
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of C2-tetrasubstituted indolin-3-ones via Cu-catalyzed oxidative dimerization of 2-aryl indoles and cross-addition with indoles†
Anoop Singh‡
a,
Satheeshvarma Vanaparthi‡ a,
Sachin Choudharya,
Rangan Krishnanb and
Indresh Kumar*a
a,
Sachin Choudharya,
Rangan Krishnanb and
Indresh Kumar*a
aDepartment of Chemistry, Birla Institute of Technology and Science, Pilani 333 031, Rajasthan, India. E-mail: indresh.chemistry@gmail.com; indresh.kumar@pilani.bits-pilani.ac.in
bDepartment of Chemistry, BITS Pilani, Hyderabad Campus, Secunderabad, India
First published on 2nd August 2019
Abstract
An efficient protocol for the synthesis of 2,2-disubstituted indolin-3-ones under mild conditions has been developed. This reaction involves the copper-catalyzed in situ oxidative de-aromatization of 2-arylindoles to indol-3-one, followed by self-dimerization as well as cross-addition with indoles under mild conditions. The result generates a wide variety of C2-tetrasubstituted indolin-3-ones with good to high yields (62–82%).
Introduction
Indolin-3-ones are privileged scaffolds that function as intermediates for the synthesis of medicinally important compounds.1 In particular, 2,2-disubstituted 1,2-dihydro-3H-indol-3-one, also known as pseudoindoxyls bearing C2 stereocenters have continually appeared in natural products such as austamide (I), strobilanthoside A (II), isatisine A (III), and halichrome A (IV), as well as in many other bioactive synthetic compounds (Fig. 1).2 Moreover, compounds with this skeleton have also exhibited exciting applications in the areas of fluorescence labeling and optoelectronic materials in recent years.3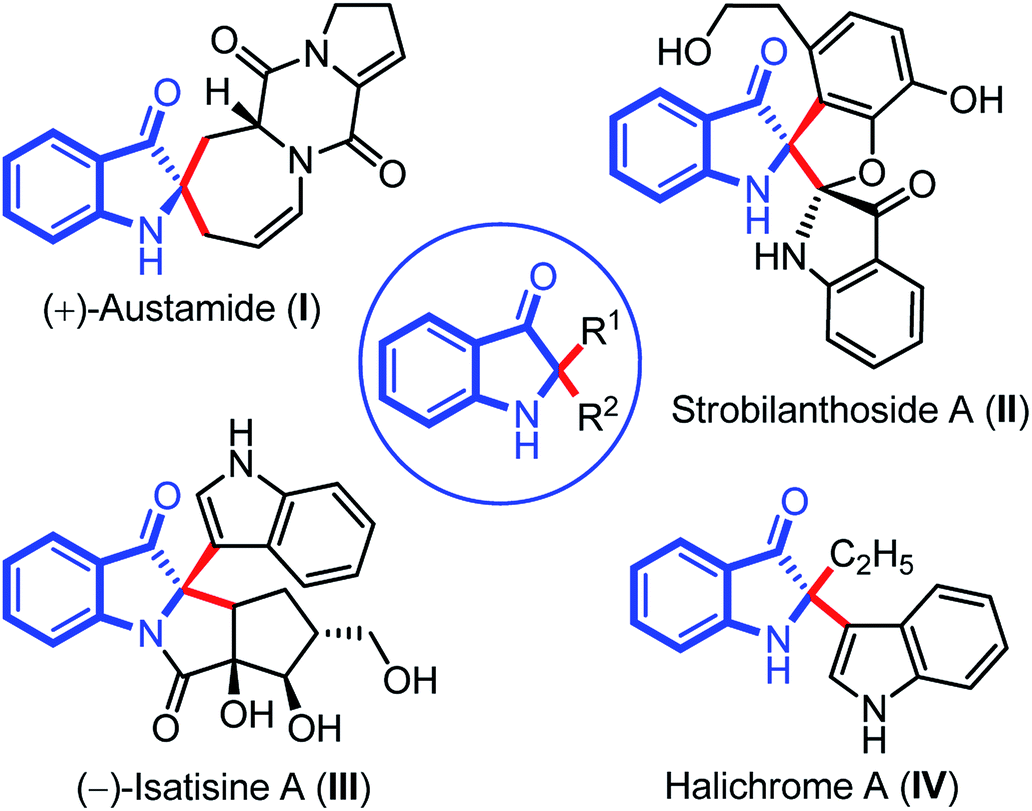 | ||
| Fig. 1 2,2-disubstituted indolin-3-one as basic core in important natural products. | ||
Owing to the wide utility of 2,2-disubstituted indolin-3-ones, several methods had been developed in the past few years which include; transition-metal catalyzed annulation reactions,4 the cascade Fischer indolization/Claisen rearrangement,5 and photooxidative rearrangements,6 along with other methods.7 Besides these methods, chemoselective addition of various nucleophiles to 2-aryl-indol-3-one, an activated cyclic C-acylimine, is another exciting way to access 2,2-disubstituted indolin-3-one derivatives.8 However, the synthesis of 2-aryl-3H-indol-3-ones requires troublesome multistep syntheses and are not easily accessible.9 To overcome this problem, some attention has recently been given to the chemistry of dearomative cascade reactions of 2-substituted indoles for the direct construction of C2-quaternary indolin-3-ones (Scheme 1a). In this context, self-dimerization of 2-substituted indoles have been explored either through Cu-catalysis (Scheme 1a(i))10 or other metal-catalysis (Scheme 1a(ii)).11 However, these methods required high temperature and sometimes hazardous components. Likewise, oxidative cross-addition of indole to 2-substituted indole could be another way to achieve C2-quaternary indolin-3-ones; however, that is a difficult task to accomplish in terms of selectivity. Guchhait and coworkers developed an exciting and the very first protocol for the cross-addition of indoles to 2-substituted indoles to access 2,2-disubstituted indolin-3-ones with a chiral center under Pd-catalysis in a chemoselective fashion (Scheme 1a(iii)).12 Very recently, Yu and coworkers developed a metal-free approach for the cross-addition of indoles with a series of 2-substituted indoles to rapidly access 2,2-disubstituted indolin-3-ones (Scheme 1a(iv)).13
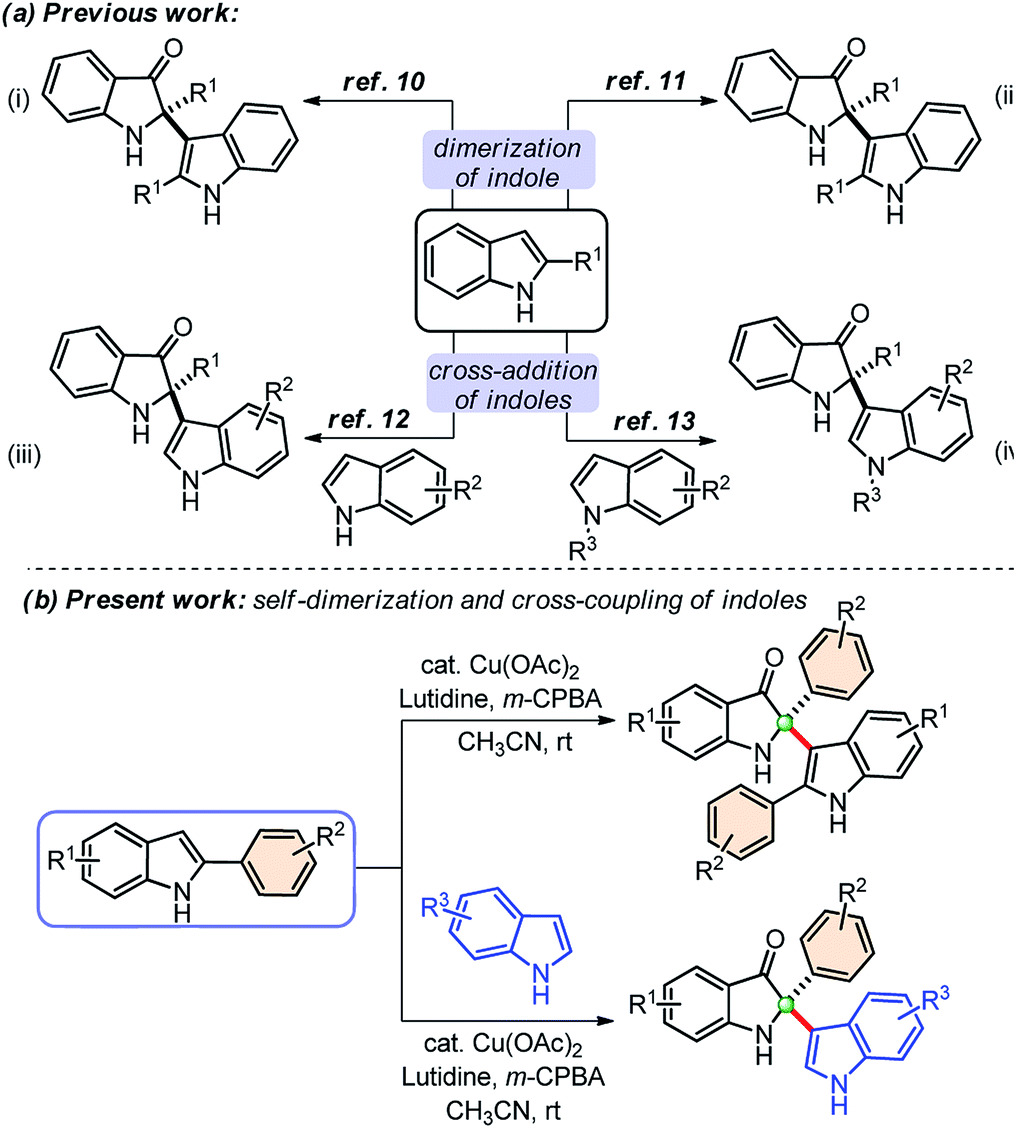 | ||
| Scheme 1 Synthetic approaches from 2-aryl indoles to access 2,2-disubstituted indolin-3-ones. | ||
Due to the high significance of C2-quaternary indolin-3-ones, and limitations of the existing methods, finding direct access to these compounds through the self-dimerization of 2-substituted indoles or the cross-addition of indoles with 2-substituted indoles under mild conditions, is still highly desirable. Here, we present such a general and straightforward finding for the synthesis of 2,2-disubstituted indolin-3-ones from 2-aryl indoles through copper-catalyzed self-dimerization and cross-addition with indoles at room temperature (Scheme 1(b)).
Results and discussion
Copper-catalyzed transformations are one of the most studied methods in synthetic chemistry due to their efficiency, good functional group tolerance.14 In this context, Cu-catalyzed tandem oxidative reactions of 2-aryl indol-3-ones, in situ generated from 2-arylindoles, have been explored to synthesize 2-arylbenzoxazinone,15 and polyhydropyrido[1,2-a]indoles/tetracyclic quinazolinones.16 Encouraged by these relevant precedents, we envisaged that a general copper-catalyzed method could be developed for the self-dimerization of 2-substituted indoles and cross-addition with indoles through the in situ generations of indol-3-ones under mild conditions. Herein, we describe the successful implementation of our protocol.We begin this study for the oxidative cross-dimerization of 2-phenyl indole 1a as model substrate to prepare 2-phenyl-2-(2-phenyl-1H-indol-3-yl)indolin-3-one 2a. In this context, optimization of the reaction conditions was carried out by employing several bases, oxidizing agents, catalysts and solvents, and the results are shown in Table 1. Initially, reaction failed to work, when 1a was treated with catalysts CuCl (30 mol%), pyridine with; K2S2O8 (entry 1, Table 1), oxone (entry 2, Table 1). Trace amount (<10%) of the product was obtained with air as oxidants in toluene (entry 3, Table 1) and DMSO (entry 4, Table 1) as solvents, respectively. However, product 2a was obtained with low yield (34%), when the reaction was carried out with CuCl (cat.), pyridine, and TBHP (tert-butyl hydroperoxide) in CH3CN (entry 5, Table 1) at room temperature. Additional efforts were made to improve the reaction yields either by changing the oxidants, base, and catalysts (entries 6–11, Table 1). An improvement in the reaction yields was observed by employing lutidine, in place of pyridine, with TBHP (45%) (entry 8, Table 1), and with m-CPBA (meta-chloro perbenzoic acid) (52%) (entry 10, Table 1). The dimerized product 2a was obtained with moderate yield (63%) when Cu(OAc)2 (entry 11, Table 1) was employed in place of CuCl as a catalyst with TBHP as oxidant, which was again improved to 75% yield by using m-CPBA as oxidant (entry 12, Table 1). Any additional change in the reaction conditions either; by changing oxidant (entry 12, Table 1) or lowering the catalyst loading (entry 14, Table 1) failed to improve the reaction yield. The reaction failed to produce any dimerization product in the absence of catalyst (entry 15, Table 1), and base (entry 16, Table 1). Thus, we preferred to perform this reaction to yield cross-dimerized product 2a under the standardized conditions (entry 12, Table 1). Moreover, reaction only furnished 2-indoles substituted 3-oxindole 2a through the addition of indole as a nucleophile at the C2-position of in situ generated indol-3-one in a chemoselective fashion.
|
||||||
|---|---|---|---|---|---|---|
| Entry | Solvent | Catalyst | Base | Oxidant | Time (h) | Yieldb (%) |
| a Unless otherwise indicated, the reaction was carried out with 2-phenylindole 1a (0.5 mmol), solvent (3.0 mL), catalyst (30 mol%), base (1.0 mmol), oxidant (0.3 mmol), reaction time (h) at rt.b Isolated yield of 2a refer to 1a.c Cu(OAc)2 (15 mmol%) was used. | ||||||
| 1 | Toluene | CuCl | Py | K2S2O8 | 24 | n.r. |
| 2 | Toluene | CuCl | Py | Oxone | 24 | n.r. |
| 3 | Toluene | CuCl | Py | Air | 24 | <10 |
| 4 | DMSO | CuCl | Py | Air | 24 | <10 |
| 5 | CH3CN | CuCl | Py | TBHP | 24 | 34 |
| 6 | CH3CN | CuCl | Py | Oxone | 24 | 25 |
| 7 | CH3CN | CuCl | Py | Air | 24 | 30 |
| 8 | CH3CN | CuCl | Lutidine | TBHP | 22 | 45 |
| 9 | CH3CN | CuCl | K2CO3 | TBHP | 24 | 20 |
| 10 | CH3CN | CuCl | Lutidine | m-CPBA | 20 | 52 |
| 11 | CH3CN | Cu(OAc)2 | Lutidine | TBHP | 20 | 63 |
| 12 | CH3CN | Cu(OAc)2 | Lutidine | m-CPBA | 18 | 75 |
| 13 | CH3CN | Cu(OAc)2 | Lutidine | H2O2 | 18 | 43 |
| 14c | CH3CN | Cu(OAc)2 | Lutidine | m-CPBA | 24 | 65 |
| 15 | CH3CN | — | Lutidine | m-CPBA | 24 | n.r. |
| 16 | CH3CN | Cu(OAc)2 | — | m-CPBA | 24 | n.r. |
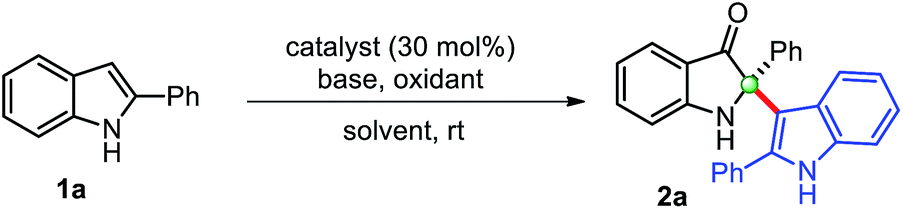
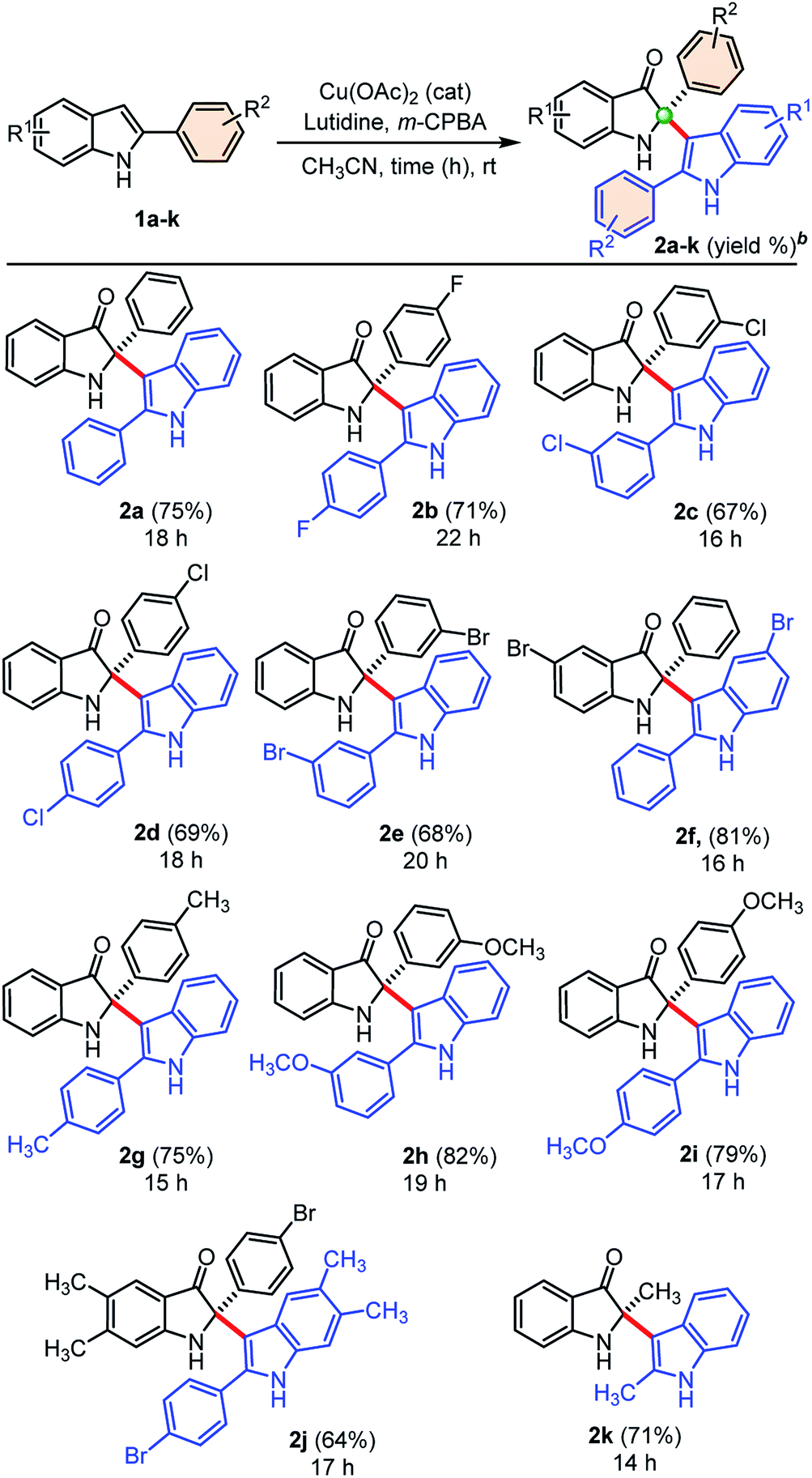
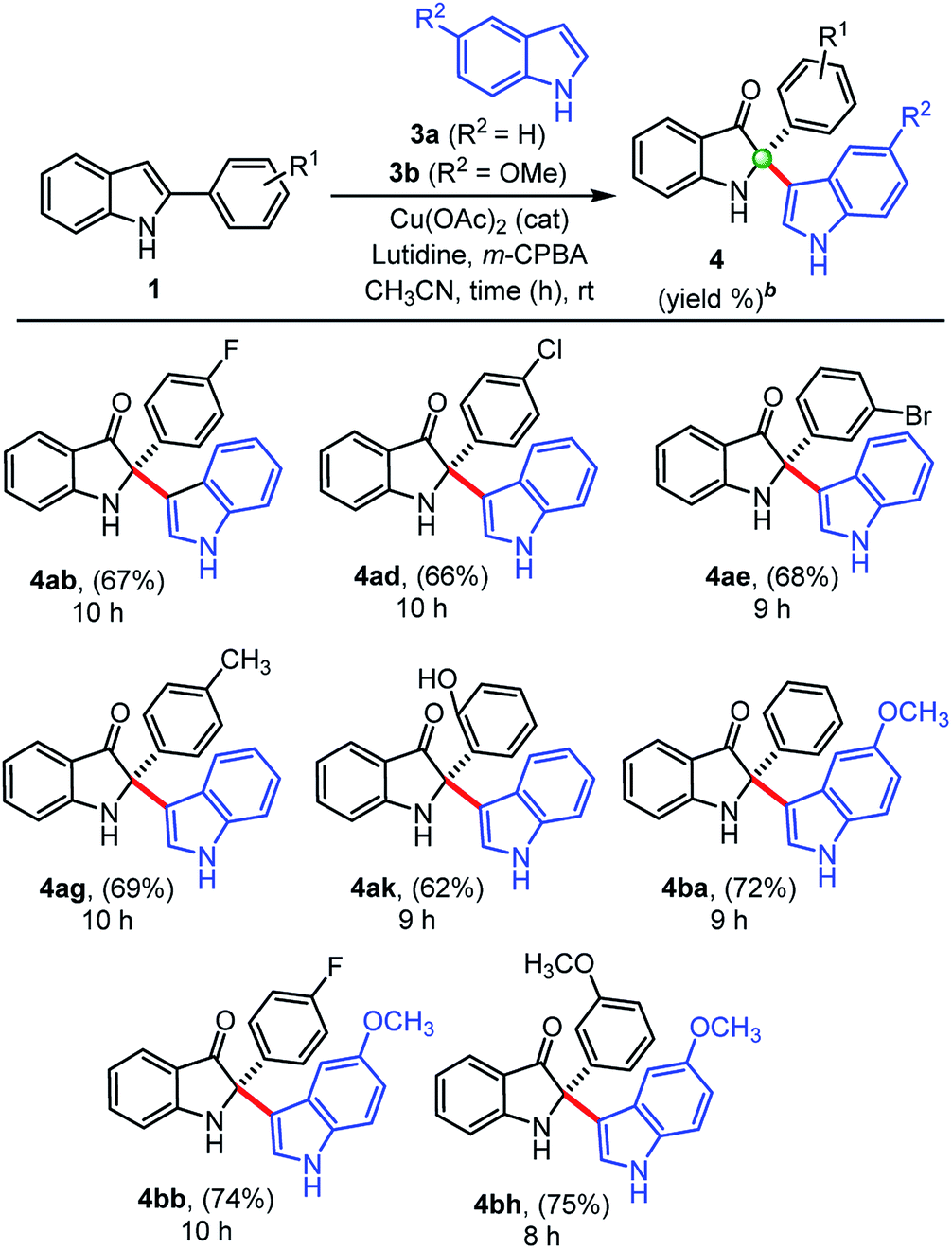
Next, we explored the generality of our developed cross-dimerization protocol with variously substituted 2-aryl indoles 1a–k under standardized conditions, and results are shown in Table 2. The reaction was found to be quite general concerning the substituents on both the aryl-rings of 2-aryl-indole 1 and accomplished within 14–22 h at room temperature to the furnish corresponding cross-dimerized product, i.e., 2-indoles substituted 3-oxindoles 2 in good to high yields (68–82%). Initially, electron-withdrawing substituents like 4-F, 3-Cl, 4-Cl, 3-Br substitutions on the phenyl ring of 2-aryl indole 1b, 1c, 1d, and 1e gave corresponding products 2b (71%), 2c (67%), 2d (69%), and 2e (68%), respectively. The electron-donating substituents like –CH3 and –OCH3 on the phenyl ring of 2-aryl indoles furnished similar products 2g, 2h, and 2i with relatively higher yields (75–82%) than the substrates with electron-withdrawing groups. The product 2f obtained in high yields (81%) when 5-bromo-2-phenyl-1H-indole 1f was employed under standardized conditions. Moreover, densely substituted 2-aryl indole 1j could furnish corresponding cross-dimerized product 2j with moderate (64%) yields, due to the presence of internal steric hindrance. In case of 2-methyl indole as substrate, corresponding dimerized product 2k was also obtained with good yield (71%). The developed protocol was found to be a general w.r.t. the alkyl/aryl substituents at the C2-position of indoles.
We also extended this developed protocol for the cross addition of indoles 3 at the C2-position of 2-arylindole 1, and the results are shown in Table 3. Pleasingly, cross-addition of indoles furnished similar products in good to high yields, in all the cases, under similar reaction conditions. Initially, indole 3a reacted with 2-aryl indoles (1b, 1d, 1e) substituted with electron-withdrawing groups (–F, –Cl, –Br) and furnished corresponding cross-addition products 4ab (67%), 4ad (66%), and 4ae (68%), respectively. Similarly, products 4ag (69%) and 4ak (62%) were obtained, when simple indole 3a reacted with 2-aryl indoles, 1g and 1k substituted with electron donating groups (–CH3, –OH), respectively. Moreover, 5-methoxy indole 3b furnished corresponding cross-addition products 4ba (72%), 4bb (74%), and 4bh (75%) with improved yields, when treated with 2-phenylindole 1a and other substituted 2-aryl indoles (1b and 1h), respectively, in the presence of Cu(OAc)2 (cat.), m-CPBA (1.0 equiv.), and lutidine (2.0 equiv.) as base at room temperature. Interestingly, the cross-addition reaction of indoles is relatively faster as compared to the cross-dimerization. In general, the self-dimerization products yields were found slightly more that than the cross-addition reactions, because in case of cross-addition of indoles we also observed a trace amount (<10%) of self-dimerized products in almost all the cases.
The practical use of this method was also demonstrated to access both cross-dimerization of 2-arylindole and cross-addition of indole with 2-arylindole products on a gram-scale without much variation in yield, as shown in Scheme 2. Pleasingly, the gram-scale reaction successfully afforded 2g with a higher yield (81%) (Scheme 2a) as compared to the small-scale response (Table 2). The single-crystal X-ray diffraction analysis confirmed the structure of cross-dimerized product 2g.17 Moreover, 2,2-disubstituted indole-3-one 4bb was isolated with 78% yield, when cross-addition of 2-substituted indole 1b was performed with 4-methoxy indole 3b, under standardized conditions (Scheme 2b).
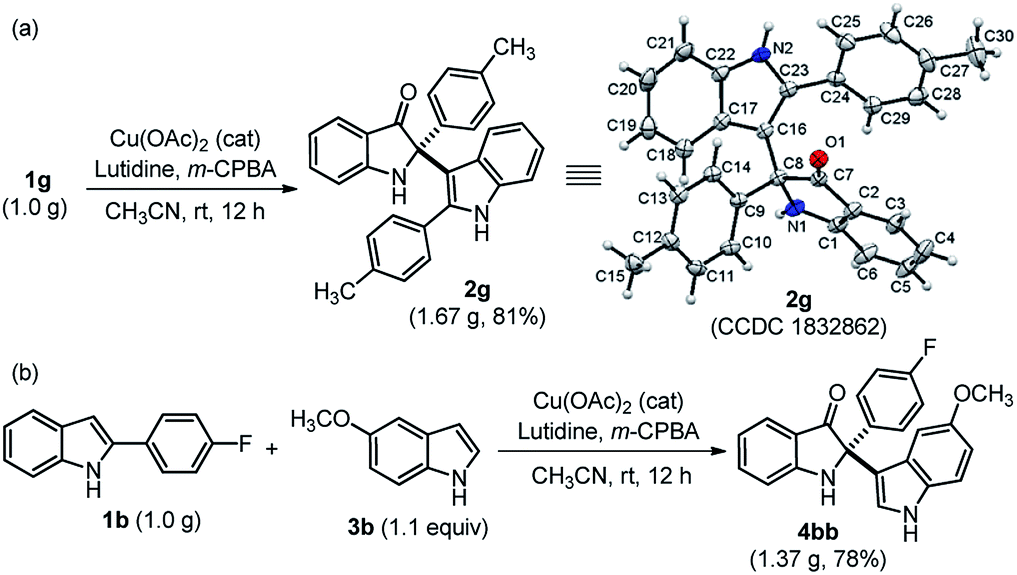 | ||
| Scheme 2 Practical utility at the gram-scale synthesis of 2g and 4bb. Single crystal X-ray structure of 2g (the thermal ellipsoids are drawn at the 50% probability level). | ||
Based on previous reports and our findings, a tentative mechanism for the synthesis of self-dimerized 2-phenyl-2-(2-phenyl-1H-indol-3-yl)indolin-3-ones 2 and cross-addition 2-(1H-indol-3-yl)-2-phenylindole-3-ones 4 is drawn, as shown in Scheme 3. Initially, the copper-catalyzed oxidation of 2-phenylindole 1a to intermediate 2-phenylindole-3-one (A), which was confirmed by in situ HRMS data. This intermediate (A) undergoes a chemoselective C2-addition with another 2-phenylindole 1a to furnished self-dimerization product 2a. Whereas, for the cross-addition product 4, intermediate (A) was trapped with another indole moiety.
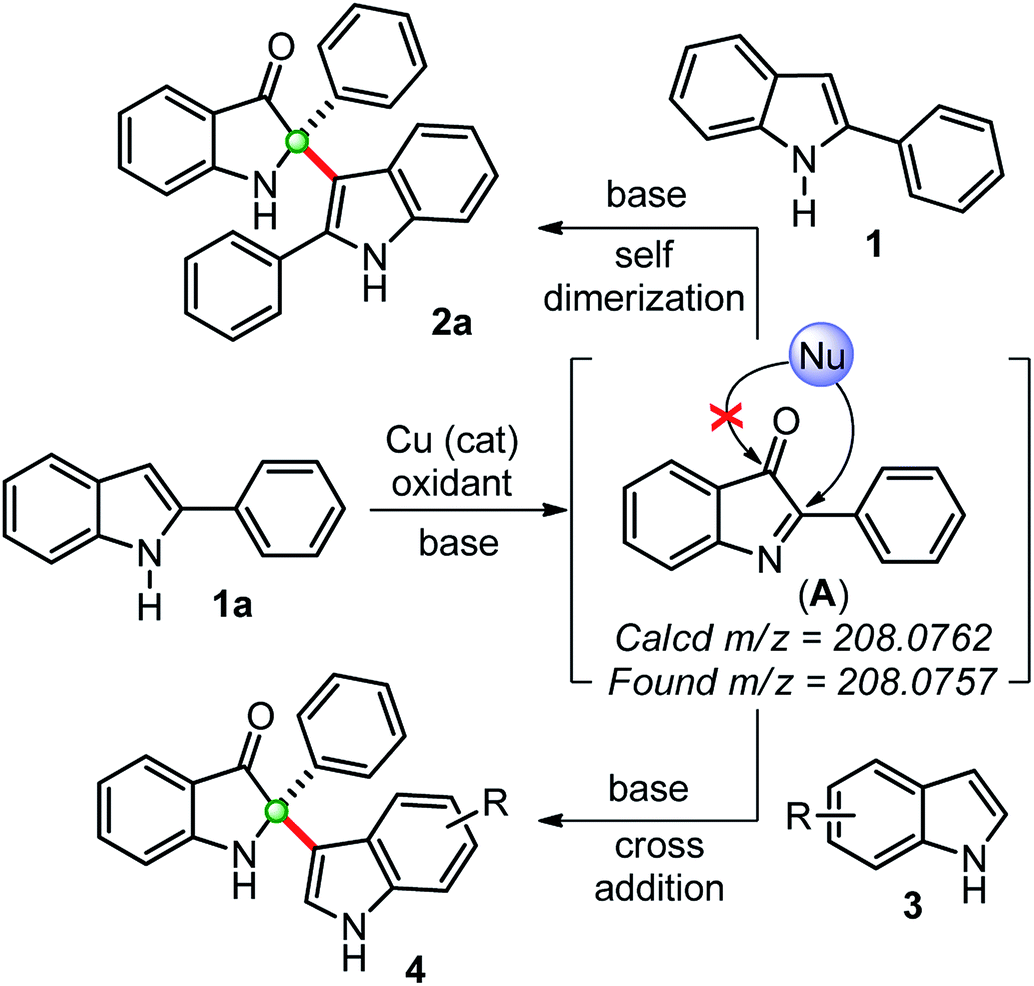 | ||
| Scheme 3 Plausible way of reaction for the synthesis of 2,2-disubstituted indole-2-ones through in situ generations of indole-3-one confirmed by HRMS. | ||
Conclusions
In summary, we have developed an efficient and general protocol for the synthesis of 2,2-disubstituted indolin-3-ones through the self-dimerization of 2-aryl indoles and cross-addition of 2-aryl indoles with indoles. This reaction proceeds with in situ generation of indol-3-one, followed by chemoselective nucleophilic addition under mild copper-catalyzed conditions. This simple strategy provides convenient and either way to access indolin-3-ones bearing C2-quaternary center in good to high yields. The developed protocol utilized nontoxic, readily available materials, and practically viable at the gram-scale synthesis.Experimental
General remarks
All reactions were observed using thin-layer chromatography (on SiO2 gel F254 plates) under standard condition. The desired compounds were purified through flash column chromatography packed with silica gel (100–200 meshes size) as the stationary phase and eluting solvent, hexane–ethyl acetate solvent mixture was used as mobile phase. Melting points were determined in open capillary tubes on an EZ-Melt Automated melting point apparatus and are uncorrected. NMR spectra were recorded on a Bruker AV 400 spectrometer. Chemical shifts were reported in parts per million (ppm) using deuterated solvent or tetramethylsilane (TMS) as an internal standard. High-resolution mass spectra (HRMS-ESI) were recorded using quadrupole time-of-flight (Q-TOF) mass spectrometer (Applied Biosystem). All the chemicals were obtained from the commercial supplier and were used without purification.Typical procedure for the synthesis of oxidative dimerized product 2
To a stirred solution of 2-phenylindole 1 (0.5 mmol) in CH3CN (3.0 mL) was added lutidine (1.0 mmol), Cu(OAc)2 (30 mol%) and m-CPBA (meta-chloroperoxybenzoic acid, 0.3 mmol) successively at room temperature. The combined reaction mixture was stirred at the same temperature until TLC confirmed the complete consumption of starting material. Subsequently, the reaction was quenched with H2O (3.0 mL) and stirred with EtOAc (10 mL). The organic layer was separated, and the aqueous layer was again extracted with EtOAc (5.0 mL). The combined organic extracts were washed with brine, dried over Na2SO4 anhydrous, and concentrated under reduced pressure. Column chromatography purification through silica gel by eluting the mixture of hexane/EtOAc gave corresponding dimerized product 2 as mainly yellow solid with 64–82% yields.Typical procedure for the synthesis of (4)
To a stirred solution of 2-phenylindole 1 (0.5 mmol) in CH3CN (3.0 mL) was added lutidine (1.0 mmol), Cu(OAc)2 (30 mol%) and m-CPBA (0.5 mmol) successively, followed by substituted indole 3 (1.1 equiv.) at room temperature and progress of the reaction was monitored by TLC. Once the reaction was over, it was quenched with water (3.0 mL) and stirred with EtOAc (10 mL). The organic layer was separated, and the aqueous layer was again extracted with EtOAc (5.0 mL). The combined organic extracts were washed with brine and dried over anhydrous Na2SO4, followed by concentrated in the vacuum after filtration. Purification through silica-gel column chromatography by eluting the mixture of hexane/EtOAc gave corresponding product 4 as mainly yellow solid with 62–75% yields.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by an OPERA-grant of BITS Pilani and DST-SERB (EMR/2016/005599). SV thanks to SERB-NPDF grant (PDF/2017/001791) for Research Fellowship. The authors are also grateful for generous support from the DST-FIST (SR/FST/CSI-270/2015) for HRMS facility to the Department of Chemistry at BITS-Pilani.Notes and references
- (a) J. H. Lee, J.-H. So, J. H. Jeon, E. B. Choi, Y.-R. Lee, Y.-T. Chang, C.-H. Kim, M. A. Bae and J. H. Ahn, Chem. Commun., 2011, 47, 7500 RSC; (b) A. Parra, R. Alfaro, L. Marzo, A. Moreno-Carrasco, J. L. García Ruano and J. Alemán, Chem. Commun., 2012, 48, 9759 RSC; (c) L. Kong, M. Wang, F. Zhang, M. Xu and Y. Li, Org. Lett., 2016, 18, 6124 CrossRef CAS PubMed; (d) Y.-J. Li, N. Yan, C.-H. Liu, Y. Yu and Y.-L. Zhao, Org. Lett., 2017, 19, 1160 CrossRef CAS PubMed.
- (a) J.-F. Liu, Z.-Y. Jiang, R.-R. Wang, Y.-T. Zeng, J.-J. Chen, X.-M. Zhang and Y.-B. Ma, Org. Lett., 2007, 9, 4127 CrossRef CAS PubMed; (b) C.-J. Tan, Y.-T. Di, Y.-H. Wang, Y. Zhang, Y.-K. Si, Q. Zhang, S. Gao, X.-J. Hu, X. Fang, S.-F. Li and X.-J. Hao, Org. Lett., 2010, 12, 2370 CrossRef CAS PubMed; (c) J. Lee and J. S. Panek, Org. Lett., 2011, 13, 502 CrossRef CAS PubMed; (d) C. V. S. Kumar, V. G. Puranik and C. V. Ramana, Chem.–Eur. J., 2012, 18, 9601 CrossRef CAS PubMed; (e) T. Abe, A. Kukita, K. Akiyama, T. Naito and D. Uemura, Chem. Lett., 2012, 41, 728 CrossRef CAS; (f) W. Gu, Y. Zhang, X.-J. Hao, F. M. Yang, Q.-Y. Sun, S. L. Morris-Natschke, K.-H. Lee, Y.-H. Wang and C.-L. Long, J. Nat. Prod., 2014, 77, 2590 CrossRef CAS PubMed.
- (a) P. N. Wyrembak and A. D. Hamilton, J. Am. Chem. Soc., 2009, 131, 4566 CrossRef CAS PubMed; (b) S. Matsumoto, D. Samata, M. Akazome and K. Ogura, Tetrahedron Lett., 2009, 50, 111 CrossRef CAS; (c) Y. Goriya and C. V. Ramana, Chem. Commun., 2013, 49, 6376 RSC.
- (a) A. Wetzel and F. Gagosz, Angew. Chem., Int. Ed., 2011, 50, 7354 CrossRef CAS PubMed; (b) P. Patel and C. V. Ramana, Org. Biomol. Chem., 2011, 9, 7327 RSC; (c) P. X. Zhou, Z. Z. Zhou, Z. S. Chen, Y. Y. Ye, L. B. Zhao, Y. F. Yang, X. F. Xia, J. Y. Luo and Y. M. Liang, Chem. Commun., 2013, 49, 561 RSC; (d) S. R. Mothe, M. L. Novianti, B. J. Ayers and P. W. H. Chan, Org. Lett., 2014, 16, 4110 CrossRef CAS PubMed; (e) N. Li, T. Y. Wang, L. Z. Gong and L. Zhang, Chem.–Eur. J., 2015, 21, 3585 CrossRef CAS PubMed; (f) T. Zhang, Z. Qi, X. Zhang, L. Wu and X. Li, Chem.–Eur. J., 2014, 20, 3283 CrossRef CAS PubMed; (g) R. Kuppusamy, P. Gandeepan and C. H. Cheng, Org. Lett., 2015, 17, 3846 CrossRef CAS PubMed; (h) R. R. Liu, S. C. Ye, C. J. Lu, G. L. Zhuang, J. R. Gao and Y. X. Jia, Angew. Chem., Int. Ed., 2015, 54, 11205 CrossRef CAS PubMed; (i) N. Marien, T. Luo and G. Verniest, Synlett, 2017, 28, 934 CrossRef CAS; (j) X. X. Zhang, P. Li, C. Lyu, W. X. Yong, J. Li, X. Y. Pan, X. B. Zhu and W. D. Rao, Adv. Synth. Catal., 2017, 359, 4147 CrossRef CAS; (k) W. Fu and Q. Song, Org. Lett., 2018, 20, 393 CrossRef CAS PubMed.
- Z. Xia, J. Hu, Y. Q. Gao, Q. Yao and W. Xie, Chem. Commun., 2017, 53, 7485 RSC.
- (a) S. Lerch, L.-N. Unkel and M. Brasholz, Angew. Chem., Int. Ed., 2014, 53, 6558 CrossRef CAS PubMed; (b) W. Ding, Q.-Q. Zhou, J. Xuan, T.-R. Li, L.-Q. Lu and W.-J. Xiao, Tetrahedron Lett., 2014, 55, 4648 CrossRef CAS; (c) L. Bu, J. Li, Y. Yin, B. Qiao, G. Chai, X. Zhao and Z. Jiang, Chem.–Asian J., 2018, 13, 2382 CrossRef CAS PubMed.
- (a) Y. Liu and W. W. Jr McWhorter, J. Org. Chem., 2003, 68, 2618 CrossRef CAS PubMed; (b) K. Higuchi, Y. Sato, S. Kojima, M. Tsuchimochi, K. Sugiura, M. Hatori and T. Kawasaki, Tetrahedron, 2010, 66, 1236 CrossRef CAS.
- (a) L.-Q. Li, M.-Y. Han, M.-X. Xiao and Z.-X. Xie, Synlett, 2011, 1727 CAS; (b) M. Rueping, S. Raja and A. Nuñez, Adv. Synth. Catal., 2011, 353, 563 CrossRef CAS; (c) Q. Yin and S.-L. You, Chem. Sci., 2011, 2, 1344 RSC; (d) M. Rueping, R. Rasappan and S. Raja, Helv. Chim. Acta, 2012, 95, 2296 CrossRef CAS; (e) S. Nakamura, N. Matsuda and M. Ohara, Chem.–Eur. J., 2016, 22, 9478 CrossRef CAS PubMed; (f) J.-S. Li, Y.-J. Liu, S. Li and J.-A. Ma, Chem. Commun., 2018, 54, 9151 RSC.
- (a) K.-Q. Ling, Synth. Commun., 1995, 25, 3831 CrossRef CAS; (b) Y. Liu and W. W. Jr McWhorter, J. Am. Chem. Soc., 2003, 125, 4240 CrossRef CAS PubMed; (c) Q. Liu, P. Chen and G. Liu, ACS Catal., 2013, 3, 178 CrossRef CAS; (d) E. Najahi, A. Valentin, P.-L. Fabre, K. Reybier and F. Nepveu, Eur. J. Med. Chem., 2014, 78, 269 CrossRef CAS PubMed.
- (a) P. Sang, Y. Xie, J. Zou and Y. Zhang, Adv. Synth. Catal., 2012, 354, 1873 CrossRef CAS; (b) Y.-B. Kong, J. Y. Zhu, Z.-W. Chen and L.-X. Liu, Can. J. Chem., 2014, 92, 269 CrossRef CAS.
- (a) F. Lin, Y. Chen, B. Wang, W. Qin and L. Liu, RSC Adv., 2015, 5, 37018 RSC; (b) X.-Y. Zhou, X. Chen, L.-G. Wang, D. Yang and J.-H. Li, Synlett, 2018, 835 CrossRef CAS.
- S. K. Guchhait, V. Chaudhary, V. A. Rana, G. Priyadarshani, S. Kandekar and M. Kashyap, Org. Lett., 2016, 18, 1534 CrossRef CAS PubMed.
- X. Jiang, B. Zhu, K. Lin, G. Wang, W.-K. Su and C. Yu, Org. Biomol. Chem., 2019, 17, 2199 RSC.
- For review, see: (a) W. A. Wendlandt, A. M. Suess and S. S. Stahl, Angew. Chem., Int. Ed., 2011, 50, 11062 CrossRef PubMed; (b) Y. Liu and J.-P. Wan, Org. Biomol. Chem., 2011, 9, 6873 RSC; (c) S. E. Allen, R. R. Walvoord, R. Padilla-Salinas and M. C. Kozlowski, Chem. Rev., 2013, 113, 6234 CrossRef CAS PubMed.
- M. Yamashita and A. Iida, Tetrahedron Lett., 2014, 55, 2991 CrossRef CAS.
- M. Yamashita, Y. Nishizono, S. Himekawa and A. Iida, Tetrahedron, 2016, 72, 4123 CrossRef CAS.
- The X-ray crystallographic structure for 2g has been deposited at the Cambridge Crystallographic Data Centre (CCDC), under deposition numbers CCDC 1832862.†.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1832862. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra04741g |
| ‡ Authors have contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |