
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynergetic effects of Cu/TiO2 sensitized with different cyanine dyes on hydrogen evolution†
Layla S. Almazroaia and
Rasha E. El-Mekawy *ab
*ab
aDepartment of Chemistry, Faculty of Applied Science, Umm Al-Qura University, Makkah, Saudi Arabia. E-mail: rashachemistry1@yahoo.com
bDepartment of Petrochemicals, Egyptian Petroleum Research Institute, Nasr City, Cairo, Egypt
First published on 8th August 2019
Abstract
The production of hydrogen as an alternative fuel is a major challenge for developed countries. To achieve this, photocatalysis is a promising method, which depends mainly on a renewable energy source, namely, the sun. Many derivatives of cyanine-sensitized Cu/TiO2 were prepared and characterized by XRD, SEM-EDS, TEM, BET surface area and UV-vis spectrophotometry and their photocatalytic activity was studied. The study of hydrogen production over the sensitized photocatalysts confirmed the significant role of the dyes in the enhancement of hydrogen production. Dye molecules harness light radiation to produce strong oxidizing species, such as hydroxyl radicals, which in turn form hydrogen molecules. Combining sonolysis with photocatalysis processes proved that ultrasound waves inhibit the agglomeration of particles and maintain their surface area. This synergetic process increased the efficiency of hydrogen production by 7 times using photocatalysis only.
Introduction
Since the innovative photoelectrochemical cell created by Fujishima and Honda using titanium oxide (TiO2) was used in water splitting in 1972,1 there have been many attempts to design simple, commercial, and viable photocatalytic reactions to produce hydrogen or remove pollutants. Exhaustive studies in the field of photocatalysis have shown various fascinating applications of photocatalytic reactions based on the use of semiconductors, such as TiO2, Fe2O3, ZnO, ZrO2, SnO2 and CdS.2–4 Much work has focused on TiO2 because the potentials of its valence band (VB) and conduction band (CB) are straddled between the potentials of oxygen and hydrogen production, respectively. However, its efficiency of conversion is very low due to electron/hole (e−/h+) recombination and low harvesting of solar radiation. Metal loading, metal doping, non-metal doping,5–12 and sensitizing of TiO2 with dyes13–15 are some effective ways to fabricate new colored photocatalysts with high efficiency. Many metals, such as Pt, Pd, Au, and Rh, exhibit good activity in hydrogen production. Electron spin resonance (ESR) signals confirmed that the excited electron in the CB of TiO2 transfers to the Fermi levels of the loaded metal, increasing the lifetime of the generated electron and hole.16 Attention has been focused on non-noble metals, such as Cu and Sn, which display significant enhancements in water splitting in the presence of sacrificial reagents (20 and 44 times that of TiO2, respectively7,17–20). Compared with precious noble metals, the rate of hydrogen production over Co/TiO2 reached 77.8% of that of Pt/TiO2.21 Metal-loaded TiO2 is not sufficient to afford reasonably efficient hydrogen production from pure water. In recent years, dye sensitization has become a promising way to activate TiO2 under visible light in hydrogen production.13–22 The electrons are injected into the CB of TiO2 from the lowest unoccupied molecular orbital (LUMO) of the sensitized dye, which has higher energy than the CB edge of TiO2. In contrast, the efficiency of dyes without semiconductors is very low. Dye-sensitized solar cells have higher conversion efficiencies than plant photosystems, although there is a theoretical limit of ca. 30% to their efficiency.23 The highest known conversion efficiency of 15% was achieved in a solar cell that employed an organic–inorganic hybrid perovskite (CH3NH3PbI3) as a sensitizing dye.24One of the most important types of dye is cyanine dyes. Cyanine dyes have made great impressions in many different fields internationally; hence, it is important to study their photochemical and photophysical properties.25–27 This importance is a result of the nature of the chemical structure of these dyes, as they are among the most sensitive optical materials.28 They are known as fluorophores and are fundamentally recognized by their UV-vis absorption and fluorescence spectra, molar extinction coefficients, quantum efficiencies, and Stokes shifts.28–31 In this research article, the features of pure chromophoric dyes and numerous pathways for their design towards higher efficiency as catalysts in photo- and sono-catalysis are explored.
Enhancement of hydrogen production is not limited to modification of the photocatalyst, but also focuses on the techniques used in the process. Recently, a promising technique has exhibited good results regarding the efficiency of oxidation processes on photocatalysts. The sonophotocatalysis process is a combination of photocatalysis with sonolysis.32–35 The ultrasound may decrease the particle size and increase the mass transfer. Moreover, it influences surface cleaning.36 Sonolysis implies the generation of very high heat (around 5000 K) from cavity implosion because water decomposes to H2O2 and H2 when it is irradiated with ultrasound waves.37 In contrast, the photocatalysis technique produces little or no hydrogen evolution under special conditions. Surprisingly, water was decomposed to hydrogen and oxygen simultaneously in the liquid phase by sonophotocatalysis.35 In this article, four new derivatives of cyanine that incorporate Cu/TiO2 were investigated to improve the rate of hydrogen production in the visible light region, which represents 95% of sun radiation. A comparison of the efficiencies of these samples when used in photocatalysis and sonophotocatalysis was achieved.
Results and discussion
Chemistry of cyanine derivatives
It is useful to list a new series of long-lasting cyanine dyes, which have distinct application effects and are leaving a distinctive mark in the field of renewable energy; therefore, we focused our thinking on preparing an innovative series of cyanine dyes. Initially, 3-(2-benzylpyridin-1-ium-1-yl)propane-1-sulfonate (2) was synthesized by the reaction of benzylpyridine with carefully chosen blocking groups which can be attached to the nitrogen atom of pyridine, such as propanesultone in freshly distilled toluene under reflux for 4 h under nitrogen gas. This reaction was carried out in a Dean–Stark apparatus; these conditions allowed the system to be run in the absence of air and water, as depicted in Scheme 1.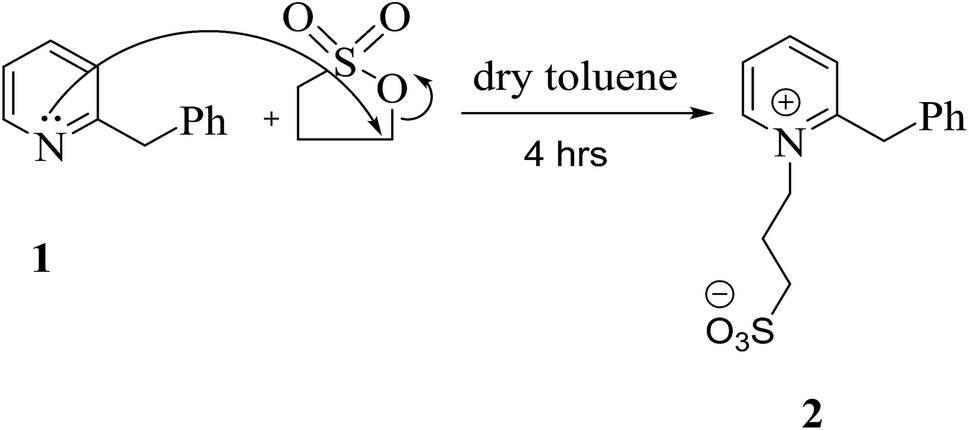 | ||
| Scheme 1 Designation of the quaternary nitrogen incorporated in the pyridine ring. | ||
The structure of quaternary ammonium salt 2 was elucidated by 1H NMR spectroscopy. This spectrum showed quintet signals at δ 1.67 ppm, which are attributable to the middle methylene protons of the sultone chain, and two triplet signals at δ 2.75 and 3.51 ppm corresponding to the 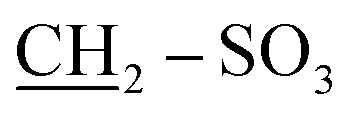 and
and 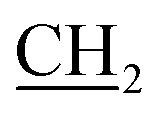 propane –N+ protons in addition to a singlet signal at δ 3.70 ppm, which is due to the CH2 protons of the benzyl group. The mass spectrum of 2 exhibited a molecular ion peak at m/z = 291 (M+, 47%) corresponding to the molecular formula C15H17NO3S. Additionally, the 13C NMR spectrum provided further confirmation of the chemical structure of 2.
propane –N+ protons in addition to a singlet signal at δ 3.70 ppm, which is due to the CH2 protons of the benzyl group. The mass spectrum of 2 exhibited a molecular ion peak at m/z = 291 (M+, 47%) corresponding to the molecular formula C15H17NO3S. Additionally, the 13C NMR spectrum provided further confirmation of the chemical structure of 2.
Modulation of cyanine dyes is possible through various strategies; for example, the symmetrical amalgamation of π-conjugated bridges such as N-[5-anilino-3-chloro-2,4-(propane-1,3-diyl)2,4-pentadiene-1-ylidene]anilinium chloride38 with two units of quaternary ammonium salt 2 yielded heptamethine cyanine dye 5, which acted as a precursor for the formation of other analogues. Near-infrared (NIR) chromophore 5 was produced by two pathways. One is a straightforward two pot-component synthesis, where compound 3 is combined with two moles of pyridinium salt 2 by refluxing for 3.5 h in the presence of absolute ethanol, acetic anhydride as the solvent, and sodium acetate as a basic catalyst under nitrogen gas. Acetic anhydride serves as a solvent and also effectively inhibits the polymerization of polyconjugated system 3. The other pathway involves heating one mole of compound 2 with 3 in the presence of n-butanol and benzene in a ratio of 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 for 2 to 5 h, resulting in hexamethine cyanine dye 5 in good yield; this is converted to 5 by refluxing with one mole of compound 2 in pyridine for 3 h, as outlined in Scheme 2. Compound 5 was elucidated based on its correct analytical and spectral data. The assignment of the 1H NMR spectrum showed four triplet signals at δ 1.79 (C4–H2 and C6–H2 of the cyclohexenyl protons), 2.11 (two CH2–SO3), 2.74 (CH2–N), and 4.1 (CH2–N+) ppm. On the other hand, the 1H-NMR spectrum revealed no singlet signal at δ 3.70 ppm due to CH2 of the benzyl group and instead showed two new singlet signals which appeared at δ 4.40 and 6.36 ppm; these are due to the two
:3 for 2 to 5 h, resulting in hexamethine cyanine dye 5 in good yield; this is converted to 5 by refluxing with one mole of compound 2 in pyridine for 3 h, as outlined in Scheme 2. Compound 5 was elucidated based on its correct analytical and spectral data. The assignment of the 1H NMR spectrum showed four triplet signals at δ 1.79 (C4–H2 and C6–H2 of the cyclohexenyl protons), 2.11 (two CH2–SO3), 2.74 (CH2–N), and 4.1 (CH2–N+) ppm. On the other hand, the 1H-NMR spectrum revealed no singlet signal at δ 3.70 ppm due to CH2 of the benzyl group and instead showed two new singlet signals which appeared at δ 4.40 and 6.36 ppm; these are due to the two ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH protons.
CH protons.
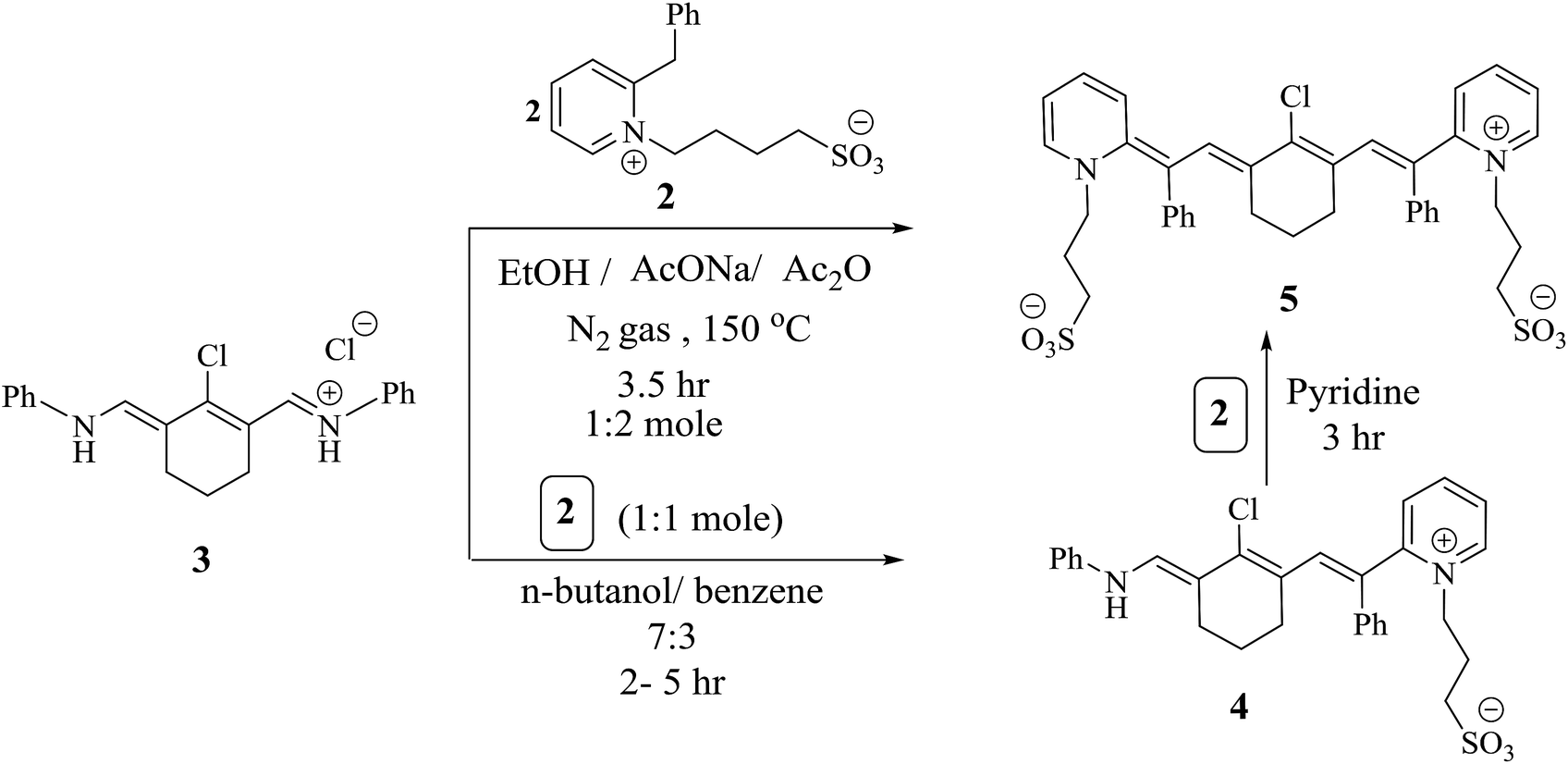 | ||
| Scheme 2 Utilizing alternate pathways for the formation of heptamethine cyanine dye 5. | ||
The chlorine atom at the meso position of conjugated system 5 underwent a nucleophilic substitution reaction with another nucleophilic moiety. The following protocols are based on the methods dedicated to or recommended for synthesizing different members of the heptamethine cyanine dye family that become excited in the NIR region. Chromophore 5 was a sensible candidate for constructing the efficient light harvesting cyanine dye 6 via heating under reflux with one molar ratio of piperazine in N,N-dimethylformamide and sodium hydroxide as a catalyst for 5 h under nitrogen gas. Two units of compound 5 were linked by reacting two units of structure 5 with one mole of piperazine via two nucleophilic substitution reactions of two chlorine atoms at the meso position of 5 to produce the heptamethine cyanine dye dimer 7. Alternatively, chromophore 6 was refluxed under nitrogen gas to avoid air bubbles or moisture from the reaction in the presence of sodium hydride and N,N-dimethylformamide to form compound 7. The chemical structure of compound 6 was confirmed based on its spectral and elemental analyses. The IR spectrum showed an absorption band at ύ = 3453 cm−1 corresponding to the NH functional group. On the other hand, the 1H NMR spectrum displayed a characteristic singlet signal at δ = 12.10 ppm due to the NH proton; however, this disappeared in the 1H NMR spectrum of compound 7, which gave evidence for its consumption in the formation of the dimer. Another cyanine dye, 8, was produced by several possible pathways; 1,6-hexadiol was reacted with compound 5 by removing HCl during refluxing in the presence of sodium hydroxide and N,N-dimethylformamide in a closed system to avoid air and moisture. However, two linked moles of compound 5 underwent a reaction with 1,6-hexadiol by two nucleophilic reactions of two hydroxyl groups and two chlorin groups, affording heptamethine cyanine dye dimer 9, which can be synthesized by an alternate method. When compound 8 reacted with one mole of compound 5 in the presence of sodium hydride as a base and N,N-dimethylformamide as the solvent under nitrogen gas, structure 9 was obtained and subjected to spectral and elemental analyses. The IR spectrum showed no absorption band at ύ = 3412 cm−1; this indicates that the hydroxyl group disappeared. Also, this fact is elucidated by the 1H NMR spectrum, which revealed no absorption band at δ = 11.63 ppm due to the OH proton. All of these data confirm the chemical structure of dimer 9 (Scheme 3).
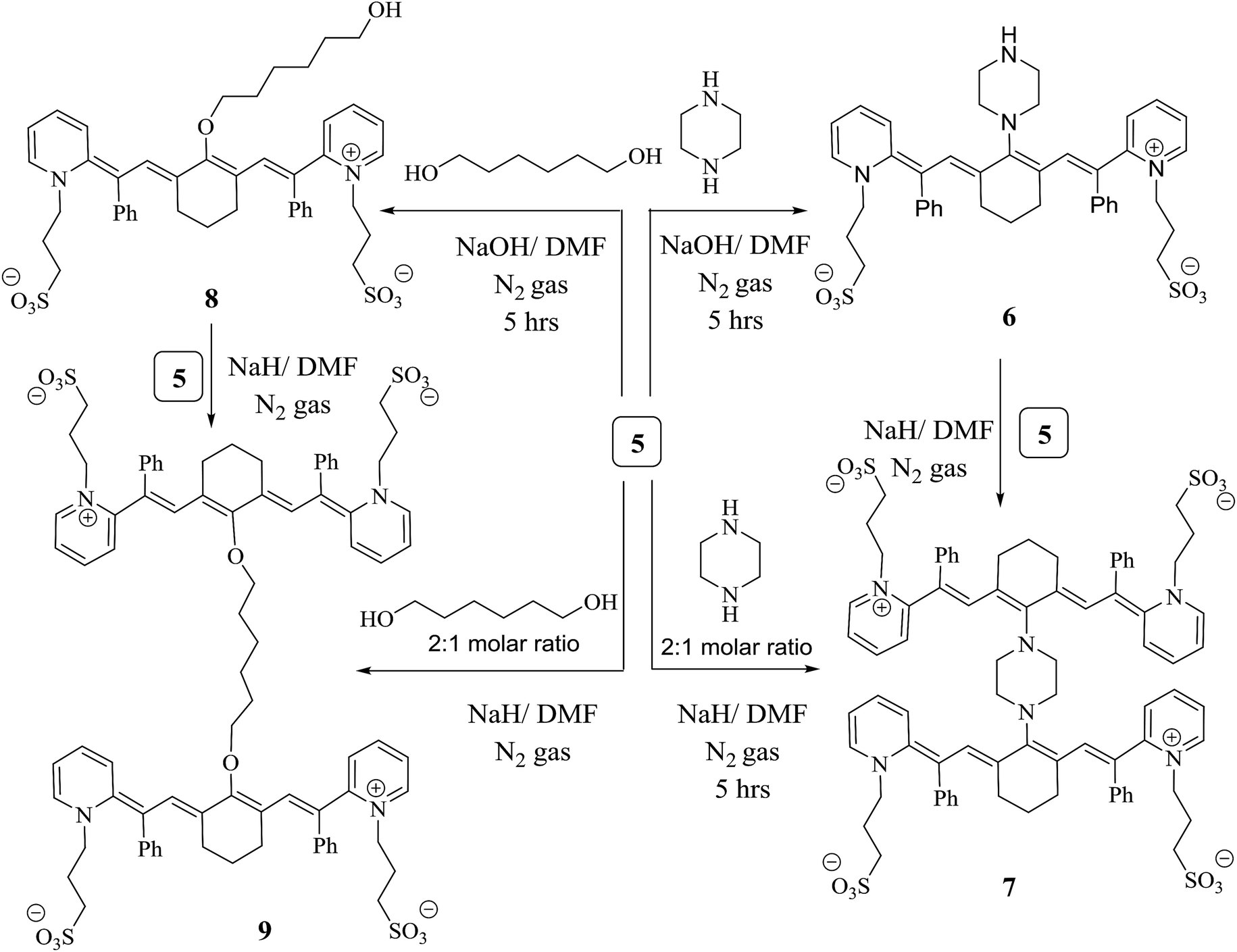 | ||
| Scheme 3 Formation of heptamethine cyanine dye dimers. | ||
Physical properties of cyanine derivatives and sensitized Cu/TiO2
Heptamethine cyanine dyes are characterized by tunable long wavelength absorption and emission due to their long π-conjugated systems. These dyes possess a wide response spectrum, high extinction coefficients (>200000 l mol−1 cm−1), good fluorescence, and high photostability. Synthetic chromophores39,40 are known to absorb between the visible and infrared zones of the electromagnetic spectrum. NIR fluorescent cyanine dyes 6, 7, 8 and 9 display absorption bands at λmax = 639, 649, 648 and 650 nm but appear in expanded emission frequencies in the NIR-region at λmax = 666, 674, 670 and 675 nm. It was observed that compounds 7 and 9 show one absorption band, which indicates the planarity of the two joint units of delocalized conjugated systems in space; in addition, the 1H NMR spectrum showed one signal for the same protons in each unit. The collected data of the UV-vis spectra showed that the photosensitization of these dyes increases as the conjugation increases. This leads to increasing delocalization of the expanded π-system because the resonance between the two nitrogen atoms of the pyridinium and pyridine rings increases, while the energy gap between the HOMO and LUMO decreases and the excitation energy decreases. This factor leads to the liberation of a large current of electrons and appears to be essential for charge transfer during excitation from the ground state to the first excited state and the triplet excited state; this generates more singlet oxygen species, which are responsible for the reactivity of the molecules. Note that compounds 7 and 9 give high absorption bands with high absorption intensities due to the presence of two units of the conjugated system, causing bathochromic shifts toward the NIR region (Fig. 1(A)) (Table 1).
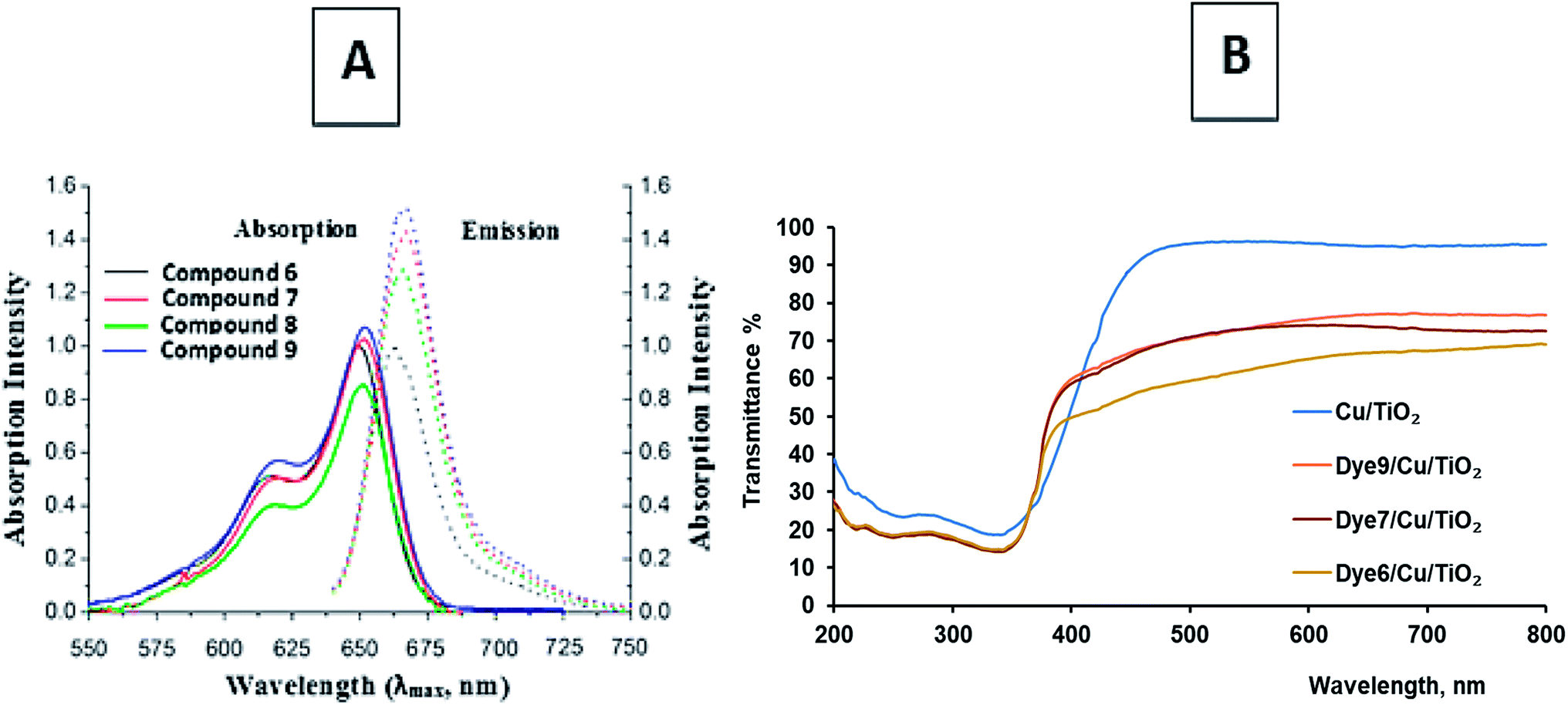 | ||
| Fig. 1 (A) Absorption and emission bands of the chromophoric heptamethine carbocyanine dyes. (B) The transmittance% spectra of dye-sensitized Cu/TiO2. | ||
| Compound no. | Absorption band | Emission band | Molar absorptivity | Stokes shift | Quantum yield (Q) |
|---|---|---|---|---|---|
| λmax (nm) | λem (nm) | logε (l mol−1 cm−1) |
λmax–λem | ||
| 6 | 639 | 660 | 5.73 | 21 | 1.032 |
| 7 | 649 | 674 | 5.63 | 25 | 1.038 |
| 8 | 648 | 670 | 5.73 | 22 | 1.033 |
| 9 | 650 | 675 | 5.73 | 25 | 1.038 |
Dye-sensitized Cu/TiO2 exhibited a much higher capacity for absorbance in the visible region than Cu/TiO2, as shown in Fig. 1(B). The difference between the absorbance spectra of the different dye-sensitized Cu/TiO2 photocatalysts was small. The stability of the synthesized photosensitizers was explained by thermal gravimetric analysis (TGA). TGA gave fruitful results regarding the excellent stability of compounds 6, 7, 8 and 9, as depicted in Fig. 2. Firstly, moisture is liberated; then weight loss starts from breakage of the substituent at the meso position at high temperatures, which does not affect the conjugated system. This fact indicates the suitability of the compounds for application in the field of renewable energy.
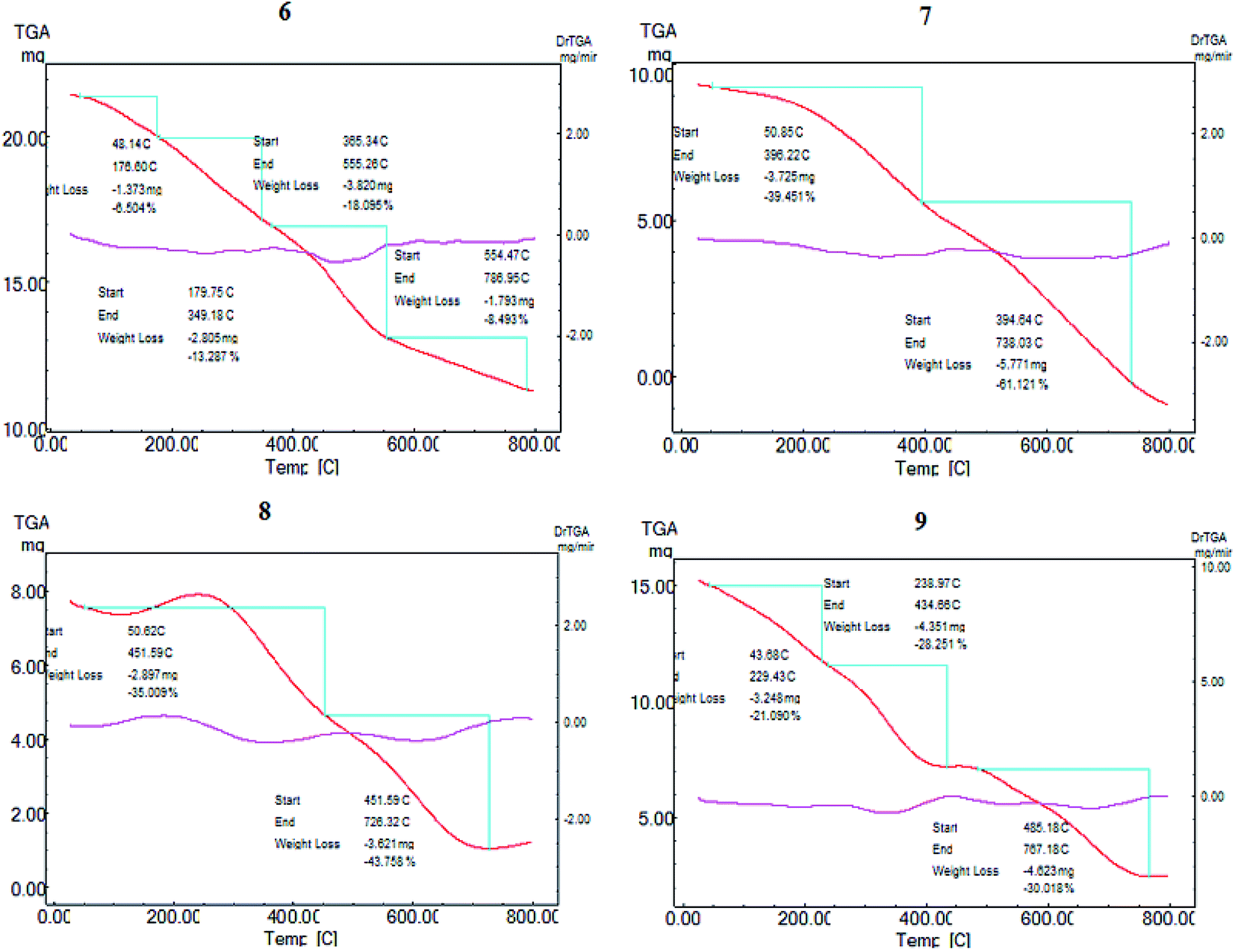 | ||
| Fig. 2 Thermal gravimetric analysis of the novel effective photosensitizers 6, 7, 8 and 9. | ||
SEM and EDS analyses of the cyanine dye derivative Cu/TiO2 and dye-sensitized Cu/TiO2 were performed. Fig. 3(A) displays the morphologies and the compositions of these dyes. The structures of the synthesized dyes 6, 7, 8 and 9 contain pores and active sites which provide areas for the delocalization of π-electrons; also, the continuous generation of a current of electrons aids the subsequent photosensitization to the triplet excited state, leading to continuous generation of singlet oxygen species. On the other hand, Cu/TiO2 was smooth, spherical and crystalline in size before and after sensitization (Fig. 3(B)). The uniform distributions of the elements of the composites were clear in the elemental maps of dye-sensitized Cu/TiO2. EDS spectra confirmed the immobilization of the dyes on Cu/TiO2.
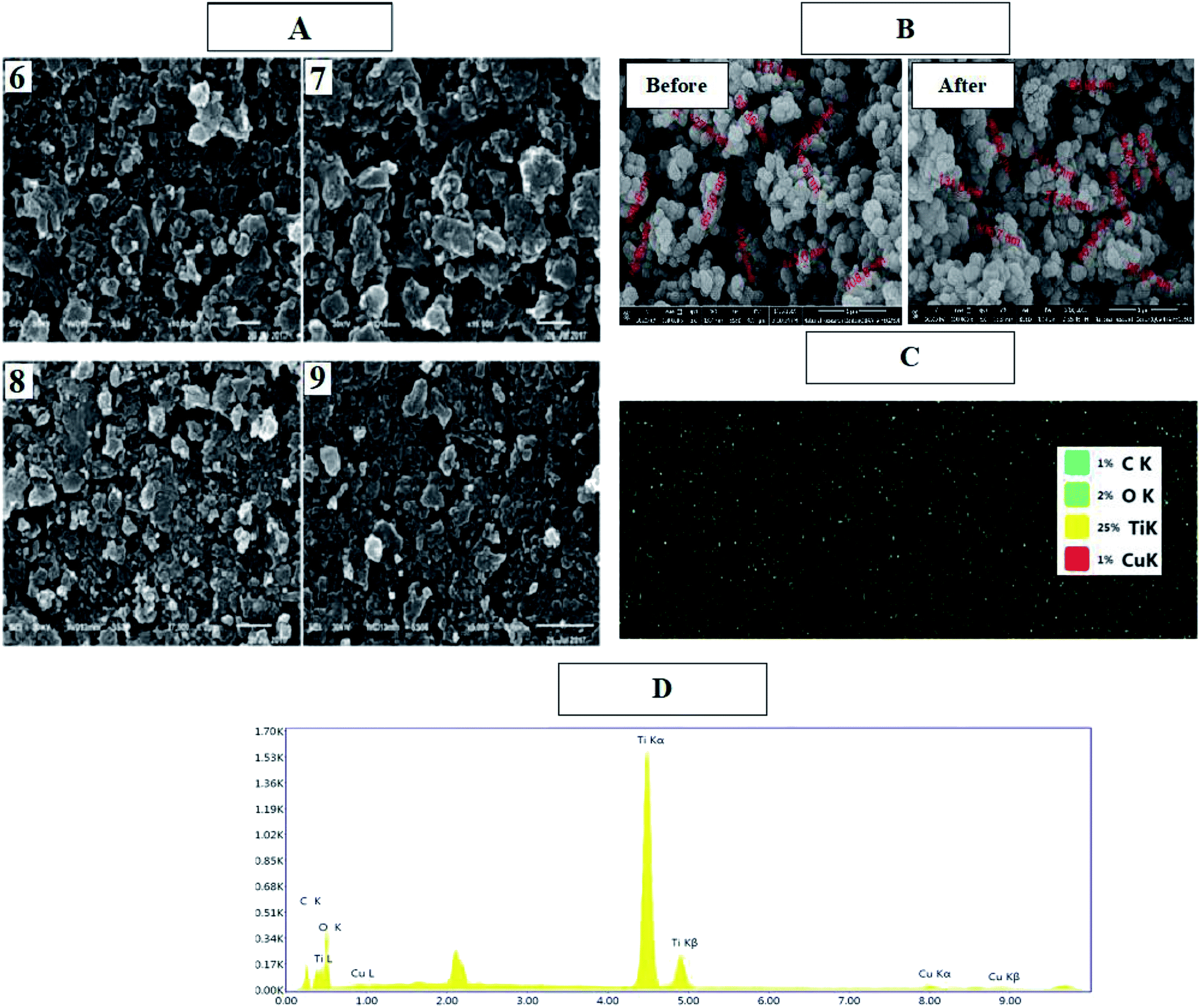 | ||
| Fig. 3 SEM images of dyes 6, 7, 8, and 9 (A) and Cu/TiO2 before and after sensitization (B), EDS mapping of dye sensitized Cu/TiO2 (C), and EDS elemental analysis of dye 9/CuTiO2 (D). | ||
The spherical particles of Cu/TiO2 before and after sensitization of the dye are also shown in the TEM images (Fig. 4). Some dark spots can be assigned to Cu due to the high electronic density of the metal. In contrast, the image after sensitization did not display a clear change because the dye molecules are organic and have low electronic density. However, EDX proved the existence of metal and dye molecules, as mentioned previously. The BET surface area of dye-sensitized Cu/TiO2 was slightly higher than that of Cu/TiO2, namely 16.1 and 14.3 m2 g−1, respectively.
 | ||
| Fig. 4 TEM images of Cu/TiO2 before and after dye sensitization. | ||
The XRD patterns of the different conjugated carbocyanine dyes 6, 7, 8 and 9 displayed excellent crystallinity of their conjugated chemical structures, and the patterns of the derivatives of dye-sensitized Cu/TiO2 are shown in Fig. 5. It is clear that the crystallinity of TiO2 did not change after doping with Cu and sensitizing of the dyes. The XRD spectra were in agreement with the standard TiO2 Degussa41 except for the rutile (110) peak at 2θ = 28° due to a phase transformation that occurred during metal doping. There were no peaks for Cu or the dye due to their low content, although EDS analysis confirmed their existence. Moreover, the positions of the peaks of TiO2 did not change, indicating that the doping of Cu occurred by a substitution process into the titania lattice instead of by interstitial incorporation. The crystalline size D was calculated by the Scherrer equation:
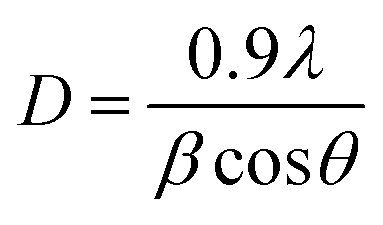
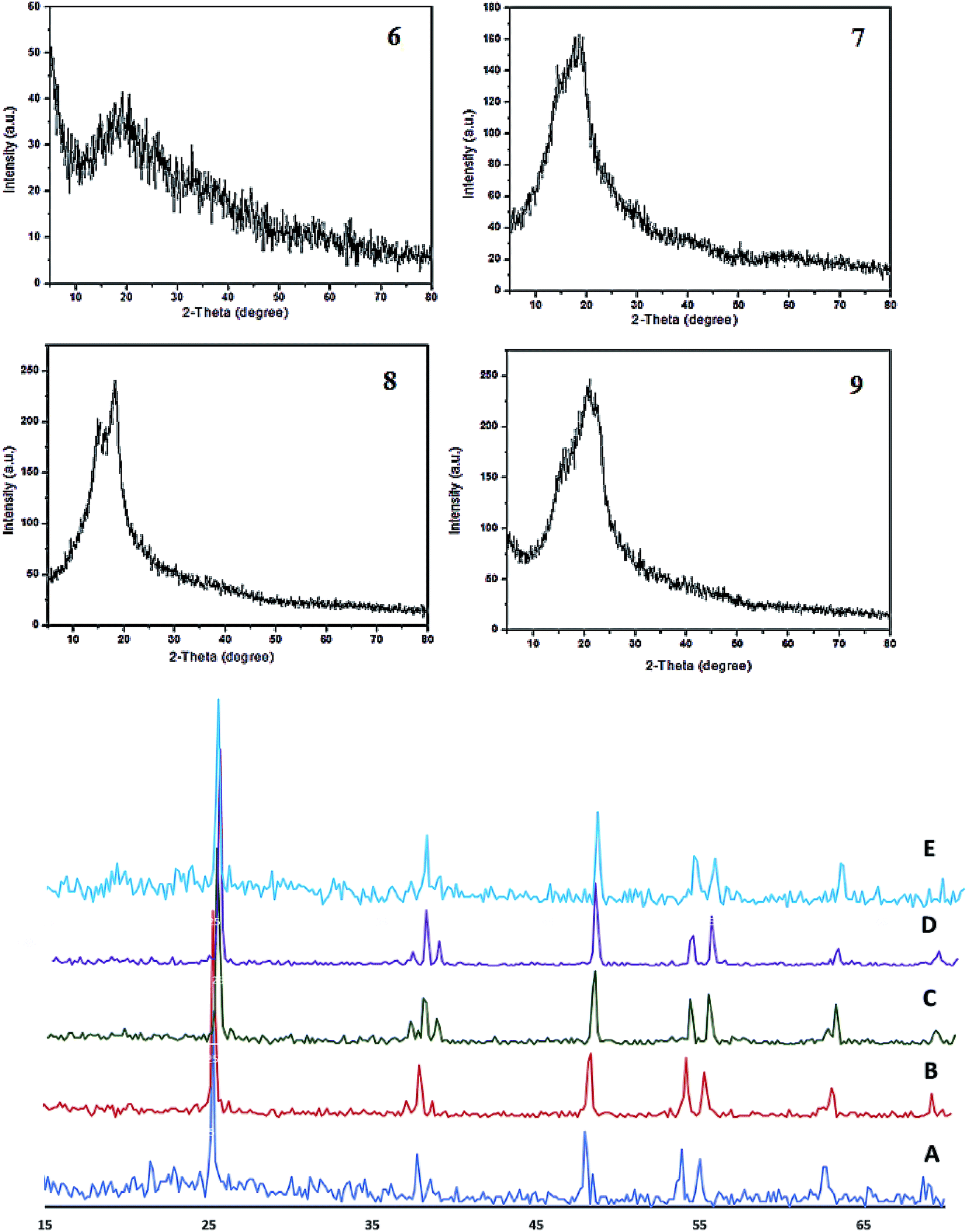 | ||
| Fig. 5 X-ray diffraction patterns of heptamethine cyanine dyes 6, 7, 8 and 9 (top) and of Cu/TiO2 (A), 9/Cu/TiO2 (B), 8/Cu/TiO2 (C), 7/Cu/TiO2 (D), and (E) 6/Cu/TiO2 (bottom). | ||
| Sample | Particle size (nm) |
|---|---|
| Cu/TiO2 | 35.7 |
| Dye 9–Cu/TiO2 | 83.6 |
| Dye 8–Cu/TiO2 | 54.4 |
| Dye 7–Cu/TiO2 | 88.7 |
| Dye 6–Cu/TiO2 | 83.5 |
Photocatalytic activity of dye-sensitized Cu/TiO2 in hydrogen evolution
Hydrogen evolution over the catalysts before and after sensitization under UV and visible light was investigated, as shown in Fig. 6. The photocatalytic activity of Cu/TiO2 was very low compared with those of the catalysts with absorbed dyes. This result agreed with previous studies.42,43 The order of photocatalytic activity of the dye derivative-absorbed Cu/TiO2 samples was 9 > 6 > 7 > 8. The structure of these sensitizers influenced their photoactivity. Although 9 and 7 had high absorption intensity due to their highly conjugated systems, 9 was more active than 7, and the latter was less active than 6. The structure of 7 suffers from steric hindrance and rigidity, which may increase the opportunity of aggregation and lead to increased possibility of aggregation of Cu/TiO2 particles in general.44–46 The particle size of the catalyst with 7 was greater than that of the catalysts with 9 and 6. Dye 8-sensitized Cu/TiO2 with a particle size of 54.4 nm had the lowest activity and lowest absorption intensity. However, the enhancement of the photoactivity of the sensitized metal oxides was attributed to the high extinction coefficients of the dyes, which is a measure of the strength of the light absorbance.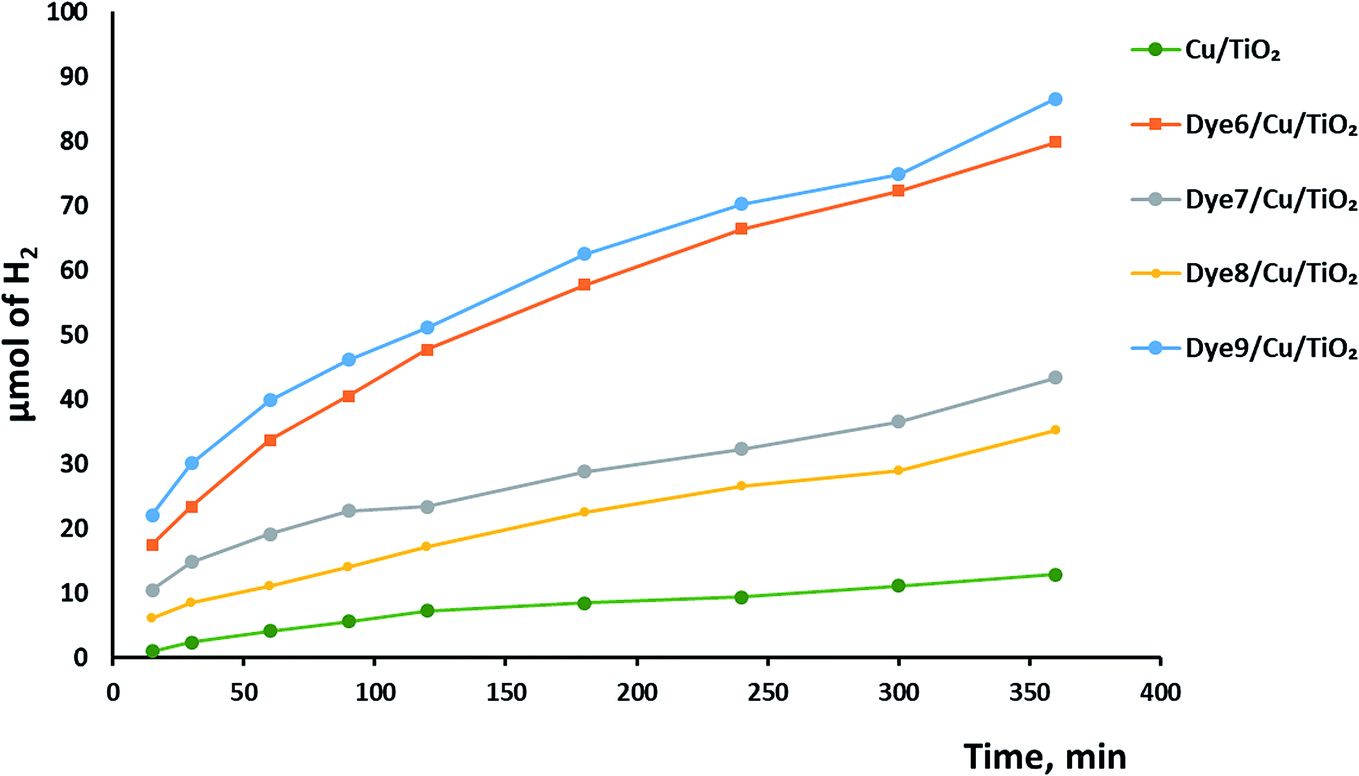 | ||
| Fig. 6 The photocatalytic hydrogen production over dye-sensitized Cu/TiO2. | ||
The mechanism of hydrogen production over sensitized catalysts has been proposed in many studies.13 The first step is excitation of the dye molecule by the irradiation of light. The LUMO energy level of the dye is usually above the CB of TiO2. Therefore, the excited electron transfers from the excited dye molecule to the CB of TiO2. Then, this electron may be trapped by a Cu atom; the SEM images show the uniform dispersion of Cu over TiO2. This process inhibits the recombination of the e−/h+ pair and increases the time of their separation.47 The excited electron forms hydroxyl radicals, leading to the production of a hydrogen gas molecule. On the other hand, the hole formed at the HOMO of the dye is consumed by the oxidation of the sacrificial reagent, forming H2 gas.48,49 Although TiO2 absorbs light, its absorbance is restricted to the UV region, which has very low intensity in the spectrum of the Xe lamp. Moreover, the bulk and surface e−/h+ recombination of TiO2 is one of the reasons that scientists tend to view the sensitization process as a promising solution. Fig. 7 shows a schematic of one of the suggested mechanisms.
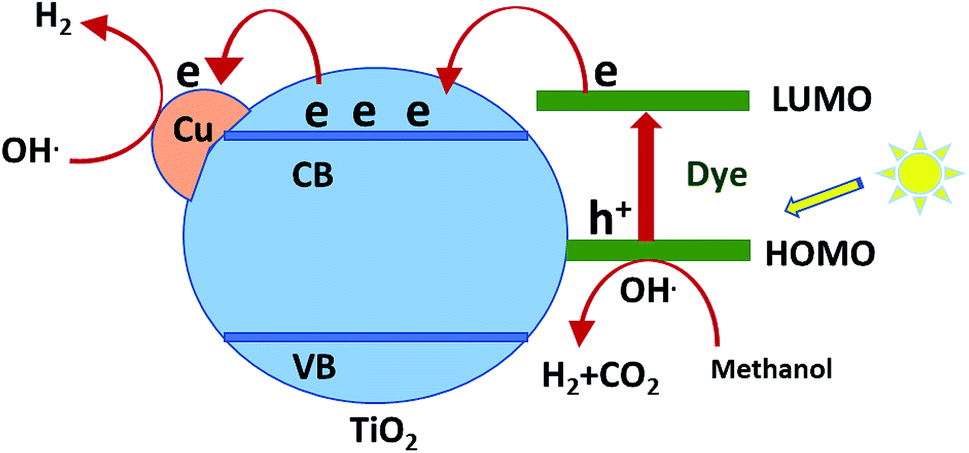 | ||
| Fig. 7 Schematic of the suggested mechanism of H2 production over sensitized Cu/TiO2. | ||
Synergistic effect on the rate of hydrogen evolution, which is a combination of ultrasound waves and light radiation, was investigated. The results shown in Fig. 8 confirm the positive effects of ultrasound waves in preventing the agglomeration or aggregation of particles over time. Moreover, hydroxyl radicals were formed by the photocatalysis process by excited electrons and by the collapse of bubbles because of the ultrasound waves. The comparison between photocatalytic and sonophotocatalytic hydrogen production after 5.5 hours is plotted in Fig. 9. The synergistic effect increased the amount of hydrogen produced after 5.5 hours by about 7 times.
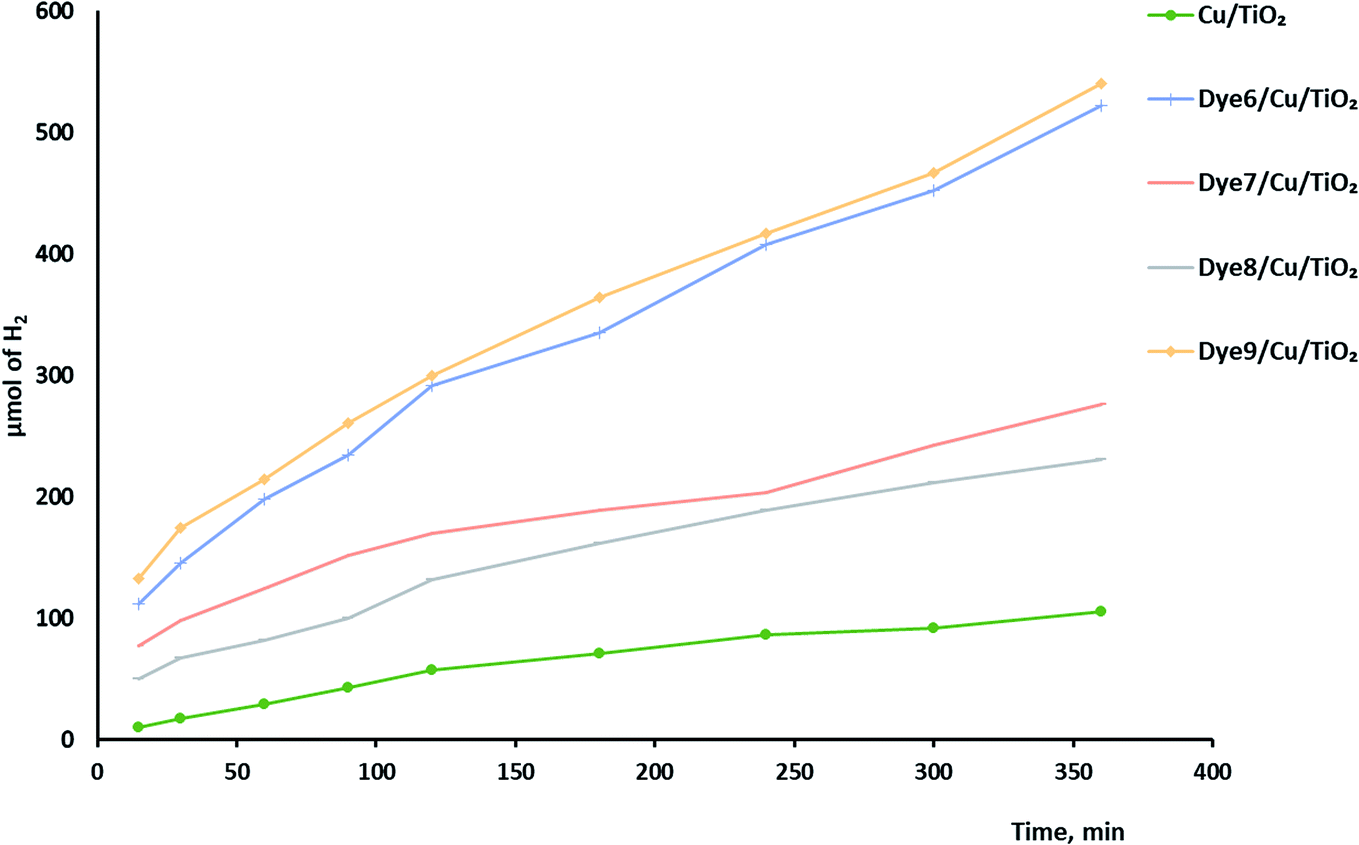 | ||
| Fig. 8 Sonophotocatalytic hydrogen production over dye-sensitized Cu/TiO2. | ||
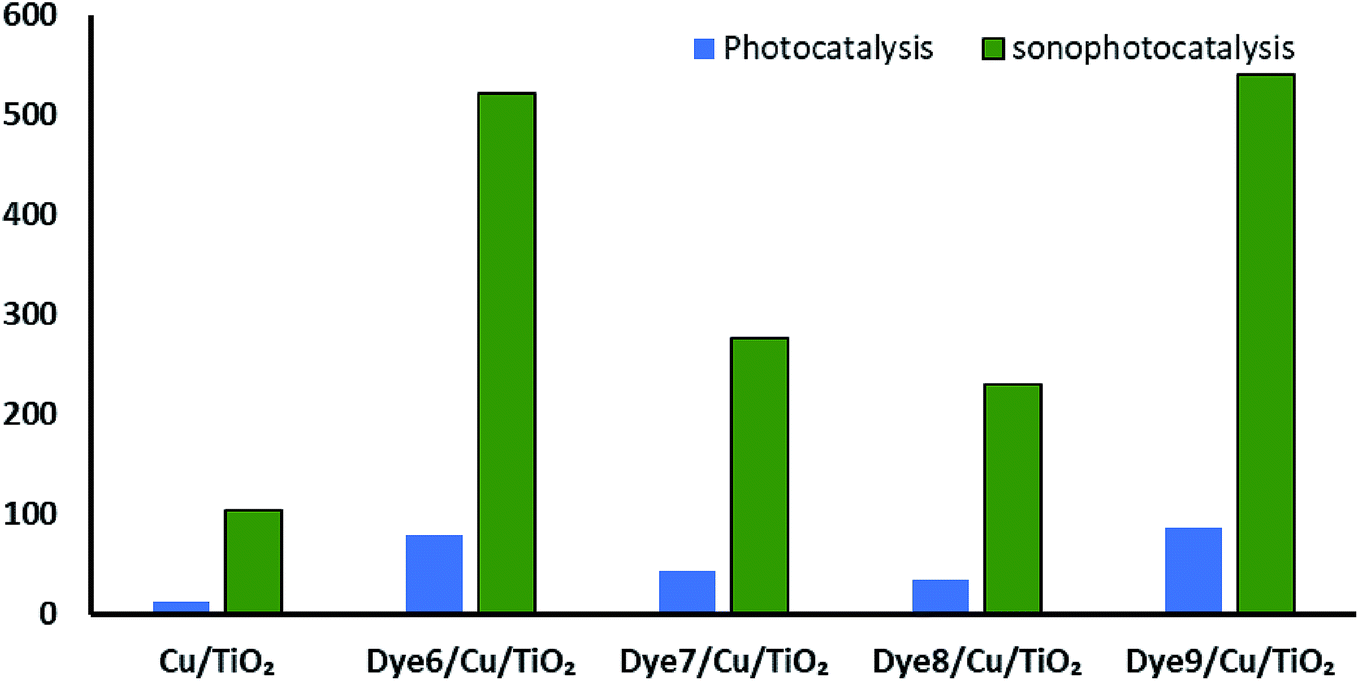 | ||
| Fig. 9 Comparison between photo- and sonophotocatalytic hydrogen production. | ||
The effects of different amounts of the catalyst on the rate of photocatalytic hydrogen production were investigated, as shown in Fig. 10. It is logical that increasing the amount of the catalyst would increase the surface area for the oxidation–reduction reaction and consequently increase hydrogen production. Irradiation of more than 0.14 g of the catalyst lowered the rate of hydrogen production due to the shielding effect by the particles.50,51
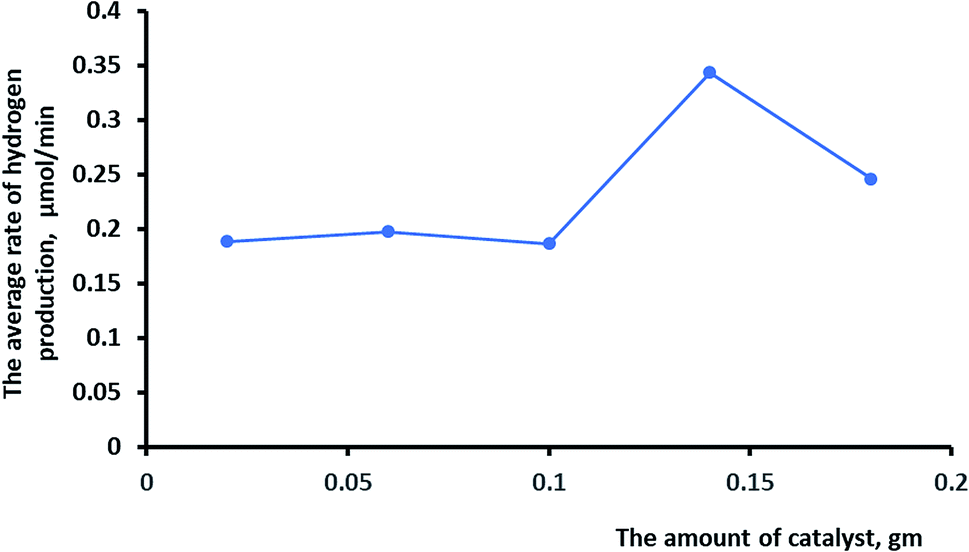 | ||
| Fig. 10 The effects of the amount of catalyst on the rate of hydrogen production. | ||
Experimental
Characterization
Melting degrees were uncorrected in degrees centigrade and tested on a Gallenkamp electric melting point instrument. The infrared (IR) spectra were detected (KBr disks) on a Mattson 5000 FTIR (Fourier transform infrared) spectrometer, the 1H-NMR and 13C NMR spectra were determined using a Bruker WPSY 200 MHz spectrometer with TMS as an internal standard, and the chemical shifts were measured in δ ppm using solvents such as DMSO-d6 and/or CDCl3 at the Faculty of Science, Mansoura University, Egypt. The mass spectroscopy was performed at 70 eV with a Varian MAT 311 instrument, and elemental analyses (C, H and N) were performed at the Microanalytical Center, Faculty of Science, Cairo University. The outcomes were in good approval (±0.03) with the studied rates. X-ray diffraction (XRD) was employed to measure the crystallinity of the polymers. XRD was performed using a Philips model PW 3710 (Philips, USA) diffractometer with CuKα radiation (λ = 0.1542 nm) in a sealed tube operated at 40 kV and 30 mA. Energy dispersive X-ray spectrometry (EDS) and scanning electron microscopy (SEM) were performed using a Philips model XL 30CP, USA. The films were cut and mounted on a brass stub with double-sided adhesive tape and were coated with 50 Å of gold vapor with an SCD-040 Balzers sputter. The specimen was finally characterized by SEM using an accelerating voltage of 30 kV and 7500× 10000 and 15000 magnification of the original specimen size.
Yield 89%; white needle crystals; mp 152 °C; IR (KBr): ύ/cm−1 = 3101 (Ar–CH), 2926 (CH2), 1621 (CN), 1367, 1151 (2 SO), 618 (S–O); 1H NMR (DMSO-d6) δ/ppm = 1.67 (q, 2H, CH2), 2.75 (t, 2H, CH2–SO3), 3.51 (t, 2H, CH2–N+), 3.70 (s, 2H, CH2 of benzyl protons), 7.01–7.24 (m, 5H, Ar–H), 8.31 (d.d, 1H, CH), 8.53 (d, 1H, CH), 8.74 (d.d, 1H, CH), 9.01 (d, 1H, CH); 13C NMR (DMSO-d6) δ/ppm = 22.60, 28.82, 39.00, 51.87, 59.61, 125.78, 126.46, 128.72, 129.80, 145.74, 146.82, 151.39; MS: (m/z, %) 291 (M+, 16), 243 (40%), 201 (63%), 124 (57%), 114 (100%), 96 (62%), 71 (72%). Anal. calcd for C15H17NO3S (291): C, 61.94; H, 5.72; N, 4.81%. Found: C, 61.92; H, 5.70; N, 4.82%.
Preparation of cyanine dye derivatives
:3) under N2 atmosphere. The solution was evaporated under reduced pressure. The collected solid product was rinsed using diethyl ether and dried to afford compound 4.Yield 68%; reddish brown crystals; mp 198 °C; IR (KBr): ύ/cm−1 = 3351 (NH), 3010 (Ar–H), 2976 (CH2), 1617 (CN), 1600 (CC), 759 (C–Cl); 1H NMR (DMSO-d6) δ/ppm = 1.25 (m, 2H, C5–H2 of cyclohexenyl ring), 1.67 (m, 2H, CH2), 1.91 (t, 4H, C4–H2 and C6–H2 of cyclohexenyl ring), 2.13 (t, 2H, CH2–SO3), 3.65 (t, 2H, CH2–N+), 6.11 (s, 1H, =CH), 7.11 (s, 1H, =CH), 7.40–7.62 (m, 10H, Ar–H), 8.41 (d.d, 1H, CH of pyridinium ring), 8.63 (d, 1H, CH of pyridinium ring), 8.86 (d.d, 1H, CH of pyridinium ring), 8.96 (d, 1H, CH of pyridinium ring), 10.51 (br s, 1H, NH, D2O exchangeable); 13C NMR (DMSO-d6) δ/ppm = 22.81, 27.75, 29.79, 29.00, 52.82, 60.06, 117.30, 119.82, 122.66, 125.62, 125.96, 127.66, 128.20, 129.61, 130.91, 136.54, 141.00, 141.13, 143.30, 144.87, 146.40; MS: (m/z, %) 520 (M+ + 2, 23%), 483 (55%), 403 (19%), 311 (32%), 269 (42%), 204 (100%), 179 (64%), 166 (18%); Anal. calcd for C29H29N2O3SCl (52): C, 66.85; H, 5.61; N, 5.38%. Found: C, 66.85; H, 5.58; N, 5.35%.
Pathway 2: in boiling pyridine, a mixture of compound 2 (2.91 g, 10 mmol) and carbocyanine dye 4 (5.2 g, 10 mmol) was heated under reflux in the presence of nitrogen gas for 3 h at 150 °C. The solvent was evaporated under reduced pressure. The resulting crystals were collected, washed with absolute ethanol and petroleum ether, and dried to form compound 5.
Yield 74%; red crystals; mp 187 °C; IR (KBr): ύ/cm−1 = 3186 (Ar–H), 3099 (CH2), 1538 (CN), 1532 (CC), 794 (C–Cl); 1H NMR (DMSO-d6) δ/ppm = 1.25 (m, 2H, C5–H2 of cyclohexenyl ring), 1.67 (q, 4H, 2CH2), 1.79 (t, 4H, C4–H2 and C6–H2 of cyclohexenyl ring), 2.11 (t, 4H, 2CH2–SO3), 2.74 (t, 2H, CH2–N), 4.10 (t, 2H, CH2–N+), 4.40 (s, 1H, CH), 6.24 (d.d, 1H, CH of pyridine ring), 6.36 (s, 1H, CH), 6.60 (d, 1H, CH of pyridine ring), 6.61 (d, 1H, CH of pyridine ring), 6.95 (d.d, 1H, CH of pyridine ring), 7.31 (d.d, 1H, CH of pyridinium ring), 7.51–7.60 (m, 12H, Ar–H), 7.80 (d.d, 1H, CH of pyridinium ring); 13C NMR (DMSO-d6) δ/ppm = 22.18, 23.63, 25.47, 27.23, 30.00, 30.18, 51.37, 51.87, 60.10, 108.58, 113.47, 122.10, 122.45, 125.24, 125.49, 126.69, 127.218, 128.78, 129.33, 136.85, 138.42, 141.00, 141.32, 144.53, 145.70, 147.21; MS: (m/z, %) 720 (M+ + 2, 15), 720 (34%), 640 (25%), 603 (29%), 651 (54%), 481 (60%), 439 (39%), 362 (26%), 285 (100%), 142 (53%), 117 (45%), 92 (40%); Anal. calcd for C38H38N2O6S2Cl (718): C, 63.54; H, 5.33; N, 3.90%. Found: C, 63.53; H, 5.34; N, 3.89%.
General procedure for the synthesis of cyanine dyes 6 and 8
In a round-bottomed flask fitted with an air condenser, 0.01 mole of piperazine and 1,6-hexadiol with 0.01 mole of compound 5 in N,N-dimethylformamide and anhydrous sodium hydroxide was heated under reflux in the presence of N2 gas for 5 h at 150 °C. The reaction mixture was cooled and poured onto ice-cooled water. The precipitated solid was collected by filtration and recrystallized from absolute ethanol, affording compounds 6 and 8.N), 1562 (CC); 1H NMR (DMSO-d6) δ/ppm = 1.90 (m, 2H, C5–H2 of cyclohexenyl ring), 2.11 (q, 4H, 2CH2), 1.79 (t, 4H, C4–H2 and C6–H2 of cyclohexenyl ring), 3.00–3.21 (m, 8H, 2CH2–SO3, 2CH2–N), 4.10 (t, 4H, 2CH2–N of piperazine ring), 4.30 (t, 2H, CH2–N+), 6.20 (s, 1H, CH), 6.50 (s, 1H, CH), 7.51–7.60 (m, 18H, Ar–H, pyridine-H and pyridinium-H), 12.10 (s, 1H, NH); 13C NMR (DMSO-d6) δ/ppm = 23.52, 24.32, 27.13, 30.00, 31.21, 51.38, 52.87, 61.10, 109.57, 112.34, 121.10, 121.45, 125.34, 126.49, 126.62, 127.212, 128.75, 129.63, 136.75, 138.22, 141.10, 141.52, 144.23, 145.71, 147.28; MS: (m/z, %) 767 (M+, 85), 682 (34%), 602 (25%), 651 (54%), 481 (60%), 439 (39%), 362 (26%), 285 (100%), 142 (53%), 117 (45%), 92 (40%); Anal. calcd for C42H47N4O6S2 (767): C, 65.69; H, 6.17; N, 7.30%. Found: C, 65.67; H, 6.19; N, 7.32%.N), 1600 (CC); 1H NMR (DMSO-d6) δ/ppm = 0.8–1.20 (m, 8H, 4CH2), 1.70–1.91 (m, 6H, 3CH2 of cyclohexenyl ring), 2.40–2.81 (m, 12H, 2CH2, 2CH2–SO3, 2CH2–N, CH2–N+), 4.70 (t, 2H, CH2–O), 5.00 (t, 2H, OH), 6.34 (s, 1H,CH), 6.85 (s, 1H,CH), 7.00–8.31 (m, 18H, Ar–H, pyridine-H and pyridinium-H), 11.63 (s, 1H, OH); 13C NMR (DMSO-d6) δ/ppm = 23.57, 24.00, 26.23, 31.10, 32.24, 52.38, 52.87, 61.10, 109.47, 112.31, 121.14, 123.45, 125.34, 126.459, 126.67, 128.21, 128.76, 129.61, 134.75, 133.22, 140.13, 142.53, 143.23, 144.71, 146.18; MS: (m/z, %) 799 (M+, 75), 719 (34%), 639 (25%), 597 (54%), 555 (60%), 454 (39%), 438 (26%), 361 (100%), 284 (53%), 217 (45%), 152 (40%); Anal. calcd for C42H47N4O6S2 (799): C, 66.06; H, 6.43; N, 3.50%. Found: C, 66.03; H, 6.40; N, 3.48%.General methodology for the synthesis of carbocyanine dyes 7 and 9
Pathway 1: under nitrogen gas, a mixture of 0.02 mol of piperazine and 1,6-hexadiol and 0.01 mol of compound 5 was refluxed in a two-necked round-bottomed flask fitted with an air condenser in N,N-dimethylformamide and anhydrous sodium hydride (0.025 mol) for 5 h. The reaction content was allowed to cool overnight and was then poured onto ice-cold water and acidified with dil. HCl. The isolated solid was collected by filtration and purified by column chromatography with CH2Cl2:hexane (3:1) to afford compounds 7 and 9.
Pathway 2: under nitrogen gas, a mixture of 0.01 mol of compound 5 and 0.01 mol of compounds 6 and 8 was refluxed in a two-necked round-bottomed flask fitted with an air condenser in N,N-dimethylformamide and anhydrous sodium hydride (0.025 mol) for 5 h. The reaction mixture was allowed to cool overnight and then poured onto ice-cold water and acidified with dil. HCl. The isolated solid was collected by filtration and purified by column chromatography with CH2Cl2: hexane (3:1) to afford compounds 7 and 9.
N), 1541 (CC); 1H NMR (DMSO-d6) δ/ppm = 1.10–1.50 (m, 2H, C5–H2 of cyclohexenyl ring, 8H, 4CH2, 4H, C4–H2 and C6–H2 of cyclohexenyl ring), 1.89 (s, 4H, 2CH2–N of piperazine ring), 2.10 (t, 2CH2–N), 3.02 (t, 8H, 4CH2–SO3), 4.17 (t, 4H, 2CH2–N+), 5.72 (s, 1H, CH), 6.41 (s, 1H, CH), 6.71–8.25 (m, 36H, Ar–H, pyridine-H and pyridinium-H),; 13C NMR (DMSO-d6) δ/ppm = 25.51, 26.31, 27.23, 30.80, 33.28, 50.37, 52.12, 59.10, 108.52, 111.37, 122.11, 121.45, 124.34, 125.49, 12.62, 127.71, 128.76, 129.61, 135.75, 137.22, 140.12, 142.52, 143.23, 144.71, 148.28; Anal. calcd for C80H84N6O12S4 (1449): C, 66.28; H, 5.84; N, 5.80%. Found: C, 66.27; H, 5.82; N, 5.78%.N), 1535 (CC); 1H NMR (DMSO-d6) δ/ppm = 0.7–1.30 (m, 8H, 4CH2), 1.80 (t, 8H, 4CH2 of cyclohexenyl ring), 1.83 (t, 4H, 2CH2 of cyclohexenyl ring), 2.49 (t, 8H, 4CH2–N), 2.40–2.93 (m, 8H, 4CH2–SO3), 4.71 (t, 8H, 4CH2–N+), 5.1 (t, 4H, 2CH2–O), 6.10 (s, 1H, CH), 6.55 (s, 1H,CH), 6.80–8.40 (m, 36H, Ar–H, pyridine-H and pyridinium-H); 13C NMR (DMSO-d6) δ/ppm = 21.14, 23.10, 25.43, 30.12, 32.26, 50.18, 52.84, 60.12, 110.47, 114.31, 120.14, 124.41, 125.64, 127.45, 127.67, 128.28, 128.71, 129.43, 133.15, 136.22, 141.13, 142.59, 153.13, 154.71, 156.18; Anal. calcd for C82H88N4O14S4 (1481): C, 66.46; H, 5.99; N, 3.78%. Found: C, 66.46; H, 5.99; N, 3.78%.Preparation of dye-sensitized Cu/TiO2
Wet impregnation was used to prepare Cu/TiO2. 1 g of TiO2 (P25) was mixed with 3 ml of a solution of copper nitrate (Cu(NO3)2) under ultrasonication. The slurry was dried in air overnight at 120 °C and then calcined at 500 °C. Then, 0.5 g of Cu/TiO2 was stirred with a mixture of dye and anhydrous ethanol for 24 h in the dark and then filtered, washed with ethanol and dis. water, and dried at 100 °C for 4 h.Photo- and sonocatalytic hydrogen production
The water splitting reaction was carried out in a closed circulation quartz reactor system at room temperature and under atmospheric pressure. The system was bubbled with nitrogen for 20 min to remove unwanted gases, such as air or oxygen. In the experiment, 0.1 g of the catalyst was dispersed by a magnetic stirrer in 100 ml water, and methanol was used as a sacrificial agent. During the experiment, and under constant stirring, the reaction mixture was irradiated by a 500 W Xe lamp. The concentration of the evolved hydrogen was measured using an Agilent GC 7890A system equipped with a thermal conductivity detector (TCD) using argon as a carrier gas. The experiments were repeated in the presence of ultrasonic waves; a Ti probe connected to an ultrasonic wave generator was immersed in the mixture to investigate the samples sonophotocatalytically.Conclusion
Derivatives of cyanine-sensitized Cu/TiO2 were prepared, characterized, and investigated in hydrogen production. The cyanine dyes played significant roles in enhancing the photoactivity of metal-loading metal oxides due to their high absorbance intensity and chemical stability. The dye molecules adsorbed on the surface of Cu/TiO2 served as an initiator in the photocatalysis process rather than TiO2. The transfer of the excited electrons from the dye molecules to TiO2 and then to the loaded Cu inhibits the recombination of e−/h+ pairs despite the small surface area. Combining ultrasound waves with photo irradiation remarkably increased the rate of hydrogen production over the dye-sensitized Cu/TiO2. Sonophotocatalysis is a useful technique for high energy conversion efficiency; it supports continuous dispersion of the particles in the reactor and contributes to the formation of powerful radical species. The optimum amount of photocatalyst was 0.14 g. Above this amount, the rate of hydrogen production decreased.Conflicts of interest
The authors declare that there is no conflict of interest.Acknowledgements
The authors are deeply grateful to the Deanship of Scientific Research at Umm Al-Qura University, which supported this work financially (Grant no 15SCI-3-3-0024).Notes and references
- A. Fujishima and K. Honda, Nature, 1972, 238(5358), 37 CrossRef CAS PubMed.
- T. Feng, et al., Int. J. Photoenergy, 2014, 1 CrossRef.
- R. Lamba, et al., Talanta, 2015, 131, 490–498 CrossRef CAS PubMed.
- X. F. Lei, X. X. Xue and H. Yang, Appl. Surf. Sci., 2014, 321, 396 CrossRef CAS.
- M. Bellardita, et al., Chem. Phys., 2007, 339(1–3), 94 CrossRef CAS.
- D. Chen, et al., Appl. Surf. Sci., 2013, 284, 921 CrossRef CAS.
- W. Chen, et al., Appl. Surf. Sci., 2018, 445, 527 CrossRef CAS.
- K.-H. Chung, et al., Int. J. Hydrogen Energy, 2018, 43(24), 11422 CrossRef CAS.
- G. Malekshoar and A. K. Ray, Chem. Eng. Sci., 2016, 152, 35 CrossRef CAS.
- H.-Y. Lin and C.-y. Shih, J. Mol. Catal. A: Chem., 2016, 411, 128 CrossRef CAS.
- K. Villa, et al., Sol. Energy, 2012, 86(1), 558 CrossRef CAS.
- J.-H. Yan, et al., J. Alloy. Comp., 2009, 472(1), 429 CrossRef CAS.
- J. Li, et al., Int. J. Hydrogen Energy, 2013, 38(25), 10746 CrossRef CAS.
- L. Sang, et al., Int. J. Hydrogen Energy, 2017, 42(50), 29686 CrossRef CAS.
- Y. Yuan, et al., Chem. Eng. J., 2015, 27, 8 CrossRef.
- M. Anpo and M. Takeuchi, J. Catal., 2003, 216, 505 CrossRef CAS.
- N. L. Wu and M. S. Lee, Front. Chem. Sci. Eng., 2004, 29(15), 1601 CAS.
- S. Xu, et al., Int. J. Hydrogen Energy, 2010, 35(11), 5254 CrossRef CAS.
- Y. Zou, et al., Int. J. Hydrogen Energy, 2014, 39(28), 15403–15410 CrossRef CAS.
- D. Gao, et al., ACS Sustain. Chem. Eng., 2019, 7(11), 10084–10094 CrossRef CAS.
- W. Chen, et al., Mater. Res. Bull., 2019, 116, 16–21 CrossRef CAS.
- A. A. Nada, et al., Int. J. Hydrogen Energy, 2008, 33(13), 3264 CrossRef CAS.
- H. J. Snaith, Adv. Funct. Mater., 2010, 20(1), 13 CrossRef CAS.
- J. Burschka, et al., Nature, 2013, 499(7458), 316 CrossRef CAS PubMed.
- A. Levitz, F. Marmarchi and M. Henary, Molecules, 2018, 23(2), 226 CrossRef PubMed.
- T. L. Dost, M. T. Gressel and M. Henary, Anal. Chem. Insights, 2017, 12, 1177390 Search PubMed.
- C. P. Toseland, J. Chem. Biol., 2013, 6(3), 85 CrossRef PubMed.
- X. Liu, Z. Xu and J. M. Cole, J. Phys. Chem. C, 2013, 117(32), 16584 CrossRef CAS.
- R. E. El-Mekawy and A. A. Fadda, Bioorg. Med. Chem. Lett., 2018, 28(10), 1747 CrossRef CAS PubMed.
- R. E. El-Mekawy and A. A. Fadda, RSC Adv., 2017, 7(86), 54706 RSC.
- H. Hyun, et al., Angew. Chem., Int. Ed. Engl., 2015, 54(30), 8648 CrossRef CAS PubMed.
- M. Ahmad, et al., Ultrason. Sonochem., 2014, 21(2), 761 CrossRef CAS PubMed.
- S. Anandan and M. Ashokkumar, Ultrason. Sonochem., 2009, 16(3), 316 CrossRef CAS PubMed.
- S. Chakma and V. S. Moholkar, Ultrason. Sonochem., 2015, 22, 287 CrossRef CAS PubMed.
- H. Harada, C. Hosoki and A. Kudo, J. Photochem. Photobiol. A Chem., 2001, 141(2–3), 219 CrossRef CAS.
- H. Harada, Ultrason. Sonochem., 2001, 8(1), 55 CrossRef CAS PubMed.
- C. G. Joseph, et al., Ultrason. Sonochem., 2009, 16(5), 583 CrossRef CAS PubMed.
- A. A. Fadda and R. E. El-Mekawy, Dyes Pigments, 2013, 99(2), 512 CrossRef CAS.
- N. M. M. Hamada and E. M. Sharshira, Molecules, 2011, 16(3), 2304 CrossRef CAS PubMed.
- I. Timtcheva, et al., J. Photochem. Photobiol. A Chem., 2000, 130(1), 7 CrossRef CAS.
- S. Ko, et al., Mater. Sci. Eng., B, 2010, 166, 127 CrossRef CAS.
- V. Vinesh, A. R. M. Shaheer and B. Neppolian, Ultrason. Sonochem., 2019, 50, 302 CrossRef CAS PubMed.
- P. L. Gentili, et al., Int. J. Hydrogen Energy, 2009, 34(22), 9042 CrossRef CAS.
- H. Guo, et al., Electrochim. Acta, 2014, 120, 23 CrossRef CAS.
- S. Nachimuthu, et al., Phys. Chem. Chem. Phys., 2016, 18(2), 1071 RSC.
- G. Janssens, et al., Chem. Phys. Lett., 2001, 344(1), 1 CrossRef CAS.
- X.-j Yang, et al., Trans. Nonferrous Metals Soc. China, 2015, 25(2), 504 CrossRef CAS.
- A. A. Nada, et al., Int. J. Hydrogen Energy, 2005, 30(7), 687 CrossRef CAS.
- A. Tiwari and U. Pal, Int. J. Hydrogen Energy, 2015, 40(30), 9069 CrossRef CAS.
- X. Wang, K. Shih and X. Y. Li, Water Sci. Technol., 2010, 61(9), 2303 CrossRef CAS PubMed.
- A. Mehta, et al., New J. Chem., 2017, 41, 4573 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra04696h |
| This journal is © The Royal Society of Chemistry 2019 |