
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhosphonic acid mediated practical dehalogenation and benzylation with benzyl halides†
Jing Xiao *a,
Yonghao Maa,
Xiaofang Wua,
Jing Gaoa,
Zilong Tanga and
Li-Biao Han*b
*a,
Yonghao Maa,
Xiaofang Wua,
Jing Gaoa,
Zilong Tanga and
Li-Biao Han*b
aKey Laboratory of Theoretical Organic Chemistry and Functional Molecule of Ministry of Education, School of Chemistry and Chemical Engineering, Hunan University of Science and Technology, Xiangtan 411201, China
bNational Institute of Advanced Industrial Science and Technology (AIST), Tsukuba, Ibaraki 305-8565, Japan. E-mail: libiao-han@aist.go.jp; Fax: +81-29-861-6344; Tel: +81-298-61-4855
First published on 18th July 2019
Abstract
For the first time, by using H3PO3/I2 system, various benzyl chlorides, bromides and iodides were dehalogenated successfully. In the presence of H3PO3, benzyl halides underwent electrophilic substitution reactions with electron-rich arenes, leading to a broad range of diarylmethanes in good yields. These transformations feature green, cheap reducing reagents and metal-free conditions. A possible mechanism was proposed.
Dehalogenation and reactions of benzylation with benzyl halides are important transformations in synthetic organic chemistry. Herein we report a simple and green method for dehalogenation and benzylation with benzyl halides under metal-free conditions. Thus, under the H3PO3/I2 system, dehalogenation took place readily with benzyl chlorides, bromides and iodides to produce the corresponding benzyl derivatives in high yields. In addition, in the absence of I2, benzyl halides underwent electrophilic substitution reactions with electron-rich arenes, generating diarylmethanes efficiently.
Over the past few decades, significant efforts have been devoted to the reduction of halides.1 Among them, a variety of reducing reagents were applied to dehalogenation reactions successfully. Some of the most representative reducing agents include metal hydrides such as tri-n-butyltin hydrides,2 organosilanes,3 sodium borohydrides4 and lithium aluminium hydride.5 Besides, catalytic hydrogenation is also an important approach to the reduction of halides. For example, dehalogenation catalyzed by transition-metals such as nickle,6 palladium,3b,7 ruthenium,8 rhodium,9 indium,2a,10 iron11 and cobalt12 etc. have gained increased attention. Although great progress has been achieved in dehalogenation reactions, nontoxic, green, simple and cheap reducing systems still need to be explored (Scheme 1).
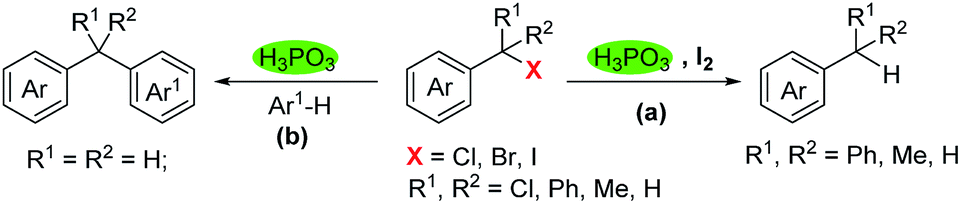 | ||
| Scheme 1 Phosphonic acid mediated dehalogenation and benzylation with benzyl halides. | ||
Recently, our group was interested in the use of H3PO3 in synthetic methodologies due to its low-toxicity, ready availability and cheap cost.13 It is estimated that a million tons of phosphonic acid was generated as the side-product from the process of electroless nickle plating using H3PO2 and other chemical processes. Although most of the H3PO3 was lain idle as waste, we are still fascinated by the great potential ability in reduction reactions since H3PO3 has a reactive P(O)H bond that might serve as a green H source. Thus, for the first time, the H3PO3/I2 combination was used for the reduction of benzyl halides, generating a variety of benzyl derivatives in good yields. This reducing system has the advantages of being green, cheap and metal-free.
Our investigation began with the reduction of benzyl bromide in the presence of H3PO3 and I2. As shown in Table 1, reduction of benzyl bromide took place in benzene at 100 °C for 22 h, generating the desired product toluene in 48% GC yield (entry 1). Next, the temperature of the reaction was examined (entry 2 and 3). The suitable temperature was 120 °C and gave the product in 76% yield. Further screening of the amount of I2 or H3PO3 failed to improve the reaction efficiency (entry 4–6). Among the solvents examined, polar solvents such as chlorobenzene, CH3CN and dioxane gave poor results (entry 7–9). To our delight, a weak polar solvent DCE (1,2-dichloroethane) was a relatively good solvent with a yield of 69% (entry 10). When prolonging the reaction time to 36 h, DCE gave a better result than benzene with a yield of 92% (entry 11 and 12). The yield of the product could not be increased by elevating the temperature to 130 °C in DCE (entry 13).
|
|||||
|---|---|---|---|---|---|
| Entry | H3PO3 (equiv.) | I2 (equiv.) | Solvent | Temp/°C | Yield (%) |
| a Conditions: a mixture of 1a (0.3 mmol), H3PO3, and I2 in solvent (0.6 mL) was stirred at indicated temperature for the 22 h. Yield based on GC yield using dodecane as an internal standard.b 36 h. | |||||
| 1 | 2.0 | 10% | Benzene | 100 | 48% |
| 2 | 2.0 | 10% | Benzene | 120 | 76% |
| 3 | 2.0 | 10% | Benzene | 130 | 75% |
| 4 | 2.0 | 15% | Benzene | 120 | 74% |
| 5 | 2.0 | 5% | Benzene | 120 | 70% |
| 6 | 3.0 | 10% | Benzene | 120 | 64% |
| 7 | 2.0 | 10% | PhCI | 120 | 21% |
| 8 | 2.0 | 10% | CH3CN | 120 | 7% |
| 9 | 2.0 | 10% | Dioxane | 120 | 26% |
| 10 | 2.0 | 10% | DCE | 120 | 69% |
| 11 | 2.0 | 10% | DCE | 120 | 92%b |
| 12 | 2.0 | 10% | Benzene | 120 | 61%b |
| 13 | 2.0 | 10% | DCE | 130 | 82% |
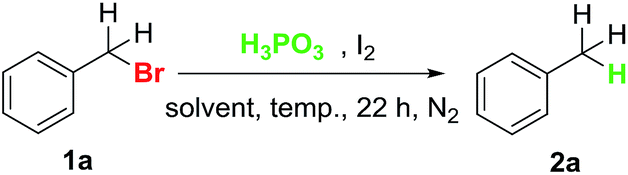
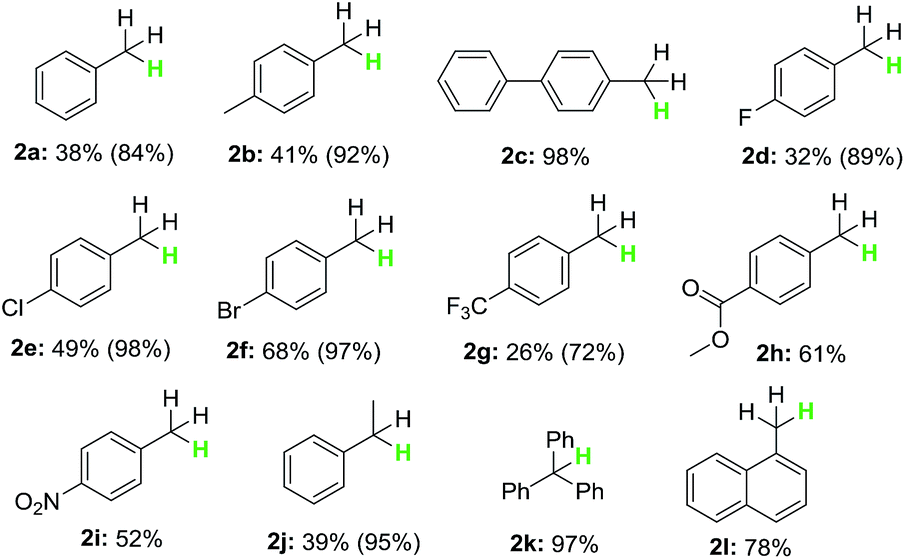
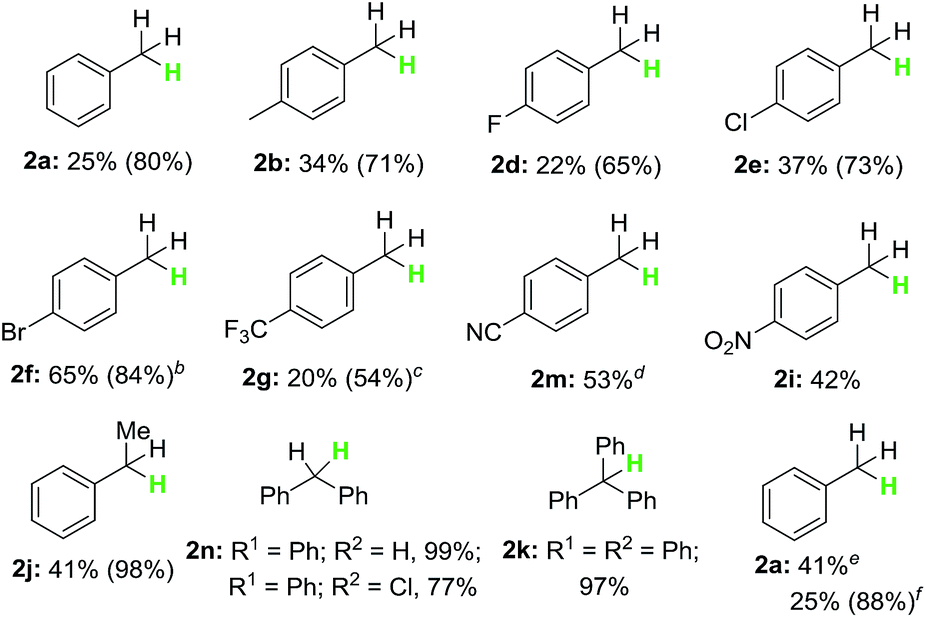
With the optimized reaction conditions in hand, the scope of the reaction with respect to benzyl halides was examined (Table 2). Benzyl bromide was reduced to the corresponding product in 84% GC yield, albeit 38% isolated yield was obtained due to its low boiling point (2a). Bromides bearing methyl or phenyl groups at the para-position on benzene were reduced smoothly to afford the product in high yields (2b and 2c). Interestingly, substrates containing halogens such as F, Cl and Br at the para-position on benzene stayed intact during the dehalogenation (2d–2f). Other electron-deficient bromides having CF3, C(O)OMe and NO2 groups were also tolerated well in this reductive system (2g–2i). Secondary and tertiary benzyl bromides such as (1-bromoethyl)benzene and (bromomethanetriyl)tribenzene also underwent this reduction successfully generating the product in 95% and 97% yield, respectively (2j and 2k). When 1-(bromomethyl)naphthalene was subjected into the reaction, 78% yield of the product was obtained (2l). Unfortunately, alkyl and aryl bromides did not go through this dehalogenation reaction.
|
|---|
| a Conditions: a mixture of 1 (0.6 mmol), H3PO3, and I2 in DCE was stirred at 120 °C for 36 h. Isolated yield based on three parallel reactions. Yield in parenthesis are GC yield. |
 |
We next extended the scope of benzyl halides under similar conditions. As shown in Table 3, benzyl chloride also proceeded smoothly (2a), and various reduction products were obtained. Chlorides bearing substituents (Me, F, Cl and Br) were suitable for this reduction reaction, generating the corresponding product in good yields (2b, 2d–2f). With regard to electron-withdrawing group under H3PO3/I2 system, we observed that CF3, CN and NO2 substituents were tolerated in good yields (2g, 2m and 2i). Furthermore, secondary chlorides such as (1-chloroethyl)benzene and chlorodiphenylmethane were also worked well in this transformation, providing the product in high yields (2j and 2n). Gratefully, large steric hindrance triphenylmethane chloride was reduced readily under the present condition in 97% yield (2k). Finally, we conducted the dehalogenation of benzotrichloride and benzyl iodide to explore possible reduction of polyhalogenated substrates and iodides. Indeed, 2a was formed in 41% and 88% yield, respectively.
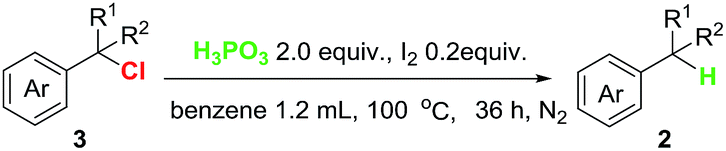
During the above studies of dehalogenation of benzyl halides, we found that interestingly, when the reaction was performed in the absence of I2, electrophilic substituent reaction took place to affording diarylmethane in excellent yields. Diarylmethane are widely used in organic synthesis, pharmaceutical development and material science.14 Among the methods reported, Friedel–Crafts (FC) benzylation of arenes15 and transition-metal catalysed cross coupling reactions16 are the important approaches to preparing these compounds. However, those methods are usually need equivalent expensive Lewis acids, metal and not readily available materials. Although various strategies for the metal-free access to diarylmethanes have been explored,17 a more simple, practical, and greener method for their preparation are highly desirable.
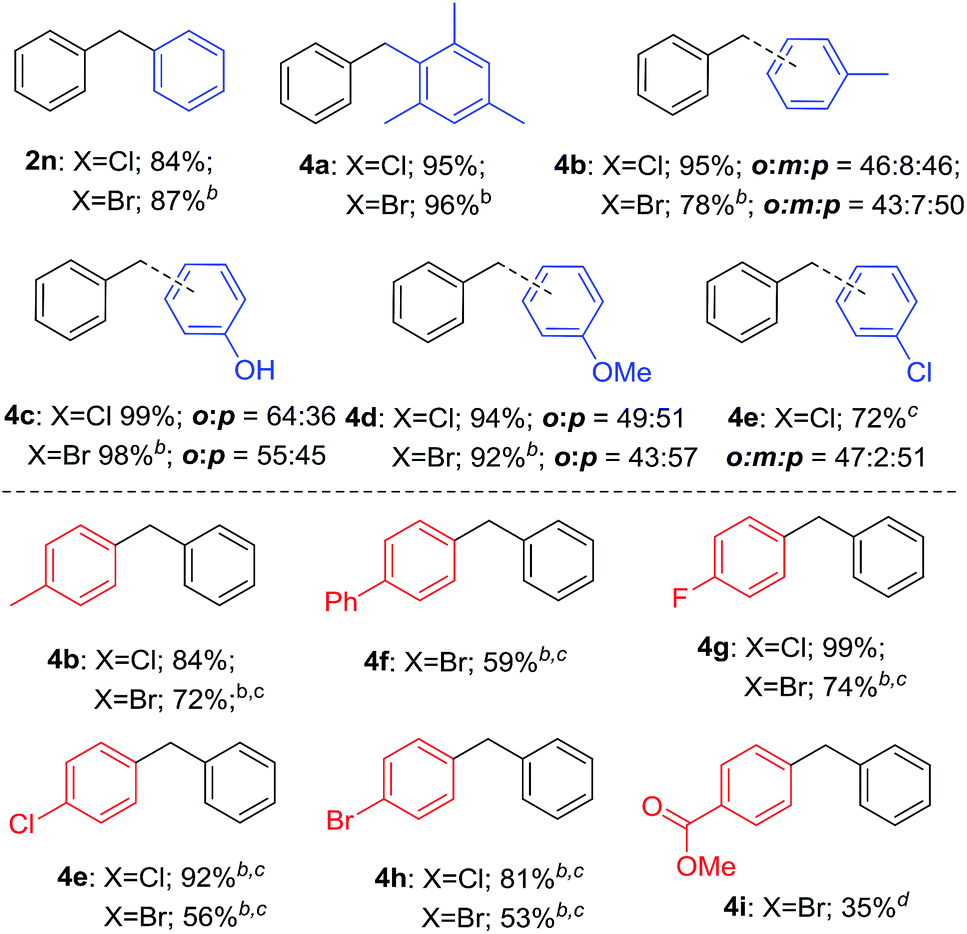
As shown in Table 4, various arenes were subjected into this electrophilic substitution reaction. Thus, in the presence of H3PO3, benzyl chlorides and bromides underwent this transformation successfully with benzene or mesitylene to afford the corresponding diarylmethanes in good to excellent yields (2n and 4a). Gratifying, electron-rich arenes such as toluene, phenol and anisole were also compatible in this reaction conditions with isomer products generated (4b–4d). In addition, when chlorobenzene was used as the substrate, good yield of the corresponding product was observed (4e). Subsequently, a serious of benzyl halides with benzene were investigated in the presence of H3PO3. Benzyl halides bearing methyl, phenyl, fluoride, chloride and bromide led to good to high yield of the products (4b, 4f–4h). It is worth noting that benzyl bromide with electron-drawing group such as methyl ester was also applicable in this reaction, albeit in moderate yield of the product (4i). Unfortunately, the reaction gave trace product with benzyl halides having strong electron-withdrawing groups such as nitro or trifluoromethyl group.
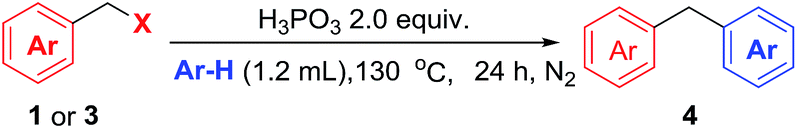
To highlight the synthetic utility of these transformation, some gram–scale reactions were conducted. As shown in Scheme 2, 12 mmol (chloromethylene)dibenzene 3n was reduced smoothly in the presence of 2.0 equiv. H3PO3 and 10 mol% I2 in benzene at 130 °C for 36 h, generating 1.956 g product in 97% isolated yield (eqn (1)). Besides, we were pleased to find that the gram scale synthesis of diphenylmethane by reaction of benzyl chloride with benzene was also achieved (eqn (2)). The above results revealed that these reactions were practical and have the potential to scale up.
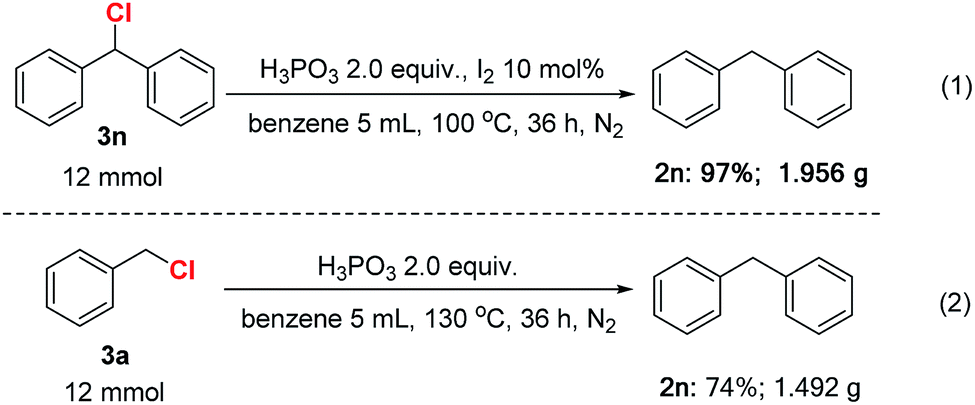 | ||
| Scheme 2 Gram-scale reactions. | ||
Finally, some control experiments were conducted to evaluate the different halides and electron effect of these reactions. 4-Methylbenzyl chloride and 4-methylbenzyl bromide were reduced to the product with 20% and 31% substrate conversion, respectively (eqn (3)). Benzyl bromides bearing electron-donating group methyl, H and electron-withdrawing group CF3 were also subjected into the reaction in one pot, and the corresponding product were obtained in 17%, 9% and trace yield with a large amount of substrate recovered, respectively (eqn (4)). Similarly, the benzylation of benzene by the above three bromides affording the corresponding diarylmethanes in a downward trend in yields (eqn (5)). The above results revealed that benzyl bromides were more reactive than benzyl chloride in dehalogenation reactions. Besides, in the same type of halides, electron-donating group having more positive influence than electron-deficient whether in reduction or benzylation reactions, which may suggest that these reactions were involving a carbon cation mechanism (Scheme 3).
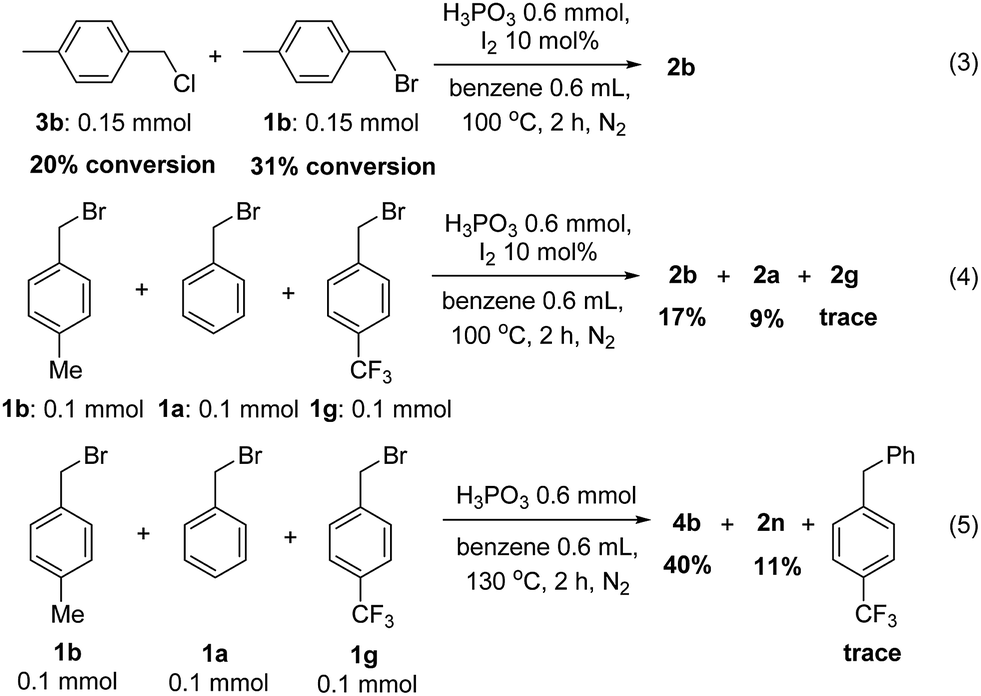 | ||
| Scheme 3 Control experiments. | ||
Based on the above results and previous reports,13 a possible mechanism was proposed in Scheme 4. HI was generated in situ by reaction of H3PO3 and I2, halogen exchange between benzyl halides and HI afforded intermediate A, further reduction of A by HI to give the corresponding product (eqn (a)). For benzylation of arenes by halides, we deduced that carbon cation intermediate B was generated via C–X bond cleavage in the presence of H3PO3, B underwent electrophilic substitution reaction with arenes to afford the corresponding diarylmethanes (eqn (b)).
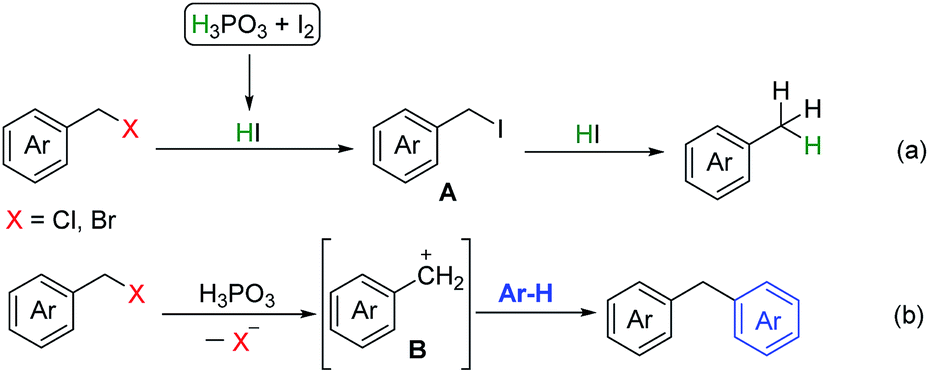 | ||
| Scheme 4 Possible mechanism. | ||
Conclusions
In summary, we report a practical method for dehalogenation of benzyl halides and preparation of diarylmethanes by using a H3PO3 system. Various benzyl halides could be readily reduced by the H3PO3/I2 combination for the first time. In the absence of I2, electrophilic substitution reactions between benzyl halides and arenes occurred smoothly, furnishing diarylmethanes in good yields. This method provides a simple, cheap and green approach for the reduction of benzyl halides and synthesis of diarylmethanes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
JX is thankful for a postdoc fellowship from AIST and financial support from the National Natural Science Foundation of China (21703061) and the Natural Science Foundation of Hunan province (2017JJ3081).Notes and references
- (a) F. Alonso, I. P. Beletskaya and M. Yus, Chem. Rev., 2002, 102, 4009–4092 CrossRef CAS; (b) G. Chelucci, S. Baldino, G. A. Pinna and G. Pinna, Curr. Org. Chem., 2012, 16, 2921–2945 CrossRef CAS; (c) C. E. Castro, Reviews of Environmental Contamination and Toxicology, Springer, New York, 1998, 155, pp. 1–67 Search PubMed; (d) G. L. Larson, Org. React., 2008, 71, 5–119 Search PubMed; (e) T. Hennebel, J. Benner, P. Clauwaert, L. Vanhaecke, P. Aelterman, R. Callebaut, N. Boon and W. Verstraete, Biotechnol. Lett., 2011, 33, 89–95 CrossRef CAS; (f) Á. F. Cañete, C. O. Salas and F. C. Zacconi, Molecules, 2013, 18, 398–407 CrossRef.
- (a) K. Inoue, A. Sawada, I. Shibata and A. Baba, Tetrahedron Lett., 2001, 42, 4661–4663 CrossRef CAS; (b) A. Rahm, R. Amardeil and M. Degueil-Castaing, J. Organomet. Chem., 1989, 371, 4–8 CrossRef; (c) M. Pereyre, J. P. Quintard and A. Rahm, Reduction of Organic Halides in Tin in Organic Synthesis, Butterworth, London, 1986 Search PubMed.
- (a) C. Chatgilialoglu, Acc. Chem. Res., 1992, 25, 188–194 CrossRef CAS; (b) R. Boukherroub, C. Chatgilialoglu and G. Manuel, Organometallics, 1996, 15, 1508–1510 CrossRef CAS; (c) J. Yang and M. Brookhart, Adv. Synth. Catal., 2009, 351, 175–187 CrossRef CAS.
- S. Krishnamurthy and H. C. Brown, J. Org. Chem., 1980, 45, 849–856 CrossRef CAS.
- S. Krishnamurthy and H. C. Brown, J. Org. Chem., 1982, 47, 276–280 CrossRef CAS.
- (a) A. F. Barrero, E. J. Alvarez-Manzaneda, R. Chahboun, R. Meneses and J. L. Romera, Synlett, 2001, 4, 485–488 CrossRef; (b) S. Zinovyev, A. Perosa, S. Yufit and P. Tundo, J. Catal., 2002, 211, 347–354 CrossRef CAS; (c) J. M Khurana, S. Kumar and B. Nand, Can. J. Chem., 2008, 86, 1052–1054 CrossRef.
- (a) T. Hara, K. Mori, M. Oshiba, T. Mizugaki, K. Ebitania and K. Kaneda, Green Chem., 2004, 6, 507–509 RSC; (b) G. L. Larson and J. L. Fry, Org. React., 2008, 71, 1–737 Search PubMed; (c) T. Hara, T. Kaneta, K. Mori, T. Mitsudome, T. Mizugaki, K. Ebitanic and K. Kaneda, Green Chem., 2007, 9, 1246–1251 RSC.
- (a) A. Jana, J. Mondal, P. Borah, S. Mondal, A. Bhaumik and Y. Zhao, Chem. Commun., 2015, 51, 10746–10749 RSC; (b) M. C. Haibach, B. M. Stoltz and R. H. Grubbs, Angew. Chem., Int. Ed., 2017, 56, 15123–15126 CrossRef CAS.
- K. Fujita, M. Owaki and R. Yamaguchi, Chem. Commun., 2002, 2964–2965 RSC.
- Á. F. Cañete, C. O. Salas and F. C. Zacconi, Molecules, 2013, 18, 398–407 CrossRef PubMed.
- H. Guo, K.-i. Kanno and T. Takahashi, Chem. Lett., 2004, 33, 1356–1357 CrossRef CAS.
- (a) B. Sahoo, A. E. Surkus, M. M. Pohl, J. Radnik, M. Schneider, S. Bachmann, M. Scalone, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2017, 56, 11242–11247 CrossRef CAS; (b) F. Ungváry and L. Markó, J. Organomet. Chem., 1980, 193, 379–382 CrossRef.
- J. Xiao and L.-B. Han, Tetrahedron, 2019, 75, 3510–3515 CrossRef CAS.
- (a) J. S. Kim and D. T. Quang, Chem. Rev., 2007, 107, 3780–3799 CrossRef CAS; (b) Y.-Q. Long, X.-H. Jiang, R. Dayam, T. Sanchez, R. Shoemaker, S. Sei and N. Neamati, J. Med. Chem., 2004, 47, 2561–2573 CrossRef CAS PubMed; (c) W. N. Washburn, J. Med. Chem., 2009, 52, 1785–1794 CrossRef CAS; (d) T. Brotin and J.-P. Dutasta, Chem. Rev., 2009, 109, 88–130 CrossRef CAS.
- (a) C. Friedel and J. M. Crafts, Compt. Rend., 1877, 84, 1450 Search PubMed; (b) G. A. Olah, S. J. Kuhn and S. H. Flood, J. Am. Chem. Soc., 1962, 84, 1688–1695 CrossRef CAS; (c) M. Rueping and B. J. Nachtsheim, Beilstein J. Org. Chem., 2010, 6, 6, DOI:10.3762/bjoc.6.6.
- (a) H. Jiang, S.-C. Sha, S. A. Jeong, B. C. Manor and P. J. Walsh, Org. Lett., 2019, 21, 1735–1739 CrossRef CAS; (b) J. Xiao, T. Chen and L.-B. Han, Org. Lett., 2015, 17, 812–815 CrossRef CAS; (c) J. Xiao, a. J. Yang, T. Chen and L.-B. Han, Adv. Synth. Catal., 2016, 358, 816–819 CrossRef CAS; (d) B. Liégault, J. L. Renaud and C. Bruneau, Chem. Soc. Rev., 2008, 37, 290–299 RSC.
- (a) R.-J. Tang, T. Milcent and B. Crousse, J. Org. Chem., 2018, 83, 14001–14009 CrossRef CAS; (b) G. Schafer and J. W. Bode, Angew. Chem., Int. Ed., 2011, 50, 10913–10916 CrossRef; (c) J. Zhu, M. Perez and D. W. Stephan, Angew. Chem., Int. Ed., 2016, 55, 8448–8451 CrossRef CAS; (d) P. A. Champagne, Y. Benhassine, J. Desroches and J. F. Paquin, Angew. Chem., Int. Ed., 2014, 53, 13835–13839 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra04770k |
| This journal is © The Royal Society of Chemistry 2019 |