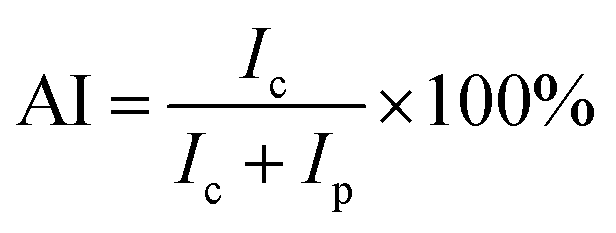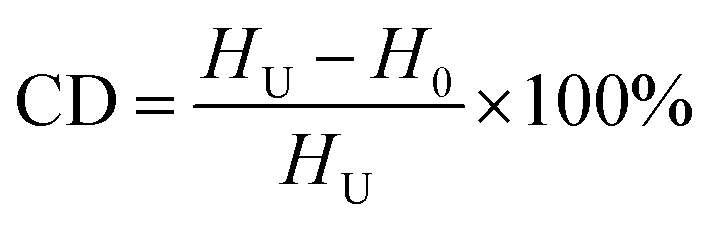DOI:
10.1039/C9RA04732H
(Paper)
RSC Adv., 2019,
9, 28146-28152
Effect of graphene oxide coatings on the structure of polyacrylonitrile fibers during pre-oxidation process†
Received
24th June 2019
, Accepted 19th August 2019
First published on 6th September 2019
Abstract
In this paper, graphene oxide (GO) was successfully prepared by the modified Hummers’ method and then uniformly dispersed in an aqueous solution containing a small amount of polyvinyl alcohol (PVA) as an adhesive. The solution was uniformly coated on the surface of polyacrylonitrile (PAN) fibers and then the fibers were pre-oxidized at 240 °C for 20 min in the air. The pre-oxidation degree of PAN fibers and fibers coated with different contents of GO was analyzed by the Fourier transform infrared (FT-IR) spectroscopy, differential scanning calorimetry (DSC), X-ray diffraction (XRD) and thermogravimetric analysis (TGA). In addition, the surface and cross-section of PAN fibers before and after pre-oxidation were observed by scanning electron microscopy (SEM). The experimental results showed that the presence of GO coatings significantly improved the pre-oxidation degree of PAN fibers, at the same time, the pre-oxidation degree of PAN fibers increased with the increase of GO contents from 0.2 to 1.0 mg ml−1. The cross-section morphology of the pre-oxidized PAN fibers revealed that the degree of pre-oxidation inside fibers was uniform. This was because the GO coatings acted as a medium to transfer heat, removing the heat released during the pre-oxidation process and increasing the pre-oxidation degree.
1. Introduction
Composite materials refer to a new type of material composed of at least two materials such as organic polymer, inorganic non-metal, metal and other materials through the composite processes including the winding, compression molding, autoclave molding and other processes.1–4 They retain the main characteristics of the original component materials and can be processed through structural design. Among the composite materials, carbon fiber reinforced composites are indispensable strategic materials widely used in aerospace, military, automotive, civil aircraft, construction, bridge building, sports goods manufacture and other fields5–7 because of their light weight and high strength.8 Carbon fiber is a microcrystalline graphite material obtained by carbonizing and graphitizing the organic fibers, the most important of which are polyacrylonitrile (PAN), pitch and rayon.9–11 High-performance carbon fibers made from the PAN precursors possess a relatively simple production process, and the output of PAN-based carbon fibers accounts for more than 90% of the total global carbon fiber production.
The preparation process of PAN-based carbon fibers mainly includes four processes, which are the preparation of raw silk, pre-oxidation, low-temperature carbonization and high-temperature carbonization. The pre-oxidation process occurs within a certain temperature range; however, it will release a lot of heat, and the fusing phenomenon will occur when the temperature is too high. The melting phenomenon of PAN fibers during the pre-oxidation process will greatly reduce the production efficiency and increase the cost of carbon fibers. Therefore, the entire pre-oxidation process requires strict control of the temperature and heat released from the fibers, indicating that the pre-oxidation process is time-consuming, energy-consuming and hard to control, thereby increasing the manufacturing cost of carbon fibers. Many experts have used various methods to reduce the manufacturing cost of carbon fibers by increasing the pre-oxidation rate, such as by gamma ray irradiation, microwave plasma assistance and other methods.12
Graphene oxide (GO) is generally acquired by oxidation of graphite through the methods such as Brodie method,13 modified Staudenmaier method14 and Hummers’ method.15 Among them, the Hummers’ method is the most widely used one owing to the good timeliness and the safe preparation process. The GO can be well dispersed in the water due to the abundant functional groups on its surface. The GO as one kind of carbon materials has a relatively high thermal conductivity. Nowadays, many researchers dispersed the GO in the water as a heat exchange medium to increase the heat transfer coefficient of the fluid. Milad and Ramin et al. studied the effect of the nano-fluids on the thermal conductivity with different concentrations of GO, showing that the thermal conductivities of the nano-fluids increased by 56% and 42.2%, respectively.16,17
In order to solve the problem that the PAN fibers are easy to fuse during cyclization owing to the excessive heat release, the GO with large thermal conductivity is applied to transfer the heat during pre-oxidation process. In this article, we intend to study the effect of the GO coatings on the pre-oxidation process of PAN fibers. The GO is used to quickly remove the heat released during the pre-oxidation process, thereby reducing the melting phenomenon and accelerating the pre-oxidation process.
2. Materials and methods
2.1. Materials
PAN fibers with 24![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 filaments per fiber bundle were wet-spun from a co-polymer of acrylonitrile/acrylamide/methyl acrylate and supplied by Weihai Fiber Development Co., Ltd. (Weihai, China). The PVA with the Mw of 8000 was acquired from Sinopharm Chemical Reagent Beijing Co., Ltd., China. The GO was prepared from the graphite powder (99.5%, Jiangsu Legend Technology Co., Ltd., China) by the modified Hummers’ method.18
000 filaments per fiber bundle were wet-spun from a co-polymer of acrylonitrile/acrylamide/methyl acrylate and supplied by Weihai Fiber Development Co., Ltd. (Weihai, China). The PVA with the Mw of 8000 was acquired from Sinopharm Chemical Reagent Beijing Co., Ltd., China. The GO was prepared from the graphite powder (99.5%, Jiangsu Legend Technology Co., Ltd., China) by the modified Hummers’ method.18
2.2. Preparation of the GO/PVA solution
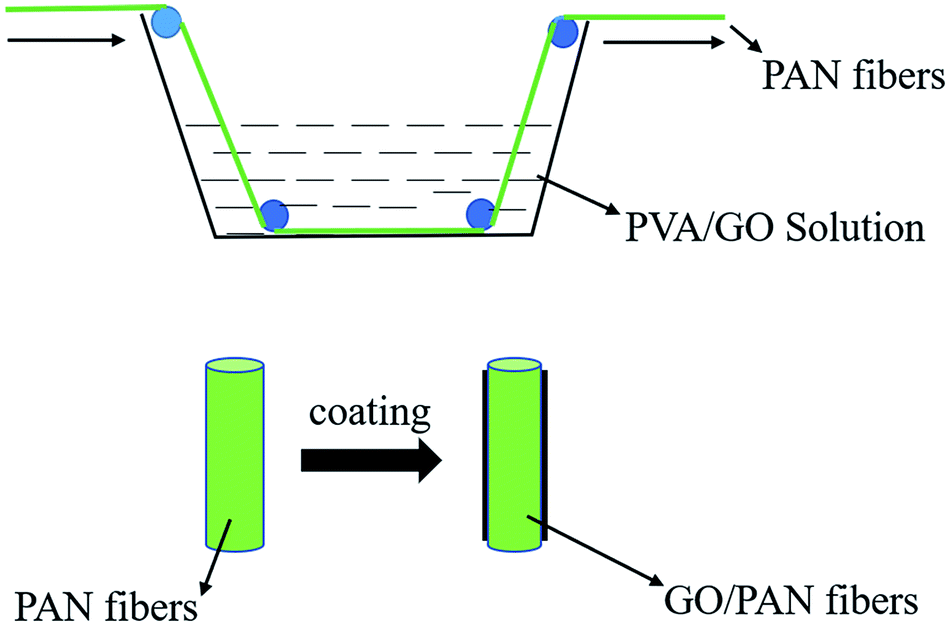
The PVA was selected as an adhesive in consideration of the adhesion effect of the GO coatings on fiber surface. However, the monofilaments were bonded to each other to affect the heat dissipation when the PVA content was too high. So, an experiment was carried out to determine the content of PVA in advance. 0.07, 0.09, 0.11 and 0.13 g of PVA were added to 30 ml of deionized water, respectively, followed by stirring the solution in an oil bath at 95 °C for 5 h until completely dissolved, and then the solution was coated on the surface of PAN fibers. It was found that there were no differences of the morphologies between the pure PAN fibers and fibers coated with 2.3 mg ml−1 of the PVA aqueous solution after drying in an oven at 110 °C for 1 h. So, the 0.07 g of PVA was added to 15 ml of deionized water and followed by magnetically stirring in an oil bath at 95 °C until completely dissolved. The 6, 12, 18, 24 and 30 mg of the homemade GO were added to 15 ml of deionized water and ultrasonic treatment for 1 h, respectively. The completely dissolved PVA solution and the uniformly dispersed GO solution were mixed and ultrasonicated for 2 h to obtain a well-distributed PVA/GO solution. At the same time, the PVA solution was prepared with 0.07 g of PVA and 30 ml of deionized water as a comparison. The preparation process of GO/PVA solution was shown in Fig. 1.
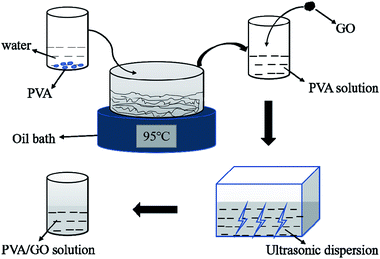 |
| | Fig. 1 Preparation of the GO/PVA solution. | |
2.3. Preparation of PAN fibers coated with the GO/PVA solution
The PAN fibers were immersed in the uniformly dispersed GO/PVA solution with different contents of GO until evenly coated, after this, the fibers were dried in an oven at 100 °C for 2 h to obtain the PAN fibers coated with GO, named GO/PAN fibers. Similarly, the PAN fibers with PVA named PAN fibers were also prepared. The coating process of PAN fibers was shown in Fig. 2.
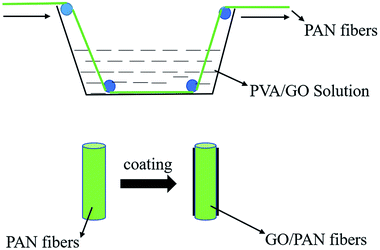 |
| | Fig. 2 Preparation process of PAN fibers coated with GO/PVA and PVA solutions. | |
2.4. Pre-oxidation of the PAN fibers
The dried PAN fibers and GO/PAN fibers with different contents of GO were pre-oxidized in a pre-oxidation oven at 240 °C for 20 min in the air, respectively.
2.5. Characterizations
FT-IR characterization of the pre-oxidized PAN fibers was recorded on a Nicolet 6700 FT-IR spectrophotometer using the method of KBr troches with a scanning wavenumber range of 4000–400 cm−1. The data was analyzed by OMNIC 8.2 (USA, Thermo Fisher Nicolet company) and all the IR spectra in our paper were normalized during the analysis. The relative cyclization rate (RCR) calculations were performed based on the normalized IR spectra according to formula (1):| |
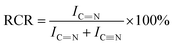 | (1) |
where IC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N: transmission peak intensity of CN; IC
N: transmission peak intensity of CN; IC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N: transmission peak intensity of CN.
N: transmission peak intensity of CN.
X-ray diffraction (XRD) studies of the pre-oxidized PAN fibers were carried out on a diffractometer (D/max 2550 VB, Bruker Co., Japan). The fibers were cut into powders before the test. The range of the scan angle was from 5 to 60° in a step width of 0.02. The aromatic index (AI) of the fibers was calculated according to formula (2):
| |
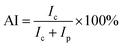 | (2) |
where
Ic: diffraction intensity of PAN fibers at 2
θ = 29°;
Ip: diffraction intensity of PAN fibers at 2
θ = 16.5°.
The thermal behaviors of the pre-oxidation PAN fibers were studied by differential scanning calorimeter (DSC, Q100, TA instrument) with a heating rate of 10 °C min−1 within the temperature range from 30 to 400 °C in the nitrogen. The cyclization degree (CD) of the PAN fibers was calculated according to formula (3):
| |
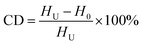 | (3) |
where
HU: heat release from the pure PAN fibers;
H0: heat release from the pre-oxidized PAN fibers.
The thermal stability of the PAN fibers was investigated by a thermogravimetric analysis (TGA, TG 209F1, Netzsh, Germany) instrument. The curves were recorded with a ramp of 10 °C min−1 from room temperature to 900 °C under a working N2 flux equal to 50 ml min−1 during the test.
The changes of the density of PAN fibers were measured by a liquid weight balance (PZ-B-5, China) using the floatation method. The dried fibers were cut into 0.5 mm or less and then placed in the formulated equilibrium solution which was a mixture of carbon tetrachloride and N-heptane. The fibers should be suspended for more than 4 h in the mixture by adjusting the proportions of the heavy liquid and light liquid. The density of the fibers was acquired according to the density of the solution measured by the liquid weight balance.
The skin-core structures of the pre-oxidation PAN fibers were observed using a scanning electron microscope (SEM, TM-1000, Hitachi). The samples were coated with gold using a sputter coater (Desk-II; Denton Vacuum) in advance and then placed on a table in the chamber for observation.
3. Results and discussion
3.1. FT-IR analysis of the PAN fibers with different GO contents
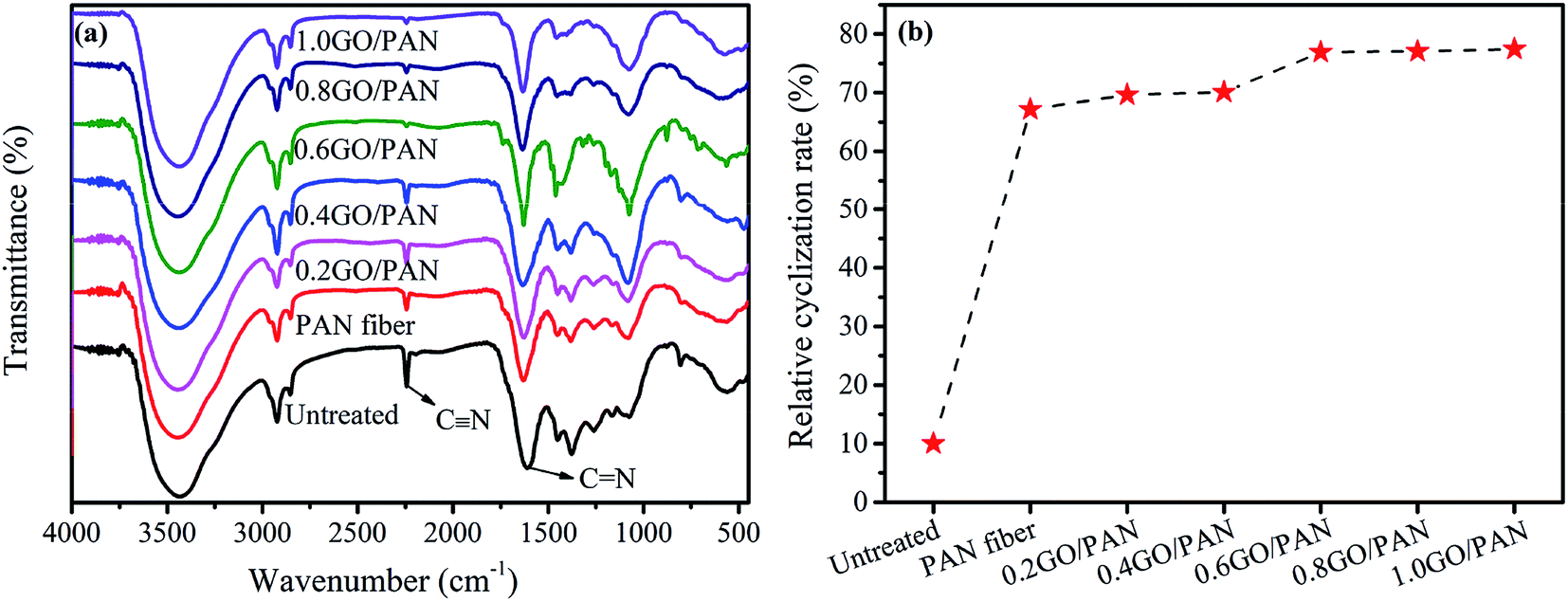
As we all know that the changes in the concentration and shrinkage of the nitrile group can be used to analyze the process of pre-oxidation reaction. The changes of the infrared spectra of PAN fibers coated with different contents of GO are given in Fig. 3. From the FT-IR test results, the changes of the transmission peak intensity of PAN fibers during the pre-oxidation process can be obtained. The following bands in PAN fibers are identified: the –CN stretching of acrylonitrile unit at 2240 cm−1, the double bond stretching vibration zone at 1690–1500 cm−1, the C–H stretching vibration zone at 1475–1000 cm−1. The changes of the bands at 1600 and 2240 cm−1 are significantly important during the pre-oxidation process. Compared with the untreated PAN fibers, the stretching vibration peak at 2240 cm−1 of pre-oxidation PAN fibers is more shaper while the band at 1600 cm−1 becomes weaker. These changes indicate that the cyclization reaction mainly occurs among the CN bonds in PAN fibers, resulting in the generation of CN bonds. Comparing the FT-IR spectra of PAN fibers with different GO contents, the peak intensity of CN gradually decreases with the increase of GO content, while the CN bond increases gradually, indicating an increase of the degree of cyclization. The pre-oxidation process of PAN fibers is an exothermic process, and a large amount of heat causes the fibers to melt, so it is necessary to quickly remove the heat released during the reaction. As one kind of carbon materials, the GO with high heat conduction can be used in the pre-oxidation process to carry the heat released during the reaction away. Therefore, the GO on the surface of PAN fibers can quickly remove the heat released during the cyclization process, thereby accelerating the reaction process of pre-oxidation, increasing the degree of cyclization and improving the reaction efficiency.
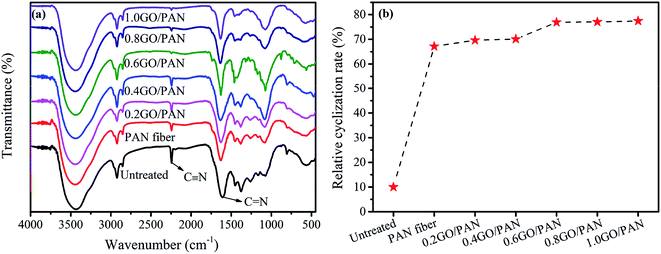 |
| | Fig. 3 FT-IR spectra (a) and RCR (b) of the untreated PAN fibers and PAN fibers with different contents of GO. | |
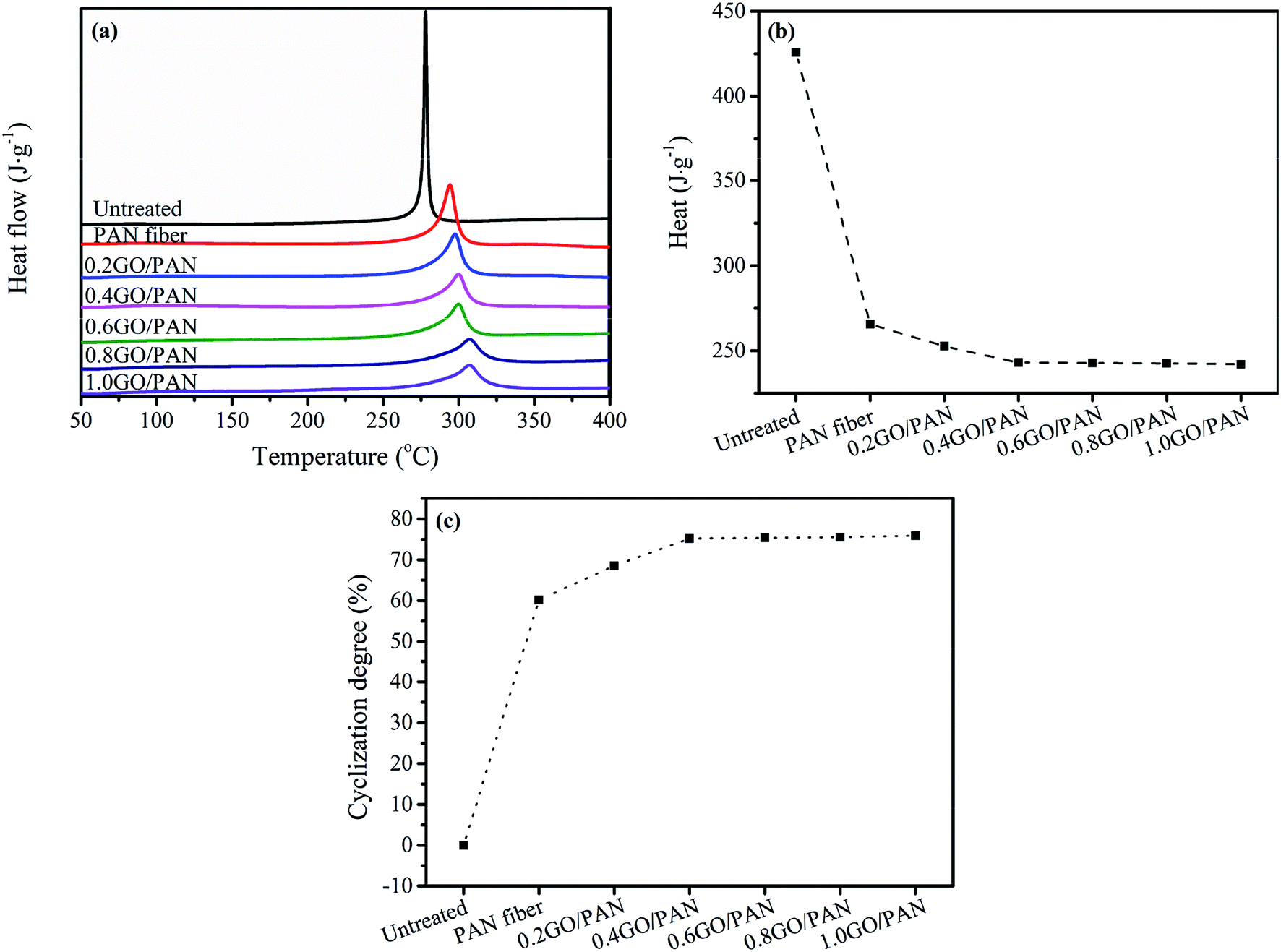
3.2. DSC analysis of the PAN fibers with different GO contents
Fig. 4a shows the DSC curves of the untreated PAN fibers and PAN fibers with different GO contents. It is well known that the pre-oxidized PAN fibers form a complex ladder network structure, which is an anti-decomposition structure without releasing heat during the heating process. Since the cyclization reaction of PAN fibers is an exothermic process, the influence of different GO contents on the degree of cyclization can be analyzed according to the exotherm during the heating process. Therefore, the degree of pre-oxidation of PAN fibers can be judged according to the difference in the released heat of the fibers during heating. It can be seen from Fig. 4b that as the GO content increases, the heat released by PAN fibers gradually decreases, indicating that the cyclized structure of PAN fibers increases and the degree of cyclization increases shown in Fig. 4c.
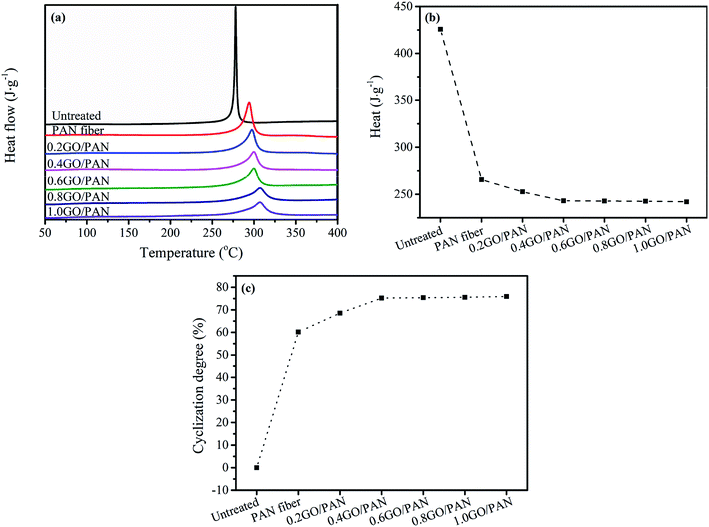 |
| | Fig. 4 DSC curves (a), heat release (b) and (c) CD of the untreated PAN fibers and PAN fibers with different contents of GO. | |
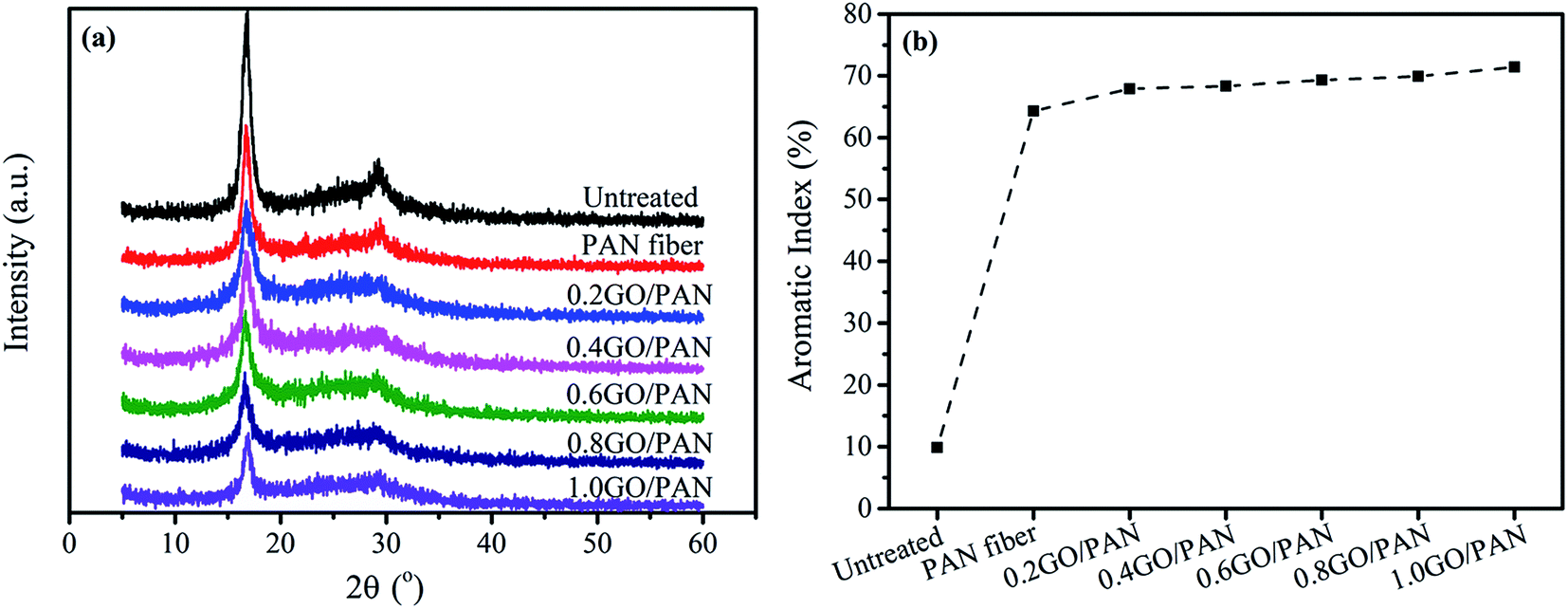
3.3. XRD analysis of the PAN fibers with different contents of GO
Fig. 5a and b show the XRD patterns and aromatic index of the untreated and PAN fibers with different contents of GO, respectively. There are two typical diffraction peaks at 2θ = 16.5° and 2θ = 29.0° in XRD patterns of PAN fibers, reflecting the ordered crystal structure in PAN linear macromolecules, as shown in Fig. 5a. The strong diffraction peak at 2θ = 16.5° corresponding to the (100) crystal plane reflects the spacing among the molecular chains. At the same time, the other weak diffraction peak at 2θ = 29.0° corresponding to the (110) crystal plane is existed, which reflects the distance between the near parallel molecular pieces. In addition, there is a wide diffuse reflection region between the two diffraction peaks of 2θ = 16.5° and 29.0°, indicating that the disordered phase is distributed throughout the structure in PAN fibers. The crystal structure is destroyed during the pre-oxidation reaction due to the formation of the cross-linked ladder structures in PAN fibers, leading to a decrease in the crystallinity of PAN fibers. From the XRD spectra, we can get that the intensity of the crystallization peak of treated PAN fibers is lower than that of the untreated after pre-oxidation, indicating that the treated PAN fibers have a certain degree of cyclization and cyclization structure to some extent. At the same time, with the increase of GO content, the crystallinity of the treated PAN fibers decreases, indicating that the degree of cyclization is gradually increasing, as depicted in Fig. 5b. Overall, the presence of GO can promote the pre-oxidation process of PAN fibers.
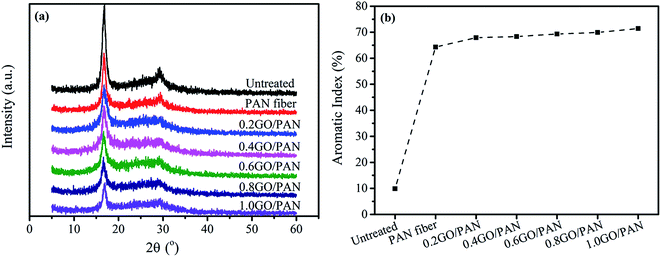 |
| | Fig. 5 XRD patterns (a) and AI (b) of the untreated PAN fibers and PAN fibers with different content of GO. | |
3.4. TGA and DTG analysis of the PAN fibers with different contents of GO
During the pre-oxidation reaction, the molecular chains of PAN fibers are cyclized and crosslinked to form a decomposition-resistant body-shaped cyclic structure. Therefore, the decomposition process and the residual quality of fibers with different degrees of pre-oxidation are different with the increasing temperature due to the differences of the contents of the decomposition-resistant body-shaped annular structure. We can evaluate the pre-oxidation degree of the pre-oxidized PAN fibers according to the decomposition and the residual quality during the heating process. Fig. 6 gives the TGA and DTG curves of PAN fibers with different contents of GO, from which we can get that the PAN fibers have three main decomposition intervals. The mass loss of PAN fibers at the first interval from room temperature to 150 °C is due to the evaporation of moisture in the fibers. The quality of PAN fibers changes drastically in the second decomposition interval from 275 to 600 °C, which is a severe decomposition interval. The internal structure and quality loss of PAN fibers have small changes during the third zone from 600 to 900 °C. Comparing the different thermal decomposition curves, the residual qualities (Table 1) and DTG curves, it can be concluded that the residual quality of PAN fibers is significantly increased after pre-oxidation, and the thermal decomposition process is relatively moderate. Comparing the effects of different contents of GO on the thermal decomposition process, it can be concluded that the addition of GO can increase the residual quality of pre-oxidized fibers, slow down the decomposition process and increase the cyclization process of PAN fibers. These circumstances all indicate that the degree of cyclization of PAN fibers increases as the increase of GO contents.
Table 1 Initial decomposition temperature and residual mass of the untreated and PAN fibers with different contents of GO
| Sample |
Initial decomposition temperature |
Residual quality (%) |
| Untreated |
267.47 |
35.30 |
| PAN fiber |
280.75 |
43.29 |
| 0.2 GO/PAN |
289.39 |
47.95 |
| 0.4 GO/PAN |
290.94 |
50.36 |
| 0.6 GO/PAN |
294.06 |
50.90 |
| 0.8 GO/PAN |
301.91 |
56.80 |
| 1.0 GO/PAN |
302.80 |
57.74 |
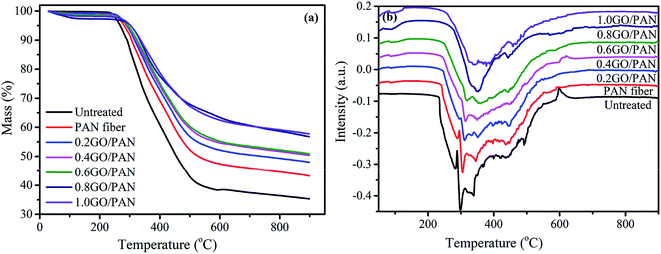 |
| | Fig. 6 The TGA (a) and DTG (b) curves of the untreated and PAN fibers with different contents of GO. | |
3.5. The density of the PAN fibers with different contents of GO
The PAN fibers undergo cyclization, dehydrogenation, oxidation and cross-linking reactions during the pre-oxidation process. During the reaction, the molecular chains cross-link to form a trapezoidal structure as well as the release of the lighter element of H and the addition of the higher mass element of O, which will increase the density of the fibers. Therefore, we can judge the degree of pre-oxidation of the fibers based on the density of the fibers. The density test results in Table 2 show that the density of PAN fibers increases after pre-oxidation, and the density of fibers increases with the increase of GO content, indicating that the internal cyclization structure of the fibers is increasing, which are consistent with the FT-IR, DSC, XRD, and TG results.
Table 2 The density changes of PAN fibers
| Sample |
Density (g cm−3) |
| Untreated |
1.171 |
| PAN fiber |
1.298 |
| 0.2 GO/PAN |
1.311 |
| 0.4 GO/PAN |
1.319 |
| 0.6 GO/PAN |
1.326 |
| 0.8 GO/PAN |
1.329 |
| 1.0 GO/PAN |
1.334 |
3.6. SEM images of the PAN fibers with different contents of GO
The PAN fibers will produce a “sheath core structure” in the radial direction during the pre-oxidation process, which leads to the generation of internal defects of the fibers and affects the final mechanical properties of the PAN-based carbon fibers. More importantly, the cyclization reaction of cyano group is a first-order reaction, and the temperature effect has a decisive influence on the reaction rate. Therefore, it is especially important to control the temperature during the reaction process to reduce the generation of the sheath-core structure. Comparing the different fiber surfaces shown in Fig. 7a–d, it can be seen that the gully on the surface of pre-oxidized PAN fibers coated with GO is significantly shallower than that of the uncoated, which may be due to the presence of the GO coatings. It can be seen from the cross-sectional images shown in Fig. 7e and f that the pre-oxidized PAN fibers have substantially no core-sheath structure after coating with GO, and the degree of pre-oxidation of the entire cross-section of fibers is very uniform. This may be due to the fact that the GO can carry the heat away quickly, so that the fibers will not generate defects owing to local overheating, which can improve the cyclization uniformity of the pre-oxidized fibers.
 |
| | Fig. 7 SEM images of PAN fibers, (a) untreated PAN fibers; (b) pre-oxidated PAN fibers coated with PVA; (c) and (d) pre-oxidated PAN fibers coated with GO/PVA; (e) cross-section of pre-oxidated PAN fibers coated with PVA; (f) cross-section of pre-oxidated PAN fibers coated with GO/PVA. | |
4. Conclusions
In this paper, the GO was successfully prepared by the modified Hummers’ method and uniformly dispersed in an aqueous solution containing a small amount of PVA in advance. The solution was uniformly coated on the surface of the PAN fibers and then pre-oxidized at 240 °C for 20 min in the air. The results showed that the pre-oxidation degree of PAN fibers coated with GO was higher than that of the uncoated fibers, and the degree of pre-oxidation increases with the increase of GO content. The FT-IR results showed that the degree of cyclization of PAN fibers increased from 67.13% to 77.45% when coated with 1.0 mg ml−1 of the GO, and this change trend was the same as the results of the XRD, DSC, TG and density tests. Meanwhile, the TG results indicated that the PAN fibers coated with GO had a higher residual quality and had a higher cyclization structure.
Author contributions
M. Q. performed experiments and wrote the paper; X. D. and Z. L. analyzed the data and collected the resources; H. K. and M. Y. designed the experiments and revised the paper.
Funding
This work was financially supported by the State Key Laboratory for Modification of Chemical Fibers and Polymer Materials, Donghua University (LK1602).
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
The authors would like to express sincere thanks to State Key Laboratory for Modification of Chemical Fibers and Polymer Materials.
Notes and references
- F. Henninger and K. Friedrich, Composites, Part A, 2002, 33, 1479–1486 CrossRef.
- M. K. Kang and W. I. Lee, Polym. Compos., 1999, 20, 293–304 CrossRef CAS.
- A. K. Bledzki and O. Faruk, Polym.-Plast. Technol. Eng., 2004, 43, 871–888 CrossRef CAS.
- S. W. Kim, K. J. Lee, J. C. Seferis and J. D. Nam, Adv. Polym. Technol., 1997, 16, 185–197 CrossRef CAS.
- L. Sun and Z. Deng, in Material and Manufacturing Technology II, Pts 1 and 2, 2012, vol. 341–342, pp. 173–176 Search PubMed.
- W. Feifei, Mater. Sci. Forum, 2017, 893, 31–34 Search PubMed.
- N. Li, Y. Li, J. Jelonnek, G. Link and J. Gao, Composites, Part B, 2017, 122, 61–70 CrossRef CAS.
- J.-H. Lu and J. P. Youngblood, Composites, Part B, 2015, 82, 221–225 CrossRef CAS.
- H. Zhang, G. Liang, A. Gu and L. Yuan, Ind. Eng. Chem. Res., 2014, 53, 2684–2696 CrossRef CAS.
- X. F. Zhao, Y. F. Wen, T. Qi, S. Q. Lai and L. J. Gao, Adv. Mater. Res., 2014, 3326, 194–198 Search PubMed.
- Y. G. Ko, U. S. Choi, D. J. Ahn, J. S. Kim and T. Y. Kim, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 3875–3883 CrossRef CAS.
- L. Tan and A. Wan, Mater. Lett., 2011, 65, 3109–3111 CrossRef CAS.
- A. Lerf, A. Buchsteiner, J. Pieper, S. Schottl, I. Dekany, T. Szabo and H. P. Boehm, J. Phys. Chem. Solids, 2006, 67, 1106–1110 CrossRef CAS.
- H. S. Toh, A. Ambrosi, C. K. Chua and M. Pumera, J. Phys. Chem. C, 2011, 115, 17647–17650 CrossRef CAS.
- W. S. H. Jr and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef.
- M. R. Esfahani, E. M. Languri and M. R. Nunna, Int. Commun. Heat Mass Transfer, 2016, 76, 308–315 CrossRef CAS.
- R. Ranjbarzadeh, A. H. M. Isfahani, M. Afrand, A. Karimipour and M. Hojaji, Appl. Therm. Eng., 2017, 125, 69–79 CrossRef CAS.
- D. C. Marcano, D. V. Kosynkin, J. M. Berlin, A. Sinitskii, Z. Sun, A. S. Slesarev, L. B. Alemany, W. Lu and J. M. Tour, ACS Nano, 2018, 12, 2078 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra04732h |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a and
Muhuo Yu*a
a and
Muhuo Yu*a