
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A turn-on fluorescent probe with a dansyl fluorophore for hydrogen sulfide sensing†
Yehan Yan‡
 a,
Lijuan Chen‡a,
Renyong Liua,
Yu Zhenga and
Suhua Wang*b
a,
Lijuan Chen‡a,
Renyong Liua,
Yu Zhenga and
Suhua Wang*b
aWest Anhui University, Luan, Anhui 237012, China
bSchool of Environmental Science and Engineering, North China Electric Power University, Beijing 102206, China. E-mail: wangsuhua@ncepu.edu.cn
First published on 3rd September 2019
Abstract
Hydrogen sulfide (H2S) is a biologically relevant molecule that has been newly identified as a gasotransmitter and is also a toxic gaseous pollutant. In this study, we report on a metal complex fluorescent probe to achieve the sensitive detection of H2S in a fluorescent “turn-on” mode. The probe bears a dansyl fluorophore with multidentate ligands for coordination with copper ions. The fluorescent “turn-on” mode is facilitated by the strong bonding between H2S and the Cu(II) ions to form insoluble copper sulfide, which leads to the release of a strongly fluorescent product. The H2S limit of detection (LOD) for the proposed probe is estimated to be 11 nM in the aqueous solution, and the utilization of the probe is demonstrated for detecting H2S in actual lake and mineral water samples with good reproducibility. Furthermore, we designed detector vials and presented their successful application for the visual detection of gaseous H2S.
1. Introduction
Hydrogen sulfide (H2S) is a highly toxic air pollutant that is widely generated from industrial production processes, involving sulfuric acid, sulfur, dyes, and cosmetics, as well as the microbial degradation processes by which anaerobic bacteria reduce inorganic sulfates and organic sulfides.1 Prolonged exposure to H2S may lead to respiratory paralysis, olfactory fatigue, vagueness of consciousness, and even permanent cerebral injury.2,3 In addition, H2S is a newly identified endogenous gaseous transmitter molecule like NO and CO, which has been associated with the regulation of cardiovascular, vasodilation, central nervous, respiratory, and immune systems.4–10 The level of H2S in humans has been documented to be between 10 and 100 μM, and an abnormal concentration of H2S has been related with Down syndrome, diabetes, Alzheimer's disease, and arterial and pulmonary hypertension.11–13 Therefore, analytical approaches capable of conducting selective and sensitive H2S detection in complicated environmental and biological systems are essential.Current techniques employed for H2S analysis include chemical titration,14,15 colorimetry,16 electrochemical assay,17 gas chromatography,18 and inductively coupled plasma atomic emission spectroscopy (ICP-AES).19 Compared to these methods, fluorescence analysis has proven to be a promising option because of its high sensitivity, high temporal and spatial detection resolutions, and the ability for conducting in situ monitoring of reactive and transient target analytes.20–27 Generally, the fluorescence probes for H2S mainly based on reduction reactions,28–32 nucleophilic addition reactions,33–35 and thiolysis reactions.36–38 In addition, metal displacement approach utilizes the strong affinity of metal ions with sulfides ions to rapidly attain reaction equilibrium, and achieve the real-time detection of H2S.39,40
Based on metal displacement approach of copper metal complexes, some colorimetric or fluorescent sensors for H2S have been emerging in recent years, owning to the low solubility product of CuS (Ksp = 6.3 × 10−36). For example, Li et al. synthesized a Cu-complex probe (BODIPY–DPA–Cu) by attaching di-(2-picolyl)amine (DPA) and BODIPY dye, in the presence of H2S, the fluorescence at 546 nm enhanced 19-fold in the PBS buffer (10 mM, pH 7.4). It showed a high sensitivity and selectivity for sulfide.41 Kim J. Y. and co-workers developed a Cu-cyclen-dansyl (Cu-CD) probe for the quantification of H2S in PBS buffer containing 10% DMSO with good selectivity among competitive anions.42 Kaushik and co-workers designed a probe for selective detection of H2S by copper complex embedded in vesicles, different from the MDA mechanism, in which both metal and indicator get displaced upon binding of H2S with metal center.43 Other relevant probes for H2S detection based copper metal complexes are listed in Table S1.†44–51
The present work capitalizes on this property by combining a dansyl fluorophore with a Cu(II) metal complex to design a displacement-reaction-based probe for H2S detection in a fluorescent “turn-on” mode. The fluorescent metal complex probe includes a multidentate ligand that can coordinate with Cu2+ ions to produce a stable dansyl fluorophore Cu-complex (DNS–Cu). The DNS–Cu complex exhibits a very weak background fluorescence because the empty d orbital of paramagnetic Cu(II) can accept the excited state electrons in the DNS fluorophore, and hence, block the process of ligand fluorescence generation. In addition, H2S strongly binds with the Cu2+ ions to form CuS and releases a free fluorescent DNS fluorophore, resulting in a greatly enhanced probe fluorescence. The H2S limit of detection (LOD) for the proposed probe (DNS–Cu) is determined to be 11 nM, and the probe demonstrates good H2S selectivity, reproducibility, and anti-interference performance. In addition, the probe was used to develop detector vials for visualizing gaseous H2S, which demonstrates the applicability of the proposed fluorescent probe for sensing H2S gas in the environment.
2. Experimental
2.1 Materials
All of reagents were received from commercial suppliers. Sodium sulfide (Na2S), sodium nitrate (NaNO3), sodium sulfate (Na2SO4), sodium thiocyanate (NaSCN), sodium thiosulfate (Na2S2O3), sodium fluoride (NaF), sodium chloride (NaCl), sodium nitrate (NaNO3), sodium nitrite (NaNO2), sodium bromide (NaBr), potassium iodide (KI), sodium sulfite (Na2SO3), sodium hypochlorite (NaClO), trisodium phosphate (Na3PO4), copper(II) chloride dihydrate (CuCl2·2H2O), reduced glutathione (GSH), L-cysteine (L-Cys), N,N-dimethyl formamide, ethyl acetate (EA), petroleum ether (PE), dichloromethane, anhydrous potassium carbonate, 5-dimethylamino-1-naphthalenesulfonyl chloride, triethylamine, methanol and 2-chloroethylamine hydrochloride. Adenosine 5′-diphosphate disodium salt (ADP), adenosine 5′-triphosphate disodium salt (ATP), adenosine 5′-monophosphate (AMP), pyrophosphoric acid (PPi), the ultrapure water (18.2 MΩ cm) was used throughout the experiments.2.2 Instrumentation
The fluorescence spectra were obtained on a PerkinElmer LS-55 luminescence spectrometer. Mass spectra were recorded on a Thermo Proteome X-LTQ MS. 1H-NMR spectrum was acquired on a Brucker Avance 400 MHz, using CDCl3 as solvent and tetramethylsilane (TMS) as internal standard. Fluorescence photos were taken under a UV lamp, with a Canon 350D digital camera.2.3 Synthesis of the dansyl fluorophore and its Cu(II) metal complex (DNS–Cu)
The synthesis processes of DNS and the DNS–Cu complex were illustrated in Scheme 1. Firstly, we added 2-chloroethylamine hydrochloride (65 mg) to a dichloromethane (10 mL) solution of dansyl chloride (150 mg) and triethylamine (Et3N; 117 μL). After stirring for 1 h, a pale-yellow oil was obtained by removing the solvent, and then purified via column chromatography with a PE![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :EA ratio of 4:1. The resulting product is herein denoted as compound 1 (107 mg, 61%). For the synthesis of DNS, we added 62.5 mg of compound 1, 37.6 mg of compound 2, and 27.6 mg of anhydrous K2CO3 into 3 mL of a dimethylformamide (DMF) solution. After stirring the mixture for 24 h, 15 mL of ice water was poured into the mixture to generate a precipitate. The solid crude product was collected through filtration, and the desired pale yellow oil DNS product (40 mg, 43.1%) was obtained by column chromatography using PE:EA ratios from 4:1 to 1:1. The DNS–Cu probe complex was easily synthesized by mixing equal molar amounts of DNS and CuCl2·2H2O solutions in C2H5OH/H2O (1:1, v/v). The structures of the DNS and DNS–Cu complex were both characterized by mass spectrometry (MS). Electrospray ionization (ESI) MS (ESI-MS) of DNS (m/z): calculated for 464.19, found 465.19 (M + H+), as shown in Fig. S1 in the ESI† and its proton nuclear magnetic resonance (1H-NMR; Fig. S2,† 400 MHz, CDCl3). 1H-NMR (400 MHz, CDCl3) δ 8.56–8.51 (m, 1H), 8.43 (d, J = 8.5 Hz, 1H), 8.32 (d, J = 8.7 Hz, 1H), 7.53 (td, J = 7.7, 1.8 Hz, 1H), 7.45–7.39 (m, 1H), 7.36 (dd, J = 8.6, 7.6 Hz, 1H), 7.21 (dd, J = 1.8, 0.8 Hz, 1H), 7.13–7.03 (m, 3H), 6.69 (s, 1H), 6.15 (dt, J = 9.9, 4.9 Hz, 1H), 5.94 (d, J = 2.9 Hz, 1H), 3.56 (s, 2H), 3.20 (s, 2H), 2.82–2.92 (m, 2H), 2.75 (s, 6H), 2.53 ppm (t, J = 4 Hz, 2H).
:EA ratio of 4:1. The resulting product is herein denoted as compound 1 (107 mg, 61%). For the synthesis of DNS, we added 62.5 mg of compound 1, 37.6 mg of compound 2, and 27.6 mg of anhydrous K2CO3 into 3 mL of a dimethylformamide (DMF) solution. After stirring the mixture for 24 h, 15 mL of ice water was poured into the mixture to generate a precipitate. The solid crude product was collected through filtration, and the desired pale yellow oil DNS product (40 mg, 43.1%) was obtained by column chromatography using PE:EA ratios from 4:1 to 1:1. The DNS–Cu probe complex was easily synthesized by mixing equal molar amounts of DNS and CuCl2·2H2O solutions in C2H5OH/H2O (1:1, v/v). The structures of the DNS and DNS–Cu complex were both characterized by mass spectrometry (MS). Electrospray ionization (ESI) MS (ESI-MS) of DNS (m/z): calculated for 464.19, found 465.19 (M + H+), as shown in Fig. S1 in the ESI† and its proton nuclear magnetic resonance (1H-NMR; Fig. S2,† 400 MHz, CDCl3). 1H-NMR (400 MHz, CDCl3) δ 8.56–8.51 (m, 1H), 8.43 (d, J = 8.5 Hz, 1H), 8.32 (d, J = 8.7 Hz, 1H), 7.53 (td, J = 7.7, 1.8 Hz, 1H), 7.45–7.39 (m, 1H), 7.36 (dd, J = 8.6, 7.6 Hz, 1H), 7.21 (dd, J = 1.8, 0.8 Hz, 1H), 7.13–7.03 (m, 3H), 6.69 (s, 1H), 6.15 (dt, J = 9.9, 4.9 Hz, 1H), 5.94 (d, J = 2.9 Hz, 1H), 3.56 (s, 2H), 3.20 (s, 2H), 2.82–2.92 (m, 2H), 2.75 (s, 6H), 2.53 ppm (t, J = 4 Hz, 2H).
 | ||
| Scheme 1 The scheme shows the synthetic procedure of DNS and its copper complex (DNS–Cu) probe. The DNS–Cu complex probe can combine with S2− to generate a very stable CuS precipitation and release the DNS fluorophore. | ||
2.4 Determination of the binding constant of the DNS–Cu complex
The stability constant K of the DNS–Cu complex was calculated from a Benesi–Hildebrand plot according to the following Benesi–Hildebrand equation.52,53Here, Fmax is the fluorescence intensity of the free DNS, F is the fluorescence intensity of DNS with Cu2+, Fmin stands for the fluorescence intensity of DNS in the presence of excessive Cu2+

2.5 Measurement of quantum yields (QYs) of the DNS–Cu complex
The QY was measured by using fluorescein (Φs = 0.95 in 0.1 M NaOH) as reference and calculated using the following equation.where ΦF stands for the fluorescence quantum yields, Abs and ∑F denote the absorbance and the measured integrated fluorescence intensity at the excitation wavelength, and η is the refractive index of the solvent used. The refractive index of H2O and C2H5OH/H2O (1
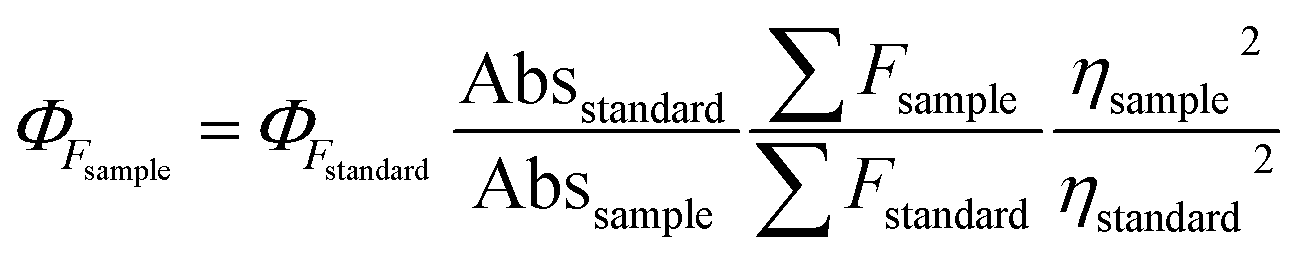 :1, v/v) mixture solvent was 1.33 and 1.35, respectively.
:1, v/v) mixture solvent was 1.33 and 1.35, respectively.
2.6 Detection sensitivity of the DNS–Cu complex probe for H2S
Firstly, 2.0 μL of the DNS–Cu probe solution (1.0 mM) was added to 2.0 mL of a C2H5OH/H2O (1:1, v/v) solution, yielding a 1.0 μM DNS–Cu concentration in the probe solution. The Na2S solution was freshly prepared used as the H2S source in the aqueous solution. The added concentrations of sulfur ion (S2−) in the probe solution were 0, 0.125, 0.250, 0.375, 0.500, 0.625, 0.750, 0.875, 1.000, 1.125, 1.250, and 1.375 μM. The fluorescence spectra were obtained between the range of 400 nm and 700 nm under an excitation wavelength of 338 nm. The primary fluorescence peak of the DNS–Cu probe obtained at 534 nm was employed for S2− sensitivity testing. The fluorescence ratio F/F0 of the DNS–Cu probe, where F0 is the value of F obtained with no added S2− ions, was plotted versus the S2− ions concentration for quantitative analysis. All data were performed three times under equivalent conditions, and the average values are calculated.
2.7 Detection selectivity of the DNS–Cu complex probe for H2S
The selective responses of the DNS–Cu probe for other related anions and small molecules containing thiol groups were carefully examined using the same testing procedure as was employed for S2−. The stock solutions of these species (1 mM of NO3−, NO2−, SO42−, SO32−, SCN−, S2O32−, F−, Cl−, Br−, I−, PO43−, and ClO−) were prepared in ultrapure water.To further illustrate the practical application of the proposed probe, anti-interference experiments were conducted by adding NO3−, NO2−, SO42−, or SO32− (50 equiv.), and SCN−, S2O32−, F−, Cl−, Br−, I−, PO43−, or ClO− (10 equiv.) into the DNS–Cu probe (1.0 μM) solution. Then, 1 equiv. of S2− ions (1.0 μM) was added into the mixture solution, followed by recording the fluorescence intensity at 534 nm.
The selectivity of the DNS–Cu probe was also examined for H2S and other different common gases. Gaseous H2S was obtained by slowly dropping phosphoric acid on sodium sulfide. Carbon monoxide (CO) gas was achieved from the reaction of formic acid with concentrated sulfuric acid. Carbon dioxide (CO2) gas was prepared from the titration reaction between dilute sulfuric acid and sodium bicarbonate. Sulfur dioxide (SO2) was got through a quantitative reaction of sodium hydrosulfide and concentrated sulfuric acid. Ammonia (NH3) gas was generated through the chemical reaction between NH4Cl and Ca(OH)2. Nitric oxide (NO) and nitrogen dioxide (NO2) gas were obtained from pure gas. Then, different of these gas samples were injected into the DNS–Cu probe solution using a syringe, recording the fluorescence spectra subsequently and taking the fluorescence photos by a digital camera.
3. Results and discussion
3.1 Characterization of DNS and DNS–Cu
The structure of the DNS confirmed by the analysis of MS and 1H-NMR (Fig. S1 and S2†). As illustrated in Scheme 1, the DNS fluorophore exhibits a bright fluorescence at 534 nm, after coordinating with Cu2+, DNS–Cu displays a very weak fluorescence (Fig. S3†). The MS analysis verified the formation of the DNS–Cu complex. ESI-MS of DNS–Cu (m/z): calculated 526.1100 found 526.0400 (Fig. S4†). The value of stability constant (K) for the DNS–Cu complex was determined to be 2.7 × 104 based on Benesi–Hildebrand method, as seen in Fig. S5.†3.2 Stability and sensitivity of the DNS–Cu probe for H2S
We firstly investigated the stability of the DNS–Cu complex probe before the sensitivity experiment. As noted from Fig. S6,† the fluorescence intensity of the probe at 534 nm exhibits no distinct changes after six consecutive irradiations for 60 min, indicating that the DNS–Cu complex probe is stable against photobleaching. When the probe solution (1.0 μM) is exposed to S2− ions, as seen in Fig. 1A, the fluorescence intensity of the probe greatly increased as the S2− ions concentration increased from 0 to 1.375 μM. This can be attributed to the strong binding of S2− with the Cu(II) metal center of the DNS–Cu complex to form stable CuS precipitation, subsequently releases the DNS fluorophore. The MS and 1H-NMR analysis proved the release of the DNS in Fig. S7,† and the quantum yields were increased from 1.8% to 25.5% after the DNS–Cu probe reaction with S2−(Fig. S8†). In addition, the fluorescence intensity exhibits a dose–response with increasing S2− concentration up to an S2− dose of 1.0 μM, after which further increases in the S2− concentration produce no further increase in the fluorescence intensity. This indicates that the stoichiometric reaction between the DNS–Cu probe and S2− was 1:1. The plot of F/F0 versus the S2− concentration in Fig. 1B exhibits a good linear relationship with a coefficient of determination R2 = 0.9988 in the S2−concentration range of 0 to 1.0 μM. The LOD was estimated to be 11 nM based on the definition of LOD = 3 × S.D./k, where k is the slope of the curve in Fig. 1B, and 3 × S.D. stands for 3 times standard deviation of the blank signal.
 | ||
| Fig. 1 (A) Fluorescence of the DNS–Cu probe solution (1.0 μM) in the presence of different concentrations of S2− ions. (B) Plot of the fluorescence ratio F/F0 of the probe with respect to the S2− concentration, where F0 and F represent the fluorescence intensity of the DNS–Cu probe at 534 nm without and with the addition of S2− ions, respectively. | ||
3.3 Selectivity of the DNS–Cu probe for H2S
As shown in Fig. 2A, the fluorescence intensity of the DNS–Cu probe solution increased sharply after the addition of S2− (1.0 μM), whereas no apparent changes in the fluorescence intensity were observed after adding the NO3−, NO2−, SO42−, SO32−, SCN−, S2O32−, F−, Cl−, Br−, I−, PO43−, and ClO− anionic species (1.0 μM), indicating a high selectivity for S2− ions. For further practical application, the anti-interference experiments of the probe were conducted by adding NO3−, NO2−, SO42−, SO32− (50 equiv.), and SCN−, S2O32−, F−, Cl−, Br−, I−, PO43− ClO− (10 equiv.) into the probe solution, respectively. Then 1 equiv. of S2− ions (1.0 μM) were added subsequently into the mixture solution, followed by recording the fluorescence intensity at 534 nm. The results of the anti-interference experiments shown in Fig. 2B, indicating that the probe exhibits a good anti-interference against other anionic species. | ||
| Fig. 2 (A) Fluorescence spectra of the DNS–Cu probe solution (1.0 μM) for S2− ions (1.0 μM) and other anion species (1.0 μM). (B) F/F0 values at 534 nm of the probe to various anions. The sparse bars represent the fluorescence responses of the probe in the presence of other anions (50 equiv. of NO3−, NO2−, SO42−, SO32− and 10 equiv. of SCN−, S2O32−, F−, Cl−, Br−, I−, PO43− and ClO−), the dense bars represent the subsequent addition of 1 equiv. of S2− (1.0 μM) into the mixture solution. | ||
In order to verify the practicability of probe in complex environments, some biological phosphates (PPi, ATP, ADP, AMP) and endogenous biomolecule (GSH, L-Cys) were investigated with DNS–Cu probe. The results show that the PPi, ATP, ADP and AMP could not turn on the fluorescence of the probe system (Fig. S9†). Even in the presence of 10 equiv. of PPi, ATP, ADP and AMP (10 μM), H2S (1.0 μM) also can enhance the fluorescence of the DNS–Cu probe in the same manner as in the absence of these analytes. These results suggest that biological phosphates dose not interference the detection system. However, it is noted that GSH and cysteine could enhance the fluorescence of the DNS–Cu probe, which may be attributed to the affinity of Cu2+ with –SH groups. Fortunately, their interference with these thiol-containing molecules can be readily eliminated through treatment with dimethyl sulfoxide (DMSO), which oxidizes the thiol groups to form disulfides that have less affinity with Cu(II), which minimizes their interference effect (Fig. S10†).
3.4 Potential reusability of the DNS–Cu probe
The fluorescence of the DNS–Cu probe can be activated by the addition of S2−, quenched by the addition of 1 equiv. of Cu(II), and then recovered again by adding another 1 equiv. of S2−. As shown in Fig. 3, such fluorescence on–off cycles with S2− and Cu(II) could be repeated for 4 times with little degradation in the fluorescence intensity (Fig. 3B), and their corresponding fluorescent images recorded in Fig. 3A. The good reusability of the fluorescent probe for the alternate detection of S2− and Cu2+ can be developed as a logic gate by operating the two inputs In(S2−) and In(Cu2+), as indicated in Fig. 3C. Optical logic gates, such as the YES,54 NOT,55 AND,56 OR,57 NOR,58 XOR,59 and INHIBIT gates60 have been investigated widely in recent years. Here, the two inputs can be set as 0 and 1 to represent the absence and presence of fluorescence, respectively, where exposure to S2− results in a fluorescence on state, and the output readout is 1, while the fluorescence would be turned off by exposure to Cu2+ ions, leading to an output of 0. When there are no inputs of H2S and Cu2+ to the initial logic solution of the probe DNS–Cu complex, it shows no fluorescence, then the output signal is 0. While both of H2S and Cu2+ exist at the same time, it still no fluorescence, the output signal is 0 as well. Briefly, no fluorescence results from both inputs set as 0 or 1 simultaneously, the output signals were both 0, which match with the INHIBIT logic function.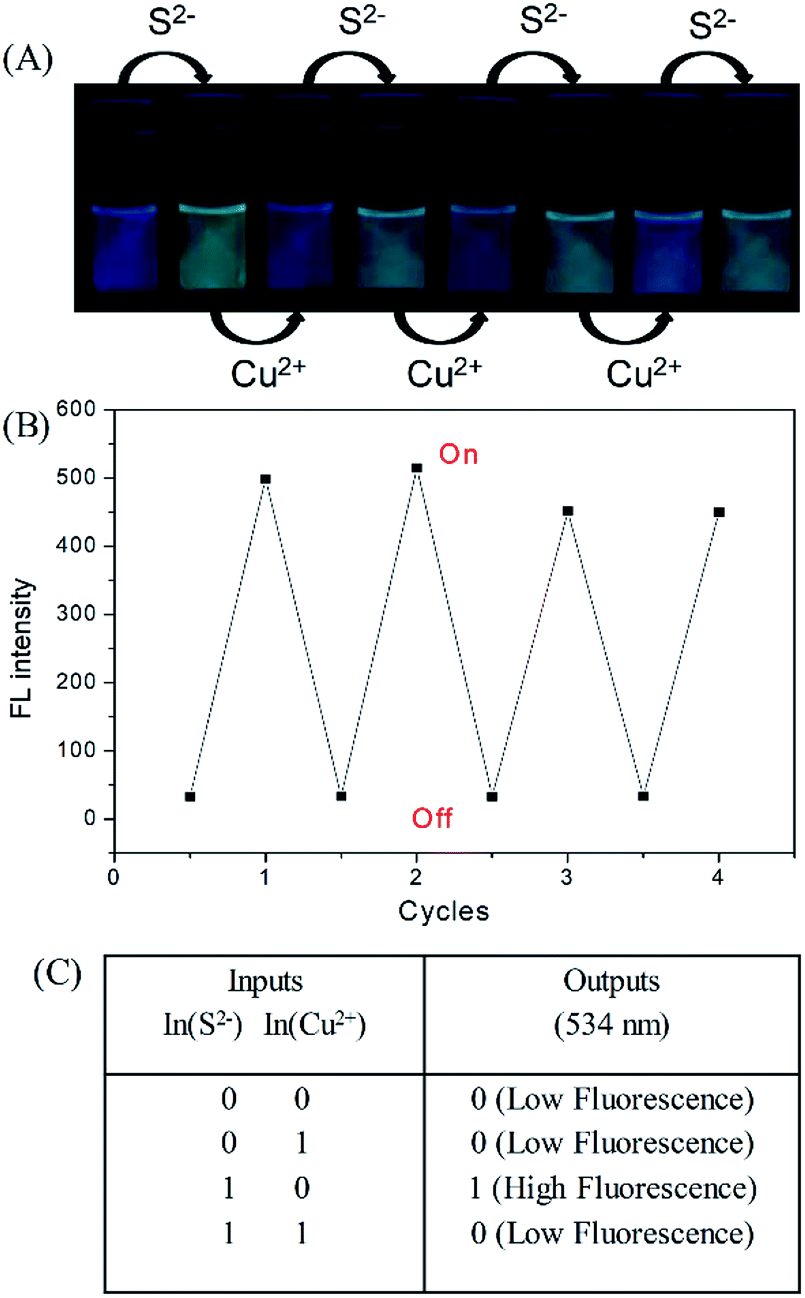 | ||
| Fig. 3 Fluorescence images (A) and fluorescence intensity (B) of the DNS–Cu probe solution with the alternate addition of S2− and Cu2+. The photographs were recorded under an ultraviolet (UV) lamp. (C) Logic table of an INHIBIT logic gate. | ||
3.5 Application of the DNS–Cu probe to actual water samples
Spike and recovery experiments were conducted using actual lake and mineral water samples. The lake water samples were collected from Shushan lake and filtered through a 0.45 μm microporous filter to remove insoluble particles. The mineral water samples were bought from a local supermarket and used directly without any pretreatment. The original concentrations of S2− in lake water and mineral water samples were firstly confirmed to be 4.2 nM and 5.3 nM by ICPMS (iCAP RQ). These values were much lower than the maximum allowable level of S2− (15 μM) set by World Health Organization (WHO) for drinking water. Thus, they do not pose any health concerns. Then the spiked recovery tests were conducted with three different S2− concentrations (0, 500, and 1000 nM) in a mixed solution of water/ethanol (v/v = 1:1). The average concentration and standard deviations of S2− in the spiked lake and mineral water samples are presented in Table 1. The concentrations of sulfide estimated in the non-spiked lake and mineral water samples were less than the LOD of the method. The two values are lower than the detection limit, suggesting that the sensitivity of the method has its own limitation and cannot be directly used to real water samples with trace contents, but can be used in the further recovery calculation. The recovery results in the lake water samples are slightly greater than 100%, which may be attributed to the microbial degradation process. Generally, the recovery rates ranged from 98.2% to 102.3% for the lake and mineral water samples, which are statistically near 100%, and therefore validates the ability of the proposed probe for S2− ions sensing in complex samples.
| Add S2− concentration (nM) | Lake water | Mineral water | ||
|---|---|---|---|---|
| Found (nM) | Recovery (%) | Found (nM) | Recovery (%) | |
| 0 | 6.2 | 7.8 | ||
| 500 | 508 | 101.6 ± 3.09 | 491 | 98.2 ± 1.03 |
| 1000 | 1023 | 102.3 ± 1.48 | 997 | 99.7 ± 2.14 |
3.6 Visualization of H2S gas using detector vials
Since S2− and H2S can quickly reach equilibrium in aqueous solution, this method can be applied to detect gaseous H2S in aqueous solution. For this measurement, we fabricated detector vials to achieve on-site and rapid detection of gaseous H2S. The vials itself has no fluorescence with a rubber stopper, different levels of H2S gas (0, 0.5, 1.0, 5.0, 10, and 20 ppm) were syringed into the vials. The images as presented in Fig. 4 indicate that the fluorescence color intensity in the vials increased with increasing H2S concentration. The limit of detection of this method was determined as 0.5 ppm, based on the minimum amount of H2S to produce a slightly different fluorescent color, which could be visible by five persons. The fluorescence intensity of the detector vial achieved a maximum level at an H2S concentration of 20 ppm.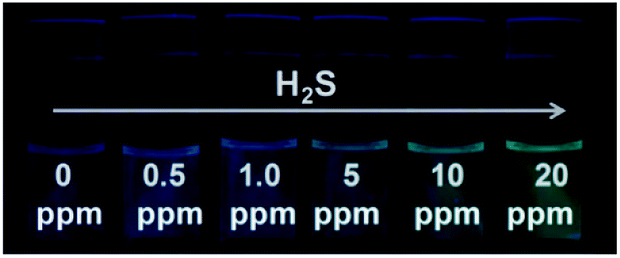 | ||
| Fig. 4 Visual detection of different concentrations of H2S gas under a UV light illumination in the dark using detector vials. | ||
In addition, the selectivity of the proposed probe for H2S gas was investigated by evaluating the incidence of fluorescence when filling the detector vials with other common gases, such as N2, NH3, NO2, SO2, CO2, CO, and NO (100 ppm). As presented in Fig. 5. The results indicated that the injection of these other gaseous compounds has no substantial effect on the fluorescence intensity of the probe, and that only H2S gas activated the probe fluorescence. These results demonstrated a sensitivity and selectivity of this method for H2S determination in the gas state.
 | ||
| Fig. 5 (A) Fluorescence spectra of the DNS–Cu probe solution for gaseous H2S. (B) The fluorescence images of the detector vials taken under a 365 nm UV lamp. The concentration of H2S gas was 20 ppm, N2, NH3, NO2, SO2, CO2, CO and NO was 100 ppm. | ||
4. Conclusions
We have fabricated a dansyl-based copper complex for use as a “turn-on” fluorescence probe in H2S sensing. The subsequent addition of H2S effectively snatches Cu(II) from the DNS–Cu complex, and thus releases a free dansyl moiety, leading to the fluorescence enhancement of the probe. The LOD for this method was determined to be 11 nM in aqueous solutions. In addition, the probe provided good H2S detection results with actual water samples. Moreover, the good reusability of the fluorescent probe for cyclical detection of H2S and Cu(II), which can be used to develop “INHIBIT” logic circuit.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial aid was supported by the National Natural Science Foundation of China (Grant No. 21804100), the National Key Research and Development Program of China (2017YFA0207000), the Natural Science Foundation of Anhui (KJ2018A0416, 1808085QB44), and the Science Foundation of West Anhui University (No. WXZR201801, WGKQ201702020, WGKQ201702002).References
- Y. H. Yan, K. Zhang, H. Yu, H. J. Zhu, M. T. Sun, T. Hayat, A. Alsaedi and S. H. Wang, Talanta, 2017, 174, 387–393 CrossRef CAS PubMed.
- L. Li, M. Bhatia, Y. Z. Zhu, Y. C. Zhu, R. D. Ramnath, Z. J. Wang, F. B. Anuar, M. Whiteman, M. Salto-Tellez and P. K. Moore, FASEB J., 2005, 19, 1196–1198 CrossRef CAS PubMed.
- K. Abe and H. Kimura, J. Neurosci., 1996, 16, 1066–1071 CrossRef CAS PubMed.
- E. Culotta and D. E. Koshland, Science, 1992, 258, 1862–1865 CrossRef CAS PubMed.
- T. Morita, M. A. Perrella, M. E. Lee and S. Kourembanas, Proc. Natl. Acad. Sci. U.S.A., 1995, 92, 1475–1479 CrossRef CAS PubMed.
- C. Y. Miao and Z. Y. Li, Br. J. Pharmacol., 2012, 165, 643–658 CrossRef CAS PubMed.
- G. D. Yang, L. Y. Wu, B. Jiang, W. Yang, J. S. Qi, K. Cao, Q. H. Meng, A. K. Mustafa, W. T. Mu, S. M. Zhang, S. H. Snyder and R. Wang, Science, 2008, 322, 587–590 CrossRef CAS PubMed.
- D. Boehning and S. H. Snyder, Annu. Rev. Neurosci., 2003, 26, 105–131 CrossRef CAS PubMed.
- H. Kimura, Mol. Neurobiol., 2002, 26, 13–19 CrossRef CAS PubMed.
- A. Martelli, L. Testai, M. C. Breschi, C. Blandizzi, A. Virdis, S. Teddei and V. Calderone, Med. Res. Rev., 2012, 32, 1093–1130 CrossRef CAS PubMed.
- P. Kamoun, M. C. Belardinelli, A. Chabli, K. Lallouchi and B. C. Vekemans, Am. J. Med. Genet., Part A, 2003, 116, 310–311 CrossRef PubMed.
- L. L. Yang, J. P. Wang, L. Yang, C. Zhang, R. L. Zhang, Z. Z. Zhang, B. H. Liu and C. L. Jiang, RSC Adv., 2016, 6, 56384–56391 RSC.
- K. Eto, T. Asada, K. Arima, T. Makifuchi and H. Kimura, Biochem. Biophys. Res. Commun., 2002, 293, 1485–1488 CrossRef CAS PubMed.
- M. Wronski, Anal. Chem., 1971, 43, 606–607 CrossRef CAS PubMed.
- S. Balasubramanian and V. Pugalenthi, Water Res., 2000, 34, 4201–4206 CrossRef CAS.
- M. G. Choi, S. Cha, H. Lee, H. L. Jeon and S. K. Chang, Chem. Commun., 2009, 47, 7390–7392 RSC.
- B. Spilker, J. Randhahn, H. Grabow, H. Beikirch and P. Jeroschewski, J. Electroanal. Chem., 2008, 612, 121–130 CrossRef CAS.
- P. R. Bérubé, P. D. Parkinson and E. R. Hall, J. Chromatogr. A, 1999, 830, 485–489 CrossRef.
- M. Colon, M. Iglesias, M. Hidalgo and J. L. Todoli, J. Anal. At. Spectrom., 2008, 23, 416–418 RSC.
- D. Y. Gong, X. T. Zhu, Y. J. Tian, S. C. Han, M. Deng, A. Iqbal, W. S. Liu, W. W. Qin and H. C. Guo, Anal. Chem., 2017, 89, 1801–1807 CrossRef CAS PubMed.
- H. H. Li, H. Yu, M. T. Sun, K. A. Alamry, A. M. Asiri and S. H. Wang, Int. J. Environ. Sci. Technol., 2018, 15, 1193–1200 CAS.
- M. T. Sun, H. Yu, H. H. Li, H. D. Xu, D. J. Huang and S. H. Wang, Inorg. Chem., 2015, 54, 3766–3772 CrossRef CAS PubMed.
- J. L. Yao, K. Zhang, H. J. Zhu, F. Ma, M. T. Sun, H. Yu, J. Sun and S. H. Wang, Anal. Chem., 2013, 85, 6461–6468 CrossRef CAS PubMed.
- M. Yang, M. T. Sun, Z. P. Zhang and S. H. Wang, Talanta, 2013, 105, 34–39 CrossRef CAS PubMed.
- F. L. Meng, Z. Guo and X. J. Huang, TrAC, Trends Anal. Chem., 2015, 68, 37–47 CrossRef CAS.
- F. L. Meng, H. X. Zheng, Y. F. Sun, M. Q. Li and J. H. Liu, Sensors, 2017, 7, 1478 CrossRef PubMed.
- X. G. San, G. D. Zhao, G. S. Wang, Y. B. Shen, D. Meng, Y. J. Zhang and F. L. Meng, RSC Adv., 2017, 7, 3540–3549 RSC.
- H. J. Peng, Y. F. Cheng, C. F. Dai, A. L. King, B. L. Predmore, D. J. Lefer and B. H. Wang, Angew. Chem., Int. Ed., 2011, 50, 9672–9675 CrossRef CAS PubMed.
- A. R. Lippert, E. J. New and C. J. Chang, J. Am. Chem. Soc., 2011, 133, 10078–10080 CrossRef CAS.
- B. F. Chen, W. Li, C. Lv, M. M. Zhao, H. Jin, H. F. Jin, J. B. Du, L. R. Zhang and X. J. Tan, Analyst, 2013, 138, 946–951 RSC.
- K. Zhang, J. Zhang, Z. Xi, L. Y. Li, X. X. Gu, Q. Z. Zhang and L. Yi, Chem. Sci., 2017, 8, 2776–2781 RSC.
- Y. H. Yan, H. Yu, Y. J. Zhang, K. Zhang, H. J. Zhu, T. Yu, H. Jiang and S. H. Wang, ACS Appl. Mater. Interfaces, 2015, 7, 3547–3553 CrossRef CAS PubMed.
- Y. Qian, J. Karpus, O. Kabil, S. Y. Zhang, H. L. Zhu, R. Banerjee, J. Zhao and C. He, Nat. Commun., 2011, 2, 495 CrossRef PubMed.
- H. Li, Q. Yao, J. Fan, N. Jiang, J. Wang, J. Xia and X. Peng, Chem. Commun., 2015, 51, 16225–16228 RSC.
- S. Singha, D. Kim, H. Moon, T. Wang, K. H. Kim, Y. H. Shin, J. Jung, E. Seo, S. J. Lee and K. H. Ahn, Anal. Chem., 2014, 87, 1188–1195 CrossRef PubMed.
- C. Zhang, L. Wei, C. Wei, J. Zhang, R. Wang, Z. Xi and L. Yi, Chem. Commun., 2015, 51, 7505–7508 RSC.
- Y. L. Pak, J. Li, K. C. Ko, G. Kim, J. Y. Lee and J. Yoon, Anal. Chem., 2016, 88, 5476–5481 CrossRef CAS PubMed.
- Y. Zhang, H. Chen, D. Chen, D. Wu, X. Chen, S. H. Liu and J. Yin, Org. Biomol. Chem., 2015, 13, 9760–9766 RSC.
- K. Sasakura, K. Hanaoka, N. Shibuya, Y. Mikami, Y. Kimura, T. Komatsu, T. Ueno, T. Terai, H. Kimura and T. Nagano, J. Am. Chem. Soc., 2011, 133, 18003–18005 CAS.
- F. P. Hou, L. Huang, P. X. Xi, J. Cheng, X. F. Zhao, G. Q. Xie, Y. J. Shi, F. J. Cheng, X. J. Yao, D. C. Bai and Z. Z. Zeng, Inorg. Chem., 2012, 51, 2454–2460 CrossRef CAS.
- X. Li, Y. Gong, K. Wu, S. H. Liang, J. Cao, B. Yang, Y. Hu and Y. Han, RSC Adv., 2014, 4, 36106–36109 RSC.
- J. Y. Kim, S. Sarkar, K. N. Bobba, P. T. Huynh, A. Bhise and J. Yoo, Org. Biomol. Chem., 2019 10.1039/c9ob00948e.
- R. Kaushik, R. Sakla, A. Ghosh, G. T. Selvan, P. M. Selvakumar and D. A. Jose, ACS Sens., 2018, 3, 1142–1148 CrossRef CAS PubMed.
- R. Kaushik, A. Ghosh and D. A. Jose, Coord. Chem. Rev., 2017, 347, 141–157 CrossRef CAS.
- S. Wang, H. C. Ding, Y. S. Wang, C. B. Fan, Y. Y. Tu, G. Liu and S. Z. Pu, RSC Adv., 2018, 8, 33121–33128 RSC.
- M. G. Choi, S. Cha, H. Lee, H. L. Jeon and S. K. Chang, Chem. Commun., 2009, 47, 7390–7392 RSC.
- X. Gu, C. Liu, Y. C. Zhu and Y. Z. Zhu, Tetrahedron Lett., 2011, 52, 5000–5003 CrossRef CAS.
- F. Hou, L. Huang, P. Xi, J. Cheng, X. Zhao, G. Xie, Y. Shi, F. Cheng, X. Yao, D. Bai and Z. Zeng, Inorg. Chem., 2012, 51, 2454–2460 CrossRef CAS PubMed.
- S. Mirra, S. Milione, M. Strianese and C. Pellecchia, Eur. J. Inorg. Chem., 2015, 2272–2276 CrossRef CAS.
- H. Fang, P. C. Huang and F. Y. Wu, Spectrochim. Acta, Part A, 2018, 204, 568–575 CrossRef CAS.
- X. F. Shanga, X. J. Yueb, Y. M. Chena, C. S. Lia, H. L. Chenc and T. Y. Wang, Inorg. Chem. Commun., 2019, 99, 1–10 CrossRef.
- H. A. Benesi and J. H. Hildebrand, J. Am. Chem. Soc., 1949, 71, 2703–2707 CrossRef CAS.
- S. Zhang, T. Yu, M. T. Sun, H. Yu, Z. P. Zhang, S. H. Wang and H. Jiang, Talanta, 2014, 126, 185–190 CrossRef CAS PubMed.
- Y. Shiraishi, Y. Tokitoh and T. Hirai, Chem. Commun., 2005, 42, 5316–5318 RSC.
- T. B. Pittman, M. J. Fitch, B. C. Jacobs and J. D. Franson, Phys. Rev. A, 2003, 68, 032316 CrossRef.
- S. Uchiyama, N. Kawai, A. P. de Silva and K. Iwai, J. Am. Chem. Soc., 2004, 126, 3032–3033 CrossRef CAS PubMed.
- P. Ghosh, P. K. Bharadwaj, S. Mandal and S. Ghosh, J. Am. Chem. Soc., 1996, 118, 1553–1554 CrossRef CAS.
- X. L. Feng, X. R. Duan, L. B. Liu, F. D. Feng, S. Wang, Y. L. Li and D. B. Zhu, Angew. Chem., Int. Ed., 2009, 48, 5316–5321 CrossRef CAS PubMed.
- A. Credi, V. Balzani, S. J. Langford and J. F. Stoddart, J. Am. Chem. Soc., 1997, 119, 2679–2681 CrossRef CAS.
- R. K. Pathak, K. Tabbasum, A. Rai, D. Panda and C. P. Rao, Anal. Chem., 2012, 84, 5117–5123 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra04790e |
| ‡ Yehan Yan and Lijuan Chen contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |