Sr-0.09 compound for a field change of 50 kOe.37 However, in the present case, we have achieved a near room temperature (310 K) MCE with a significant ΔSM value (4.61 J kg−1 K−1) for Sr-0.18 compound which is larger than that reported for numerous perovskite manganites29,31,35,45,47 Apart from the spin, charge and orbital ordering, the conventional DE interaction is the only favourable mechanism behind the entropy change in the pristine compound.37 However, from the magnetization and magnetocaloric properties, it can be understood that the presence of Mn2+ ions in deficient compounds enhances the ferromagnetism and ΔSM due to the multiple DE interaction via Mn3+–O2−–Mn2+–O2−–Mn4+ path. In addition to that, the Mn2+/Mn3+/Mn4+ ratio is the key parameter that determines the strength of the multiple DE interactions and hence, the ΔSM of the deficient compounds. For practical applications at room temperature, the magnetocaloric materials should have a maximum ΔSM value within the range of 300 K ≤ T ≤ 310 K. Hence Sr-0.18 compound is found to be an ideal candidate for room temperature magnetocaloric applications. Thus, the present investigation reveals that a significant magnetic entropy change near room temperature can be achieved by the creation of deficiency at the Sr-site in La0.67Sr0.33−yMnO3−δ compound and the composition with a deficiency of y = 0.18, i.e., La0.67Sr0.15□0.18MnO3−δ could be a potential material for room temperature magnetic refrigeration applications.
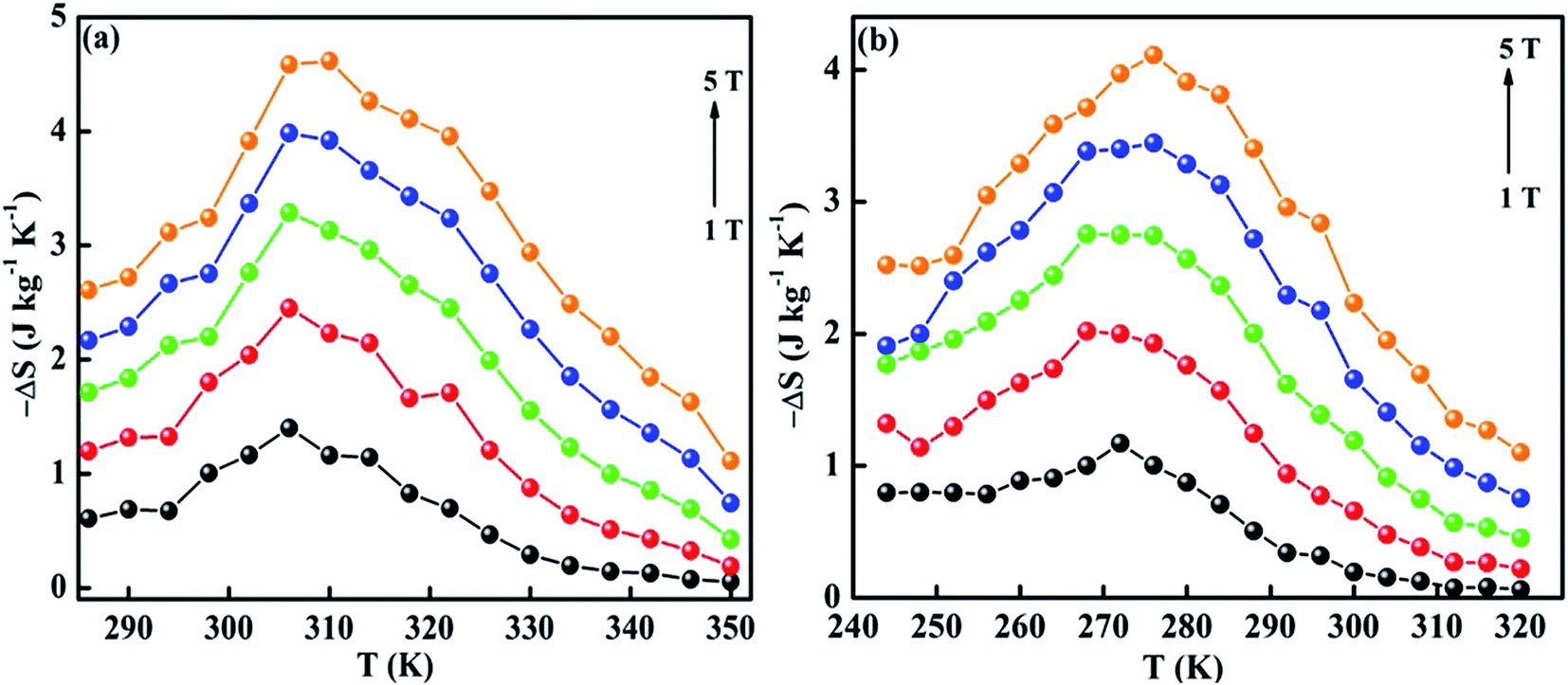 | ||
| Fig. 6 Magnetic entropy change of (a) La0.67Sr0.15□0.18MnO3−δ and (b) La0.67Sr0.06□0.27MnO3−δ compounds. | ||
Conclusion
The effect of Sr-site deficiency on La0.67Sr0.33−yMnO3−δ (y = 0.18 and 0.27) compounds and a detailed investigation on their structural, magnetic and magnetocaloric properties were carried out. The Rietveld refinement of the X-ray patterns confirms that both the compounds belong to rhombohedral crystal symmetry with Rc space group. Deficiency at the Sr-site compels to form Mn2+ states in addition to Mn3+/Mn4+, and the Mn2+ ions at the A-site favor multiple DE interactions via Mn3+–O2−–Mn2+–O2−–Mn4+ path and hence the ferromagnetic nature and magnetization enhanced. La0.67Sr0.15□0.18MnO3−δ was found to be ferromagnetic at 300 K and exhibit a ΔSM of 4.61 J kg−1 K−1 at 310 K for a 50 kOe field. The present study indicates that the creation of deficiency at the Sr-site of La0.67Sr0.33MnO3 helps to tune the TC towards room temperature with a significant magnetic entropy change and the compound could be a potential candidate for room temperature magnetic refrigeration applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge the financial support received from the Council of Scientific and Industrial Research (CSIR), Govt. of India, sponsored project MLP0031 and Department of Science and Technology sponsored project number, GAP232339. B. Arun and V. R. Akshay are thankful to the Council of Scientific and Industrial Research (CSIR), Government of India for granting the Senior Research Fellowship and also grateful to the Academy of Scientific and Innovative Research (AcSIR), CSIR.References
- K. A. Gschneidner Jr and V. K. Pecharsky, Annu. Rev. Mater. Res., 2000, 30, 387–429 CrossRef.
- V. Franco, J. S. Blazquez, B. Ingale and A. Conde, Annu. Rev. Mater. Res., 2012, 42, 305–342 CrossRef CAS.
- P. Sande, L. E. Hueso, D. R. Miguens, J. Rivas, F. Rivadulla and M. A. Lopez-Quintela, Appl. Phys. Lett., 2001, 79, 2040–2042 CrossRef CAS.
- A. M. Tishin and I. Spichkin, The Magnetocaloric Effect and Its Applications, Institute of Physics Publishing, Bristol, 2003 Search PubMed.
- M. H. Phan and S. C. Yu, J. Magn. Magn. Mater., 2007, 308, 325–340 CrossRef CAS.
- K. A. Gschneidner Jr, V. K. Pecharsky and A. O. Tsokol, Rep. Prog. Phys., 2005, 68, 1479–1539 CrossRef.
- K. A. Gschneidner Jr and V. K. Pecharsky, Int. J. Refrig., 2008, 31, 945–961 CrossRef.
- V. K. Pecharsky and K. A. Gschneidner Jr, Phys. Rev. Lett., 1997, 78, 4494–4497 CrossRef CAS.
- H. Wada and Y. Tanabe, Appl. Phys. Lett., 2001, 79, 3302–3304 CrossRef CAS.
- F. X. Hu, B. G. Shen, J. R. Sun, Z. H. Cheng, G. H. Rao and X. X. Zhang, Appl. Phys. Lett., 2001, 78, 3675–3677 CrossRef CAS.
- J. Shen, B. Gao, Q. Y. Dong, Y. X. Li, F. X. Hu, J. R. Sun and B. G. Shen, J. Phys. D: Appl. Phys., 2008, 41, 245005 CrossRef.
- O. Tegus, E. Bruck, K. H. J. Buschow and F. R. deBoer, Nature, 2002, 415, 150–152 CrossRef CAS PubMed.
- Q. Y. Dong, B. G. Shen, J. Chen, J. Shen, F. Wang, H. W. Zhang and J. R. Sun, J. Appl. Phys., 2009, 105, 053908 CrossRef.
- A. Giguere, M. Foldeaki, W. Shcnelle and E. Gmelin, J. Phys.: Condens. Matter, 1999, 11, 6969–6981 CrossRef CAS.
- T. Krenke, E. Duman, M. Acet, E. Wassermann, X. Moya, L. Manosa and A. Planes, Nat. Mater., 2005, 4, 450–454 CrossRef CAS PubMed.
- L. W. Li, G. Hu, Y. Qi and I. Umehara, Sci. Rep., 2017, 7, 42908 CrossRef CAS PubMed.
- Y. Yalin, L. W. Li, S. Kunpeng, Q. Yang and H. Dexuan, Intermetallics, 2017, 80, 22–25 CrossRef.
- A. Boutahar, R. Moubah, E. K. Hlil, H. Lassri and E. Lorenzo, Sci. Rep., 2017, 7, 13904 CrossRef CAS PubMed.
- B. Arun, M. V. Suneesh and M. Vasundhara, J. Magn. Magn. Mater., 2016, 418, 265–272 CrossRef.
- B. Arun, V. R. Akshay, G. R. Mutta, Ch. Venkatesh and M. Vasundhara, Mater. Res. Bull., 2017, 94, 537–543 CrossRef CAS.
- B. Sudakshina, B. Arun, K. D. Chandrasekhar, H. D. Yang and M. Vasundhara, Phys. B, 2018, 539, 14–20 CrossRef CAS.
- K. McBride, S. Bennington-Gray, J. Cook, L. Stella, S. Felton and D. Poulidi, CrystEngComm, 2017, 19, 3776–3791 RSC.
- M. Abassi, Za. Mohamed, J. Dhahri and E. K. Hlil, Dalton Trans., 2016, 45, 4736–4746 RSC.
- M. Jeddi, H. Gharsallah, M. Bejar, M. Bekri, E. Dhahri and E. K. Hlil, RSC Adv., 2018, 8, 9430–9439 RSC.
- B. Arun, M. Athira, V. R. Akshay, B. Sudakshina, G. R. Mutta and M. Vasundhara, J. Magn. Magn. Mater., 2018, 448, 322–331 CrossRef CAS.
- B. Sudakshina, K. D. Chandrasekhar, H. D. Yang and M. Vasundhara, J. Phys. D Appl. Phys., 2017, 50, 065004 CrossRef.
- B. Sudakshina, B. Arun and M. Vasundhara, J. Magn. Magn. Mater., 2018, 448, 250–256 CrossRef CAS.
- M. Oumezzine, A. C. Galca, I. Pasuk, C. F. Chiril, A. Leca, V. Kuncser, L. C. Tanase, A. Kuncser, C. Ghica and M. Oumezzine, Dalton Trans., 2016, 45, 15034–15040 RSC.
- N. Kallel, S. Kallel, A. Hagaza and M. Oumezzine, Phys. B, 2009, 404, 285–288 CrossRef CAS.
- C. P. Reshmi, S. Savitha Pillai, K. G. Suresh and M. R. Varma, Solid State Sci., 2013, 19, 130–135 CrossRef CAS.
- S. Hcini, M. Boudard, S. Zemni and M. Oumezzine, Ceram. Int., 2014, 40, 16041–16050 CrossRef CAS.
- M. A. Zaidi, J. Dhahri, I. Zeydi, T. Alharbi and H. Belmabrouk, RSC Adv., 2017, 7, 43590–43599 RSC.
- A. B. Hassine, A. Dhahri, M. L. Bouazizi, M. Oumezzine and E. K. Hlil, Ceram. Int., 2017, 43, 1390–1393 CrossRef.
- I. Sfifir, A. Ezaami, W. Cheikhrouhou-Koubaa and A. Cheikhrouhou, J. Alloys Compd., 2017, 696, 760–767 CrossRef CAS.
- M. Chebaane, R. Bellouz, Ma. Oumezzine, E. K. Hlil and A. Fouzri, RSC Adv., 2018, 8, 7186–7195 RSC.
- B. Arun, M. V. Suneesh, B. Sudakshina, V. R. Akshay, K. D. Chandrasekhar and M. Vasundhara, J. Phys. Chem. Solids, 2018, 123, 327–335 CrossRef CAS.
- B. Arun, V. R. Akshay and M. Vasundhara, Dalton Trans., 2018, 47, 15512–15522 RSC.
- W. Boujelben, A. Cheikh-Rouhou, J. Pierre and J. C. Joubert, Phys. B, 2002, 321, 37–44 CrossRef CAS.
- M. Ellouze, W. Boujelben, A. Cheikhrouhou, H. Fuess and R. Madar, J. Alloys Compd., 2003, 352, 41–47 CrossRef CAS.
- L. Malavasi, J. Mater. Chem., 2008, 18, 3295–3308 RSC.
- W. Cheikh-Rouhou Koubaa, M. Koubaa, A. Cheikh-Rouhou, W. Boujelben and A. M. Haghiri-Gosnet, J. Alloys Compd., 2008, 455, 67–72 CrossRef CAS.
- M. Khlifi, A. Tozri, M. Bejar, E. Dhahri and E. K. Hlil, J. Magn. Magn. Mater., 2012, 324, 2142–2146 CrossRef CAS.
- R. Bellouz, M. Oumezzine, A. Dinia, G. Schmerber, E. K. Hlil and M. Oumezzine, RSC Adv., 2015, 5, 64557–64565 RSC.
- M. Wali, R. Skini, M. Khlifi, E. Dhahri and E. K. Hlil, Dalton Trans., 2015, 44, 12796–12803 RSC.
- R. Skini, M. Khlifi and E. K. Hlil, RSC Adv., 2016, 6, 34271–34279 RSC.
- J. Makni-Chakroun, I. Sfifir, W. Cheikhrouhou-Koubaa, M. Koubaa and A. Cheikhrouhou, J. Magn. Magn. Mater., 2017, 432, 484–493 CrossRef CAS.
- S. Choura-Maatar, R. M’nassri, W. Cheikhrouhou-Koubaa, M. Koubaa, A. Cheikhrouhou and E. K. Hlil, RSC Adv., 2017, 7, 50347–50357 RSC.
- B. Arun, V. R. Akshay, K. D. Chandrasekhar, G. R. Mutta and M. Vasundhara, J. Magn. Magn. Mater., 2019, 472, 74–85 CrossRef CAS.
- B. Arun, V. R. Akshay, D. C. Kakarla and M. Vasundhara, J. Magn. Magn. Mater., 2019, 489, 165418 CrossRef CAS.
- A. V. Pashchenko, V. P. Pashchenko, N. A. Liedienov, V. K. Prokopenko, Yu. F. Revenko, N. E. Pismenova, V. V. Burhovetskii, V. Y. Sycheva, A. V. Voznyak, G. G. Levchenko, V. P. Dyakonov and H. Szymczak, J. Alloys Compd., 2017, 709, 779–788 CrossRef CAS.
- A. V. Pashchenko, V. P. Pashchenko, V. K. Prokopenko, V. A. Turchenko, Yu. F. Revenko, A. S. Mazur, V. Ya. Sycheva, N. A. Liedienov, V. G. Pitsyuga and G. G. Levchenko, J. Exp. Theor. Phys., 2017, 124, 100–113 CrossRef CAS.
- K. Guidara, N. Abdelmoula, J. Dhahri and E. Dhahri, Phase Transitions, 2000, 70, 243–252 CrossRef CAS.
- W. Boujelben, A. Cheikh-Rouhou and J. C. Joubert, Eur. Phys. J. B, 2001, 24, 419–423 CrossRef CAS.
- M.-H. Phan, S. C. Yu and N. H. Hur, J. Magn. Magn. Mater., 2003, 262, 407–411 CrossRef CAS.
- R. Skini, A. Omri, M. Khlifi, E. Dhahri and E. K. Hlil, J. Magn. Magn. Mater., 2014, 364, 5–10 CrossRef CAS.
- P. Orgiani, A. Galdi, C. Aruta, V. Cataudella, G. De Filippis, C. A. Perroni, V. Marigliano Ramaglia, R. Ciancio, N. B. Brookes, M. Moretti Sala, G. Ghiringhelli and L. Maritato, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 205122 CrossRef.
- D. Abou-Ras, W. Boujelben, A. Cheikh-Rouhou, J. Pierre, J.-P. Renard, L. Reversat and K. Shimizu, J. Magn. Magn. Mater., 2001, 233, 147–154 CrossRef CAS.
- M. F. Hundley and J. J. Neumeier, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 55, 11511–11515 CrossRef CAS.
- A. V. Pashchenko, V. P. Pashchenko, V. K. Prokopenko, Yu. F. Revenko, Yu. S. Prylipko, N. A. Ledenev, G. G. Levchenko, V. P. Dyakonov and H. Szymczak, Acta Mater., 2014, 70, 218–227 CrossRef CAS.
- A. Tkach, A. Almeida, J. Agostinho Moreira, T. M. Correia, M. R. Chaves, O. Okhay, P. M. Vilarinho, I. Gregora and J. Petzelt, Appl. Phys. Lett., 2011, 98, 052903 CrossRef.
- N. A. Liedienov, A. V. Pashchenko, V. A. Turchenko, V. Ya. Sycheva, A. V. Voznyak, V. P. Kladko, A. I. Gudimenko, D. D. Tatarchuk, Y. V. Didenko, I. V. Fesych, I. I. Makoed, A. T. Kozakov and G. G. Levchenko, Ceram. Int., 2019, 45, 14873–14879 CrossRef CAS.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. M. Lau, A. R. Gerson, R. St and C. Smart, Appl. Surf. Sci., 2011, 257, 2717–2730 CrossRef CAS.
- D. R. Baer, D. L. Blanchard, M. H. Engelhard and J. M. Zachara, Surf. Interface Anal., 1991, 17, 25–30 CrossRef CAS.
- C. Doroftei and L. Leontie, RSC Adv., 2017, 7, 27863–27871 RSC.
- X. Xie, M. Chen, T. Liu, H. Jiang, H. Zhang and A. Chang, J. Mater. Sci.: Mater. Electron., 2017, 28, 8655–8661 CrossRef CAS.
- R. Natarajan, N. Palaniswamy, M. Natesan and V. S. Muralidharan, Open Corros. J., 2009, 2, 114–124 CrossRef CAS.
- B. K. Banerjee, Phys. Lett., 1964, 12, 16–17 CrossRef.
- M. H. Phan, S. B. Tian, S. C. Yu and A. N. Ulyanov, J. Magn. Magn. Mater., 2003, 256, 306–310 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2019 |